 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of the IGF-II-like hormone vesiculin using regioselective formation of disulfide bonds
Geoffrey M.
Williams
a,
Garth J. S.
Cooper
b,
Kathryn
Lee
b,
Lynda
Whiting
b and
Margaret A.
Brimble
*a
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland, New Zealand. E-mail: m.brimble@auckland.ac.nz; Fax: +64 9 3737422; Tel: +64 9 9238259
bSchool of Biological Sciences, The University of Auckland, 3a Symonds Street, Auckland, New Zealand
First published on 22nd March 2013
Abstract
Diabetes mellitus, characterised by hyperglycemia and altered β-cell function, is an increasingly common disorder affecting millions of individuals world-wide. While therapeutic regimens exist to manage the condition, diabetic individuals remain prone to complications that are detrimental to both their length and quality of life. An improved understanding of the disease which may then enable development of new treatments is therefore a desirable goal. Vesiculin, a novel IGF-II-like protein was recently isolated from the secretory granules of murine β-cells, and preliminary studies indicate it is capable of signalling via the insulin receptor (IR)/insulin-like growth factor receptor 1(IGF1R) family giving it the potential to elicit both metabolic and mitogenic responses in the beta-cell. In order to facilitate further studies on this new member of the insulin-family of hormones, we undertook a chemical synthesis of the protein using regioselective disulfide bond formation.
Introduction
Diabetes mellitus type 2 is an increasingly common metabolic disorder characterised by the development of insulin resistance in peripheral tissues and consequent elevated blood glucose levels. While many treatments exist to manage the condition, long-term hyperglycemia can lead to severe physiological complications including eventual pancreatic β-cell failure and dependence on external sources of insulin.Identification of proteins that regulate metabolic processes involving β-cells enable an improved understanding of the disease and may lead to new treatments. Buchanan et al. reported1 the identification and preliminary characterisation of murine vesiculin, a novel IGF-II-like protein isolated from the secretory granules of a murine β-cell line. Elucidation of the primary structure showed it to be comprised of two so-called A- and B-peptide chains cross-linked by disulfide bonds, and ostensibly generated from IGF-II itself by excision of a tetrapeptide corresponding to residues 37–40 (Fig. 1).
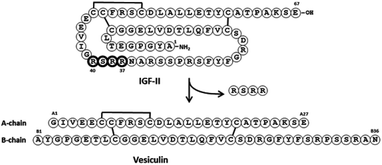 | ||
| Fig. 1 Vesiculin generated from IFG-II by excision of residues 37–40 (bold). | ||
Furthermore, preliminary in vitro experiments showed that vesiculin stimulated glycogen synthesis in muscle tissue with the same or better efficacy as native IGF-II, which led the authors to suggest1 that it may be an important β-cell regulatory hormone and thus warranted further investigation. However, any such studies would necessarily be hampered by supply of material. Isolation of murine vesiculin from β-cell granules is extremely laborious and cannot afford sufficient quantities for useful, large-scale physiological studies. Also, access to the human form of vesiculin2 using this approach would be impossible. The use of recombinant techniques to produce the material also presents considerable challenges owing to the post-translationally processed nature of the peptide.
The two-chain structure of vesiculin possesses the characteristic arrangement of crosslinking disulfide bonds that places it neatly into the ever-expanding insulin superfamily of peptide hormones,3 all members of which exhibit the identical motif. Fortunately, the chemical synthesis of these insulin-type A–B systems – such as insulin itself4 and the structurally related relaxins5 – is well-documented and provides a repertoire of methods by which relatively large amounts of vesiculin could be produced. Herein a synthesis of vesiculin is described in which firstly the constituent A- and B-peptide chains were synthesised separately with the cysteine residues orthogonally protected. A series of regioselective cysteine deprotection and disulfide bond formation steps then ensued, affording a useful quantity of the required compound. The insulin-like activity of synthetic vesiculin was confirmed using an ex vivo model system.
Results and discussion
The resemblance between the arrangement of disulfide bonds in the primary structures of vesiculin and insulin led us to examine the various synthetic approaches to insulin4,6–16 with a view to applying the most suitable to the present case. Based on this it was decided that the technique of regioselective disulfide synthesis would be most likely to produce a satisfactory outcome and furnish useful amounts of vesiculin. This strategy would entail synthesis of the individual A and B chains that comprise vesiculin separately then generation of the disulfide bonds in a controlled sequence of three regioselective deprotection/oxidation steps. Judicious choice of protecting groups enables firstly the formation of the intramolecular CysA6–CysA11 disulfide, which is followed by an activation of the A20 cysteine and reaction with the B-chain to form the first of the intermolecular disulfides. Finally the remaining thiol protecting groups of CysA7 and CysB9(Acm) are removed under oxidative conditions to afford the final disulphide bond.The synthesis is summarised in Scheme 1. Early attempts at producing the A-chain established that it was very poorly soluble and chromatographically intractable, giving elution profiles consistent with severe aggregation. Thus it was proposed incorporating a pentalysine tag17 at the C-terminus via an HMBA-linker14 in an effort to render the peptide both more soluble and amenable to chromatography. Carrying the tag through the remainder of the synthesis presumably would also facilitate handling and purification of subsequent intermediates.
 | ||
| Scheme 1 Schematic overview of the synthesis of vesiculin (1). Reagents and conditions: (i) Fmoc-SPPS; (ii) TFA–DODT–water–TIPS; (iii) DPDS (1.1 equiv.), water–MeOH; (iv) DPDS (excess), TFA–triflic acid; (v) Boc-SPPS; (vi) HF–p-cresol; (vii) 4 in 6 M Gn HCl, pH 8.1 then 5; (viii) AcOH–I2; (ix) 0.1 M NaOH (aq.). | ||
Thus, using Fmoc SPPS chemistry the A-chain (2) was synthesised by incorporating Cys(tBu) at position A20, Cys(Acm) at position A7 and Cys(trityl) at positions A6 and A11. A 5% piperazine solution was used for deprotection as this effectively eliminated the small but significant amount of aspartimide that was otherwise generated under the microwave conditions when 20% piperidine was used. Following cleavage from the resin, the resulting free thiols at CysA6 and CysA11 of the crude peptide were converted to an intramolecular disulfide bond using a dipyridyl disulphide (DPDS)-mediated oxidation.18 The resulting peptide 3 was easily purified by HPLC, enabled by the C-terminal pentalysine tag.
A number of attempts to synthesise the B-chain using the Fmoc approach afforded very complex crude mixtures with little product evident, whereas the Boc-SPPS format gave a much improved product profile. On cleavage from resin using HF the crude peptide, only sparingly soluble in water–TFA mixtures, was unexpectedly found to dissolve very readily in water–0.1% formic acid, which enabled more straightforward handling for chromatographic purification.
The tert-butyl protecting group of CysA20 was converted to an S-pyridyl group by treating the A-chain peptide 3 with a mixture of trifluoroacetic and triflic acids in the presence of a 9-fold excess of dipyridyl disulfide19 to afford 4. Since this reaction proceeded fairly cleanly, only the most cursory purification was performed so as to minimise handling of this moderately unstable intermediate. This entailed absorbing the components of the reaction on to a C4 HPLC column then generating a suitable HPLC gradient until the excess DPDS was eluted, then switching to an eluent with a high organic component to quickly strip out the absorbed peptide from the column in a small volume, which was then lyophilised. This material in its crude form was then reacted under denaturing, buffered conditions with a stoichiometric amount of the B-chain (5), the free thiol group of which induced rapid thiolysis of the S-pyridyl group of CysA20 to form 6, with its constituent chains now being crosslinked by the newly formed CysA20–CysB21 disulfide bond. The progress of the reaction was monitored by HPLC (Fig. 2a) and if necessary extra quantities of 5 were titrated into the reaction to drive it to completion (as based on consumption of the more valuable constituent, A-chain 4).
![(a) Reaction of A-chain [Cys7(Acm), Cys20(SPyr)] (4) with B-chain [(Cys9(Acm)] (5) to form the A20–B21 disulfide bond of 6. (b) Removal of CysA7,B9 Acm groups from purified 6 and formation of A7–B9 disulfide of 7. (c) Hydrolysis of pentalysine tag from purified 7, giving vesiculin (1). (d) LCMS of purified vesiculin (1). HPLC analysis was performed using a DIONEX Acclaim C18 4.6 × 150 mm column and a gradient of 1% MeCN in water (0.1% TFA) to 49% MeCN in water (0.1% TFA) over 24 min.](/image/article/2013/OB/c3ob40322j/c3ob40322j-f2.gif) | ||
| Fig. 2 (a) Reaction of A-chain [Cys7(Acm), Cys20(SPyr)] (4) with B-chain [(Cys9(Acm)] (5) to form the A20–B21 disulfide bond of 6. (b) Removal of CysA7,B9 Acm groups from purified 6 and formation of A7–B9 disulfide of 7. (c) Hydrolysis of pentalysine tag from purified 7, giving vesiculin (1). (d) LCMS of purified vesiculin (1). HPLC analysis was performed using a DIONEX Acclaim C18 4.6 × 150 mm column and a gradient of 1% MeCN in water (0.1% TFA) to 49% MeCN in water (0.1% TFA) over 24 min. | ||
Following purification, a dilute solution of 6 in aqueous acetic acid was treated with an excess of iodine, which removed the two Acm protecting groups11 from CysA7 and CysB9 and concomitantly generated the second inter-chain disulfide bond very cleanly (Fig. 2b). Rather than first attempting to isolate the highly diluted crude product, the reaction mixture was instead simply passed directly through a C18 HPLC column and the absorbed material purified by gradient HPLC. This afforded 7 in excellent yield.
Finally, the pentalysine tag was hydrolysed (Fig. 2c) by exposure to dilute NaOH17 for a few minutes giving the less polar vesiculin (1) in an overall yield of 40% based on pure A-chain (3). Furthermore, despite losing the solubilising tag, the natural product remained water soluble suggesting that the hydrophobic residues of the individually very poorly soluble constituent A- and B-chains were now buried within the tertiary structure of 1.
The insulin-like activity of synthetic murine vesiculin was investigated by assaying the peptide-stimulated incorporation of 14C-labelled glucose into muscle glycogen in ex vivo rat soleus muscle.20 The peptide was assayed at four concentrations, which were drawn from the upper end of a previously determined dose response curve1 and chosen specifically to investigate maximal activity. After a two hour incubation of muscle tissue samples in solutions containing radiolabelled glucose and peptide, the radiolabelled glycogen content of the tissue was measured. Results revealed that significant stimulation of muscle glycogen synthesis had occurred in all doses compared to a no-peptide control and that no significant differences in the magnitude of the effect were seen between vesiculin and insulin (Fig. 3), based on a two-way ANOVA. This indicates that vesiculin is a full insulin agonist in this model.
 | ||
| Fig. 3 Peptide-stimulated conversion of 14C-labelled glucose into muscle glycogen in isolated rat soleus muscle. Recombinant human insulin (Actrapid) open bars, synthetic mouse vesiculin hatched bars. | ||
Conclusion
Murine vesiculin was synthesised using a combined Fmoc/Boc solid phase synthesis strategy. Orthogonal protection of cysteine residues allowed the specific and regioselective formation of both intra- and inter-molecular disulfide bonds in good yield (40% based on purified constituent A- and B-chains). Experiments conducted with this synthetic vesiculin successfully reproduced the insulin-like responses originally observed with material obtained from a mouse-derived, immortalised pancreatic β-cell line.Having now established a means of producing vesiculin in useful quantities and confirming the biological activity of the synthetic material, a full characterisation of this novel peptide hormone – and particularly its role in β-cell autocrine regulation – is now possible.
Experimental section
Materials
9-Fluorenylmethoxycarbonyl (Fmoc) protected L-α-amino acids, Fmoc-RINK-OH linker, 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) and hydroxymethylbenzoic acid (HMBA) were purchased from GL Biochem (Shanghai, China). Boc-L-α-Asparagine-PAM-OH (PAM = p-hydroxymethylphenylacetic acid) was purchased from PolyPeptide Group (Strasbourg, France). Dimethylformamide (DMF) and acetonitrile were purchased from Global Science (Auckland, NZ); 3,6-dioxa-1,8-octanedithiol (DODT), triisopropylsilane (TIPS), diisopropylethylamine (DIPEA), and trifluoromethanesulfonic acid (TFMSA) were purchased from Sigma Aldrich. Trifluoroacetic acid (TFA) was obtained from Oakwood Products, Inc. (West Colombia, SC) and 2,2′-dipyridyl disulfide (DPDS) from Fluka (Switzerland). H-Rink-ChemMatrix resin was obtained from PCAS BioMatrix, Inc. (Quebec) and aminomethyl polystyrene resin was manufactured in-house21 from Bio-Beads (Bio-Rad Laboratories, Inc., USA). Deoxy-D-glucose 2-[14C(U)]: 300 mCi mmol−1; 0.1 μCi μL−1 was purchased from American Radiolabeled Chemicals, Inc. Low glucose DMEM was purchased from Invitrogen (11885-076) and Actrapid® Recombinant Human Insulin was purchased from Novo Nordisk, Denmark.Purifications were performed using a Dionex Ultimate 3000 HPLC system, with water–0.1% TFA as eluent A and MeCN–0.1% TFA eluent B and a Phenomenex Gemini C18 5μ 110 Å 10 × 250 mm column unless otherwise indicated. Mass spectra were recorded using an Agilent 1100MSD spectrometer.
Peptide synthesis
All syntheses were conducted on a 0.1 mmol scale, with peptide couplings employing a 5-fold molar excess of the protected amino acid in DMF activated by a 4.8 molar excess of HCTU in the presence of a 10 molar excess of DIPEA.A-chain Fmoc synthesis was carried out on ChemMatrix Rink-resin using a CEM Liberty microwave peptide synthesiser (AI Scientific, Queensland, Australia) with the Fmoc group being removed using 5% piperazine in DMF (3 minutes at 75 °C, 60 W irradiation). The exceptions were the HMBA linker itself and first residue attached to this linker (FmocGlu(tBu), corresponding to A27), which were incorporated manually: firstly, the deprotected resin was treated with 2 equivalents of each of 4-hydroxymethylbenzoic acid, DIC, and HOBt in DMF for 1 hour. This coupling was repeated, then the Fmoc amino acid was attached using the symmetrical anhydride method in which the resin was twice treated with a mixture Fmoc amino-acid (10 equiv.), DIC (10 equiv.) and 4-DMAP (0.1 equiv.) in DMF for 3 hours.
The side-chains of the amino acids were protected where necessary with TFA-labile groups, except for acetamidomethyl (Acm)-protected Cys (position A7) and tert-butyl (tBu)-protected Cys (position A20). Cleavage and deprotection of the peptide was achieved by incubating the resin in 100 mL mmol−1 of 94% TFA, 2.5% water, 2.5% DODT, 1% TIPS for 3 hours. The crude peptide was recovered by firstly filtering and then diluting the filtrate with chilled diethyl ether and pelleting the precipitate by centrifugation. The pellet was then washed several times with ether.
B-chain synthesis was carried out on in-house manufactured aminomethyl polystyrene resin21 using in situ neutralization Boc synthesis, with all deprotections (neat TFA, 10 minutes) and couplings (40 minutes) being carried out manually in a glass reaction vessel equipped with a glass filtration sinter. With the exception of S-acetamidomethyl-protected Cys (position B9) all side-chain protecting groups were HF labile. The peptide was deprotected and cleaved from the resin support by treatment with anhydrous HF containing p-cresol (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v) for 1 h at 0 °C. After evaporation of the HF under reduced pressure, crude peptide products were triturated and precipitated with chilled diethyl ether, dissolved in 50% aqueous acetonitrile containing 0.1% formic acid and lyophilized. The material was then dissolved in water–0.1% formic acid, purified by semi-preparative RP-HPLC and lyophilised to give 5. LCMS (ESI+) gave m/z 1324.9 [M + 3H+]. Calc'd [M + 3H+] requires 1324.95.
:10, v/v) for 1 h at 0 °C. After evaporation of the HF under reduced pressure, crude peptide products were triturated and precipitated with chilled diethyl ether, dissolved in 50% aqueous acetonitrile containing 0.1% formic acid and lyophilized. The material was then dissolved in water–0.1% formic acid, purified by semi-preparative RP-HPLC and lyophilised to give 5. LCMS (ESI+) gave m/z 1324.9 [M + 3H+]. Calc'd [M + 3H+] requires 1324.95.
Formation of the A-chain Cys6–Cys11 intramolecular disulfide bond: compound 3
Following completion of the Fmoc synthesis, cleavage from the resin afforded crude A-chain [CysA6,A11(H), CysA7(Acm), CysA20(tBu)] 2 (243 mg, 63 μmol) which was not purified but dissolved in deionised water (240 mL) and DPDS (16.6 mg, 76 μmol) in methanol (16 mL) added in one portion. After stirring for 1 hour analysis by HPLC showed formation of the more polar disulfide was complete. Neat TFA (0.5 mL) was then added and the solution passed through a Phenomenex Gemini C18 5μ 110 Å 10 × 250 mm column equilibrated in 1% B. The eluent was then switched to 65% B and the eluted, concentrated material collected and lyophilised. The ca. 240 mg of crude peptide was then purified in a single portion by RP-HPLC to afford 37 mg (15% yield) of 3 after lyophilisation. LCMS (ESI+) gave m/z 1284.6 [M + 3H+]. Calc'd [M + 3H+] requires 1284.0.Conversion of A-chain Cys20(tBu) to Cys20(SPyr): compound 4
A-chain [CysA7(Acm), CysA20(tBu)] 3 (37 mg, 9.6 μmol), anisole (141 μL) and DPDS (20 mg, 91 μmol) were combined and dissolved in TFA (1.2 mL) then cooled to 0 °C on ice-water. A similarly chilled mixture of TFMSA–TFA (1:4 v/v, 700 μL) was then added and the solution agitated gently at 0 °C for 3 minutes. The reaction mixture was diluted with cold diethyl ether (5 mL) and the precipitated peptide pelleted by centrifugation. The pellet was dissolved in water–0.1% TFA and loaded on to a Phenomenex Jupiter C4 RP-HPLC column and a gradient generated until the excess DPDS had been eluted (approx. 25% B), whereupon the column was ‘stripped’ by switching to 60% B to elute the A-chain [CysA7(Acm), CysA20(SPyr)] which was lyophilised to afford 4 (31 mg, 84% yield). LCMS (ESI+) gave m/z 1302.3 [M + 3H+]. Calc'd [M + 3H+] requires 1301.6. This material was not purified further but used directly in the next step.
Combination of A-chain [Cys7(Acm), Cys20(SPyr)] with B-chain [Cys9(Acm), Cys21(H)] – formation of CysA20–CysB21 disulfide bond: compound 6
The A-chain [Cys7(Acm), Cys20(SPyr)] 4 (26.5 mg, 6.8 μmol) was dissolved in 6 M aqueous guanidine·HCl (6 mL) and 1 M tris·HCl buffer (1.33 mL, pH 8.1) added. The vigorously stirred solution was cooled on ice and the B-chain [Cys9(Acm), Cys21(H)] 5 (27.5 mg, 7 μmol) in 6 M guanidine·HCl (4 mL) was added dropwise over ca. 10 minutes. After stirring for 30 minutes the reaction was judged complete by RP-HPLC (Fig. 2a) based on disappearance of the A-chain component from the elution profile. Neat TFA (150 μL) was then added and aliquots of the resulting solution purified by RP-HPLC to afford 6 (31.7 mg, 60% yield) after lyophilisation. LCMS (ESI+) gave m/z 1553.9 [M + 5H+]. Calc'd [M + 5H+] requires 1553.15.Removal of Acm protecting groups and generation of the CysA7–CysB21 disulfide bond: compound 7
The A–B peptide [CysA7,B9(Acm)] 6 (31 mg, 4 mmol) was dissolved in chilled 4:1 v/v acetic acid–water (20 mL) and cooled in ice-water. A solution of iodine (560 μL of a 0.5 M solution in MeOH, 280 μmol) was added and the solution mixed thoroughly. The reaction was incubated at 0 °C for 50 minutes with agitation at 10 minute intervals, by which time the reaction was nearly complete (Fig. 2b). Sodium ascorbate (560 μL of a 0.5 M solution in water, 280 μmol) was then added and the solution mixed thoroughly, causing the dark colour to fade to pale-brown. Water (40 mL) was added, causing complete decolourisation, followed by TFA (400 μL). Aliquots of the resulting solution were purified by RP-HPLC, which, after pooling of fractions and lyophilisation, afforded the desired material 7 (24.2 mg, 80% yield). LCMS (ESI+) gave m/z 1525.2 [M + 5H+]. Calc'd [M + 5H+] requires 1524.33.
Vesiculin 1
The A–B peptide 7 (24 mg, 3.1 μmol) was dissolved in deionised water (3.8 mL) and cooled on ice. Aqueous 0.2 M NaOH, also at 0 °C (3.8 mL) was added and the resulting solution incubated on ice for 5 minutes then acidified with TFA (60 μL) to pH 3. Aliquots of this solution were then purified by RP-HPLC to afford vesiculin (1) (18.0 mg, 85% yield) after lyophilisation. LCMS (ESI+) gave m/z 1712.4 [M + 4H+]. Calc'd [M + 4H+] requires 11711.78.Stimulation of ex vivo muscle glycogen synthesis
Soleus muscles were excised from fasted male SD rats 207 g (193–221 g) under anaesthetic and were each split longitudinally into three strips. The strips were distributed between 6 flasks (5–6 strips per flask, from 5–6 different rats) containing 10 mL carbogen-saturated low glucose DMEM media with or without peptide (0, 23.7, 71, 237 and 510 nM) and supplemented with 0.5 μCi of deoxy-D-glucose 2-[14C(U)]. The flasks were then incubated in a shaking waterbath with continuous carbogen bubbling for two hours at 30 °C before being removed and the muscle tissue samples processed to extract glycogen and measure 14C. Using already published1 methods, muscle strips were individually blotted to remove excess liquid, freeze-dried then weighed. Strips were then solubilised in 60% KOH at 70 °C for 45 minutes and glycogen precipitated in ice-cold ethanol and pelleted by centrifugation. Each pellet was then washed in ethanol, dried and resuspended in scintillation fluid before measuring cpm using a 1450 MicroBeta® TriLux scintillation counter. Each peptide concentration was tested in 2–4 separate experiments, with 5–6 muscle strips per experiment.Acknowledgements
The authors gratefully acknowledge the support of Auckland Uniservices Ltd and The Maurice Wilkins Centre for Molecular Biodiscovery.References
- C. M. Buchanan, A. R. B. Phillips and G. J. S. Cooper, Growth Horm. IGF Res., 2010, 20, 360–366 CrossRef CAS.
- C. M. Buchanan and G. J. S. Cooper, WO2006112737, 2006 Search PubMed.
- J. Piñero-Gonzáles and A. Gonzáles-Pérez, OMICS: J. Integr. Biol., 2011, 15, 439–447 CrossRef.
- A. Belgi, M. A. Hossain, G. W. Tregear and J. D. Wade, Immun., Endoc. & Metab. Agents in Med. Chem., 2011, 11, 40–47 CAS.
- J. D. Wade, F. Lin, M. A. Hossain, F. Shabanpoor, S. Zhang and G. W. Tregear, Ann. N. Y. Acad. Sci., 2009, 1160, 11–15 CrossRef CAS.
- J. Meienhofer, E. Schnabel, H. Bremer, O. Brinkhoff, R. Zabel, W. Sroka, H. Klostermayer, D. Brandenburg, T. Okuda and H. Zahn, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1963, 18, 1120–1121 CAS.
- Y.-T. Kung, W. T. Huang, C. C. Chen and L. T. Ke, Sci. Sin. (Engl. Ed.), 1965, 14, 1710–1716 CAS.
- P. G. Katsoyannis, A. Tometsko and C. Zalut, J. Am. Chem. Soc., 1966, 88, 166–167 CrossRef CAS.
- A. Marglin and R. B. Merrifield, J. Am. Chem. Soc., 1966, 88, 5051–5052 CrossRef CAS.
- P. Sieber, B. Kamber, A. Hartmann, A. Joehl, B. Riniker and W. Rittel, Helv. Chim. Acta, 1974, 57, 2617–2621 CrossRef CAS.
- K. Akaji, K. Fujino, T. Tatsumi and Y. Kiso, J. Am. Chem. Soc., 1993, 115, 11384–11392 CrossRef CAS.
- A. P. Tofteng, K. J. Jensen, L. Schäffer and T. Hoeg-Jensen, ChemBioChem, 2008, 9, 2989–2996 CrossRef CAS.
- Y. Sohma and S. B. H. Kent, J. Am. Chem. Soc., 2009, 131, 16313–16318 CrossRef CAS.
- M. A. Hossain, A. Belgi, F. Lin, S. Zhang, F. Shabanpoor, L. Chan, C. Belyea, H.-T. Truong, A. R. Blair, S. Andrikopoulos, G. W. Tregear and J. D. Wade, Bioconjugate Chem., 2009, 20, 1390–1396 CrossRef CAS.
- Y. Sohma, Q.-X. Hua, J. Whittaker, M. A. Weiss and S. B. H. Kent, Angew. Chem., Int. Ed., 2010, 49, 5489–5493 CrossRef CAS.
- F. Liu, E. Y. Luo, D. B. Flora and J. P. Mayer, Org. Lett., 2013, 15, 960–963 CrossRef CAS.
- A. Kato, K. Maki, T. Ebina, K. Kuwajima, K. Soda and Y. Kuroda, Biopolymers, 2007, 85, 12–18 CrossRef CAS.
- K. Maruyama, H. Nagasawa and A. Suzuki, Peptides, 1999, 20, 881–884 CrossRef CAS.
- K. Maruyama, K. Nagata, M. Tanaka, H. Nagasawa, A. Isogai, H. Ishizaki and A. Suzuki, J. Protein Chem., 1992, 11, 1–12 CrossRef CAS.
- G. J. S. Cooper, B. Leighton, G. D. Dimitriadis, M. Parry-Billings, J. M. Kowalchuk, K. Howland, J. B. Rothbard, A. C. Willis and K. B. M. Reid, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7763–7766 CrossRef CAS.
- P. W. R. Harris, S. H. Yang and M. A. Brimble, Tetrahedron Lett., 2011, 52, 6024–6026 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |