 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new family of bioorthogonally applicable fluorogenic labels†‡
András Hernera, Ivana Nikićb, Mihály Kállayc, Edward A. Lemkeb and Péter Kele*a
aInstitute of Chemistry, Eötvös Loránd University, H-1117, Pázmány Péter sétány 1a, Budapest, Hungary. E-mail: kelep@elte.hu; Fax: (+36)1 372 2909; Tel: (+36)1 209 0555 ext. 1610
bStructural and Computational Biology Unit, EMBL, Meyerhofstrasse 1, D-69117 Heidelberg, Germany
cDepartment of Physical Chemistry and Materials Science, Budapest University of Technology and Economics, P. O. Box 91, H-1521, Budapest, Hungary
First published on 5th March 2013
Abstract
Synthetic procedures for the construction of fluorogenic azido-labels were developed. Photophysical properties were elaborated by experimental and theoretical investigations. Of the newly synthesized fluorogenic and bioorthogonally applicable dyes two were selected on the basis of their fluorogenic performance and further subjected to in vitro and in vivo studies. Both tags exhibited excellent fluorogenic properties as in aqueous medium, the azide form of the selected dyes is virtually non-fluorescent, while their “clicked” triazole congeners showed intense fluorescence. One of these labels showed a very large Stokes shift. To the best of our knowledge this is the first reported case of mega-Stokes type of fluorogenic labels. These studies have justified that these two fluorogenic tags are remarkably suitable for bioorthogonal tagging schemes. The developed synthetic approach together with the theoretical screen of possible fluorogenic tags will enable the generation of libraries of such tags in the future.
Introduction
Small molecular modulation of biomatter is of great importance in modern chemical biological research. The development of bioorthogonal chemistry schemes has greatly facilitated the exploration of biochemical processes. Continuous efforts have been made lately for the development of new bioorthogonal reagents that offer selective, often site-specific, and high-yielding reactions with biological targets. Up to now, these efforts have resulted in the development of several bioorthogonal reagents e.g. (cyclo)alkynes, azides or tetrazines.1 Non-natural biomolecular building blocks containing one or more of these bioorthogonal functions offer easy and specific incorporation of these chemical reporters into target structures by taking advantage of gene technology or the metabolic machinery of living organisms.2 Of the biomolecular labeling techniques fluorescent modification of biological matter is by far the most popular one. Its popularity can be owed to the highly sensitive and relatively cheap detectability of the fluorescent signal with remarkable spatial and temporal resolution and their potential for multichannel imaging. Indeed, labeling of amino acids, proteins, lipids, nucleic acids, sugars, or even cells and viruses with synthetic fluorescent dyes has become an indispensible tool in bioanalytical sciences.3One advantage of applying synthetic fluorophores is the ability to employ chemical approaches to control the photophysical properties and chemical reactivity of fluorescent tags. Applying modern synthetic organic chemistry strategies enables efficient tailoring of the chemical structure to obtain probes for specific needs. Fluorescence tagging schemes often follow a two (or more)-step procedure, where excessive amounts of the fluorophore are administered to the sample to be labeled. After the incubation time the unreacted reagent should be removed. However, this task is not easy in many instances and the remaining, non-specifically bound fluorescent labels contribute to very high levels of background fluorescence therefore compromising the sensitivity of detection (low signal-to-noise ratio). Solution to this problem is offered by the use of so-called fluorogenic (switchable, “turn-on”) probes where the reagents are weakly or non-fluorescent at the wavelength of detection.4 The fact that these labels are introduced as virtually non-fluorescent derivatives eliminates the need for the separation of excessive reagents without compromising sensitivity. Many fluorogenic dyes have been developed to sense for example differences in microenvironments, however, very few fluorogenic reagents are available for covalent modification of biomatter.4,5
Bioorthogonal tagging schemes have benefited greatly from the use of the azide group. The significance of this functional group lies behind its high energetic feature together with its stability under a wide variety of reaction conditions.6 Noteworthy is its selective reactivity that enables reactions solely with particular counterparts in the presence of several other functional groups present in biological media. Its participation in conventional bioorthogonal modulation schemes e.g. copper-catalyzed or strain promoted cycloaddition with alkynes or the Staudinger ligation technique with phosphanes as reaction partners has just raised the azide group to the most popular one.7 Another less emphasized feature of the azide group is the fact that its incorporation into fluorescent scaffolds often results in dramatic drop in fluorescence intensity. These fluorogenic compounds remain non-fluorescent as long as the azide function is intact. They become intensely fluorescent, however, when the azide group is converted into a different functionality. Examples show that conversion of azides into amines or triazoles results in changes in the electronic system and the originally quenched fluorescence reinstates.4,5 Quite simply, the azide moiety offers a bioorthogonally applicable, switchable two-in-one combination under certain circumstances. Despite their obviously advantageous features azido quenched fluorescent tags are rarely reported. Design of fluorogenic labels mostly relies on empirical trial and error methods and very few examples use theoretical prediction of such features. Moreover, the so-far presented fluorogenic azido-tags lack the possibility of fine-tuning e.g. emission wavelengths, as simple fluorogenic scaffolds bear the azide moiety. Herein we introduce an approach that could be used in the design of more complex fluorogenic units with theoretically predictable photophysical features (Fig. 1).
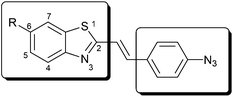 | ||
| Fig. 1 Modular setup of fluorogenic tags. | ||
Results and discussion
Design
We have chosen the 1,3-benzothiazole (Fig. 1) scaffold as a starting module for its appealing synthetic and fluorescent features. 1,3-Benzothiazoles are easily modified at their 2nd and 6th positions; therefore we have also intended to use these sites for further modifications. On the one hand we planned to shift the emission wavelength of the benzothiazole core towards the red regime by means of extending the conjugated system. On the other hand we intended to modulate the emission intensity of the fluorescent scaffold by incorporating an azide moiety into the framework. Based on former studies and theoretical investigations,5,8 we have anticipated that incorporation of a 4-azidostyryl moiety that can either be incorporated directly or in the form of its nitro-precursor will satisfy both needs. We will show that position 6 is suitable for tuning the spectral parameters by incorporating electron donating (methoxy) or withdrawing (nitro) groups. Based on our earlier work with polymethine-fluorophores we also hoped that some of these dyes will exhibit large Stokes shifts in their fluorescent form.9Synthesis
Commercially available benzothiazole scaffolds 2a and 2b were condensed with 4-nitrobenzaldehyde in molten p-TsOH in a minimum amount of refluxing toluene providing exclusively the trans-styryl products (4a–b) in moderate to good yields (Scheme 1, Method A). Reduction of the nitro-group with tin(II)chloride under acidic conditions and subsequent diazotization based azide group incorporation resulted in fluorogenic dyes 1a–b in medium yields. In the case of 2-methyl-6-nitro-1,3-benzothiazole this reaction route was not applicable for 2c bearing two nitro groups. 2c was therefore reacted with 4-azidobenzaldehyde (6) in DMSO at r.t. in the presence of NaOMe giving rise to 7c in medium yield. This intermediate was then subjected to acid catalyzed elimination effected by TFA to obtain fluorogenic dye 1c in moderate yield (Scheme 1, Method B). Method B was found to be less efficient for the synthesis of 1a as it gave a much lower overall yield (6%). This method was also not suitable for the production of 1b as it resulted in a mixture of products in low yields.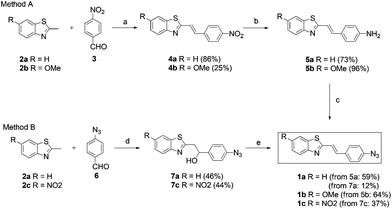 | ||
| Scheme 1 Synthesis of fluorogenic dyes. (a) 1.1 eq. p-TsOH × H2O, toluene, 110 °C, 18 h; (b) 5 eq. Sn(II)Cl2 × 2H2O, ccHCl, r.t./45 °C, 5–20 h; (c) i. NaNO2, ccHCl, 0 °C, ii. NaN3, 0 °C→r.t., 3 h; (d) 1.2 eq. NaOMe, DMSO, r.t., 6–20 h; (e) catalytic TFA, 3 Å MS, toluene, 60–70 °C, 6–16 h. | ||
The conditions of the elimination step in Method B were found to be strongly dependent on the nature of the substituents. For example, 1a and 1c were not formed when the basic medium was applied (NaOMe-DMSO at r.t.) to 7a and 7c. Products in these cases were only formed under acid catalyzed conditions. On the other hand, the electron-donating methoxy substituent facilitated the base catalyzed elimination step but resulted in an inseparable mixture of products. It is noteworthy to mention that in the case of Method B when the addition–elimination steps were carried out in a sequential manner, separation and the purification of the desired products were greatly facilitated by the intermediate primer adduct (7).
Method A can easily be generalized and extended for a large variability of substrates for modular approaches. For example, several heterocyclic derivatives with relatively acidic methyl groups (picolynium, lepidinium, (benz)oxazoles, etc.) could be applied and even libraries of fluorogenic dyes become accessible in a reasonable number of synthetic steps. Work elaborating this option is currently underway in our lab and results will be reported in due course.
To our delight, all fluorogenic dyes showed very low emission intensities as a result of the quenching effect of the azide moiety. Next, we were curious, whether it is possible to turn on the fluorescence of the fluorogenic cores as a result of chemical reaction with appropriate reaction partners. Therefore we have selected N-(prop-2-ynyl)pivalamide probe (8) as a reaction partner for 1a–c in a copper catalyzed azide–alkyne click reaction (Scheme 2).
![(a) cat. [Cu(PPh3)2]NO3, cat. TEA, CH2Cl2, r.t., 10 h.](/image/article/2013/OB/c3ob40296g/c3ob40296g-s2.gif) | ||
| Scheme 2 (a) cat. [Cu(PPh3)2]NO3, cat. TEA, CH2Cl2, r.t., 10 h. | ||
Compound 8 was selected on the basis of not having any aromatic moieties that could affect fluorescence of the products by any means. The click reactions were performed in a dichloromethane solvent with a drop of triethylamine and a catalytic amount of Cu(I)-catalyst10 at room temperature. Products (9a–c) were then collected by filtration and analyzed for their fluorescent properties.
Spectroscopic measurements
We have studied the fluorescent properties of the click-products, 9a–c, in comparison to their parent azides, 1a–c, in DMSO, CH2Cl2 and water (Table 1). In DMSO very low intensity of fluorescence was observed for all azides. Triazoles on the other hand showed orders of magnitudes stronger fluorescence, with 9b showing the best performance. In all cases hydrogen bond formation with water decreased the fluorescence intensities (ESI‡).| Fluorophore | 1a | 9a | 1b | 9b | 1c | 9c |
|---|---|---|---|---|---|---|
| a In DMSO.b Measured at λmax (abs).c In CH2Cl2.d Relative to quinine sulfate in 0.5 M H2SO4.11e In water.f Not detectable, in the background. | ||||||
| λmax (abs)/nma | 360 | 368 | 371 | 368 | 388 | 355 |
| ε/104 M−1 cm−1a,b | 4.00 | 5.30 | 3.60 | 4.22 | 3.21 | 2.27 |
| λmax (exc)/nma | 362 | 346 | 357 | 361 | 392 | 385 |
| λmax (em)/nma | 455 | 417 | 446 | 445 | 586 | 530 |
| Stokes-shift/nma | 93 | 71 | 89 | 84 | 194 | 145 |
ΦF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,d a,d | 0.001 | 0.045 | 0.002 | 0.439 | 0.047 | 0.068 |
| ΦF (triazole)/ΦF (azide)a | 45 | 220 | 1.5 | |||
| λmax (exc)/nmc | 350 | 343 | 352 | 352 | 390 | 381 |
| λmax (em)/nmc | 410 | 414 | 427 | 437 | 522 | 489 |
| λmax (exc)/nme | n.d.f | 335 | 357 | 355 | n.d.f | 350 |
| λmax (em)/nme | n.d.f | 421 | 455 | 453 | n.d.f | 540 |
| Stokes-shift/nme | n.d.f | 86 | 98 | 98 | n.d.f | 190 |
In fact, in the case of 1b and 1c practically no fluorescence could be detected in aqueous solutions therefore the fluorescence of the corresponding triazoles (9b, 9c) implies remarkable increase in the fluorescence signal (Fig. 2 and 3). These results suggest that 1b and 1c are particularly suitable to be applied in aqueous solutions. Excitation wavelengths were found to be very similar for all fluorogenic dyes but emission maxima were shifted towards the red regime in the order of 9a < 9b ≪ 9c. Noteworthy is to mention the spectral properties of 9c. It exhibits a large Stokes-shift (called “mega-Stokes”10) of 190 nm in water providing us with the possibility of more sensitive detection by eliminating the interfering scattered light and self-absorption. Moreover, to the best of our knowledge, such a combination of fluorogenic and mega-Stokes properties is unprecedented in the literature. Dyes of this kind are exceptionally useful in Förster resonance energy transfer (FRET) applications as the dye emission does not interfere with the spectral bands of the second fluorophore, which is a common problem in FRET technology.
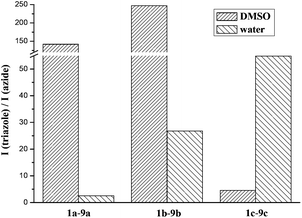 | ||
| Fig. 2 Enhancement of fluorescence intensity at λmax (em) of the triazoles in DMSO and water solvent. The concentration of azides and the corresponding triazoles were the same. | ||
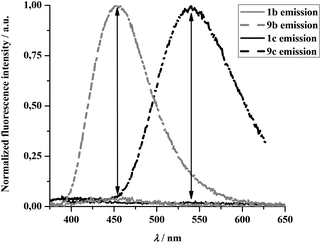 | ||
| Fig. 3 Normalized emission spectra of 1b–9b and 1c–9c. | ||
Regarding the emission wavelengths, we observed a small but persistent blue shift in all cases for the triazoles compared to their corresponding azides. This is most likely in connection with the change in molecular dipole moments associated with the evolution of the triazole motif.
Quantum chemical calculations
To interpret the experimentally observed behavior of the fluorophores we performed quantum chemical calculations using the Gaussian 09 package.12 To accelerate the calculations we employed propyne as a model system instead of the click reagent N-(prop-2-ynyl)pivalamide used in the experiments. The geometries for all the compounds were optimized at the density functional theory (DFT) level choosing the PBE0 functional13,14 and the 6-311++G** basis set. All the DFT calculations were performed with dichloromethane as a solvent invoking the polarized continuum model.15 Though the experiments were carried out in different solvents, we chose dichloromethane since the polarity of this solvent and the probability of the formation of specific solute–solvent interactions are low enabling the simplified treatment of the solvent effects. Excited-state properties were calculated using the time-dependent DFT method16 with the same functional, basis set, and solvation model. To simulate the experimental absorption (excitation) spectra vertical excitation energies as well as oscillator strengths were calculated at the optimized ground-state geometries. For the calculation of fluorescence wavelengths and intensities the geometries of the first excited singlet state were optimized and vertical excitation energies and oscillator strengths were calculated at the minima of the excited-state surfaces.We note that several isomers (conformers) exist for the investigated molecules. First, stereoisomerism about the central double bond can be observed. Since NMR experiments revealed that only the trans isomers were formed in the synthesis, we considered the latter in the calculations. Second, because of the restricted rotation about other bonds further cis and trans isomers can be identified. We calculated the energies for all the possible isomers and only retained the most stable one for each molecule for the excited-state calculations. As we tested for the H-substituted compound the excited-state properties are largely independent of which isomer is considered, thus the above restriction does not influence our conclusions.
The optimized ground-state geometries for the most stable isomers are presented (see ESI‡). The geometries are planar for the azide derivatives, while for the triazoles the triazole ring forms an angle of about 30 degrees with the plane of the phenyl ring.
We found that the rotational barrier of the rotation around the bond connecting the azide/triazole moiety to the phenyl ring is low, consequently the rotation is allowed at room temperature. Since the molecules are expected to have several low-lying excited states, and upon the rotation around a bond the excited-state potential energy surfaces (PESs) may cross each other and an internal conversion may take place, we mapped the excited-state PESs in more detail and started the geometry optimization from several initial geometries differing in the torsion angle between the azide/triazole and phenyl groups.
Provided that the excited-state geometry optimization is started from a geometry close to the optimized ground-state structure, the relaxation of the geometry is moderate for all molecules, and the optimized S1-state geometries are close to the ground-state ones. Remarkable conformational change takes place only at the triazole moiety of the triazole derivatives, which is perpendicular to the phenyl ring in the S1 state of the unsubstituted molecule, while the excited-state conformation is planar for the other compounds. The calculated transition wavelengths and oscillator strengths are collected in Table 2.
| Substituent | Absorption | Emission | ||
|---|---|---|---|---|
| λ/nm | Osc. strength | λ/nm | Osc. strength | |
| a Numbers in parentheses show the experimental results in CH2Cl2. | ||||
| Azides | ||||
| –H | 393 (350) | 1.76 | 451 (410) | 1.78 |
| –OMe | 407 (352) | 1.78 | 467 (427) | 1.83 |
| –NO2 | 445 (390) | 1.51 | 512 (522) | 1.60 |
| Triazoles | ||||
| –H | 388 (343) | 1.78 | 442 (414) | 1.78 |
| –OMe | 402 (352) | 1.71 | 458 (437) | 1.81 |
| –NO2 | 422 (381) | 1.58 | 497 (489) | 1.66 |
The agreement of the experimental and theoretical absorption and emission wavelengths is satisfactory and justifies the selection of the applied theoretical model. However, the oscillator strengths for the emissions are practically identical for the azide and the triazole derivatives suggesting that for the azides the fluorescence from the minima found in the optimizations is not the dominant decay channel.
We found that upon the rotation of the azide group the first excited state interchanges with a non-emissive state. In the case the excited-state geometry optimization is started from the distorted structure, we obtain a minimum on the potential energy surface of the latter state, which is very close to the ground-state potential surface (ca. 0.5 eV above the ground state energy) and the transition probability of the excited state → ground state transition is zero. The corresponding geometries are displayed (see ESI‡). Consequently, these results suggest the existence of an alternative, radiationless decay channel explaining the low fluorescence intensities observed for the azide derivatives. Due to the conformation change of the azide group, a state-switch takes place which opens up the possibility of the internal conversion of the two lowest excited states followed by another internal conversion process to the ground state.
In vitro peptide labeling
Following model reactions and photophysical characterization we were curious to test the performance of our new fluorogenic labels in action. For this we have chosen a cyclooctyne (CyO) bearing peptide sequence (CyO-Gly-Pro-Leu-Gly-Val-Arg-DL-Pra-NH2; DL-Pra = racemic propargyl-glycine) from our previous studies.17 The cyclooctyne moiety enables rapid in situ copper-free tagging of the peptide e.g. with fluorogenic azides under physiological conditions (pH = 7.20 phosphate buffer). Due to their performance in the model reactions we have selected 1b and 1c for in vitro labeling reactions. The tagging reaction was monitored in time by following fluorescence intensity changes at 455 and 540 nm, respectively, while exciting at 355 nm. In both cases rapid increase in fluorescence intensity could be observed (Fig. 4).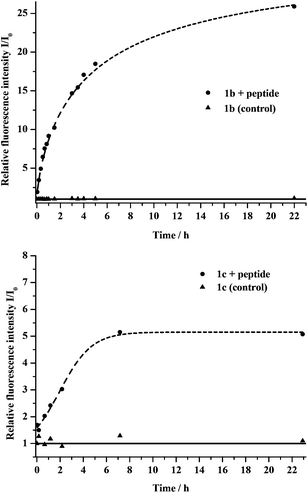 | ||
| Fig. 4 Top: coupling reaction of 1b and CyO-pep. The dots show the datapoints of the labelled peptide, the triangles belong to the control sample (without peptide). Bottom: coupling reaction of 1c and CyO-pep. The dots show the datapoints of the labelled peptide, the triangles belong to the control sample (without peptide). | ||
Reaction with 1b was somewhat faster than in the case of 1c. The final fluorescence intensity was much higher for the former fluorogenic tag as well. Control reactions of both fluorogenic tags in the absence of the peptide showed no change in fluorescence intensities, implying that there are no e.g. nitrene involved decomposition processes that would result in fluorescent species. The fact that the non-fluorescent azides of 1b and 1c show very low fluorescence intensities means that the evolving signal can be detected with excellent s/n ratios, a feature greatly desired in biological tagging schemes.
In vivo protein labeling
We were also curious to explore the performance of selected fluorogenic azido-tags under in vivo conditions where the dyes have to stand more challenging conditions. In our previous work we have genetically encoded cyclooctyne (SCO) bearing unnatural amino acid 10 using a rationally engineered tRNApyl/PylRSAF pair.2a,b An Amber (TAG) mutated Green Fluorescent Protein (GFP) reporter construct was expressed in E. coli cells together with a pEvol PylRSAF plasmid in the presence of 10, leading to site-specific incorporation of SCO into GFP. Living cells were treated with 1b, 1c and lysed subsequently. The lysates were loaded onto an SDS PAGE gel for whole cell lysate analysis. The gel was analysed for fluorescence by exciting the samples in the UV-range (365 nm) and detecting the emission intensities. Afterwards the gels were stained with Coomassie (Fig. 5).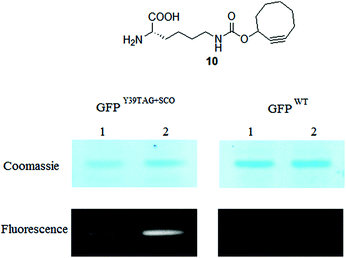 | ||
| Fig. 5 Fluorescent and corresponding Coomassie stained images of GFPY39TAG+SCO and GFPwt labeling reactions with compounds 1b, 1c (lanes 1 and 2, respectively) inside live E. coli. | ||
Analysis of the gel under UV excitation revealed that both fluorogenic labels efficiently labelled the SCO-tagged GFP. In the control experiment where wild type (WT) GFP was expressed in the cells no labeling of the protein took place. Furthermore, it was also obvious that 1b and 1c can stand the harsh conditions applied during lysis and no fluorescent decomposition products were formed (for full details and full gel images see ESI‡). These results served as evidence that the two new fluorogenic labels can be used in bioorthogonal fluorescent tagging schemes resulting in background fluorescence free images.
Experimental
General
Unless otherwise indicated, all starting materials were obtained from commercial suppliers (Sigma-Aldrich, Fluka, Merck, Alfa Aesar, Reanal, Molar Chemicals, Romil) and used without further purification. Dichloromethane stabilized with ethanol was used, unless otherwise mentioned. SCO was purchased from Sichem (Bremen, Germany). Tamra-azide was ordered from www.baseclick.eu. Analytical thin-layer chromatography (TLC) was performed on silica gel 60 F254 precoated aluminium TLC plates from Merck. Column chromatography was carried out with flash silica gel (0.040–0.063 mm) from Molar Chemicals. Visualisation of TLC samples was performed with a 254/365 nm UV lamp or using aqueous KMnO4 solution (1.5 g KMnO4, 10 g K2CO3 and 1.25 ml 10 m% NaOH in 200 ml of water). The aromatic azides (1a–c, 6, 7a, 7c) could be visualised as a yellow spot after 0.5–1 minute irradiation with a 254 nm UV lamp on a TLC plate. NMR spectra were recorded on a Bruker DRX-250 or a Varian VNMRS 600 MHz spectrometer. Chemical shifts (δ) are given in parts per million (ppm) using solvent signals as the reference. Coupling constants (J) are reported in Hertz (Hz). Splitting patterns are designated as s (singlet), d (doublet), t (triplet), qr (quadruplet), qv (quintuplet), m (multiplet), dd (doublet of a doublet), td (triplet doublet), dt (doublet triplet), br s (broad singlet). The exact masses were determined with an Agilent 6230 time-of-flight mass spectrometer. Samples were introduced by the Agilent 1260 Infinity LC system and the mass spectrometer was operated in conjunction with a JetStream source in positive/negative ion mode. All melting points were measured on a Bibby Scientific SMP10 Melting Point Apparatus and are uncorrected. IR spectra were obtained on a Bruker IFS55 spectrometer on a single-reflection diamond ATR unit. The fluorescence measurements were carried out on a Varian Cary Eclipse spectrofluorimeter, the UV-VIS measurements on a Jasco V-660 spectrophotometer.All reactions involving aromatic azides were carried out in the dark. CAUTION! Working with azide salts in the presence of halogenated solvents could lead to the formation of hazardous diazomethane.
After 1 month we have observed a slow decomposition of 1a–c fluorogenic dyes even storing it under an inert atmosphere at −20 °C in the dark. Although the amount of the decomposition product is not significant it is advisable to purify it before usage by passing through a short plug of silica using CH2Cl2 as an eluent.
Synthesis of dyes
:1); mp >218 °C (decompose); IR (neat): νmax/cm−1 1517 and 1340 (NO2); δH(600 MHz, DMSO-d6) 8.28 (2 H, d, J = 8.5 Hz, Ar-H), 8.14 (1 H, d, J = 7.9 Hz, Ar-H), 8.07 (2 H, d, J = 8.5 Hz, Ar-H), 8.03 (1 H, d, J = 7.9 Hz, Ar-H), 7.88 (1 H, d, J = 16.1 Hz, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.82 (1 H, d, J = 16.1 Hz, CHCH), 7.55 (1 H, t, J = 7.6 Hz, Ar-H), 7.48 (1 H, t, J = 7.2 Hz, Ar-H); δC(150 MHz, DMSO-d6) 165.5, 153.3, 147.2, 141.8, 134.7, 134.3, 128.6, 126.7, 125.9, 125.8, 123.9, 122.8, 122.3; HRMS (ESI) [M + H]+ calcd for [C15H10N2O2S + H]+: 283.0536, found: 283.0542.:1); mp >178 °C (decompose); IR (neat) νmax/cm−1 3316 br and 1598 (NH2); δH(250 MHz, CDCl3) 7.96 (1 H, d, J = 8.1 Hz, Ar-H), 7.83 (1 H, d, J = 7.9 Hz, Ar-H), 7.44–7.38 (4 H, m, 3 × Ar-H, CHCH), 7.33 (1 H, t, J = 7.5 Hz, Ar-H), 7.21 (1 H, d, J = 16.3 Hz, CHCH), 6.68 (2 H, d, J = 8.0 Hz, Ar-H), 3.92 (1.6 H, brs, NH2); δC(62.5 MHz, CDCl3) 167.9, 153.9, 147.9, 138.0, 134.1, 129.0, 126.1, 125.8, 124.9, 122.5, 121.4, 118.2, 115.0; HRMS (ESI) [M + H]+ calcd for [C15H12N2S + H]+: 253.0794, found: 253.0795.
CH), 7.82 (1 H, d, J = 16.1 Hz, CHCH), 7.55 (1 H, t, J = 7.6 Hz, Ar-H), 7.48 (1 H, t, J = 7.2 Hz, Ar-H); δC(150 MHz, DMSO-d6) 165.5, 153.3, 147.2, 141.8, 134.7, 134.3, 128.6, 126.7, 125.9, 125.8, 123.9, 122.8, 122.3; HRMS (ESI) [M + H]+ calcd for [C15H10N2O2S + H]+: 283.0536, found: 283.0542.:1); mp >178 °C (decompose); IR (neat) νmax/cm−1 3316 br and 1598 (NH2); δH(250 MHz, CDCl3) 7.96 (1 H, d, J = 8.1 Hz, Ar-H), 7.83 (1 H, d, J = 7.9 Hz, Ar-H), 7.44–7.38 (4 H, m, 3 × Ar-H, CHCH), 7.33 (1 H, t, J = 7.5 Hz, Ar-H), 7.21 (1 H, d, J = 16.3 Hz, CHCH), 6.68 (2 H, d, J = 8.0 Hz, Ar-H), 3.92 (1.6 H, brs, NH2); δC(62.5 MHz, CDCl3) 167.9, 153.9, 147.9, 138.0, 134.1, 129.0, 126.1, 125.8, 124.9, 122.5, 121.4, 118.2, 115.0; HRMS (ESI) [M + H]+ calcd for [C15H12N2S + H]+: 253.0794, found: 253.0795.Method A (from 5a). (E)-2-(4-Aminostyryl)-1,3-benzothiazole (5a) (100 mg, 0.3963 mmol, 1 eq.) was dissolved in a mixture of 2 ml cc. HCl(aq.), 2 ml water and 1 ml MeOH. To this solution was added an NaNO2 solution (33 mg, 0.4783 mmol, 1.2 eq. in 200 μl water, +100 μl washing water) at 0 °C, dropwise. Covering the reaction flask with aluminium foil is necessary from this point! The temperature was kept at 0 °C, and after 10 minutes an NaN3 solution (31 mg, 0.4768 mmol, 1.2 eq. in 200 μl water, +100 μl washing water) was added dropwise, slowly. After the addition, the mixture was allowed to warm to r.t. and stirred for a further 3 h. Next, the reaction mixture was poured onto ice-water (20–30 ml), extracted with CH2Cl2 (4 × 15 ml) and washed with brine (1 × 15 ml). After drying over MgSO4 the drying agent was removed by filtration and the filtrate was concentrated in vacuo. The product was purified by column chromatography (SiO2, hexanes–EtOAc = 10
:1 → 7:1) to result in a pale yellow solid that was further washed with hexanes to obtain a 65 mg (59%) product. Rf = 0.62 (hexanes–EtOAc 5:1); mp >170 °C (decompose); IR (neat) νmax/cm−1 2115 (N3); δH(600 MHz, CDCl3) 7.99 (1 H, d, J = 7.7 Hz, Ar-H), 7.86 (1 H, d, J = 8.5 Hz, Ar-H), 7.56 (2 H, d, J = 8.5 Hz, Ar-H), 7.48 (1 H, d, J = 16.3 Hz, CHCH), 7.47 (1 H, t, J = 8.5 Hz, Ar-H), 7.37 (1 H, t, J = 7.0 Hz, Ar-H), 7.34 (1 H, d, J = 16.3 Hz, CHCH), 7.06 (2 H, d, J = 8.5 Hz, Ar-H); δC(62.5 MHz, CDCl3) 166.7, 153.9, 141.0, 136.3, 134.4, 132.3, 128.8, 126.3, 125.3, 123.0, 121.8, 121.5, 119.5; HRMS (ESI) [M + H]+ calcd for [C15H10N4S + H]+: 279.0699, found: 279.0695.Method B (from 7a). 1-(4-Azidophenyl)-2-(1,3-benzothiazol-2-yl)ethanol (7a) (263 mg, 0.8875 mmol) was stirred in dry toluene (7 ml) in the presence of 3 Å molecular sieves. After wrapping the flask with an aluminium foil the reaction mixture was heated to 70 °C. During warming the reactant was dissolved completely. Trifluoroacetic acid was added (2 drops) and the reaction mixture was stirred for 16 h. After cooling to r.t. it was diluted with CH2Cl2 (8 ml) and the organic phase was washed with water (2 ml) and dried over MgSO4. The mixture was then filtered and the solvent was evaporated. The residue was purified by column chromatography (SiO2, hexanes–EtOAc = 10
:1 → 5:1). The pale yellow residue (44 mg) was transferred onto a filter funnel and washed with hot hexanes (3–5 ml). A pale yellow powder was obtained (30 mg, 12%).:1); mp >256 °C (decompose); IR (neat) νmax/cm−1 1506 and 1334 (NO2), 1218 (Caryl–O–Calkyl); δH(600 MHz, DMSO-d6) 8.27 (2 H, d, J = 8.9 Hz, Ar-H), 8.04 (2 H, d, J = 8.5 Hz, Ar-H), 7.91 (1 H, d, J = 8.9 Hz, Ar-H), 7.82 (1 H, d, J = 16.2 Hz, CHCH), 7.72 (1 H, d, J = 2.7 Hz, Ar-H), 7.70 (1 H, d, J = 16.2 Hz, CHCH), 7.14 (1 H, dd, J1 = 8.9 Hz, J2 = 2.5 Hz, Ar-H), 3.86 (3 H, s, CH3); δC(150 MHz, DMSO-d6) 165.8, 157.8, 147.1, 142.0, 136.0, 133.5, 128.4, 126.1, 123.4, 124.0, 119.6, 116.2, 104.7, 55.7; HRMS (ESI) [M + H]+ calcd for [C16H12N2O3S + H]+: 313.0641, found: 313.0643.:0 → 10:1) to obtain the product as a pale yellow solid (89 mg, 96%). Rf = 0.07 (hexanes–EtOAc 5:1); mp >183 °C (decompose); IR (neat) νmax/cm−1 3316 br and 1622 (NH2), 1218 (Caryl–O–Calkyl); δH(250 MHz, CDCl3) 7.83 (1 H, d, J = 8.8 Hz, Ar-H), 7.38 (2 H, d, J = 8.4 Hz, Ar-H), 7.31–7.25 (2 H, m, CHCH and Ar-H), 7.17 (1 H, d, J = 16.1 Hz, CHCH), 7.04 (1 H, dd, J1 = 9.0 Hz, J2 = 2.5 Hz, Ar-H), 6.68 (2 H, d, J = 8.5 Hz, Ar-H), 3.89 (1.6 H, brs, NH2), 3.88 (3 H, s, CH3); δC(62.5 MHz, CDCl3) 165.5, 157.6, 148.5, 147.7, 137.0, 135.5, 128.8, 126.0, 123.0, 118.4, 115.2, 115.1, 104.2, 55.8; HRMS (ESI) [M + H]+ calcd for [C16H14N2OS + H]+: 283.0900, found: 283.0906.Method A (from 5b). (E)-6-Methoxy-2-(4-aminostyryl)-1,3-benzothiazole (5b) (77 mg, 0.2727 mmol, 1 eq.) was dissolved in a mixture of 1.5 ml cc. HCl(aq.), 1.5 ml water and 2 ml MeOH. To this solution was added NaNO2 solution (23 mg, 0.3333 mmol, 1.2 eq. in 300 μl water, +100 μl washing water) at 0 °C, dropwise. Covering the reaction flask with aluminium foil is necessary from this point! The temperature was kept at 0 °C, and after 10 minutes an NaN3 solution (21 mg, 0.3230 mmol, 1.2 eq. in 300 μl water, +100 μl washing water) was added dropwise. After the addition the mixture was allowed to warm to r.t. and stirred for a further 3 h. Next, the reaction mixture was poured onto ice-water (15–20 ml), extracted with CH2Cl2 (3 × 10 ml) and washed with brine (1 × 10 ml). The organic phase was dried over MgSO4 and filtered. The solvent was evaporated on a rotary evaporator and the residue purified by column chromatography (SiO2, CH2Cl2) to give a pale yellow solid that was transferred onto a filter funnel and washed with a little amount of hexanes (3–5 ml), and dried (54 mg, 64%). Rf = 0.50 (hexanes–EtOAc 5
:1); mp >145 °C (melt and decompose); IR (neat) νmax/cm−1 2114 (N3), 1266 (Caryl–O–Calkyl); δH(250 MHz, CDCl3) 7.86 (1 H, d, J = 8.2 Hz, Ar-H), 7.54 (2 H, d, J = 8.2 Hz, Ar-H), 7.44–7.27 (3 H, m, CHCH and 2 × Ar-H), 7.12–6.98 (3 H, m, Ar-H and CHCH), 3.89 (3 H, s, CH3); δC(62.5 MHz, CDCl3) 164.3, 158.0, 148.4, 140.7, 135.8, 135.3, 132.5, 128.6, 123.5, 121.9, 119.5, 115.6, 104.1, 55.8; HRMS (ESI) [M + H]+ calcd for [C16H12N4OS + H]+: 309.0805, found: 309.0807.:1 → 3:1). An orange solid was obtained (273 mg, 46%). The product was sufficiently pure and was used directly in the next step without further purification. Rf = 0.33 (hexanes–EtOAc 3:1); δH(250 MHz, CDCl3) 8.01 (1 H, d, J = 8.2 Hz, Ar-H), 7.86 (1 H, d, J = 8.8 Hz, Ar-H), 7.45 (2 H, d, J = 8.8 Hz, Ar-H), 7.49 (1 H, t, J = 7.6 Hz, Ar-H), 7.39 (1 H, t, J = 7.5 Hz, Ar-H), 7.03 (2 H, d, J = 8.8 Hz, Ar-H), 5.29 (1 H, t, J = 6.0 Hz, CH-OH), 4.44 (0.9 H, br s, OH), 3.43 (2 H, d, J = 6.3 Hz, CH2); δC(62.5 MHz, CDCl3) 168.5, 152.7, 139.5, 134.6, 128.8, 127.3, 126.2, 125.2, 122.7, 121.5, 119.1, 72.1, 42.8.Method B (from 7c). 1-(4-Azidophenyl)-2-(6-nitro-1,3-benzothiazol-2-yl)ethanol (7c) (231 mg, 0.6767 mmol) was stirred in dry toluene (5 ml) in the presence of 3 Å molecular sieves. After covering with aluminium foil the reaction vessel was warmed to 70 °C. Meanwhile, the reactant was dissolved completely. Trifluoroacetic acid was added (3 drops) and the reaction mixture was stirred for 7 h. During the reaction the product precipitated as an orange powder. After cooling to 3–5 °C (in fridge) the precipitate was filtered and washed with hot hexanes (2–3 ml). A yellow powder was obtained (82 mg, 37%). Rf = 0.48 (hexanes–EtOAc 3
:1); mp >178 °C (decompose); IR (neat) νmax/cm−1 2133 (N3), 1510 and 1340 (NO2); δH(250 MHz, CDCl3) 8.79 (1 H, d, J = 1.9 Hz, Ar-H), 8.35 (1 H, dd, J1 = 8.8 Hz, J2 = 2.5 Hz, Ar-H), 8.05 (1 H, d, J = 8.8 Hz, Ar-H), 7.61 (1 H, d, J = 15.8 Hz, CHCH), 7.60 (2 H, d, J = 9.5 Hz, Ar-H), 7.35 (1 H, d, J = 16.4 Hz, CHCH), 7.09 (2 H, d, J = 8.8 Hz, Ar-H); δC(62.5 MHz, CDCl3) 172.3, 157.7, 144.9, 141.9, 139.1, 134.7, 131.5, 129.3, 122.9, 122.0, 120.7, 119.7, 118.1; HRMS (ESI) [M + H]+ calcd for [C15H9N5O2S + H]+: 324.0555; found: 324.0557.:1 → 2:1). An orange-yellow viscous compound was obtained (231 mg, 44%). The product was sufficiently pure and was used directly in the next step without further purification. Rf = 0.15 (hexanes–EtOAc 3:1); δH(250 MHz, CDCl3) 8.75 (1 H, d, J = 2.2 Hz, Ar-H), 8.32 (1 H, dd, J1 = 8.9 Hz, J2 = 2.3 Hz, Ar-H), 8.04 (1 H, d, J = 9.0 Hz, Ar-H), 7.40 (2 H, d, J = 8.4 Hz, Ar-H), 7.00 (2 H, d, J = 8.5 Hz, Ar-H), 5.28 (1 H, t, J = 6.2 Hz, CH-OH), 4.01 (0.7 H, br s, OH), 3.49 (2 H, d, J = 6.0 Hz, CH2); δC(62.5 MHz, CDCl3) 174.7 and 173.3, 156.9 and 156.2, 144.8 and 144.6, 139.6 and 139.1, 135.8 and 135.2, 127.15 and 127.13 (CH), 122.7 and 122.4 (CH), 121.5 and 121.4 (CH), 119.1 (CH), 118.0 and 117.8 (CH), 71.8 (CH), 43.5 ppm (CH2). Duplication of peaks is most probably due to the chelate effect between OH and N. The orders of the carbons were determined by DEPT135 measurement.Synthesis of click conjugates
:1); mp >258 °C (decompose); IR (neat) νmax/cm−1 3311 br (aromatic ring) 1627 and 1541 (CONHR); δH(600 MHz, DMSO-d6) 8.59 (1 H, s, triazole-H), 8.12 (1 H, d, J = 8.2 Hz, Ar-H), 8.07 (1 H, t, J = 5.6 Hz, NH), 8.03–7.98 (5 H, m, 5 × Ar-H), 7.76 (1 H, d, J = 16.4 Hz, CHCH), 7.73 (1 H, d, J = 16.4 Hz, CHCH), 7.54 (1 H, t, J = 8.0 Hz, Ar-H), 7.46 (1 H, t, J = 7.6 Hz, Ar-H), 4.39 (2 H, d, J = 5.9 Hz, CH2), 1.13 (9 H, s, CH3); δC(150 MHz, DMSO-d6) 177.4, 166.1, 153.4, 146.8, 136.8, 135.9, 135.2, 134.1, 129.0, 126.5, 125.5, 122.7, 122.5, 122.2, 120.5, 120.0, 37.9, 34.5, 27.3; HRMS (ESI) [M + H]+ calcd for [C23H23N5OS + H]+: 418.1696, found: 418.1701.:1); mp >214 °C (decompose); IR (neat) νmax/cm−1 3331 br (aromatic ring), 1624 and 1536 (CONHR).1224 (Caryl–O–Calkyl); δH(600 MHz, DMSO-d6) 8.58 (1 H, s, triazole-H), 8.07 (1 H, t, J = 5.5 Hz, NH), 7.98 (4 H, s, Ar-H), 7.88 (1 H, d, J = 9.0 Hz, Ar-H), 7.71–7.65 (2 H, m, CHCH and Ar-H), 7.64 (1 H, d, J = 16.2 Hz, CHCH), 7.12 (1 H, dd, J1 = 9.0 Hz, J2 = 1.7 Hz, Ar-H), 4.39 (2 H, d, J = 5.5 Hz, CH2), 3.86 (3 H, s, O–CH3), 1.13 (9 H, s, CH3); δC(150 MHz, DMSO-d6) 177.4, 163.5, 157.6, 147.8, 146.7, 136.6, 135.6, 135.4, 134.7, 128.8, 123.1, 122.9, 120.5, 120.0, 115.8, 104.7, 55.7, 37.9, 34.5, 27.3; HRMS (ESI) [M + H]+ calcd for [C24H25N5O2S + H]+: 448.1802, found: 448.1802.:1); mp >240 °C (decompose); IR (neat) νmax/cm−1 3293 (aromatic ring), 1516 and 1336 (NO2), 1628 (CONHR); δH(600 MHz, DMSO-d6) 9.20 (1 H, d, J = 2.3 Hz, Ar-H), 8.60 (1 H, s, triazole-H), 8.34 (1 H, dd, J1 = 8.8 Hz, J2 = 2.3 Hz, Ar-H), 8.16 (1 H, d, J = 9.4 Hz, Ar-H), 8.08 (1 H, t, J = 5.6 Hz, NH), 8.05 (2 H, d, J = 8.8 Hz, Ar-H), 8.02 (2 H, d, J = 8.8 Hz, Ar-H), 7.92 (1 H, d, J = 15.8 Hz, CHCH), 7.82 (1 H, d, J = 16.4 Hz, CHCH), 4.39 (2 H, d, J = 5.9 Hz, CH2), 1.13 (9 H, s, CH3); δC(150 MHz, DMSO-d6) 177.4, 172.5, 157.2, 146.8, 144.2, 138.2, 137.2, 134.8, 129.5, 122.7, 122.2, 121.9, 120.6, 120.0, 119.3, 37.9, 34.4, 27.3; HRMS (ESI) [M + H]+ calcd for [C23H22N6O3S + H]+: 463.1547, found: 463.1553.Model reactions with CyO-peptide
The reaction mixture contained 1 × 10−4 M azido-dye (1b, 1c) and 2 × 10−4 M CyO-peptide in a 2 ml phosphate buffered solution. For fluorescence measurements aliquots were drawn from the reaction mixture: a 25 μl reaction mixture and a 2475 μl phosphate buffer were mixed (final concentration 1 × 10−6 M for the starting amount of dye) and emission was monitored at emission maxima using 355 nm excitation.The control experiment contained 650 μl 3.24 × 10−4 M 1b (DMSO solution) and 1350 μl phosphate buffer (pH 7.20) and incubated at the same temperature.
The control experiment contained 497 μl 4.021 × 10−4 M 1b (DMSO solution) and 1503 μl phosphate buffer (pH 7.20) and incubated at the same temperature.
Live cell labeling and lysate analysis of cyclooctyne expressing E. coli
Top 10 E. coli cells containing pEvol PylRSAF and pBAD plasmids for expressing GFPY39TAG or GFPWT were grown in the presence of ampicillin and chloramphenicol in a Terrific broth medium. GFPY39TAG was grown in the presence (GFPY39TAG+SCO) and absence of SCO (GFPY39TAG−SCO) and GFPWT was grown also in the presence of SCO.2a,b Cells were induced with arabinose at OD 0.4 and allowed to grow overnight. After harvesting, the cells were resuspended in 1× phosphate buffered saline (PBS, pH 7.4) and the OD was adjusted to ∼1.7. A total volume of 1.5 ml of this suspension was used in further steps. The cells were centrifuged and washed 3 times with PBS. Pellets were afterwards resuspended in 1 ml PBS and incubated with 500 μM compounds 1b, 1c (both 50 mM stock in DMSO) or 50 μM of TAMRA-azide (40 mM stock in DMSO) at 37 °C in the dark shaking for 4.5 h. After labeling, the cells were centrifuged and pellets were washed rigorously with PBS containing 5% DMSO. Subsequently samples were lysed and loaded onto an SDS PAGE gel for whole cell lysate analysis. The gel was analysed for fluorescence by exciting the samples in the UV-range (365 nm) and detecting the emission with an ethidium bromide filter setting on a commercially available gel documentation system (Alpha Innotech, CA). Afterwards the gel was stained with Coomassie.Conclusion
In conclusion, we have developed synthetic procedures for the construction of fluorogenic tags. In addition, by using theoretical chemical calculations we are able to predict and screen for potential fluorogenic azides that are worth to be synthesized. We have selected and synthesized three new fluorogenic and bioorthogonally applicable dyes, 1a–c. In aqueous media the azides 1a–c are virtually non-fluorescent. Although “clicked” triazole derivative 9a exhibits moderately increased emission intensity, 9b and 9c show intense fluorescence. Photophysical studies have justified 1b and 1c as remarkably suitable for bioorthogonal tagging schemes. The performance of these two fluorogenic tags was tested in the fluorescence tagging of a peptide sequence by means of a strain-promoted azide–alkyne cycloaddition reaction. Both tags exhibited excellent performance. Furthermore, the applicability of these fluorogenic tags was further demonstrated in in vivo cell labeling experiments. Analysis of the gel-electrophoresis chromatograms of the cell lysates revealed that both tags can be used in background fluorescence free bioorthogonal tagging schemes.The synthetic approach and the theoretical screen of possible fluorogenic tags will allow the generation of libraries of such tags. Fast screening of these libraries greatly facilitates the selection of fluorogenic tags for certain needs (e.g. excitation and emission wavelengths). Work aiming at the synthesis of such designed libraries is underway in our laboratory and results will be reported in due course. Further aims are to fine tune the photophysical properties of such tags and generate fluorogenic azides that are excitable in the red-far-red, near infrared region as these regimes are more suitable for biological tagging and detection schemes.
Acknowledgements
P. K. acknowledges the support of the Hungarian Academy of Sciences for the János Bolyai Fellowship (BO/00148/11/7). Financial support of the Hungarian Scientific Research Fund (OTKA), Grant No. K100134 is also greatly appreciated. M. K. acknowledges the financial support by the European Research Council (ERC) under the European Community's Seventh Framework Programme (FP7/2007–2013), ERC Grant Agreement No. 200639. I. N. acknowledges support by an EMBO Long-Term Fellowship and E. A. L. funding of the DFG Emmy Noether and SPP1623 program.Notes and references
- (a) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC; (b) C. R. Becer, R. Hoogenboom and U. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS; (c) K. V. Reyna and Q. Lin, Chem. Commun., 2010, 46, 1589 RSC; (d) K. E. Beatty, Mol. BioSyst., 2011, 7, 2360 RSC; (e) M. F. Debets, S. S. van Berkel, J. Dommerholt, A. T. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805 CrossRef CAS; (f) B. R. Varga, M. Kállay, K. Hegyi, Sz. Béni and P. Kele, Chem.–Eur. J., 2012, 18, 822 CrossRef CAS.
- (a) T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878 CrossRef CAS; (b) T. Plass, S. Milles, C. Koehler, J. Szymański, R. Mueller, M. Wießler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166 CrossRef CAS; (c) S. J. Luchansky and C. R. Bertozzi, ChemBioChem, 2004, 5, 1706 CrossRef CAS; (d) N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644 CrossRef CAS; (e) K. E. Beatty, F. Xie, Q. Wang and D. A. Tirrell, J. Am. Chem. Soc., 2005, 127, 14150 CrossRef CAS; (f) A. Borrmann, S. Milles, T. Plass, J. Dommerholt, J. M. M. Verkade, M. Wießler, C. Schultz, J. C. M. van Hest, F. L. van Delft and E. A. Lemke, ChemBioChem, 2012, 13, 2094 CrossRef CAS; (g) G. Charbon, E. Brustad, K. A. Scott, J. Wang, A. Løebner-Olesen, P. G. Schultz, C. Jacobs-Wagner and E. Chapman, ChemBioChem, 2011, 12, 1818 CrossRef CAS; (h) A. R. Katritzky and T. Narindoshvili, Org. Biomol. Chem., 2009, 7, 627 RSC.
- (a) M. S. T. Gonçalves, Chem. Rev., 2009, 109, 190 CrossRef; (b) S. Chen, X. Li and H. Ma, ChemBioChem, 2009, 10, 1200 CrossRef CAS; (c) J. H. Lee, J.-H. So, J. H. Jeon, E. B. Choi, Y.-R. Lee, Y.-t. Chang, C.-H. Kim, M. A. Bae and J. H. Ahn, Chem. Commun., 2011, 47, 7500 RSC; (d) T. Ehrenschwender, B. R. Varga, P. Kele and H.-A. Wagenknecht, Chem.–Asian J., 2010, 5, 1761 CrossRef CAS; (e) M. Sato, Y. Ito, N. Arima, M. Baba, M. Sobel, M. Wakao and Y. Suda, J. Biochem., 2009, 146, 33 CrossRef CAS; (f) P. Kele, X. Li, M. Link, K. Nagy, A. Herner, K. Lőrincz, Sz. Béni and O. S. Wolfbeis, Org. Biomol. Chem., 2009, 7, 3486 RSC.
- (a) A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS; (b) C. Le Droumaguet, C. Wang and Q. Wang, Chem. Soc. Rev., 2010, 39, 1233 RSC; (c) K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603 CrossRef CAS; (d) J. Qi, M.-S. Han, Y.-C. Chang and C.-H. Tung, Bioconjugate Chem., 2011, 22, 1758 CrossRef CAS; (e) G. Lukinavičius and K. Johnsson, Curr. Opin. Chem. Biol., 2011, 15, 768 CrossRef; (f) J. C. Jewett and C. R. Bertozzi, Org. Lett., 2011, 13, 5937 CrossRef CAS.
- P. Shieh, M. J. Hangauer and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 17428 CrossRef CAS.
- (a) M. F. Debets, Ch. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168 CrossRef CAS; (b) J. M. Baskin and C. R. Bertozzi, Aldrichimica. Acta, 2010, 43, 15–23 CAS.
- (a) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS; (b) P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821 CrossRef CAS; (c) C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840 RSC.
- (a) Z. Zhou and C. J. Fahrni, J. Am. Chem. Soc., 2004, 126, 8862 CrossRef CAS; (b) L. Yongjun, L. Ying, K. Xianghe, X. Min and L. Chengbu, Theor. Exp. Chem., 2000, 36, 303 CrossRef.
- (a) G. B. Cserép, K. N. Enyedi, A. Demeter, G. Mező and P. Kele, Chem.–Asian J., 2013, 8, 494 CrossRef; (b) K. Nagy, E. Orbán, Sz. Bősze and P. Kele, Chem.–Asian J., 2010, 5, 773 CrossRef CAS.
- (a) C. G. Bates, P. Saejueng, J. M. Murphy and D. Venkataraman, Org. Lett., 2002, 4, 4727 CrossRef CAS; (b) Zs. Gonda and Z. Novák, Dalton Trans., 2010, 39, 726 RSC.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS ; Erratum: 1997, 78, 1396.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS.
- P. Kele, G. Mező, D. Achatz and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2009, 48, 344 CrossRef CAS.
- N. Willand, M. Desroses, P. Toto, B. Dirie, Z. Lens, V. Villeret, P. Rucktooa, C. Locht, A. Baulard and B. Deprez, ACS Chem. Biol., 2010, 5, 1007 CrossRef CAS.
Footnotes |
| † This work is dedicated to Roger M. Leblanc on the occasion of his birthday. |
| ‡ Electronic supplementary information (ESI) available: 1H- and 13C-NMR spectra for small molecules and fluorescence spectra for 1a–c and 9a–c, further images of live cell labeling experiments and optimized geometries are provided. See DOI: 10.1039/c3ob40296g |
| This journal is © The Royal Society of Chemistry 2013 |