 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal-catalyzed functionalization of pyrazines
Nicolai I.
Nikishkin
,
Jurriaan
Huskens
and
Willem
Verboom
*
Laboratory of Molecular Nanofabrication, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: w.verboom@utwente.nl
First published on 9th April 2013
Abstract
Transition metal-catalyzed reactions are generally used for carbon–carbon bond formation on pyrazines and include, but are not limited to, classical palladium-catalyzed reactions like Sonogashira, Heck, Suzuki, and Stille reactions. Also a few examples of carbon–heteroatom bond formation in pyrazines are known. This perspective reviews recent progress in the field of transition metal-catalyzed cross-coupling reactions on pyrazine systems. It deals with the most important C–C- and C–X-bond formation methodologies.
 Nicolai I. Nikishkin | Nicolai I. Nikishkin was born in Minsk, Belarus, in 1980. He received his M.Sc. from the Belarussian State University, Minsk, in 2002. After several jobs, he came to the University of Twente, Enschede, The Netherlands in 2008, where he earned his Ph.D. in 2013. |
 Jurriaan Huskens | Jurriaan Huskens was born in Sittard, The Netherlands, in 1968. He studied chemical engineering at the Eindhoven University of Technology. He obtained his Ph.D. from the Delft University of Technology in 1994. Following a postdoctoral stay at the University of Texas at Dallas, USA and an EU Marie Curie fellowship at the Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany, he began his academic career at the University of Twente, Enschede, The Netherlands in 1998, where he is now a full professor of “Molecular Nanofabrication”. The research in his group is focused on fundamental and applied studies of assemblies and patterning. |
 Willem Verboom | Willem Verboom was born in Goes, The Netherlands, in 1954. He studied chemistry at Utrecht University, where he also obtained his Ph.D. (1980). Subsequently, he went to the University of Twente, where he is now associate professor of organic chemistry. He is the (co-)author of about 345 scientific publications. His current research interests involve the design of specific ligands for different applications and the use of micro reactors. |
Introduction
Transition-metal-catalyzed cross-coupling reactions are a well-established tool for carbon–carbon and carbon–heteroatom bond formation.1–3 However, most of the publications focus on the development of novel, more efficient catalysts relative to existing methods. Heterocyclic examples remain rare in these studies and are typically restricted to readily available pyridine derivatives.4,5 Pyrazines represent an important class of heterocyclic compounds, since the pyrazine scaffold is found in many vital pharmaceuticals6,7 and biologically active compounds.8 Functionalized pyrazines are being applied as selective extractants for f-block metals,9 photovoltaic devices,10 and effective catalysts,11 as well as ligands for catalysis.12 However, the chemistry of pyrazines is still relatively unexplored. Examples of cross-coupling reactions on pyrazine derivatives are mainly limited to reactions leading to prominent target compounds for pharmaceutical applications; however, there are only few methodological studies. On the other hand, the highly electron-deficient nature of the pyrazine system allows the successful use of the aromatic nucleophilic substitution reaction for the functionalization of halogenated pyrazines,13,14 so that the cross-coupling chemistry of pyrazines was given naturally much less attention. Although these factors led to a limited number of cross-coupling examples in this heterocyclic series, interesting reactions and applications were introduced in recent years and may give rise to further improvement and broadening of the scope of this methodology within the area of pyrazine chemistry. The aim of this perspective is to summarize existing methods for transition metal-catalyzed functionalization of pyrazines.Suzuki coupling
The popularity of the Suzuki reaction15 as a powerful and straightforward tool for carbon–carbon bond formation is undisputable among synthetic and industrial chemists. Like many other palladium-catalyzed couplings, the Suzuki reaction utilizes aryl halides and pseudohalides, such as triflates, as substrates. Special attention was paid to “activated” electron-deficient hetaryl halides. The reactivity of hetaryl chlorides and their commercial availability (wide abundance) predefined their eligibility for palladium-catalyzed coupling reactions. At the same time the presence of the pyrazine scaffold in a variety of natural biologically active compounds has made halogenated pyrazines popular hetaryl substrates for natural and pharmaceutical products synthesis. One of the very first examples of a Suzuki coupling performed on halogenated pyrazines was published by McKillop et al.,16 who successfully attempted the coupling of chloropyrazine with aryl boronic acids (Scheme 1) using conventional palladium–phosphine catalysts in good to excellent yields.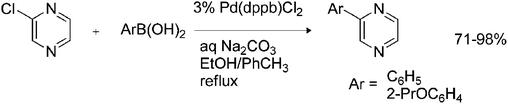 | ||
| Scheme 1 Suzuki coupling of chloropyrazine. dppb = 1,4-bis(diphenylphosphino)butane. | ||
It is interesting to note that Pd(PPh3)4, used as the catalyst in the original Suzuki reaction,17 failed to give any coupled product in the reaction of chloropyrazine with a variety of aryl boronic acids. However, when Pd(dppb)Cl2 was used as a catalyst, chloropyrazine was smoothly converted into coupled products with good to excellent yields and the presence of an electron-withdrawing or -donating group on the boronic acid did not adversely affect the reaction. These results indicate the greater effectiveness of Pd(dppb)Cl2 in this type of cross-coupling.
Using the McKillop catalyst (Pd(dppb)Cl2), Jones et al.18 extended the scope of hetaryl coupling to highly functionalized bromopyrazines (Scheme 2).
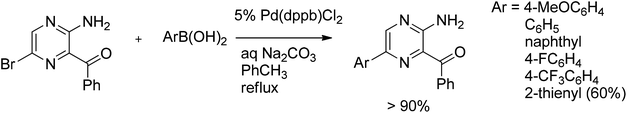 | ||
| Scheme 2 Suzuki coupling of bromopyrazines. | ||
Coupling of highly functionalized bromopyrazines with most of the aryl boronic acids gave the heterobiaryl products in very high yields of >90%. Introduction of an electron-withdrawing group on the aryl boronic acid moiety had no considerable impact on the coupled product yield as well as on the reaction rate. The only exception was thiophene-2-boronic acid, giving only 60% of the coupled product after 48 hours. However, the reaction was still clean forming no side products.18
Cavalier et al.19,20 extended the McKillop methodology further and successfully applied it for the monoarylation of aminobromopyrazine as well as the simultaneous diarylation of aminodibromopyrazine in 30–80% yields (Scheme 3). Remarkably, direct Suzuki coupling of unprotected phenols was also accomplished in moderate to good yields revealing no influence of the phenol group.
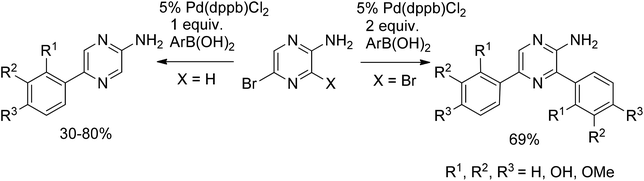 | ||
| Scheme 3 Arylation of aminobromopyrazine and aminodibromopyrazine. | ||
Thompson et al.21 have reported the first systematic study of a Suzuki reaction in the presence of an unprotected amino group on a coupling substrate scaffold. 2-Amino-5-bromopyrazine smoothly reacted with methoxy- and chloropyridyl boronic acids to give the coupled products in moderate yields, independent of the base used. Twofold couplings of 2-amino-5-bromopyrazine with arylene diboronic acid derivatives proceeded in 50–70% yields, demonstrating the general applicability of the protocol developed by McKillop not only to the synthesis of pyrazine-containing heterobiaryls, but also to extended amino-substituted tri- and tetraarylene systems (Scheme 4).
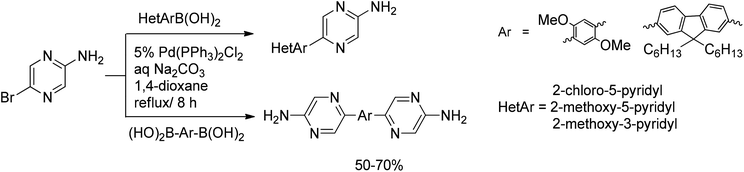 | ||
| Scheme 4 McKillop methodology extended to hetaryl coupling partners. | ||
Tapolcsanyi et al.22 examined the reactivity of chloropyrazine towards 2-pivaloylaminophenyl boronic acid. In contrast to the results obtained by McKillop, who reported Pd(PPh3)4 to be incapable of catalyzing cross-coupling of chloropyrazine with phenyl boronic acid (vide supra), 2-pivaloylaminophenyl boronic acid reacted with chloropyrazine at room temperature in the presence of Pd(PPh3)4 affording the coupled product in 53% yield (Scheme 5). In this case the apparent reactivity can be ascribed to a higher nucleophilicity of the coupling partner due to the presence of the donor pivaloylamino group that enhances the polarization of the carbon–boron bond and facilitates transmetallation of the aryl boronic acid.
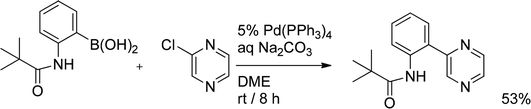 | ||
| Scheme 5 Reaction of 2-pivaloylaminophenyl boronic acid with chloropyrazine. | ||
Suzuki coupling has also been reported for activated bromopyrazines. Highly functionalized 6-bromopyrazines were subjected to Suzuki coupling conditions. Reaction with biphenyl boronic acid in the presence of cesium carbonate as a base and Pd(dppf)Cl2 as a catalyst gave the 6-arylpyrazines in 85–100% yields (Scheme 6). However, the reaction utilizes a high excess of a coupling partner compared to the coupling of regular aromatic halides.23
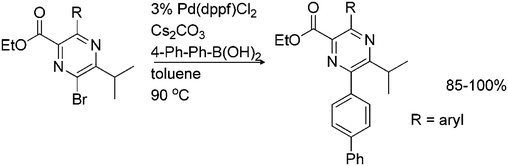 | ||
| Scheme 6 Suzuki coupling of activated bromopyrazines. | ||
In the course of the preparation of tetra- and pentaarylenes, incorporating a pyrazine core, the Suzuki approach was envisaged. Cross-coupling of 2,5-dibromo-3,6-dimethylpyrazine with 2-methoxybenzeneboronic acid and 4-tert-butylbenzeneboronic acid using Pd(PPh3)4 as a catalyst gave the desired coupled products in 76% and 39% yields, respectively (Scheme 7).
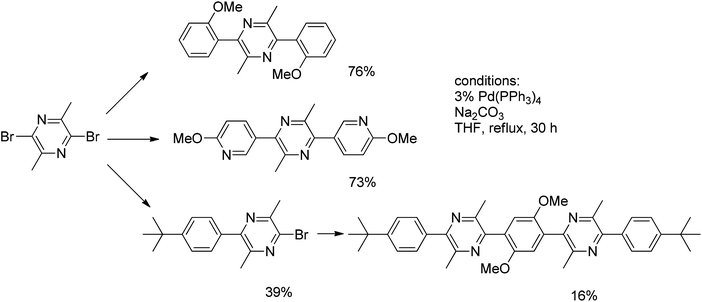 | ||
| Scheme 7 Cross-coupling of 2,5-dibromo-3,6-dimethylpyrazine. | ||
Further reaction of 2-bromo-5-(4-tert-butylphenyl)-3,6-dimethylpyrazine with 2,5-dimethoxy-1,4-benzene diboronic acid gave the linear pentaarylene system in 16% yield. The analogous reaction of 2,5-dibromo-3,6-dimethylpyrazine with 2-methoxy-5-pyridylboronic acid afforded the 2,5-dipyridylpyrazine derivative in 73% yield. This last case illustrates that Suzuki reactions enable a range of heteroaryl moieties to be incorporated into the polymer chain.24 It is worth noting that due to the lability of halogens on the electron-deficient pyrazine core, the monoarylation of 2,5-dibromo-3,6-dimethylpyrazine with 4-tert-butylbenzeneboronic acid required a considerable excess of the former.
The same group has explored Suzuki couplings of halopyrazines with pyrimidylboronic acids. In the case of 2-chloro-5-pyrimidylboronic acid the yield of the cross-coupled product was limited to 48% (Scheme 8), because of the concurrent formation of byproducts, i.e. the bipyrimidine derivative and higher oligomers derived from competitive self-coupling of boronic acid.
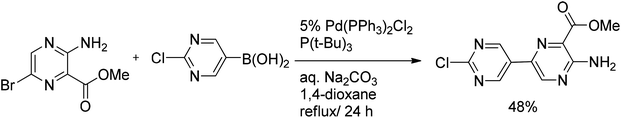 | ||
| Scheme 8 Suzuki coupling of bromopyrazine with 2-chloro-5-pyrimidylboronic acid. | ||
High biological activity of various pyrazine derivatives has resulted in numerous reports on the synthesis of various enzyme inhibitors. Developing novel phosphoinositide 3-kinase inhibitors Gonzalez et al.25 reported the Suzuki coupling of 5-bromoimidazo[1,2-a]pyrazines with imidazoleboronic acid in 75% yield (Scheme 9). The subsequent double Suzuki coupling of aminobromochloropyrazine with various organoborons was employed for the synthesis of highly potent and selective inhibitors of mitotic kinase (Scheme 10).26
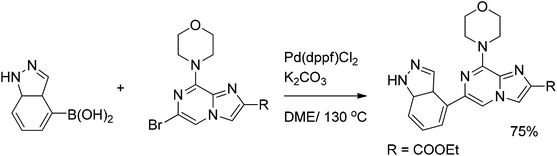 | ||
| Scheme 9 Suzuki coupling of bromoimidazopyrazines with imidazoleboronic acid. | ||
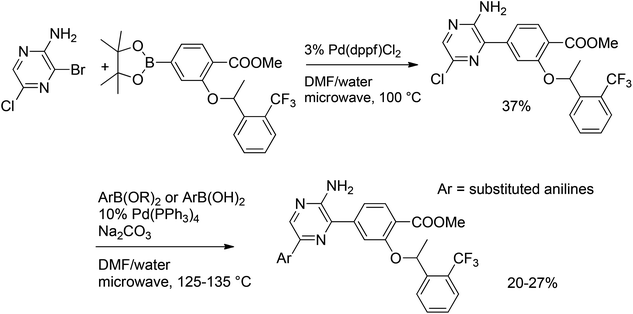 | ||
| Scheme 10 Double Suzuki coupling of bromochloropyrazine. | ||
Coupling of chloropyrazine with 2-amino-5-pyrimidylboronic acid gave the desired coupled product in a yield of 60%. The yield was increased to 72% by using iodopyrazine as a substrate and Pd2(dba)3 as a catalyst (Scheme 11). Protodeboronation of 2-amino-5-pyrimidylboronic acid to give 2-aminopyrimidine was a competing side-reaction. The overall yield of cross-coupled product was improved by the subsequent addition of a second equivalent of boronic acid to the reaction mixture after 24 h reflux.27
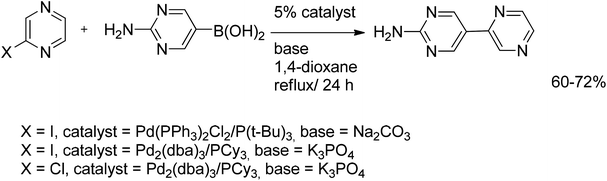 | ||
| Scheme 11 Coupling of chloropyrazine with 2-amino-5-pyrimidylboronic acid. | ||
Attempting the synthesis of the dragmacidin D scaffold, another example of the Suzuki coupling of halogenated pyrazine with hetarylboronic acids has been reported. Reacting 2,5-dibromo-3-methoxypyrazine with tosyl-protected indolylboronic acids solely yielded the 2-arylated product (Scheme 12). This regioselectivity can be attributed to the methoxy group “assistance” in the ortho-halogene displacement by coordination towards the palladium catalytical center. Introduction of the second tosyl-protected indole unit by repeating the first Suzuki step failed. A large excess of tosyl-protected indolylboronic acid in the first step also failed. No improvement occurred using stronger bases, higher temperature or the more active Pd(dppf)Cl2 catalyst. On the other hand, coupling of bromoindolylpyrazine with phenylboronic acid under standard Suzuki conditions afforded the desired coupling product in 80% yield. The latter result clearly indicates that the electronic properties of the coupling partner play an important role. Thus employing the more electron-rich TBDMS-protected indolylboronic acid as the coupling partner in the second step afforded the desired bis-indolylpyrazine in 80% yield.28
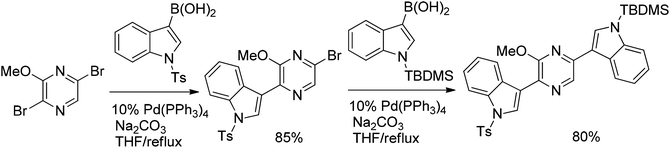 | ||
| Scheme 12 Coupling of 2,5-dibromo-3-methoxypyrazine with protected indolylboronic acids. | ||
3-Chloro-2,5-dimethylpyrazine was also found to undergo Suzuki coupling (Scheme 13). Reaction with 2-methoxynaphthylboronic acid afforded the coupled product in 61% yield. The relatively low yield and relatively long reaction time of 72 h can be explained by the presence of two electron-donating methyl groups, which deactivate the chloropyrazine towards oxidative addition.29
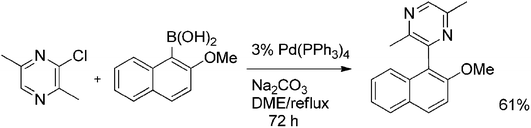 | ||
| Scheme 13 Suzuki coupling of 3-chloro-2,5-dimethylpyrazine. | ||
In a study on the preparation of asymmetrically tri- and tetrasubstituted pyrazines the application of microwave irradiation during the Suzuki coupling step was found to be highly valuable for substrate activation, speeding up the reaction (reaction time 10 min) (Scheme 14). Although significantly inactivated by the presence of multiple donor groups, pyrazines reacted with various arylboronic acids giving coupled products in excellent yields.30
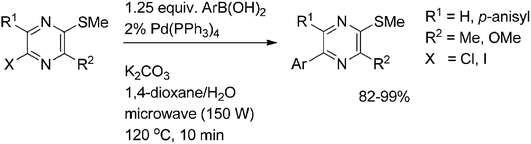 | ||
| Scheme 14 Suzuki coupling of 5-halo-2-(methylthio)pyrazines. | ||
The value of the use of trifluoroborates to improve the cross-coupling was exemplified by turning back to the reaction with chloropyrazine (Scheme 15). One major advantage of organotrifluoroborates is their considerably reduced tendency for oxidative homocoupling.31
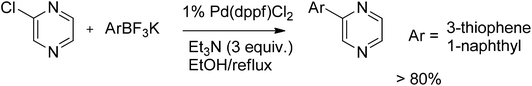 | ||
| Scheme 15 Chloropyrazine coupling with trifluoroborates. | ||
The reaction of chloropyrazine with 1-naphthyl- and 3-thiophenetrifluoroborates afforded the coupled products in >80% yield in 9 h. For comparison, the reactions involving similar boronic acid coupling partners required 3 times higher catalyst load and 5 times longer reaction times and gave lower yields.29,32 The essential advantages of organotrifluoroborates as hetaryl coupling partners over boronic acids include higher reactivity, tolerance to a wide array of electron-withdrawing and electron-donating groups in both coupling partners, and easiness of handling. In addition, organotrifluoroborates are less prone to protodeboronation.33
Another interesting approach towards regioselective Suzuki coupling was reported by Castillo et al.34 The reaction of pyridinium N-(3,5-dibromopyrazinyl)aminide and boronic acids worked effectively under standard conditions with 1 mol% of Pd(PPh3)4 catalyst to afford mono- and diarylpyrazines in good to excellent yields (Scheme 16). The regioselectivity of the monoarylation in the first step (Scheme 16) was provided by the unshared electron pair of the aminide nitrogen that coordinates to palladium and directs the oxidative addition to the ortho halogen.
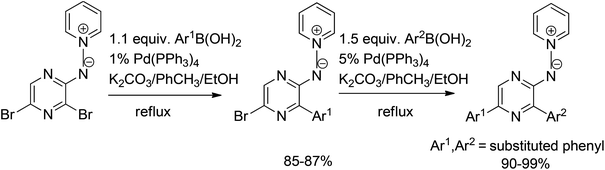 | ||
| Scheme 16 Suzuki coupling of pyridinium N-(3,5-dibromopyrazinyl)aminide with boronic acids. | ||
The Suzuki coupling was also examined with pyrazine O-triflates under standard Suzuki conditions.35 Reaction of 5-O-triflyl-3-benzyl-2-aminopyrazine with different arylboronic acids afforded the coupled products in good yields (Scheme 17) showing triflate to be a suitable leaving group. However, the procedure requires a high load (up to 10 mol%) of palladium catalyst.
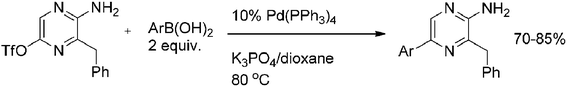 | ||
| Scheme 17 Reaction of 5-O-triflyl-3-benzyl-2-aminopyrazine with different arylboronic acids. | ||
A systematic study on the coupling of pyrazine O-tosylates with various organoboron derivatives was recently reported by Makarasen et al.36 Thus imino O-tosylates gave the highest yields reacting with aryltrifluoroborates in the presence of 4 mol% of Pd(OAc)2 and 8 mol% of biarylphosphine ligand (Scheme 18) revealing also tosylate as a good leaving group.
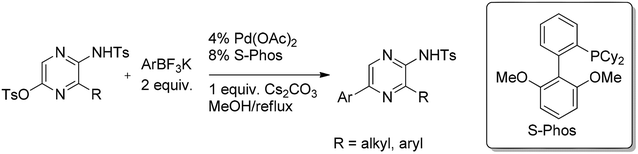 | ||
| Scheme 18 Coupling of pyrazine O-tosylates with aryltrifluoroborates. | ||
The high electrophilicity of the trifluoroborate group also happened to dominate over the electronic effects of the para-substituent in the aromatic ring, providing good to excellent yields in all cases of coupling with aryltrifluoroborates bearing highly electron-donating or -withdrawing groups at the para-position.
In summary, due to its wide scope and high tolerance towards a huge variety of functional groups, the Suzuki reaction of hetaryl organoborons as well as halides or related electrophiles provides one of the most straightforward methods for pyrazine functionalization along with the construction of pyrazine-based heterocyclic frameworks.
Stille coupling
The palladium-catalyzed Stille coupling is a versatile C–C bond forming reaction between stannanes and halides or pseudohalides with a very broad scope.37 Several examples of the Stille cross-coupling reaction have been reported in pyrazine chemistry. The Stille reaction of stannylated pyrazine and 4-methoxybenzoyl chloride afforded a mixture of the desired compound and the product of homocoupling of the tributylstannylpyrazine in 64% overall yield. The homocoupling, however, was suppressed by simply changing the order of introduction of the reagents. Mixing the aroyl chloride with the palladium catalyst prior to the introduction of the stannylated pyrazine yielded 70% of the desired compound without the formation of the homocoupling product (Scheme 19).38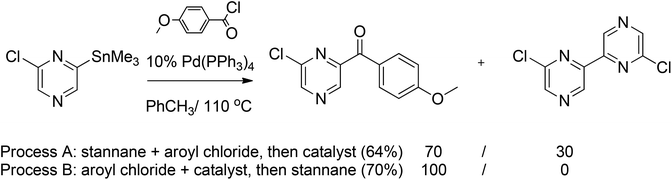 | ||
| Scheme 19 Stille coupling of stannylated pyrazine with 4-methoxybenzoyl chloride. | ||
The double Stille coupling was employed in the synthesis of a hybrid pyrazine–terpyridine ligand for self-assembled transition metal complexes. The reaction of 2,3-dichloropyrazine with 2.5 equivalents of stannylated terpyridine afforded the desired ligand in 73% yield (Scheme 20).39
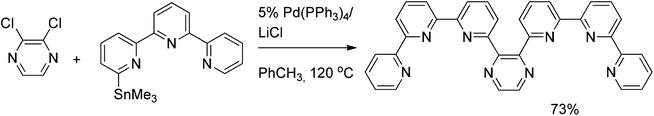 | ||
| Scheme 20 Double Stille coupling of dichloropyrazine. | ||
Bihetaryl Stille coupling with chlorotriazine starting from stannylated pyrazine was accomplished in 47% yield (Scheme 21).40 The reaction occurred selectively at the triazine chlorine leaving the chlorophenyl substituent intact.
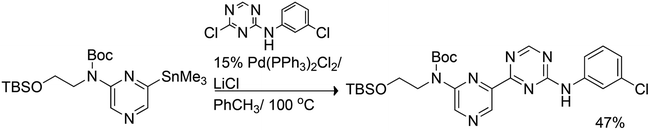 | ||
| Scheme 21 Bihetaryl Stille coupling of stannylated pyrazine with chlorotriazine. | ||
Another example of palladium-catalyzed Stille cross-coupling of the trialkylstannyl pyrazine was reported by Gündisch et al.41 The cross-coupling with azabicycloalkene triflate afforded the coupling product with a slightly reduced yield of 54% (Scheme 22).
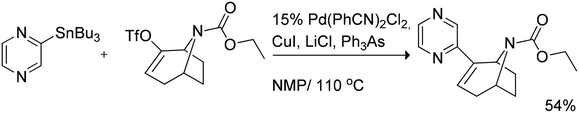 | ||
| Scheme 22 Stille cross-coupling of trialkylstannyl pyrazine with azabicycloalkene triflate. | ||
Efficient double Stille cross-coupling of distannylated pyrazines with diiododiketopyrazines gave polymers in 65–82% yields (Scheme 23). The optimal catalyst system for the polymerization of the pyrazine monomers was Pd(PPh3)2Cl2–CuI in a THF–NMP mixture. A slight excess of the stannane, used to achieve a reduction of the Pd(II) to the active Pd(0), apparently lowered the molecular weight of the obtained polymers. The same effect was observed when the PPh3 ligand was replaced by AsPh3, or when Pd-(dba)2/PPh3/CuI was used as the catalyst.42 Stille coupling of stannylated chloropyrazine with chloroiodopyrazine afforded dichlorobipyrazyl in an excellent yield of 95% (Scheme 24).43
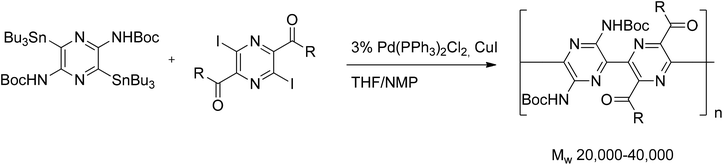 | ||
| Scheme 23 Double Stille cross-coupling of distannylated pyrazines with diiododiketopyrazines. | ||
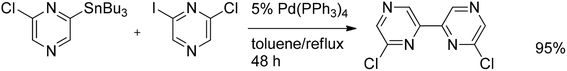 | ||
| Scheme 24 Stille coupling of stannylated chloropyrazine with iodochloropyrazine. | ||
In conclusion, the Stille cross-coupling is one of the most versatile tools for carbon–carbon bond formation within pyrazines as a result of the air- and moisture-stability of organotin reagents and the excellent functional group compatibility of the reaction. The reaction can be used for the coupling of stannylated pyrazines with a large variety of partners, i.e. aryl-, acyl-, and vinyl halides as well as triflates.
Sonogashira coupling
The palladium-catalyzed coupling of terminal alkynes with aryl/vinyl halides/pseudohalides, typically in the presence of a copper cocatalyst, is commonly known as the Sonogashira reaction.44 These coupling reactions are nowadays well-explored in pyrazine chemistry. Chloropyrazine proved to be an excellent substrate for this cross-coupling reaction. Under [Pd(allyl)Cl]2/PPh3 catalysis, using a slight excess of phenylacetylene, chloropyrazine was quantitatively converted into the corresponding diarylacetylene45 (Scheme 25).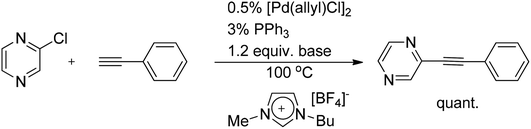 | ||
| Scheme 25 Sonogashira coupling of chloropyrazine with phenylacetylene. | ||
A Sonogashira coupling accompanied by heteroannulation was reported in a synthetic sequence towards 6-substituted-5H-pyrrolo[2,3-b]pyrazines. N-(3-Chloropyrazin-2-yl)-methanesulfonamide was subjected to the classical Sonogashira conditions with various acetylenes followed by base-induced cyclization resulting in the formation of the 6-substituted-5H-pyrrolo[2,3-b]pyrazines in 41–67% yields (Scheme 26). It is noteworthy that the reaction also tolerates amino groups as well as ketones, ortho-substituents, and carboxylic acids.46 A similar example using microwave irradiation instead of conventional heating was reported to give higher yields with shorter reaction times.47 Another improvement was made by introduction of two adjacent cyano groups into the pyrazine core, which allowed the use of unprotected aminopyrazine and significantly increased the yields of the target indoles (Scheme 26).48 The double Sonogashira reaction was easily performed on 2,3-dicyano-5,6-dichloropyrazine with a variety of alkynes, again showing the great activation effect of the cyano group (Scheme 27).49 The corresponding coupled products, which represent a class of highly demanded precursors for the synthesis of tetrapyrazinoporpyrazine-based photosensitizers, were obtained in 60–80% yields.
![Formation of 6-substituted-5H-pyrrolo[2,3-b]pyrazines under Sonogashira conditions.](/image/article/2013/OB/c3ob40460a/c3ob40460a-s26.gif) | ||
| Scheme 26 Formation of 6-substituted-5H-pyrrolo[2,3-b]pyrazines under Sonogashira conditions. | ||
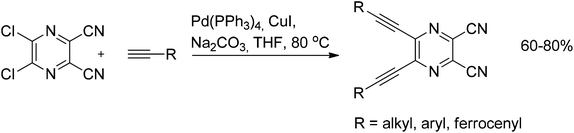 | ||
| Scheme 27 Double Sonogashira coupling of 2,3-dicyano-5,6-dichloropyrazine. | ||
In the course of a route to π-conjugated pyrazine oligomers, which are of great importance for manufacturing various electronic devices, several halogenated pyrazines were reacted with terminal acetylenes under Sonogashira conditions (Scheme 28). Since the reaction of 2,5-dibromopyrazine with a stoichiometric amount of 3,4,5-tridodecyloxyphenylacetylene afforded presumably disubstituted pyrazine (Scheme 28a), two iterative couplings with different terminal acetylenes were performed on 5-bromo-2-iodopyrazine (Scheme 28b). To further elongate the π-conjugated system and finalize the synthetic route towards an oligomer, the aforementioned pyrazine derivative was deprotected and subsequently coupled to 5-bromo-2-iodopyrazine, followed by a reaction with 3,4,5-tridodecyloxyphenylacetylene to end-cap the oligomer (Scheme 28c).50
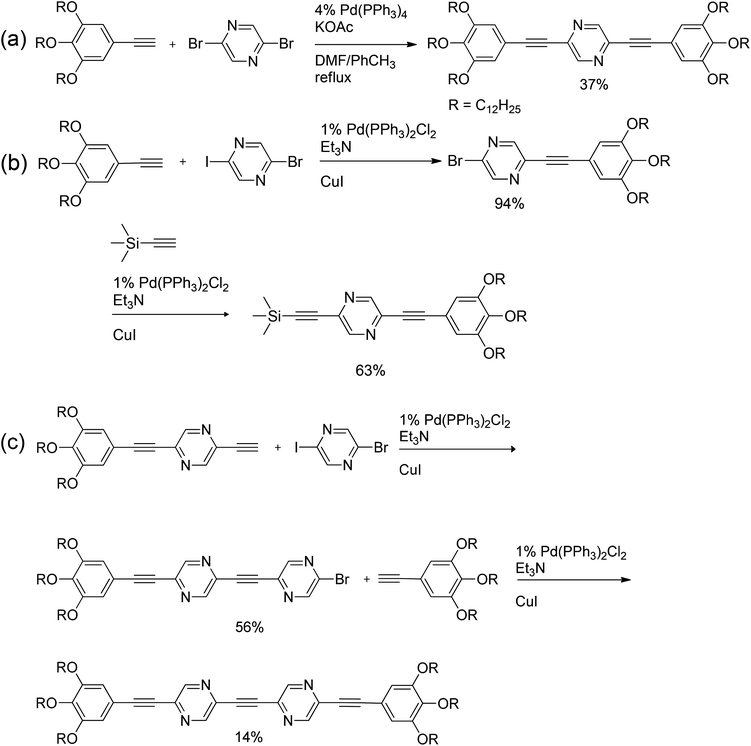 | ||
| Scheme 28 Multiple Sonogashira couplings with halogenated pyrazines. | ||
Ermolat'ev et al.51 have shown the suitability of N-4-methoxybenzyl (PMB) protected chlorinated pyrazinones for the Sonogashira reaction (Scheme 29). Selective coupling at the reactive 3-position with various acetylenes afforded the corresponding 3-ethynyl-5-chloropyrazin-2-ones in 85–93% yields.
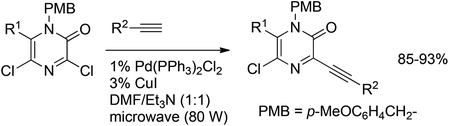 | ||
| Scheme 29 Sonogashira coupling of chlorinated pyrazinones. | ||
In general, the Sonogashira coupling represents an excellent method for the synthesis of elongated pyrazine containing π-conjugated systems. Accompanied by heteroannulation (see Scheme 26) this reaction allows the versatile synthesis of pyrazine-based fused heterocyclics.
Negishi coupling
Nickel- or palladium-catalyzed coupling of organozinc reagents with various halides/pseudohalides, commonly referred to as the Negishi reaction, represents a versatile C–C bond formation tool with a broad scope.52 Several Negishi cross-coupling examples have been reported in pyrazine chemistry. Various dichloropyrazines readily underwent direct metalations using TMPMgCl·LiCl or TMPZnCl·LiCl, where TMP is deprotonated tetramethylpiperidine and DBA is dibenzalacetone. Subsequent cross-coupling with iodobenzenes and iodothiophene gave the coupled products in good yields53 (Scheme 30).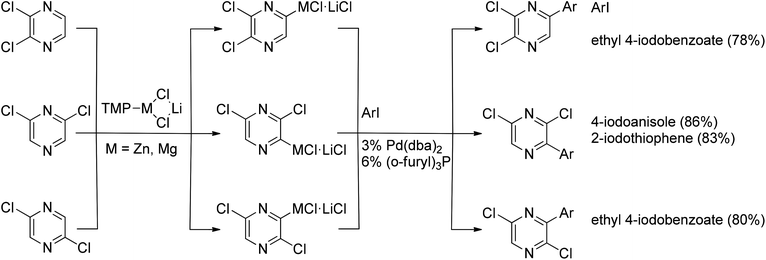 | ||
| Scheme 30 Negishi coupling of various dichloropyrazines. | ||
Hebbar et al.43 reported the Negishi reaction of 2-chloro-6-iodopyrazine with an acetylenic zinc reagent to give the coupling product in 71% yield (Scheme 31). The resulting compound was converted into the organozinc intermediate, whereupon reaction with bromopolyenes gave the coupled products in good yields.54 The same approach was successfully used for the double Negishi coupling of dichlorobipyrazyl with 1-bromo-6-substituted hexatrienes to give the corresponding coupled products in 64 and 57% yields, respectively (Scheme 32).
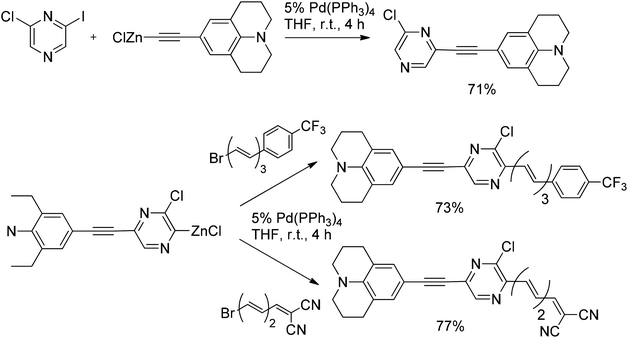 | ||
| Scheme 31 Negishi reactions of 2-chloro-6-iodopyrazine with an acetylenic zinc reagent and zinkated chloropyrazine with bromoalkenes. | ||
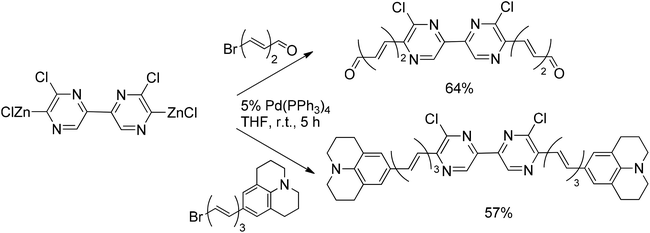 | ||
| Scheme 32 Negishi reaction of zinkated dichlorobipyrazyl with 1-bromo-6-substituted hexatrienes. | ||
Negishi coupling with sec-butylzinc halides was investigated by Fruit et al.55 (Scheme 33). Using Pd(PPh3)4 as the catalyst, partial isomerization occurred, resulting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of n-Bu and sec-Bu pyrazines. However, this was overcome by replacing the catalyst with the more selective Pd(dppf)Cl2, also increasing the overall yield up to 61%.
:1 mixture of n-Bu and sec-Bu pyrazines. However, this was overcome by replacing the catalyst with the more selective Pd(dppf)Cl2, also increasing the overall yield up to 61%.
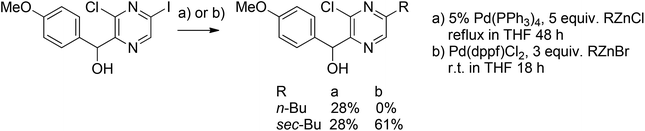 | ||
| Scheme 33 Negishi coupling of halopyrazines with alkylzinc halides. | ||
A regioselective synthesis of trialkylpyrazines via Negishi cross-coupling was reported by Pitchaiah et al.56 5-Substituted 2,3-dimethylpyrazine derivatives including ant's trail pheromone components were successfully synthesized in good yields by nickel-catalyzed Negishi cross-coupling reactions of pyrazine triflate promoted by alkyl and arylzinc halides (Scheme 34). It is interesting to note that nickel-catalyzed Negishi cross-coupling reactions with secondary alkylzinc halides, which bear a β-hydrogen and thus are prone to isomerization, produced the corresponding 5-alkylpyrazines as the only products in good yields (62–85%), without any isomerization of the alkyl groups.
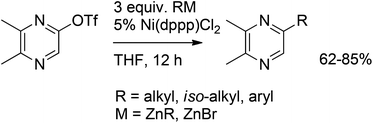 | ||
| Scheme 34 Regioselective Negishi coupling under nickel catalysis. | ||
The Negishi reaction, as the Suzuki and Stille couplings, represents a powerful tool for carbon–carbon bond formation. It is noteworthy that, due to the stability of the metallated species, halogenated pyrazines can be used as nucleophilic coupling partners as well.
Heck coupling
The palladium-catalyzed C–C coupling between aryl or vinyl halides and activated alkenes in the presence of a base is referred to as the Heck reaction.57 Malik et al.58 explored the Heck cross-coupling of 2,3-dichloropyrazine with various acrylates and styrenes. An interesting dependence of the product formation and distribution on the reaction temperature was found. Reaction of 2,3-dichloropyrazine with 2.5 equivalents of ethyl acrylate, in the presence of Pd(OAc)2 and X-Phos at 90 °C, afforded the corresponding 2,3-dialkenylpyrazine in 83% yield. Partial hydrogenation was observed when the reaction was carried out at higher temperatures (Scheme 35).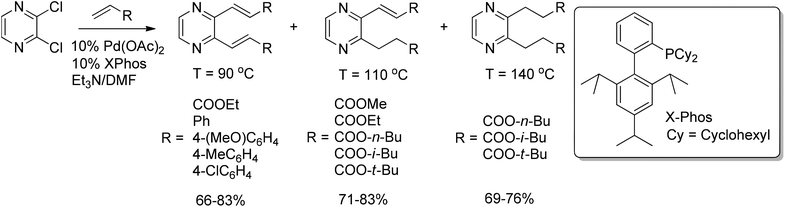 | ||
| Scheme 35 Heck cross-coupling of 2,3-dichloropyrazine with acrylates and styrenes. | ||
The partial or complete hydrogenation (caused by DMF) of the double bond of the coupled product could be explained by the presence of the strongly π-deficient pyrazine ring on one side of the double bond and the strongly electron-withdrawing ester group on the other.
A related Heck-type reaction, where the pyrazine moiety acts as an “olefin-like” coupling partner, was reported by the group of Snieckus. Thus 8-substituted imidazo[1,5-a]pyrazines were successfully arylated with a wide array of aryl halides in 45–92% yields (Scheme 36).59
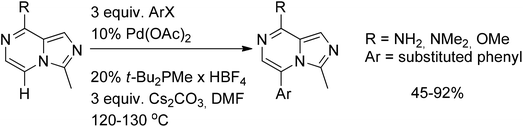 | ||
| Scheme 36 Pyrazine moiety as an “olefin-like” coupling partner in the Heck reaction. | ||
The Heck cross-coupling of pyrazines is scarcely represented in the literature, probably due to the high electrophilicity of the pyrazine ring, which makes Michael addition to the formed alkenylpyrazines very facile.
The Liebeskind–Srogl coupling
The Liebeskind–Srogl coupling is a recently discovered organic reaction that forms a coupled product from a thioester/thioether and a boronic acid using a metal catalyst.60,61 Modha et al.62 reported the successful application of Liebeskind–Srogl conditions to the coupling of p-methoxybenzylthiopyrazines with arylboronic acids. Initially, coupling of p-methoxybenzylthio-bearing pyrazine with arylboronic acid in the presence of copper(I)-thiophene-2-carboxylate (CuTC) and Pd(PPh3)4 in THF under conventional heating as well as under microwave irradiation gave poor to average yields, possibly due to decomposition of the reagents during the long reaction time at high temperature. However, the yields of the coupled products were considerably improved up to 69–93% by a simple addition of the reagents in two portions (Scheme 37).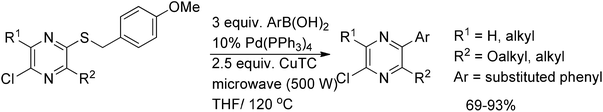 | ||
| Scheme 37 Coupling of p-methoxybenzylthiopyrazines with arylboronic acids. | ||
Earlier the same group reported the Liebeskind–Srogl coupling of methylthiopyrazines with arylboronic acids, which generally gave higher yields, but had a narrower scope being mainly limited to arylboronic reagents with donor groups. Substituted methylthiopyrazines were reacted with various boronic acids, in the presence of Pd(PPh3)4 and copper thiophene-2-carboxylate, giving the coupled products in yields ranging from 78 to 94% (Scheme 38).30
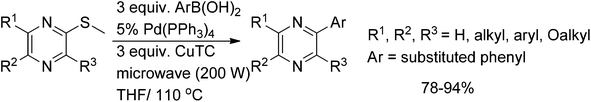 | ||
| Scheme 38 Coupling of methylthiopyrazines with arylboronic acids. | ||
These examples show that the novel Liebeskind–Srogl coupling reaction can be easily accomplished on thioether derivatives of substituted pyrazines, thus representing another method for the preparation of arylpyrazines.
C–N and C–P coupling
Examples of palladium-catalyzed C–N coupling reactions performed on halogenated pyrazines are rare, since amination of chloropyrazines is readily achievable via aromatic nucleophilic substitution of the chlorine by an appropriate primary or secondary amine in the presence of base. Nevertheless, a few interesting reactions of the so-called deactivated pyrazines will be described requiring metal catalysis.An example of the amination of chloropyrazine was recently reported by Kim et al.63 Using Pd2(dba)3 together with a bulky electron-rich phosphine ligand the corresponding phenylaminopyrazine was obtained in 59% yield (Scheme 39).
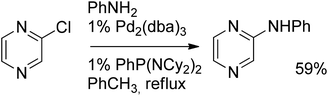 | ||
| Scheme 39 Amination of chloropyrazine. | ||
C–N cross-coupling has also been achieved with deactivated dialkylchloropyrazines (Scheme 40). The subsequent halogenation of the aminopyrazines, obtained in the first C–N coupling step, allowed a second amination, affording tetrasubstituted diaminopyrazines in 20–70% yields, demonstrating the generality, wide substrate scope, and tolerance to a variety of functional groups of the method.64
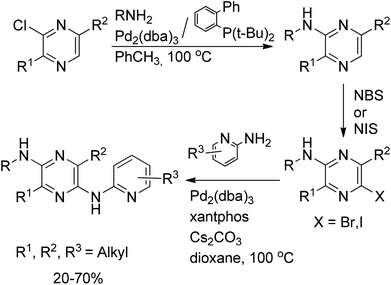 | ||
| Scheme 40 C–N cross-coupling with deactivated dialkylchloropyrazines. | ||
Recently, Hartwig et al.65 reported the high yield, selective palladium-catalyzed amination of aryl and hetaryl halides with a remarkably low catalyst loading. Chloropyrazine was coupled with n-octylamine affording the corresponding aminopyrazine in 82% yield in the presence of 0.005% of a catalyst (Scheme 41). In contrast, iodopyrazine required a higher catalyst loading and a longer reaction time. The same tendency was observed for a variety of iodo- and chloroarenes. Parallel studies revealed that the presence of the formed iodide ion considerably slows down the reaction.
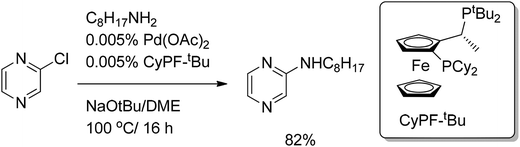 | ||
| Scheme 41 C–N cross-coupling with chloropyrazine. | ||
In a project aimed at the design of actinide selective extractants we studied the palladium-catalyzed C–P cross-coupling of chlorinated pyrazines with various phosphorus pronucleophiles. Using an appropriate base, Et3N for phosphites or DBU for less acidic phosphines and phosphine oxides, the phosphorylated pyrazines were obtained in 81–95% yields (Scheme 42).66
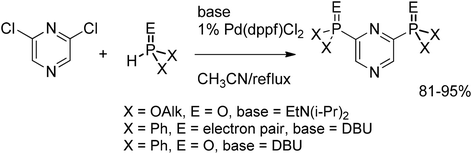 | ||
| Scheme 42 C–P cross-coupling with 2,6-dichloropyrazine. | ||
The same methodology was successfully extended to monochloropyrazines bearing various functional groups (Scheme 43).66 High tolerance of the reaction towards a variety of substituents allowed its application for the synthesis of supramolecular calix[4]arene-based multivalent actinide extractants (Scheme 44).67 It is noteworthy that cross-coupling reactions with diphenylphosphine proceeded with the same high rate in the presence of bare Pd(OAc)2 as the catalyst without an extra phosphine ligand. This increased reactivity was attributed to an autocatalytic effect of the partially phosphine-functionalized calix[4]arene, which complexes palladium, bringing the active catalyst into close proximity of the reacting functionality.
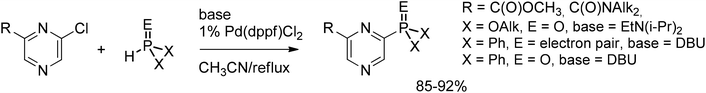 | ||
| Scheme 43 C–P cross-coupling with monochloropyrazines. | ||
![C–P cross-coupling with chloropyrazine-bearing calix[4]arenes.](/image/article/2013/OB/c3ob40460a/c3ob40460a-s44.gif) | ||
| Scheme 44 C–P cross-coupling with chloropyrazine-bearing calix[4]arenes. | ||
The palladium-catalyzed P–C cross-coupling of simple chloropyrazines with diphenylphosphine was also found to be catalyzed by Pd(OAc)2 under ligandless conditions with the same yield as in the presence of the DPPF ligand. This simplified procedure was used for the synthesis of novel hydrophilic phosphinopyrazines which revealed a high activity in the ruthenium- and rhodium-catalyzed hydrogenation of acetophenone68 and the rhodium-catalyzed polymerization of terminal arylacetylenes (Scheme 45).69
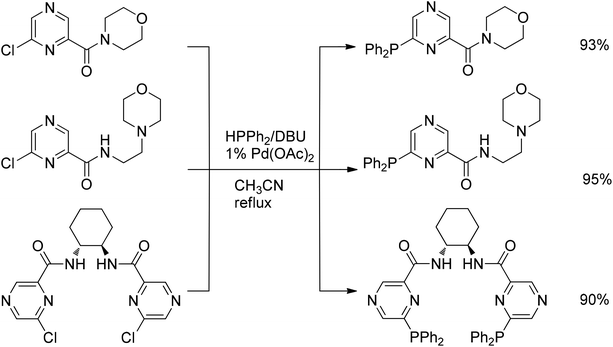 | ||
| Scheme 45 Cross-coupling of chloropyrazines with diphenylphosphine. | ||
C–H activation
A relatively new method in metal-catalyzed reactions is the direct arylation by C–H activation. In an effort at total synthesis of the natural alkaloid dragmacidin D, a method for the direct arylation of the pyrazine ring was developed. A bis-MOM-protected indole was primed for the Pd-catalyzed C–H/C–H coupling reaction with pyrazine N-oxide. Although the yield of this reaction was not superb, a 50% yield was achieved upon recycling. Despite its moderate yield, the reaction furnished the coupling product regioselectively. The second oxidative C–H/C–H coupling reaction of pyrazinone and 6-bromoindole under the influence of CF3SO3H afforded the corresponding coupling product in 57% yield with simultaneous removal of the two MOM groups (Scheme 46). Notably, the regioselectivity in these subsequent couplings was achieved by elegantly employing the “tautomeric switch” between pyrazinone and pyrazine N-oxide. Whilst pyrazine N-oxide has the most acidic, and hence more susceptible for coupling, carbon atom adjacent to the N–O moiety, the pyrazinone form of the resulting compound has the most acidic carbon atom next to a carbonyl group.70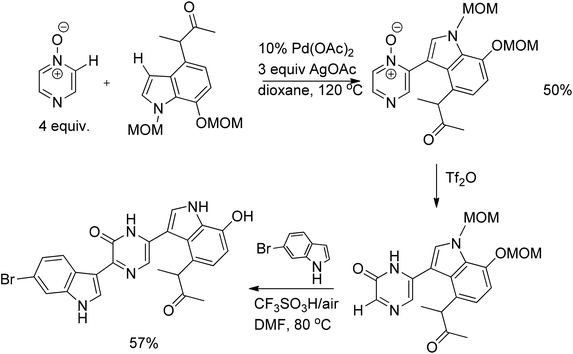 | ||
| Scheme 46 Synthesis of dragmacidin D. | ||
The regioselective C-6-arylation of 3-aminoimidazo[1,2-a]pyrazines was reported by Guchhait et al. (Scheme 47). Competing arylation at other positions was completely suppressed under concerted metallation–deprotonation conditions, using t-BuCOOK as a proton shuttle. The corresponding coupled products were obtained in 46–65% yields.71
![Regioselective arylation of 3-aminoimidazo[1,2-a]pyrazines.](/image/article/2013/OB/c3ob40460a/c3ob40460a-s47.gif) | ||
| Scheme 47 Regioselective arylation of 3-aminoimidazo[1,2-a]pyrazines. | ||
Miscellaneous couplings
The palladium-catalyzed coupling of aryl halides or triflates with Grignard reagents is generally referred to as the Kumada reaction.72,73 Despite the significance, this reaction has limited applicability due to the intolerance of Grignards towards many functional groups. An interesting example of a Kumada coupling was reported by Mongin et al.74 reacting fluoropyrazine with phenylmagnesium chloride at very mild conditions under nickel catalysis, giving the coupled product in 81% yield (Scheme 48).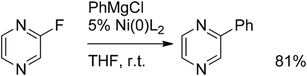 | ||
| Scheme 48 Kumada coupling of fluoropyrazine with phenylmagnesium chloride. | ||
The combination of Pd(OAc)2 and the S-Phos ligand, originally introduced by Buchwald,75 was successfully applied by the group of Knochel76 for the coupling of functionalized aryl-, benzylic- and alkylzinc reagents with various thiomethylated N-heterocycles and methylthiopyrazine in particular (Scheme 49).
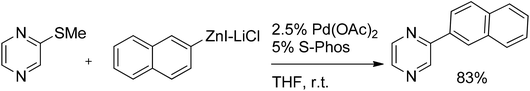 | ||
| Scheme 49 Palladium-catalyzed coupling of organozinc reagents with methylthiopyrazine. | ||
Further development of this reaction showed that methylthiopyrazine can also be cross-coupled with functionalized benzylic zinc reagents under Ni catalysis by using the inexpensive and robust system of Ni(acac)2 and DPE-Phos (Scheme 50).
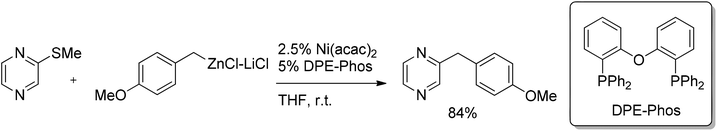 | ||
| Scheme 50 Nickel-catalyzed coupling of functionalized benzylic reagents with methylthiopyrazine. | ||
Chloropyrazines can be monoalkylated with Grignard reagents under Fe(acac)3 catalysis.77 Dickschat et al.78 reported the synthesis of mono- and dialkylpyrazines reacting chloropyrazines with alkylmagnesium bromides in the presence of Fe(acac)3 (Scheme 51).
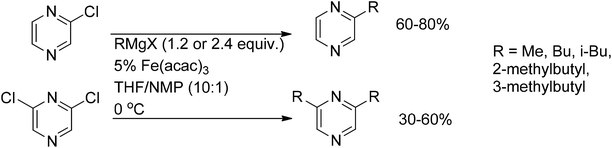 | ||
| Scheme 51 Fe(III)-catalyzed cross-coupling of chloropyrazines with Grignard reagents. | ||
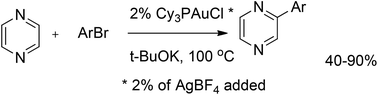
A variety of arylpyrazines were prepared by cross-coupling of pyrazine with aryl bromides in the presence of Cy3PAuCl (Scheme 52). The corresponding 2-arylated pyrazines were obtained in moderate to good yields, typically between 40% and 90%; however, 2-bromopyridine and 1-bromo-2-(trifluoromethyl)benzene gave only trace amounts of the coupled products. The low yields can be explained by the fact that electron-poor aryl bromides easily undergo hydrodebromination. Therefore, the reaction of pyrazine with electron-poor aryl bromides was performed in the presence of a catalytic amount of AgBF4 to accelerate the desired reaction and to overcome side processes. That gave the corresponding coupled products in better yields of 24% and 31%, respectively.79
Conclusions
In the past two decades remarkable progress has been made towards the development of methods for pyrazine functionalization. Starting from the very first attempts at the Suzuki coupling of chloropyrazine, the list of transition metal-catalyzed reactions, applicable for pyrazines, has included practically all coupling reactions known for regular aromatics. Moreover, the unique electronic and structural properties of pyrazine have made possible its direct functionalization via C–H activation. Thus, choosing an appropriate transition metal and ligand proved to be versatile for solving the long-standing challenge of bringing pyrazines into the family of useful substrates for transition metal-catalyzed cross-couplings. However, despite the impressive progress, a number of challenges still remain. Most of the studies described above utilize a relatively high catalyst loading. For industrial applications in particular, it will be highly desirable to develop more effective catalytic systems that require a lower use of precious metals, operate at milder conditions, and have a wider scope.References
- Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi and A. de Meijere, John Wiley & Sons, Inc., Hoboken, NJ, 2002, vol. 1 and 2 Search PubMed.
- Metal-Catalyzed Cross-Coupling Reactions, ed. A. De Meijere and F. Diederich, Wiley VCH, Weinheim, 2004, vol. 1 and 2 Search PubMed.
- J. J. Li and G. W. Gribble, Palladium in Heterocyclic Chemistry: A Guide for Synthetic Chemist, Elsevier, Amsterdam, 2nd edn, 2006 Search PubMed.
- T. J. Korn and P. Knochel, Angew. Chem., Int. Ed., 2005, 44, 2947–2951 CrossRef CAS.
- O. Baron and P. Knochel, Angew. Chem., Int. Ed., 2005, 44, 3133–3135 CrossRef CAS.
- Y. Zhang and D. Mitchison, Int. J. Tuberc. Lung Dis., 2003, 7, 6–21 CAS.
- V. Judge, B. Narasimhan and M. Ahuja, Hyg. J. D. Med., 2012, 4, 1–6 CAS.
- B. A. Ellsworth, Y. Wang, Y. Zhu, P. Annapurna, S. W. Gerritz, C. Sun, K. E. Carlson, L. Kang, R. A. Baska, Y. Yang, Q. Huang, N. T. Burford, M. J. Cullen, S. Johnghar, K. Behnia, M. A. Pelleymounter, W. N. Washburn and W. R. Ewing, Bioorg. Med. Chem. Lett., 2007, 17, 3978–3982 CrossRef CAS.
- M. Heitzmann, C. Gateau, L. Chareyre, M. Miguirditchian, M.-C. Charbonnel and P. Delangle, New J. Chem., 2010, 34, 108–116 RSC.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394–11410 RSC.
- P. Menova, F. Kafka, H. Dvorakova, S. Gunnoo, M. Sanda and R. Cibulka, Adv. Synth. Catal., 2011, 353, 865–870 CrossRef CAS.
- T. Imamoto, K. Tamura, Z. Zhang, Y. Horiuchi, M. Sugiya, K. Yoshida, A. Yanagisawa and I. D. Gridnev, J. Am. Chem. Soc., 2012, 134, 1754–1769 CrossRef CAS.
- P. J. J. Colbon, A. C. Foster and M. E. Giles, J. Heterocycl. Chem., 2008, 45, 1451–1456 CrossRef CAS.
- D. R. Carver, A. P. Komin, J. S. Hubbard and J. F. Wolfe, J. Org. Chem., 1981, 46, 294–299 CrossRef CAS.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- N. M. Ali, A. McKillop, M. B. Mitchell, R. A. Rebelo and P. J. Wallbank, Tetrahedron, 1992, 48, 8117–8126 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 36, 3437–3440 CrossRef.
- K. Jones, M. Keenan and F. Hibbert, Synlett, 1996, 509–510 CrossRef CAS.
- J.-F. Cavalier, M. Burton, C. De Tollenaere, F. Dussart, C. Marchand, J.-F. Rees and J. Marchand-Brynaert, Synthesis, 2001, 768–772 CrossRef CAS.
- J.-F. Cavalier, M. Burton, F. Dussart, C. Marchand, J.-F. Rees and J. Marchand-Brynaert, Bioorg. Med. Chem., 2001, 9, 1037–1044 CrossRef CAS.
- A. E. Thompson, G. Hughes, A. S. Batsanov, M. R. Bryce, P. R. Parry and B. Tarbit, J. Org. Chem., 2005, 70, 388–390 CrossRef CAS.
- P. Tapolcsanyi, G. Krajsovszky, R. Ando, P. Lipcsey, G. Horvath, P. Matyus, Z. Riedl, G. Hajos, B. U. W. Maes and G. L. F. Lemiere, Tetrahedron, 2002, 58, 10137–10143 CrossRef CAS.
- H. Matsushita, S.-H. Lee, K. Yoshida, B. Clapham, G. Koch, J. Zimmermann and K. D. Janda, Org. Lett., 2004, 6, 4627–4629 CrossRef CAS.
- F. Türksoy, G. Hughes, A. S. Batsanov and M. R. Bryce, J. Mater. Chem., 2003, 13, 1554–1557 RSC.
- S. M. González, A. I. Hernández, C. Varela, S. Rodríguez-Arístegui, R. M. Alvarez, A. B. García, M. Lorenzo, V. Rivero, J. Oyarzabal, O. Rabal, J. R. Bischoff, M. Albarrán, A. Cebriá, P. Alfonso, W. Link, J. Fominaya and J. Pastor, Bioorg. Med. Chem. Lett., 2012, 22, 1874–1878 CrossRef.
- P. Innocenti, K.-M. J. Cheung, S. Solanki, C. Mas-Droux, F. Rowan, S. Yeoh, K. Boxall, M. Westlake, L. Pickard, T. Hardy, J. E. Baxter, G. W. Aherne, R. Bayliss, A. M. Fry and S. Hoelder, J. Med. Chem., 2012, 55, 3228–3241 CrossRef CAS.
- K. M. Clapham, A. E. Smith, A. S. Batsanov, L. McIntyre, A. Pountney, M. R. Bryce and B. Tarbit, Eur. J. Org. Chem., 2007, 5712–5716 CrossRef CAS.
- C.-G. Yang, G. Liu and B. Jiang, J. Org. Chem., 2002, 67, 9392–9396 CrossRef CAS.
- M. McCarthy and P. J. Guiry, Tetrahedron, 1999, 55, 3061–3070 CrossRef CAS.
- V. P. Mehta, A. Sharma, K. Van Hecke, L. Van Meervelt and E. Van der Eycken, J. Org. Chem., 2008, 73, 2382–2388 CrossRef CAS.
- See: A. J. J. Lennox and G. C. Lloyd-Jones, Isr. J. Chem., 2010, 50, 664–674 CrossRef CAS.
- S. Gronowitz, A. B. Hoernfeldt, V. Kristjansson and T. Musil, Chem. Scr., 1986, 26, 305–309 CAS.
- G. A. Molander and B. Biolatto, J. Org. Chem., 2003, 68, 4302–4314 CrossRef CAS.
- R. Castillo, M. J. Reyes, M. L. Izquierdo and J. Alvarez-Builla, Tetrahedron, 2008, 64, 1351–1370 CrossRef CAS.
- W. Phakhodee, M. Toyoda, C.-M. Chou, N. Khunnawutmanotham and M. Isobe, Tetrahedron, 2011, 67, 1150–1157 CrossRef CAS.
- A. Makarasen, M. Kuse, T. Nishikawa and M. Isobe, Bull. Chem. Soc. Jpn., 2009, 82, 870–878 CrossRef CAS.
- V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1–652 CAS.
- F. Buron, N. Ple, A. Turck and G. Queguiner, J. Org. Chem., 2005, 70, 2616–2621 CrossRef CAS.
- A. R. Stefankiewicz, M. Wałęsa-Chorab, J. Harrowfield, M. Kubicki, Z. Hnatejko, M. Korabik and V. Patroniak, Dalton Trans., 2013, 42, 1743–1751 RSC.
- G.-H. Kuo, A. DeAngelis, S. Emanuel, A. Wang, Y. Zhang, P. J. Connolly, X. Chen, R. H. Gruninger, C. Rugg, A. Fuentes-Pesquera, S. A. Middleton, L. Jolliffe and W. V. Murray, J. Med. Chem., 2005, 48, 4535–4546 CrossRef CAS.
- D. Gündisch, K. Harms, S. Schwarz, G. Seitz, M. T. Stubbs and T. Wegge, Bioorg. Med. Chem., 2001, 9, 2683–2691 CrossRef.
- C. Y. Zhang and J. M. Tour, J. Am. Chem. Soc., 1999, 121, 8783–8790 CrossRef CAS.
- N. Hebbar, C. Fiol-Petit, Y. Ramondenc, G. Plé and N. Plé, Tetrahedron, 2011, 67, 2287–2298 CrossRef CAS.
- K. Sonogashira, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi and A. de Meijere, John Wiley & Sons, Inc., Hoboken, NJ, 2002, vol. 2, pp. 493–529 Search PubMed.
- S. Saleh, M. Picquet, P. Meunier and J.-C. Hierso, Tetrahedron, 2009, 65, 7146–7150 CrossRef CAS.
- C. R. Hopkins and N. Collar, Tetrahedron Lett., 2004, 45, 8087–8090 CrossRef CAS.
- C. R. Hopkins and N. Collar, Tetrahedron Lett., 2004, 45, 8631–8633 CrossRef CAS.
- A. Keivanloo, M. Bakherad, H. Nasr-Isfahani and S. Esmaily, Tetrahedron Lett., 2012, 53, 3126–3130 CrossRef CAS.
- H. Ali and J. E. van Lier, Tetrahedron Lett., 2012, 53, 4824–4827 CrossRef CAS.
- K. Pieterse, A. Lauritsen, A. P. H. J. Schenning, J. A. J. M. Vekemans and E. W. Meijer, Chem.–Eur. J., 2003, 9, 5597–5604 CrossRef CAS.
- D. S. Ermolat'ev, V. P. Mehta and E. V. Van der Eycken, QSAR Comb. Sci., 2007, 26, 1266–1273 CAS.
- E. Erdik, Tetrahedron, 1992, 48, 9577–9648 CrossRef CAS.
- M. Mosrin, T. Bresser and P. Knochel, Org. Lett., 2009, 11, 3406–3409 CrossRef CAS.
- N. Hebbar, Y. Ramondenc, G. Plé, G. Dupas and N. Plé, Tetrahedron, 2009, 65, 4190–4200 CrossRef CAS.
- C. Fruit, A. Turck, N. Plé, L. Mojovic and G. Quéguiner, Tetrahedron, 2001, 57, 9429–9435 CrossRef CAS.
- A. Pitchaiah, I. T. Hwang, J.-S. Hwang, H. Kim and K.-I. Lee, Synthesis, 2012, 1631–1636 CAS.
- G. T. Crisp, Chem. Soc. Rev., 1998, 27, 427–436 RSC.
- I. Malik, M. Hussain, A. Ali, S.-M. Tengho Toguem, F. Z. Basha, C. Fischer and P. Langer, Tetrahedron, 2010, 66, 1637–1642 CrossRef CAS.
- J.-X. Wang, J. A. McCubbin, M. Jin, R. S. Laufer, Y. Mao, A. P. Crew, M. J. Mulvihill and V. Snieckus, Org. Lett., 2008, 10, 2923–2926 CrossRef CAS.
- L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS.
- H. Prokopcova and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 2276–2286 CrossRef CAS.
- S. G. Modha, J. C. Trivedi, V. P. Mehta, D. S. Ermolat'ev and E. V. Van der Eycken, J. Org. Chem., 2011, 76, 846–856 CrossRef CAS.
- B. R. Kim, S.-D. Cho, E. J. Kim, I.-H. Lee, G. H. Sung, J.-J. Kim, S.-G. Lee and Y.-J. Yoon, Tetrahedron, 2012, 68, 287–293 CrossRef CAS.
- J. W. Corbett, M. R. Rauckhorst, F. Qian, R. L. Hoffman, C. S. Knauer and L. W. Fitzgerald, Bioorg. Med. Chem. Lett., 2007, 17, 6250–6256 CrossRef CAS.
- Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef CAS.
- N. I. Nikishkin, J. Huskens, J. Assenmacher, A. Wilden, G. Modolo and W. Verboom, Org. Biomol. Chem., 2012, 10, 5443–5451 CAS.
- N. I. Nikishkin, J. Huskens, S. A. Ansari, P. K. Mohapatra and W. Verboom, New J. Chem., 2013, 37, 391–402 RSC.
- N. I. Nikishkin, J. Huskens and W. Verboom, Tetrahedron Lett., 2013, 54, 1857–1861 CrossRef CAS.
- N. I. Nikishkin, J. Huskens and W. Verboom, Polymer DOI:10.1016/j.polymer.2013.04.028.
- D. Mandal, A. D. Yamaguchi, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2011, 133, 19660–19663 CrossRef CAS.
- S. K. Guchhait, S. Kandekar, M. Kashyap, N. Taxak and P. V. Bharatam, J. Org. Chem., 2012, 77, 8321–8328 CrossRef CAS.
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS.
- G. Bold, A. Fässler, H.-G. Capraro, R. Cozens, T. Klimkait, J. Lazdins, J. Mestan, B. Poncioni, J. Rösel, D. Stover, M. Tintelnot-Blomley, F. Acemoglu, W. Beck, E. Boss, M. Eschbach, T. Hürlimann, E. Masso, S. Roussel, K. Ucci-Stoll, D. Wyss and M. Lang, J. Med. Chem., 1998, 41, 3387–3401 CrossRef CAS.
- F. Mongin, L. Mojovic, B. Guillamet, F. Trecourt and G. Queguiner, J. Org. Chem., 2002, 67, 8991–8994 CrossRef CAS.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS.
- L. Melzig, A. Metzger and P. Knochel, Chem.–Eur. J., 2011, 17, 2948–2956 CrossRef CAS.
- A. Fürstner, A. Leitner, M. Méndez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856–13863 CrossRef.
- J. S. Dickschat, H. Reichenbach, I. Wagner-Döbler and S. Schulz, Eur. J. Org. Chem., 2005, 4141–4153 CrossRef CAS.
- M. Li and R. Hua, Tetrahedron Lett., 2009, 50, 1478–1481 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |