Open Access Article
Open Access Article
Regioselective C2-arylation of imidazo[4,5-b]pyridines†
Received
20th December 2012
, Accepted 11th February 2013
First published on 21st February 2013
Abstract
We show that N3-MEM-protected imidazo[4,5-b]pyridines undergo efficient C2-functionalisation via direct C–H arylation. Twenty-two substituted imidazo[4,5-b]pyridines are prepared and iterative, selective elaboration of functionalised imidazo[4,5-b]pyridines gives 2,7- and 2,6-disubstituted derivatives in good yields from common intermediates. Mechanistic observations are consistent with a concerted-metallation-deprotonation mechanism facilitated by coordination of copper(I)iodide to the imidazo[4,5-b]pyridine.
Introduction
Direct arylation is a highly active area of research that is increasingly applied as an alternative to the cross-coupling of an organometallic species with an aryl halide.1 The use of an unfunctionalised aromatic or heteroaromatic system as one reaction partner is a major advantage and many useful methodologies have been reported for the direct arylation of heterocycles using palladium,2 palladium and copper,3 rhodium,4 and gold5 catalytic systems.
We became interested in direct C2-functionalisation of the imidazo[4,5-b]pyridine heterocycle, a versatile purine isostere and important ring system which has seen a recent growth in medicinal chemistry application of potential therapeutic benefit; for example in protein kinase inhibitors for the treatment of cancer,6 inflammatory disease7 and diabetes.8 Recently disclosed compounds of potential therapeutic interest containing this scaffold include 1, a potent dual FLT3/Aurora kinase inhibitor targeted for the treatment of acute myeloid leukemia6a and 2, a potent inhibitor of GSK3β highlighted as a potential drug candidate for the treatment of type 2 diabetes (Fig. 1).8 Common to the imidazo[4,5-b]pyridine kinase pharmacophore is the requirement for both the N3-hydrogen bond donor and the N4-hydrogen bond acceptor which are essential for binding to the hinge region of kinases as exemplified by protein crystal structures of exemplar compounds bound to Aurora A6a and GSK3β8 respectively.
![Example imidazo[4,5-b]pyridines of potential therapeutic interest.](/image/article/2013/OB/c3ob27477b/c3ob27477b-f1.gif) |
| | Fig. 1 Example imidazo[4,5-b]pyridines of potential therapeutic interest. | |
Substitution at C2 of the imidazo[4,5-b]pyridine scaffold extends into the kinase solvent channel and is also frequently required for potent biochemical activity. Current synthetic approaches to the imidazo[4,5-b]pyridine scaffold often incorporate the C2 substituent early in the sequence through ring closure of an appropriate 2,3-disubstituted pyridine derivative, which may itself require a multistep synthesis (Scheme 1, Routes A and B).9 Recently published alternatives involve elaboration of a 2-chloro-3-aminopyridine (Scheme 1, Route C),10 and Suzuki coupling of 3-substituted-2-iodo-3H-imidazo[4,5-b]pyridine.11
![Precedented approaches to C2-substituted imidazo[4,5-b]-pyridines. Reaction partner: (A) aryl aldehyde;9a,b (B) aryl carboxylic acid;9c (C) primary aryl carboxamide.10](/image/article/2013/OB/c3ob27477b/c3ob27477b-s1.gif) |
| | Scheme 1 Precedented approaches to C2-substituted imidazo[4,5-b]-pyridines. Reaction partner: (A) aryl aldehyde;9a,b (B) aryl carboxylic acid;9c (C) primary aryl carboxamide.10 | |
Given the importance of C2-substituted imidazo[4,5-b]pyridines in medicinal chemistry, we were keen to explore the divergent synthesis of C2-aryl- and heteroaryl-substituted imidazo[4,5-b]pyridines via a C–H activation protocol. Intermolecular C–H activation has been reported on benzimidazole and purine templates using copper and palladium catalysis;12 and there are recent reports of intramolecular C–H insertion using N-substituted imidazo[4,5-b]pyridines.13 We sought an intermolecular C–H arylation protocol to directly functionalise imidazo[4,5-b]pyridines at C2, thereby circumventing the need to incorporate this substituent early in a synthetic sequence. During the preparation of this manuscript, the Langer group published the synthesis of 7-trifluoromethylimidazo[4,5-b]pyridines from 5-aminoimidazoles and 1,1,1-trifluoropentane-2,4-diones followed by subsequent C2-arylation of the resultant 7-trifluoromethylimidazo[4,5-b]pyridines using both Pd/Cu and Ni catalysis.14 Here we describe the application of C–H arylation at the C2 position of the imidazo[4,5-b]pyridine scaffold to facilitate iterative and selective elaboration to 2,7- and 2,6-disubstituted derivatives in good yield.
Results and discussion
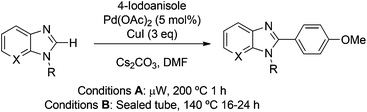
Our initial goal was the direct C2-functionalisation of imidazo[4,5-b]pyridines in the presence of the unprotected imidazole NH. C–H Activation on unprotected imidazole and benzimidazole scaffolds has been reported;12d however, application of these conditions to directly arylate the unprotected imidazo[4,5-b]pyridine scaffold at C2 with 4-iodoanisole proved unsuccessful with only recovered starting material isolated (Table 1, entry 1).
|
|
| Entry |
X |
R |
Product |
Isolated yield (%) |
Conditions |
| Heterocycle (1 eq.), 4-iodoanisole (2 eq.), Pd(OAc)2 (5 mol%), CuI (3 eq.), Cs2CO3 (2.5 eq.), DMF. |
| 1 |
N |
H |
9
|
0 |
A |
| 2 |
CH |
H |
10
|
62 |
A |
| 3 |
CH |
Me |
11
|
66 |
A |
| 4 |
N |
Me |
12
|
50 |
A |
| 5 |
N |
Me |
12
|
44 |
B |
| 6 |
N |
MEM |
13a
|
54 |
A |
| 7 |
N |
MEM |
13a
|
78 |
B |
| 8 |
N |
N1-MEM |
— |
0 |
B |
| 9 |
N |
SEM |
14
|
66 |
B |
| 10 |
N |
Boc |
— |
0 |
B |
Reaction of the corresponding unprotected benzimidazole, N-methyl benzimidazole or N3-methyl imidazo[4,5-b]pyridine (see Scheme S1† for preparation) with 4-iodoanisole proceeded smoothly in moderate yield in the presence or absence of microwave irradiation to give the corresponding C2-arylated product (Table 1, entries 2–5). We reasoned that the difference in reactivity between an unprotected imidazo[4,5-b]pyridine and benzimidazole substrate (Table 1, entries 1 and 2) may be associated with the 3,4-orientation of nitrogen atoms in the imidazo[4,5-b]pyridine acting as a bidentate coordinating ligand for the metal catalyst.
Protection of the parent imidazo[4,5-b]pyridine ring system 3 with a MEM group gave 6a, with its regioisomer 6b as a minor product separable by column chromatography and distinguishable by 2D NMR (see ESI†).15N3-Protection of 6-Br- and 7-Cl-imidazo[4,5-b]pyridines (4 and 5) was also achieved, with high regioselectivity observed in the case of the 7-chloro substituent (5) which presumably hinders competing substitution at N1 (Scheme 2).
![Preparation of MEM-protected imidazo[4,5-b]pyridines.](/image/article/2013/OB/c3ob27477b/c3ob27477b-s2.gif) |
| | Scheme 2 Preparation of MEM-protected imidazo[4,5-b]pyridines. | |
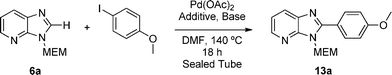
The N3-MEM isomer (6a) reacted smoothly and regioselectively under conditions previously reported on a protected purine scaffold by Čerňa et al.16 [4-iodoanisole (2 eq.), Pd(OAc)2 (5 mol%), CuI (3 eq.) and Cs2CO3 (2.5 eq.) in DMF, Table 1, entries 6 and 7] to give the desired C2-arylated product (13a). However, the corresponding N1-protected isomer (6b) failed to react (Table 1, entry 8), consistent with the hypothesis that 3,4-orientation of the two nitrogen atoms in the imidazo[4,5-b]pyridine scaffold inhibits reaction. N3-Protection with SEM (see Scheme S2† for preparation) is also compatible with C2-selective arylation (Table 1, entry 9), whilst the use of Boc as an N3-protecting group (see Scheme S2† for preparation) led to unproductive deprotection under the reaction conditions (Table 1, entry 10).
To optimise the regioselective C2-arylation of N3-MEM-protected imidazo[4,5-b]pyridine (6a), we screened a variety of reaction conditions, including those reported in the literature for C–H arylation on alternative heterocyclic systems. We found that conditions reported by Bellina which lack a carbonate base are ineffective on the imidazo[4,5-b]pyridine scaffold (Table 2, entry 2);17 in addition, no conversion is observed in the absence of CuI (Table 2, entry 3) which is required in excess for optimal conversion (Table 2, entry 4). Only limited conversion is obtained in the absence of palladium (Table 2, entry 5) and use of MgO as a C–H arylation promoter gave no conversion in our hands (Table 2, entry 6).18
Table 2 Optimisation of conditions using N3-MEM imidazo[4,5-b]pyridine (6a) and 4-iodoanisolea
|
|
| Entry |
Palladium |
Additive (eq.) |
Base |
% Conv.b/(% yield)c |
|
Conditions: heterocycle (1 eq.), 4-iodoanisole (2 eq.), Pd(OAc)2 (5 mol%), CuI (3 eq.), Cs2CO3 (2.5 eq.), DMF.
Conversion reported as ratio of starting material to product measured by LCMS.
Isolated Yield.
|
| 1 |
Pd(OAc)2 |
CuI (3.0) |
Cs2CO3 |
100 (78) |
| 2 |
Pd(OAc)2 |
CuI (3.0) |
— |
0 |
| 3 |
Pd(OAc)2 |
— |
Cs2CO3 |
0 |
| 4 |
Pd(OAc)2 |
CuI (1.0) |
Cs2CO3 |
50 |
| 5 |
— |
CuI (3.0) |
Cs2CO3 |
12 |
| 6 |
Pd(OAc)2 |
MgO (3.0) |
Cs2CO3 |
0 |
Multiple alternative direct arylation conditions have been reported in the literature, however all gave inferior results; for example, use of K2CO3 as base,19 the addition of piperidine to hinder substrate chelation of palladium,12a and a Pd(II)/Cu(II) co-catalytic system20 (Table 3, entries 1–3). Similarly application of the copper-free conditions of Larrosa21 and Fagnou2c gave limited conversion (Table 3, entries 4 and 5); however, with the addition of CuI, complete reaction was observed under Fagnou conditions (Table 3, entries 6 and 7). We observed that aryl bromides gave complete conversion under optimised reaction conditions, whilst aryl chlorides gave partial conversion (Table 3, entries 8 and 9); however an aryl boronic acid coupling partner was unreactive under a variety of conditions. In summary, optimal direct arylation conditions require the presence of Pd(OAc)2, CuI and a carbonate base (Table 2, entry 1); the addition of PivOH or PCy3·HBF4 is tolerated but not essential for reaction.
Table 3 Alternative conditions applied to N3-MEM-protected imidazo[4,5-b]pyridine 6a
|

|
| Entry |
X |
Pd |
Cu (eq.) |
Additive(s) |
Base |
Solvent |
T (°C) |
% Conv.a (% yield)b |
| Reaction times 16–40 h. % Conversion by LCMS ratio of product to starting material. Isolated yield. |
| 1 |
I |
Pd(OAc)2 |
CuI (3.0) |
— |
K2CO3 |
DMF |
140 |
100 (60) |
| 2 |
I |
Pd(OAc)2 |
CuI (2.0) |
Piperidine |
Cs2CO3 |
DMF |
150 |
81 (38) |
| 3 |
I |
Pd(OAc)2 |
Cu(OAc)2 |
PPh3 |
K2CO3 |
Toluene |
110 |
0 |
| 4 |
I |
Pd(OAc)2 |
— |
oNO2C6H4CO2H |
Ag2O |
DMF |
25 |
0 |
| 5 |
Br |
Pd(OAc)2 |
— |
PivOH, PCy3·HBF4 |
K2CO3 |
DMA |
140 |
28 (19) |
| 6 |
Br |
Pd(OAc)2 |
CuI (3.0) |
PivOH, PCy3·HBF4 |
K2CO3 |
DMF |
140 |
100 |
| 7 |
Br |
Pd(OAc)2 |
CuI (3.0) |
PivOH |
K2CO3 |
DMF |
140 |
100 |
| 8 |
Br |
Pd(OAc)2 |
CuI (3.0) |
— |
Cs2CO3 |
DMF |
140 |
100 (75) |
| 9 |
Cl |
Pd(OAc)2 |
CuI (3.0) |
— |
Cs2CO3 |
DMF |
140 |
67 |
We employed the optimal reaction conditions (Table 2, entry 1) with a variety of aryl and heteroaryl coupling partners to assess the scope and efficiency of regioselective C2-arylation (Table 4, conditions B). We observed that electron-poor aryl halides generally resulted in lower isolated yields and, in these cases, we found that addition of PCy3·HBF4,22 reduced temperature (120 °C) and use of aryl bromide substrates gave higher isolated yields although reaction times were extended (Table 4, conditions C). No competing C–H arylation at the C5 position was observed, except for product 13e, where trace amounts of a putative C2,5-bis-arylated side product were detected using conditions B.
Table 4 Synthesis of C2-substituted N3-MEM imidazo[4,5-b]pyridines
|

|
| Conditions B: ArI (2.0 eq.), Pd(OAc)2 (5 mol%), CuI (3 eq.), Cs2CO3 (2.5 eq.) DMF, 140 °C, 16–24 h. |
| Conditions C: ArBr (2.0 eq.), Pd(OAc)2 (5 mol%), PCy3·HBF4 (10 mol%) CuI (3 eq.), Cs2CO3 (2.5 eq.) DMF, 120 °C, 24–40 h. |
| Entry |
Product |
Isolated yield |
Entry |
Product |
Isolated yield |
| 1 |

|
B: 78% |
7 |
|
B: 67% |
| 2 |
|
B: 75% |
8 |
|
B: 60% |
| 3 |
|
B: 84% |
9 |
|
B: 75% |
| 4 |
|
B: 40% |
10 |
|
B: 47% |
| C: 60% |
C: 67% |
| 5 |
|
B: 35% |
11 |
|
B: 70% |
| C: 77% |
| 6 |
|
B: 46% |
12 |
|
B: 35% |
| C: 64% |
The MEM protected products 13a–l were deprotected using aq. HCl to afford the desired 2-substituted imidazo[4,5-b]pyridines, 9 and 15b–l (38–98%; see ESI, Table S1†).
We were keen to apply these optimised direct arylation conditions to the synthesis of more heavily substituted imidazo[4,5-b]pyridine scaffolds by selective, iterative functionalisation of multiple vectors. Regioselective substitution of the 6-position of 7a or the 7-position of 8a by Pd-mediated cross-coupling (see ESI Schemes S4–S7†) followed by direct C2-arylation gave the desired disubstituted compounds (Table 5, 16a to 16j). 6-Bromo-imidazo[4,5-b]pyridine 7a underwent 2,6-bis-arylation under C–H arylation conditions; however, we observed that the 7-chloro-imidazo[4,5-b]pyridine 8a underwent regioselective C2-arylation with a notable increase in rate of reaction upon the addition of PivOH (30 mol%) (Table 5, entries 7–10). This regioselective transformation affords the opportunity for subsequent C7-functionalisation of the imidazo[4,5-b]pyridine scaffold.
Table 5 Direct arylation of 6- and 7-substituted imidazo[4,5-b]pyridines
Several mechanistic rationales have been proposed for C–H arylation reactions on heteroaromatic scaffolds, two of which are most relevant to the reaction conditions described here (Scheme 3). Bellina and Rossi first proposed a classical cross-coupling mechanism involving transmetallation of an organo-copper intermediate that is in equilibrium with a substrate/Cu(I) complex (Scheme 3, mechanism A).23 This mechanism would be expected to proceed with catalytic CuI; however, our work, as well as that preceding it, has demonstrated a need for greater than stoichiometric amounts of Cu(I). A possible explanation is that a large molar excess of CuI could bias the equilibrium towards the substrate/Cu(I) complex. This mechanism is consistent with the work of Fairlamb, who proposed pre-coordination of CuI to purine nucleosides followed by deprotonation/cupration of the C8–H bond to form an organo-copper species that subsequently undergoes transmetallation into a classical Pd catalytic cycle.12a,24 Fagnou proposed an alternative Concerted-Metallation-Deprotonation (CMD) mechanism involving base-assisted C–H bond cleavage25 (Scheme 3, Mechanism B) and consistent with mechanistic studies by Macgregor.26 Recently, Gorelsky has investigated Pd(OAc)2 and CuI mediated direct arylation on azoles, and proposed that Cu does not insert into the C–H bond, but lowers the pKa of the neighbouring C2–H by coordination of an azole heteroatom, thereby facilitating a CMD-type mechanism.27
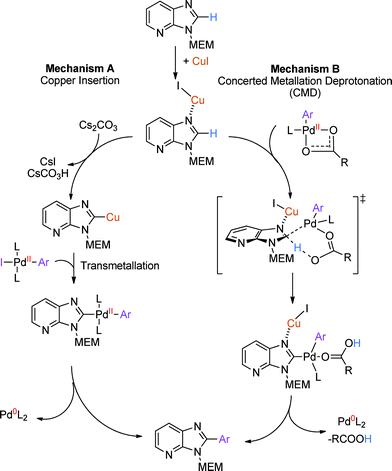 |
| | Scheme 3 Postulated mechanisms of C–H arylation. | |
A control experiment using C2-arylation conditions in the absence of CuI and Pd(OAc)2 but in the presence of acetone-d6 showed 87% incorporation of deuterium at C2 (Scheme S8†), suggesting that C2 can be deprotonated under the reaction conditions in the absence of a coordinated metal. We measured the rate of two independent parallel C–H arylation reactions using C2–H and C2–D imidazo[4,5-b]pyridine, where the steady state approximation was maintained by using a large excess of 4-iodoanisole.28 We observed a kinetic isotope effect (kH/kD = 2.6) consistent with C2–H bond cleavage occurring in the rate-determining step (ESI, Charts S1 and S2†). We therefore postulate that CuI is necessary to increase the acidity of the C2 proton, facilitating a CMD-type insertion mechanism and consistent with the proposal of Gorelsky (Scheme 3, Mechanism B).27 As pivalic acid is not essential for smooth reaction, carbonate base and/or acetate anion from Pd(OAc)2 may also serve to enhance the rate of the CMD step by acting as a proton shuttle, as previously proposed.25d
Conclusion
In summary, we have shown that the N3-MEM-protected imidazo[4,5-b]pyridine template can be regioselectively functionalised at C2 via direct C–H arylation using palladium, copper and basic carbonate conditions. The addition of a phosphine ligand increases the conversion and isolated yields in the case of electron deficient coupling partners. 22 C2-substituted imidazo[4,5-b]pyridines have been prepared. Application of these C–H arylation conditions to the iterative and regioselective elaboration of functionalised imidazo[4,5-b]pyridines gives 2,7- and 2,6-disubstituted derivatives from common intermediates in an approach amenable to the divergent synthesis of analogues and which is applicable to rapid medicinal chemistry exploration of this emerging purine isostere and ATP competitive kinase pharmacophore. There are two postulated mechanisms for this type of C–H arylation; our observations are consistent with C–H arylation of N3-MEM-protected imidazo[4,5-b]pyridines proceeding predominantly via a CMD mechanism whereby coordination of copper(I)iodide to the imidazo[4,5-b]pyridine scaffold enhances the acidity of the C2-proton.
Experimental section
All anhydrous solvents and reagents were obtained from commercial suppliers and used without any further purification unless otherwise noted. A Biotage Initiator 60 instrument was used for all microwave-assisted reactions, using sealed reaction vessels with the temperature measured by an external IR sensor. Analytical TLC was performed on pre-coated aluminum sheets of silica (60 F254 nm) and visualised by short-wave UV light at λ254. Flash column chromatography was carried out on silica gel (0.040–0.065 mm) and Flash Si-II silica gel cartridges. Purification by ion exchange was carried out using SCX-II and NH2 cartridges. Semi-automated purification was carried out on a Biotage SP1 purification system, using SNAP cartridges, or SINGLE StEP flash column cartridges. Solvent systems are reported by column volume (CV) with the solvent flow rate as stated. Melting points were determined on an EZ-Melt automated melting point apparatus. IR spectra were recorded on a Bruker Alpha P FT-IR spectrometer. Absorption maxima (νmax) are quoted in wavenumbers (cm−1). 1H NMR spectra were recorded at 500 MHz using an internal deuterium lock. Chemical shifts were measured in parts per million (ppm) using the following internal references for residual protons in the solvent: CDCl3 (δH 7.26), CD3OD (δH 3.32) and DMSO-d6 (δH 2.50). Data is presented as follows: chemical shift, multiplicity, coupling constant (J) in Hz, and integration. The following abbreviations are used for the splitting patterns: s for singlet, d for doublet, t for triplet, m for multiplet and br for broad. 13C NMR spectra were recorded at 126 MHz using an internal deuterium lock. The following internal references were used: CDCl3 (δC 77.0), CD3OD (δC 49.0) and DMSO-d6 (δC 39.5). LCMS analyses were performed using ESI/APCI, with a Purospher STAR RP-18, 30 × 4 mm column and a flow rate of 1.5 mL min−1. UV detection was at 254 nm. High Resolution MS analyses were performed using ESI/APCI. The references masses are: Caffeine [M + H]+ = 195.087652, Reserpine [M + H]+ = 609.280657, hexakis(1H,1H,3H-tetrafluoropentoxy)phosphazene [M + H]+ = 922.009798.
General procedure to MEM-protection of imidazo[4,5-b]pyridines
To a stirred suspension of the appropriate imidazo[4,5-b]pyridine derivative in toluene was added Et3N (1.5 eq.) and the mixture stirred at −10 °C for 30 min before the dropwise addition of MEM-Chloride (2 eq.) in toluene over 1 hour. Upon complete addition the mixture was heated to reflux for 4 h. On cooling the solution was concentrated in vacuo. The crude oil was purified by flash column chromatography, conditions given.
3-((2-Methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 6a.
Imidazo[4,5-b]pyridine, 3 (2.38 g, 20 mmol) was reacted following general procedure A. The crude oil was purified by flash column chromatography (cyclohexane to CH2Cl2 then gradient to 5% MeOH in CH2Cl2) to yield the product as a pale yellow oil (2.3 g, 11.1 mmol, 55%). 1H NMR: (500 MHz, CDCl3) δH 8.41 (dd, J = 4.8, 1.4 Hz, 1H), 8.21 (s, 1H), 8.07 (dd, J = 8.0, 1.4 Hz, 1H), 7.27–7.24 (m, 1H), 5.75 (s, 2H), 3.71–3.69 (m, 2H), 3.50–3.48 (m, 2H), 3.32 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 147.2, 144.7, 144.2, 135.2, 128.1, 118.7, 72.8, 71.5, 68.8, 59.0; LCMS tR = 1.61 min, m/z = 208 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C10H14N3O2 = 208.1081, found = 208.1085.
Product 6b was also isolated under these reaction conditions; regioisomers were assigned by 2D NMR (see ESI†).
1-((2-Methoxyethoxy)methyl)-1H-imidazo[4,5-b]pyridine, 6b.
Yellow oil (410 mg, 10%); 1H NMR: (500 MHz, CDCl3) δH 8.64 (dd, J = 4.8, 1.6 Hz, 1H), 8.24 (s, 1H), 7.93 (dd, J = 8.1, 1.6 Hz, 1H), 7.33–7.29 (m, 1H), 5.68 (s, 2H), 3.63–3.57 (m, 2H), 3.55–3.51 (m, 2H), 3.38 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 156.7, 145.1, 144.6, 125.8, 118.2, 118.2, 75.1, 71.3, 67.4, 58.6; LCMS tR = 1.21 min, m/z = 208 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C10H14N3O2 = 208.1081, found = 208.1090.
6-Bromo-3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 7a.
To 6-bromoimidazo[4,5-b]pyridine, 4 (1.98 g, 10.0 mmol) and Et3N (2.08 mL, 15 mmol) in toluene (200 mL) was added drop wise MEM-Chloride (2.28 mL, 20 mmol) in toluene (100 mL) over 1 hour, following general procedure A. The crude oil was purified by flash column chromatography (0–60% EtOAc in cyclohexane) to afford named product as a yellow oil (1870 mg, 6.5 mmol, 65%). 1H NMR: (500 MHz, CDCl3) δH 8.48 (d, J = 1.9 Hz, 1H), 8.25 (bs, 2H), 5.74 (s, 2H), 3.72–3.59 (m, 2H), 3.53–3.49 (m, 2H), 3.34 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 145.7, 145.4, 136.2, 130.5, 114.5, 73.1, 71.5, 69.0, 59.1, one C does not appear; LCMS tR = 2.22 min, m/z = 286, 288 (M + H)+ bromine isotopic splitting pattern; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C10H13BrN3O2 = 286.0186, found = 286.0178.
Product 7b was also isolated under these reaction conditions; regioisomers were assigned by 2D NMR.
6-Bromo-1-((2-methoxyethoxy)methyl)-1H-imidazo[4,5-b]pyridine, 7b.
Yellow oil (59 mg, 0.21 mmol, 21%). 1H NMR: (500 MHz, CDCl3) δH 8.64 (d, J = 2.0 Hz, 1H), 8.21 (s, 1H) 8.07 (d, J = 2.0 Hz, 1H), 5.64 (s, 2H), 3.60–3.58 (m, 2H), 3.54–3.52 (m, 2H), 3.36 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 155.2, 146.6, 145.8, 126.8, 121.7, 114.8, 75.7, 71.8, 68.1, 59.1; LCMS tR = 2.20 min, m/z = 286, 288 (M + H)+ bromine splitting pattern; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C10H13BrN3O2 = 286.0186, found = 286.0181.
7-Chloro-3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 8a.
To 7-chloroimidazo[4,5-b]pyridine, 5 (767 mg, 5 mmol) and Et3N (1.04 mL, 7.5 mmol) in toluene (100 mL) was added drop wise MEM-Chloride (1.14 mL, 10 mmol) in toluene (50 mL) over 1 hour, following general procedure A. The resulting oil was taken in EtOAc and washed with H2O, the aqueous layer was re-extracted with CH2Cl2, organics combined, dried (MgSO4), and concentrated in vacuo. The crude material was purified by flash column chromatography (Cyclohexane to CH2Cl2 then gradient to 5% MeOH in CH2Cl2) to yield the product as a yellow foam (1118 mg, 4.6 mmol, 92%). 1H NMR: (500 MHz, CDCl3) δH 8.33 (d, J = 5.2 Hz, 1H), 8.28 (s, 1H), 7.33 (d, J = 5.2 Hz, 1H), 5.77 (s, 2H), 3.73–3.71 (m, 2H), 3.54–3.50 (m, 2H,), 3.36 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 147.7, 145.1, 144.5, 135.1, 133.0, 119.4, 73.4, 71.5, 69.1, 59.1; LCMS tR = 2.05 min, m/z = 242 (M + H)+; purity (AUC) ≥ 95%. HRMS (M + H)+calculated for C10H13ClN3O2 = 242.0691, found = 242.0693.
Product 8b was also isolated under these reaction conditions; regioisomers were assigned by 2D NMR.
7-Chloro-1-((2-methoxyethoxy)methyl)-1H-imidazo[4,5-b]pyridine, 8b.
Yellow gum (48 mg, 0.2 mmol, 4%). 1H NMR: (500 MHz, CDCl3) δH 8.45 (d, J = 5.1 Hz, 1H), 8.27 (s, 1H), 7.27 (m, 1H), 5.87 (s, 2H), 3.67–3.60 (m, 2H), 3.56–3.50 (m, 2H), 3.33 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 158.1, 147.1, 145.6, 127.1, 123.1, 120.0, 76.1, 71.8, 67.8, 59.1; LCMS tR = 1.97 min, m/z = 242 (M + H)+; purity (AUC) ≥ 95%. HRMS (M + H)+calculated for C10H13ClN3O2 = 242.0691, found = 208.0690.
General conditions A: microwave-assisted C–H arylation conditions
The appropriate substrate (1.0 mmol), CuI (570 mg, 3.0 mmol), 4-iodoanisole (470 mg, 2.0 mmol), Cs2CO3 (812 mg, 2.5 mmol) and Pd(OAc)2 (11.2 mg, 5.0 mol%) were placed into a dried microwave vial and evacuated/filled with argon. DMF (5.0 mL) was added and the reaction mixture heated in a microwave reactor at 200 °C for 30 min. The reaction mixture was cooled to RT, diluted with EtOAc (10 mL) and stirred for 0.5 h in saturated NH4Cl solution (30 mL). After extraction with EtOAc, the organics were washed with water, brine, dried (MgSO4) and concentrated in vacuo. The crude product was purified by flash column chromatography, conditions given.
General conditions B: C–H arylation conditions
Pd(OAc)2 (5.6 mg, 0.025 mmol), CuI (285 mg, 1.5 mmol), Cs2CO3 (406 mg, 1.25 mmol) and aryl iodide (if solid, 1.0 mmol) were combined under air in a sealable reaction tube. The tube was flushed with argon and 3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 6a (104 mg, 0.5 mmol) was added, followed by anhydrous DMF (5 mL) and the appropriate aryl iodide (1.0 mmol). The tube was sealed and heated to 140 °C for 40–72 h. Upon complete conversion by LC-MS, the reaction was concentrated in vacuo. The crude mixture was purified by column chromatography, conditions given.
General conditions C: optimised C–H arylation conditions for electron poor aryl halides
Pd(OAc)2 (2.8 mg, 0.0125 mmol), CuI (143 mg, 0.75 mmol), PCy3·HBF4 (9 mg, 0.025 mmol), Cs2CO3 (203 mg, 0.625 mmol) and the appropriate aryl bromide (0.5 mmol) were combined under air in a sealable reaction tube. The tube was flushed with argon and 3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 6a (52 mg, 0.25 mmol) was added, followed by anhydrous DMF (2.5 mL) and aryl halide (if liquid, 0.5 mmol). The tube was sealed and heated to 120 °C for 40–72 h. Upon complete conversion by LC-MS, the reaction was concentrated in vacuo. The crude mixture was purified by column chromatography, conditions given.
2-(4-Methoxyphenyl)-1H-benzimidazole, 10.
Prepared using general procedure A from benzimidazole. Purification by flash column chromatography (0–5% MeOH in CH2Cl2) yielded product as a yellow solid (139 mg, 62%); m.p. 202–205 °C; literature m.p. 215–217 °C;12d1H NMR: (500 MHz, DMSO-d6) δH 12.99 (bs, 1H), 8.15 (bs, 2H), 7.58 (bs, 2H), 7.18 (bs, 2H), 7.11 (bs, 2H), 3.84 (s, 3H); 13C NMR: (126 MHz, DMSO-d6) δC 160.4, 128.1, 122.9, 122.3, 121.3, 118.6, 114.4, 110.9, 55.3; LCMS tR = 1.64 min, m/z 225 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C14H13N2O = 225.1022, found = 225.1018.
2-(4-Methoxyphenyl)-1-methyl-1H-benzimidazole, 11.
Prepared using general procedure A from N-methylbenzimidazole. Purification by flash column chromatography (0–5% MeOH in CH2Cl2) yielded product as a white solid (157 mg, 66%). Rf = 0.63 (MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 5:95); 1H NMR: (500 MHz, DMSO-d6) δH 7.80 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 7.5 Hz, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.27 (t, J = 7.5 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 3.859 (s, 3H), 3.855 (s, 3H); 13C NMR: (126 MHz, DMSO-d6) δC 160.3, 153.0, 142.5, 136.6, 130.7, 122.4, 122.0, 121.7, 118.7, 114.1, 110.3, 55.3, 31.6; LCMS tR = 1.41 min, m/z 239 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C15H15N2O = 239.1179, found = 239.1174.
:CH2Cl2 5:95); 1H NMR: (500 MHz, DMSO-d6) δH 7.80 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 7.5 Hz, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.27 (t, J = 7.5 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 3.859 (s, 3H), 3.855 (s, 3H); 13C NMR: (126 MHz, DMSO-d6) δC 160.3, 153.0, 142.5, 136.6, 130.7, 122.4, 122.0, 121.7, 118.7, 114.1, 110.3, 55.3, 31.6; LCMS tR = 1.41 min, m/z 239 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C15H15N2O = 239.1179, found = 239.1174.
2-(4-Methoxyphenyl)-3-methyl-3H-imidazo[4,5-b]pyridine, 12.
Prepared using general procedure B with N3-methylimidazo[4,5-b]pyridine (S2) in place of 3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine (6a), and 4-iodoanisole. Purification by flash column chromatography (10–100% EtOAc in cyclohexane) yielded product as a cream solid (53 mg, 44%). 1H NMR: (500 MHz, CDCl3) δH 8.40 (dd, J = 4.8, 1.4 Hz, 1H), 8.06 (dd, J = 8.0, 1.4 Hz, 1H), 7.84–7.79 (m, 2H), 7.26 (dd, J = 8.0, 4.8 Hz, 1H), 7.11–7.05 (m, 2H), 4.00 (s, 3H), 3.91 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 161.2, 154.8, 149.1, 143.4, 135.2, 130.7, 126.8, 122.3, 118.4, 114.3, 55.4, 30.5; LCMS tR = 2.27 min, m/z = 240 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C14H14N3O = 240.1131, found = 240.1139.
3-((2-Methoxyethoxy)methyl)-2-(4-methoxyphenyl)-3H-imidazo[4,5-b]pyridine, 13a.
Prepared using general procedure B and 4-iodoanisole. Purification by flash column chromatography (20–80% EtOAc in CH2Cl2) yielded the product as a brown solid (122 mg, 78%); m.p. 105–108 °C; 1H NMR: (500 MHz, CDCl3) δH 8.37 (dd, J = 4.8, 1.4 Hz, 1H), 8.14–8.09 (m, 2H), 8.06 (dd, J = 8.0, 1.4 Hz, 1H), 7.30–7.25 (m, 1H), 7.10–7.04 (m, 2H), 5.76 (s, 2H), 4.00–3.93 (m, 2H), 3.90 (s, 3H) 3.64–3.59 (m, 2H), 3.40 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 161.55, 155.4, 149.3, 143.7, 135.0, 131.2, 126.9, 121.9, 119.0, 72.0, 71.5, 69.7, 59.0, 53.4; LCMS tR = 2.63 min, m/z = 314 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C17H20N3O3 = 314.1499, found = 314.1484.
2-(4-Methoxyphenyl)-3-((2-(trimethylsilyl)ethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 14.
Prepared using general procedure B with N3-SEM-imidazo[4,5-b]pyridine (S4) in place of 3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine (6a), and 4-iodoanisole. Purification by flash column chromatography (0–50% EtOAc in cyclohexane) affords product as an orange oil (117 mg, 66%); 1H NMR: (500 MHz, CDCl3) δH 8.40 (dd, J = 4.8, 1.4 Hz, 1H), 8.17–8.10 (m, 2H), 8.09 (dd, J = 8.0, 1.4 Hz, 1H), 7.29 (dd, J = 8.0, 4.8 Hz, 1H), 7.13–7.05 (m, 2H), 5.71 (s, 2H), 3.93 (s, 3H), 3.92–3.87 (m, 2H), 1.08–1.01 (m, 2H), 0.01 (s, 9H); 13C NMR: (126 MHz, CDCl3) δC 161.2, 155.3, 148.5, 143.8, 131.3, 126.7, 121.6, 119.1, 114.3, 71.3, 67.3, 55.4, 18.1, −1.2; LCMS tR = 3.52 min, m/z = 356 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C19H26N3O2Si = 356.1789, found = 356.1790.
3-((2-Methoxyethoxy)methyl)-2-(4-tolyl)-3H-imidazo[4,5-b]pyridine, 13b.
Prepared by general procedure B using 4-iodotoluene. Purification by column chromatography (0–30% EtOAc in cyclohexane) afforded the product as a pale orange oil (112 mg, 75%). Rf = 0.51 (cyclohexane:EtOAc 1:4); 1H NMR: (500 MHz, CDCl3) δH 8.37 (dd, J = 4.8, 1.4 Hz, 1H), 8.05 (dd, J = 8.0, 1.4 Hz, 1H), 8.02 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.25 (dd, J = 8.0, 4.8 Hz, 1H), 5.75 (s, 2H), 3.92–3.96 (m, 2H), 3.57–3.60 (m, 2H), 3.37 (s, 3H), 2.43 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 155.6, 149.3, 143.9, 140.8, 134.9, 129.6, 129.6, 127.0, 126.6, 119.0, 72.0, 71.5, 68.9, 59.0, 21.5; LCMS tR = 2.35 min, m/z = 298 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C17H20N3O2 = 298.1550, found = 298.1558.
2-(4-Fluorophenyl)-3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 13c.
Prepared by general procedure B using 1-fluoro-4-iodobenzene. Purification by flash column chromatography (20–70% EtOAc in cyclohexane) yielded product as a brown solid (127 mg, 84%); m.p. 64–66 °C; 1H NMR: (500 MHz, CDCl3) δH 8.42 (dd, J = 4.8, 1.4 Hz, 1H), 8.22–8.15 (m, 2H), 8.10 (dd, J = 8.0, 1.4 Hz, 1H), 7.31 (dd, J = 8.0, 4.8 Hz, 1H), 7.30–7.22 (m, 2H), 5.77 (s, 2H), 4.02–3.95 (m, 2H), 3.64–3.60 (m, 2H) 3.39 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 163.8 (d, J = 251.7 Hz), 154.0, 148.7, 143.7, 134.3, 131.3 (d, J = 8.6 Hz), 126.8, 125.2 (d, J = 3.3 Hz), 118.7, 115.6 (d, J = 21.8 Hz), 71.4, 71.0, 68.5, 58.2; 19F NMR: (500 MHz, CDCl3) δF −109.4; LCMS tR = 2.65 min, m/z = 302 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C16H17FN3O2 = 302.1299, found = 302.1298.
2-(4-Chlorophenyl)-3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 13d.
Prepared by general procedure C using 1-chloro-4-iodobenzene. Purification by flash column chromatography (0–60% EtOAc in cyclohexane) yielded the product as a yellow solid (48 mg, 60%); m.p. 63–66 °C; 1H NMR: (500 MHz, CDCl3) δH 8.42 (dd, J = 4.8, 1.4 Hz, 1H), 8.15–8.08 (m, 3H), 7.56–7.53 (m, 2H), 7.31 (dd, J = 8.0, 4.8 Hz, 1H), 5.76 (s, 2H), 4.02–3.92 (m, 2H), 3.64–3.58 (m, 2H), 3.40 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 154.2, 149.1, 144.5, 137.1, 134.6, 131.0, 129.3, 127.8, 127.4, 119.3, 71.9, 71.5, 69.0, 59.0; LCMS tR = 2.91 min, m/z = 318 (M + H)+; purity (AUC) = 93%; HRMS (M + H)+calculated for C16H17ClN3O2 = 318.1004, found = 318.1006.
3-((2-Methoxyethoxy)methyl)-2-(4-(trifluoromethyl)phenyl)-3H-imidazo[4,5-b]pyridine, 13e.
Prepared by general procedure C using 1-bromo-4-(trifluoromethyl)benzene. Purification by flash column chromatography (20–60% EtOAc in CH2Cl2) yielded the product as a deep brown solid (68 mg, 77%); m.p. 88–96 °C; 1H NMR: (500 MHz, CDCl3) δH 8.47 (dd, J = 4.8, 1.4 Hz, 1H), 8.32 (d, J = 8.2 Hz, 2H), 8.15 (dd, J = 8.0, 1.4 Hz, 1H), 7.84 (d, J = 8.2 Hz, 2H), 7.41 (dd, J = 8.0, 4.8 Hz, 1H), 5.80 (s, 2H), 4.01–3.95 (m, 2H), 3.65–3.58 (m, 2H) 3.41 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 153.2, 148.6, 144.3, 133.3 (d, J = 228 Hz), 131.8 (d, J = 33 Hz) 129.6, 127.2, 125.3 (d, J = 4 Hz), 123.4 (d, J = 272 Hz), 119.0, 71.5, 71.0, 68.6, 58.5, one C does not appear; 19F NMR: (500 MHz, CDCl3) δF −62.9; LCMS tR = 2.93 min, m/z = 352; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C17H17F3N3O2 = 352.1267, found = 352.1253.
3-((2-Methoxyethoxy)methyl)-2-(4-(methylsulfonyl)phenyl)-3H-imidazo[4,5-b]pyridine, 13f.
Prepared by general procedure B using 1-iodo-4-(methylsulfonyl)benzene. Purification by flash column chromatography (20–60% EtOAc in CH2Cl2) yielded product as a deep brown solid (83 mg, 46%); m.p. 148–152 °C; 1H NMR: (500 MHz, CDCl3) δH 8.45 (dd, J = 4.8, 1.3 Hz, 1H), 8.39 (d, J = 8.5 Hz, 2H), 8.13–8.11 (m, 3H), 7.33 (dd, J = 8.0, 4.8 Hz, 1H), 5.78 (s, 2H), 4.00–3.94 (m, 2H), 3.63–3.57 (m, 2H) 3.38 (s, 3H), 3.12 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 153.1, 149.0, 145.1, 142.0, 134.7, 130.6, 127.9, 119.6, 71.9, 71.4, 69.1, 59.0, 44.5; LCMS tR = 2.28 min, m/z = 362 (M + H)+; purity (AUC) = 90%; HRMS (M + H)+calculated for C17H20N3O4S = 362.1175, found = 362.1190.
3-((2-Methoxyethoxy)methyl)-2-phenyl-3H-imidazo[4,5-b]pyridine, 13g.
Prepared by general procedure B using 4-iodobenzene. Purification by flash column chromatography (20% EtOAc in CH2Cl2) yielded named product as a brown oil (95 mg, 67%); 1H NMR: (500 MHz, CDCl3) δH 8.41 (dd, J = 4.8, 1.4 Hz, 1H), 8.16–8.12 (m, 2H), 8.10 (dd, J = 8.0, 1.4 Hz, 1H), 7.59–7.54 (m, 3H), 7.30 (dd, J = 8.0, 4.8 Hz, 1H), 5.78 (s, 2H), 4.00–3.94 (m, 2H), 3.64–3.58 (m, 2H) 3.40 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 155.4, 149.2, 144.2, 134.9, 130.5, 129.7, 129.5, 128.9, 127.3, 119.1, 72.0, 71.6, 69.0, 59.0; LCMS tR = 2.57, m/z = 284 (M + H)+; purity (AUC) = 88%; HRMS (M + H)+calculated for C16H18N3O2 = 284.1394, found = 284.1378.
2-(3-Fluorophenyl)-3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 13h.
Prepared by general procedure B using 1-fluoro-3-iodobenzene. Purification by flash column chromatography (0–50% EtOAc in cyclohexane) yielded product as an orange oil (90 mg, 60%); 1H NMR: (500 MHz, CDCl3) δH 8.47 (dd, J = 4.8, 1.4 Hz, 1H), 8.16 (dd, J = 8.0, 1.4 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.94 (m, 1H), 7.57 (td, J = 8.0, 5.8 Hz, 1H), 7.36 (dd, J = 8.0, 4.8 Hz, 1H), 7.29 (m, 1H), 5.82 (s, 2H), 4.03–3.95 (m, 2H), 3.66–3.60 (m, 2H), 3.41 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 163.8 (d, J = 252 Hz) 154.0, 148.7, 134.3, 131.3 (d, J = 9 Hz), 126.8, 125.2 (d, J = 4 Hz), 118.7, 115.6 (d, J = 22 Hz) 71.4, 71.0, 68.5, 58.5; 19F NMR: (500 MHz, CDCl3) δF −111.3; LCMS tR = 2.70 min, m/z = 302 (M + H)+; purity (AUC) = 94%. HRMS (M + H)+ = calculated for C16H17FN3O2 = 302.1299, found = 302.1305.
3-((2-Methoxyethoxy)methyl)-2-(o-tolyl)-3H-imidazo[4,5-b]pyridine, 13i.
Prepared by general procedure B using 1-iodo-2-methylbenzene. Purification by flash column chromatography (0–60% EtOAc in CH2Cl2) yielded product as a brown oil (111 mg, 75%); 1H NMR: (500 MHz, DMSO-d6) δH 8.43 (d, J = 4.7 Hz, 1H), 8.16 (d, J = 7.6 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H) 7.49 (t, J = 7.1 Hz, 1H) 7.43 (d, J = 7.1 Hz, 1H) 7.40–7.34 (m, 2H), 5.47 (s, 2H), 3.58–3.53 (m, 2H), 3.36–3.30 (m, 2H) 3.12 (s, 3H), 2.25 (s, 3H); 13C NMR: (126 MHz, DMSO-d6) δC 144.4, 138.4, 134.8, 131.0, 130.6, 130.5, 129.7, 127.6, 126.1, 119.4, 72.1, 71.3, 68.7, 58.4, 20.0, two quaternary C's do not appear; LCMS tR = 2.55, m/z = 298 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+ = calculated for C17H20N3O2 = 298.1550, found = 298.1545.
3-((2-Methoxyethoxy)methyl)-2-(pyridin-4-yl)-3H-imidazo[4,5-b]pyridine, 13j.
Prepared by general procedure C using 4-bromopyridine. Purification by flash column chromatography (0–6% MeOH in CH2Cl2) yielded the product as a yellow oil (48 mg, 67%); 1H NMR: (500 MHz, CDCl3) δH 9.05 (bs, 2H), 8.47 (dd, J = 4.8, 1.4 Hz, 1H), 8.29–8.08 (m, 3H), 7.34 (dd, J = 8.0, 4.8 Hz, 1H), 5.82 (s, 2H), 4.00–3.93 (m, 2H), 3.64–3.58 (m, 2H) 3.39 (s, 3H), 13C NMR: (126 MHz, CDCl3) δC 152.1, 148.6, 144.7, 136.3, 134.2, 127.5, 119.1, 73.4, 71.0, 68.6, 58.5, 2 quaternary C's do not appear; LCMS tR = 2.04 min, m/z = 285 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C15H17N4O2 = 285.1346, found = 285.1348.
3-((2-Methoxyethoxy)methyl)-2-(pyridin-3-yl)-3H-imidazo[4,5-b]pyridine, 13k.
Prepared by general procedure B using 3-iodopyridine. Purification by flash column chromatography (0–6% MeOH in CH2Cl2) yielded product as a yellow oil (99 mg, 70%); 1H NMR: (500 MHz, CDCl3) δH 9.40 (bs, 1H), 8.83 (bs, 1H), 8.51 (d, J = 8.0 Hz, 1H), 8.46 (dd, J = 4.8, 1.3 Hz, 1H), 8.14 (dd, J = 8.0, 1.3 Hz, 1H), 7.57–7.46 (m, 1H), 7.34 (dd, J = 8.0, 4.8 Hz, 1H), 5.80 (s, 2H), 4.01–3.94 (m, 2H), 3.65–3.58 (m, 2H), 3.40 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 152.0, 150.7, 149.9, 148.5, 144.2, 136.4, 134.4, 127.2, 123.1, 118.9, 71.4, 71.0, 68.6, 58.5, one quaternary C does not appear; LCMS tR = 2.12 min, m/z = 285 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C15H17N4O2 = 285.1346, found = 285.1343.
3-((2-Methoxyethoxy)methyl)-2-(1-methyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine, 13l.
Prepared by general procedure C using 4-bromo-1-methyl-1H-pyrazole. Purification by flash column chromatography (0–5% MeOH in CH2Cl2) yielded product as a yellow oil (46 mg, 64%); 1H NMR: (500 MHz, CD3OD) δH 8.44 (bs, 1H), 8.38 (d, J = 4.8 Hz, 1H), 8.25 (bs, 1H), 8.14–8.01 (m, 1H), 7.41–7.33 (m, 1H), 5.84 (s, 2H), 4.05 (s, 3H) 3.87–3.80 (m, 2H), 3.60–3.52 (m, 2H), 3.31 (s, 3H); 13C NMR: (126 MHz, CD3OD) δC 143.5, 139.3, 134.3, 132.2, 125.9, 119.0, 111.4, 71.5, 71.1, 68.2, 57.6, 38.0, 2 quaternary C's do not appear; LCMS tR = 2.12 min, m/z = 288 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C14H18N5O2 = 288.1455, found = 288.1451.
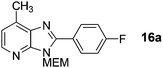
2-(4-Fluorophenyl)-3-((2-methoxyethoxy)methyl)-7-methyl-3H-imidazo[4,5-b]pyridine, 16a.
Intermediate S10 (34 mg, 0.154 mmol), Pd(OAc)2 (1.7 mg, 0.0077 mmol), CuI (88 mg, 0.462 mmol) and Cs2CO3 (125 mg, 0.385 mmol) were combined in a sealed tube and flushed with argon, before the addition of anhydrous DMF (0.8 mL) and 4-fluoroiodobenzene (35 μL, 0.308 mmol). The tube was sealed and heated to 140 °C for 14 h. The crude mixture was concentrated and purified on a Biotage SP1 (12 g SINGLE StEP column, 15 mL min−1, gradient 0–50% EtOAc in cyclohexane over 12 CV) to afford product as a cream solid (45 mg 93%). 1H NMR: (500 MHz, CDCl3) δH 8.28 (d, J = 4.8 Hz, 1H), 8.18–8.14 (m, 2H), 7.27–7.23 (m, 2H), 7.12 (dd, J = 4.8, 1.0 Hz, 1H), 5.73 (s, 2H), 3.97–3.95 (m, 2H), 3.61–3.59 (m, 2H), 3.40 (s, 3H), 2.75 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 164.2 (d, J = 251 Hz), 153.4, 148.4, 144.1, 139.2, 134.2, 132.0 (d, J = 8 Hz), 125.6, 120.3, 116.1 (d, J = 22 Hz), 72.0, 71.5, 68.9, 59.0, 16.4; 19F NMR: (500 MHz, CDCl3) δF −109.7; LCMS tR = 2.80 min, m/z = 316 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C17H19FN3O2 = 316.1461, found = 316.1459, (M + Na)+calculated for C17H18FN3O2Na = 338.1281, found = 338.1279.
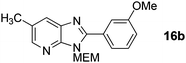
3-((2-Methoxyethoxy)methyl)-2-(3-methoxyphenyl)-6-methyl-3H-imidazo[4,5-b]pyridine, 16b.
Intermediate S8 (165 mg, 0.74 mmol), Pd(OAc)2 (8.3 mg, 0.037 mmol), CuI (422 mg, 2.2 mmol) and Cs2CO3 (601 mg, 1.85 mmol) were combined in a vial under an N2 atmosphere, DMF (3 mL) and 3-iodoanisole (176 μL, 1.48 mmol) were added and the mixture heated to 140 °C for 16 h. The mixture was concentrated in vacuo and purified by column chromatography (0–100% EtOAc in cyclohexane) to afford product as a yellow oil (166 mg, 69%); 1H NMR: (500 MHz, CDCl3) δH 8.23 (d, J = 1.8 Hz, 1H), 7.87 (d, J = 1.8 Hz, 1H), 7.71–7.65 (m, 2H), 7.43 (t, J = 8.0 Hz, 1H), 7.09–7.05 (m, 1H), 5.73 (s, 2H), 3.96–3.91 (m, 2H), 3.89 (s, 3H), 3.60–3.54 (m, 2H), 3.35 (s, 3H), 2.48 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 159.9, 155.3, 147.6, 144.9, 134.8, 130.8, 129.9, 128.6, 127.3, 121.9, 117.2, 114.2, 72.0, 71.6, 68.8, 58.9, 55.4, 18.6, LCMS tR = 2.92 min, m/z = 328 (M + H)+; purity (AUC) ≥ 95%; HRMS: (M + H)+calculated for C18H22N3O3 = 328.1656, found = 328.1663.
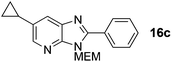
6-Cyclopropyl-3-((2-methoxyethoxy)methyl)-2-phenyl-3H-imidazo[4,5-b]pyridine, 16c.
Intermediate S9 (186 mg, 0.75 mmol), Pd(OAc)2 (8.4 mg, 0.0375 mmol), CuI (428 mg, 2.25 mmol) and Cs2CO3 (618 mg, 1.9 mmol) were combined in a vial under an N2 atmosphere, DMF (5 mL) and iodobenzene (168 μL, 1.50 mmol) were added and the mixture heated to 140 °C for 18 h. The crude mixture was concentrated in vacuo and purification by column chromatography (20%–80% EtOAc in cyclohexane) afforded product as an orange oil (152 mg, 63%). 1H NMR: (500 MHz, CDCl3) δH 8.26 (d, J = 2.1 Hz, 1H), 8.13–8.07 (m, 2H), 7.70 (d, J = 2.1 Hz, 1H), 7.56–7.48 (m, 3H), 5.72 (s, 2H), 3.98–3.87 (m, 2H), 3.62–3.53 (m, 2H), 3.37 (s, 3H), 2.06 (tt, J = 8.5, 5.1 Hz, 1H), 1.07–1.00 (m, 2H), 0.79–0.72 (m, 2H); 13C NMR: (126 MHz, CDCl3) δC 154.9, 147.2, 143.2, 134.5, 134.3, 129.9, 129.1, 128.3, 123.1, 71.5, 71.0, 68.3, 58.4, 12.7, 8.3, one quaternary C does not appear; LCMS tR = 2.49 min, m/z = 324 (M + H)+; purity (AUC) ≥ 95%; HRMS calculated for C19H22N3O2 = 324.1707, found = 324.1709.
3-((2-Methoxyethoxy)methyl)-2-(2-methoxyphenyl)-6-phenyl-3H-imidazo[4,5-b]pyridine, 16d.
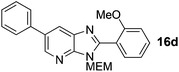
Intermediate S5 (210 mg, 0.74 mmol), Pd(OAc)2 (8.3 mg, 0.037 mmol), CuI (422 mg, 2.2 mmol) and Cs2CO3 (601 mg, 1.85 mmol) were combined in a vial under an N2 atmosphere, DMF (3 mL) and 2-iodoanisole (192 μL, 1.18 mmol) were added and the mixture heated to 140 °C for 16 h. The crude mixture was concentrated in vacuo and purification by column chromatography (10–100% EtOAc in cyclohexane) afforded product as a yellow oil (132 mg, 46%). 1H NMR: (500 MHz, CDCl3) δH 8.68 (d, J = 2.1 Hz, 1H), 8.27 (d, J = 2.1 Hz, 1H), 7.70–7.62 (m, 3H), 7.58–7.50 (m, 3H), 7.46–7.40 (m, 1H), 7.14 (td, J = 7.5, 1.0 Hz, 1H), 7.06 (dd, J = 8.4, 1.0 Hz, 1H), 5.72 (s, 2H), 3.86 (s, 3H), 3.65–3.57 (m, 2H), 3.45–3.36 (m, 2H), 3.28 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 147.4, 143.0, 138.6, 134.9, 132.4, 131.6, 131.6, 128.6, 127.1, 127.0, 125.3, 120.5, 110.7, 72.3, 70.9, 68.3, 58.4, 55.2, 3 quaternary C's not observed; LCMS tR = 2.53 min, m/z = 390 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C23H24N3O3 = 390.1812, found = 390.1823.
2-(4-Fluorophenyl)-3-((2-methoxyethoxy)methyl)-6-(1-methyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine, 16e.
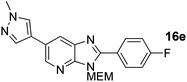
Intermediate S6 (60 mg, 0.209 mmol), Pd(OAc)2 (2.3 mg, 0.104 mmol), CuI (119 mg, 0.627 mmol), Cs2CO3 (170 mg, 0.522 mmol) were combined in a sealed tube flushed with argon gas, DMF (1 mL) and 4-fluoroiodobenzene (48 μL, 0.418 mmol) was added. The tube was heated to 140 °C for 14 h. The crude material was concentrated and purification on Biotage SP1 (12 g SINGLE StEP column, 15 mL min−1, 3 CV CH2Cl2, then gradient over 12CV of 0–100% (5% MeOH in CH2Cl2)) afforded a yellow oil of product (48 mg, 60%). 1H NMR: (500 MHz, CD3OD) δH 8.61 (s, 1H), 8.14 (s, 1H), 8.10 (dd, J = 8.4, 5.2 Hz, 2H), 8.07 (s, 1H), 7.91 (s, 1H), 7.34 (t, J = 8.4 Hz, 2H), 5.69 (s, 2H), 3.95 (s, 3H), 3.93–3.86 (m, 2H), 3.61–3.54 (m, 2H), 3.32 (s, 3H); 13C NMR: (126 MHz, CD3OD) δC 164.4 (d, J = 251.1 Hz), 142.1, 136.2, 131.8 (d, J = 8.7 Hz), 128.2, 125.3, 122.6, 120.1, 115.7 (d, J = 22.2 Hz), 72.0, 71.3, 68.6, 57.7, 37.7; 19F NMR: (500 MHz, CD3OD) δF −110.9; LCMS tR = 2.81 min, m/z = 382 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C20H21FN5O2 = 382.1674, found = 382.1672; (M + Na)+calculated for C20H20FN5O2Na = 404.1493, found = 404.1486.
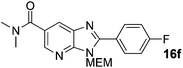
2-(4-Fluorophenyl)-3-((2-methoxyethoxy)methyl)-N,N-dimethyl-3H-imidazo[4,5-b]pyridine-6-carboxamide, 16f.
Intermediate S7 (35 mg, 0.126 mmol), Pd(OAc)2 (1.4 mg, 0.006 mmol), CuI (72 mg, 0.378 mmol), PCy3·HBF4 (4.6 mg, 0.013 mmol), and Cs2CO3 (102 mg, 0.315 mmol) were combined in a sealed tube flushed with argon. DMF (1.3 mL) and 4-fluoroiodobenzene (29 μL, 0.252 mmol) were added and the mixture heated to 140 °C for 16 h. The mixture was concentrated and purification by flash column chromatography (20–80% EtOAc in cyclohexane) afforded the product as a brown oil (34 mg, 72%). 1H NMR: (500 MHz, CDCl3) δH 8.54 (d, J = 1.8 Hz, 1H), 8.21–8.14 (m, 2H), 8.14 (d, J = 1.8 Hz, 1H), 7.30–7.23 (m, 2H), 5.77 (s, 2H), 3.99–3.93 (m, 2H), 3.64–3.58 (m, 2H), 3.40 (s, 3H), 3.19 (bs, 3H) 3.10 (bs, 3H); 13C NMR: (126 MHz, CDCl3) δC 169.6, 143.6, 131.9 (d, J = 9 Hz), 126.2, 116.2 (d, J = 21 Hz), 72.1, 71.4, 69.1, 59.1, 40.0 and 35.8, all aromatic CH's accounted for, 6 of 7 quaternary C's do not appear; 19F NMR: (500 MHz, CDCl3) δF −108.6; LCMS tR = 2.54 min, m/z = 373 (M + H)+; purity (AUC) = 93%; HRMS (M + H)+calculated for C19H22FN4O3 = 373.1670, found = 373.1667.
7-Chloro-3-((2-methoxyethoxy)methyl)-2-(4-methoxyphenyl)-3H-imidazo[4,5-b]pyridine, 16g.
Prepared according to general procedure B from intermediate 8a (as 1.0 M solution in DMF) and 4-iodoanisole, with added PivOH (15 mg, 0.15 mmol). Purification by flash column chromatography (0–50% EtOAc in CH2Cl2) yielded product as a cream solid (74 mg, 85%); 1H NMR: (500 MHz, CDCl3) δH 8.26 (d, J = 5.3 Hz, 1H), 8.16–8.11 (m, 2H), 7.31 (d, J = 5.3 Hz, 1H), 7.09–7.05 (m, 2H), 5.74 (s, 2H), 3.98–3.92 (m, 2H), 3.91 (s, 3H) 3.64–3.58 (m, 2H), 3.40 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 161.8, 156.1, 150.0, 143.8, 133.6, 133.0, 131.6, 121.3, 119.4, 114.4, 72.4, 71.5, 69.0, 59.1, 53.4, one quaternary C does not appear; LCMS tR = 2.90 min, m/z = 348 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C17H18ClN3O3 = 348.1109, found = 348.1112.
7-Chloro-2-(4-fluorophenyl)-3-((2-methoxyethoxy)methyl)-3H-imidazo[4,5-b]pyridine, 16h.
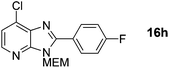
Prepared according to general procedure B from intermediate 8a (as 1.0 M solution in DMF) and 4-fluoroiodobenzene, with added PivOH (15 mg, 0.15 mmol). Crude product purified by flash column chromatography (0–50% EtOAc in cyclohexane) to afford product as a white solid (142 mg, 85%). 1H NMR: (500 MHz, CDCl3) δH 8.29 (d, J = 5.2 Hz, 1H), 8.22–8.14 (m, 2H), 7.33 (d, J = 5.2 Hz, 1H), 7.28–7.20 (m, 2H), 5.74 (s, 2H), 3.98–3.92 (m, 2H), 3.64–3.57 (m, 2H), 3.39 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 164.44 (d, J = 252.1 Hz), 155.1, 149.8, 144.3, 134.1, 132.8, 132.1 (d, J = 8.5 Hz), 125.1 (d, J = 3.5 Hz), 119.6, 116.1 (d, J = 21.8 Hz), 72.4, 71.4, 69.1, 59.1; 19F NMR: (500 MHz, CDCl3) δF −108.73; LCMS tR = 2.91 min, m/z = 336 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C16H16FClN3O2 = 336.0910, found = 336.0911.
7-Chloro-3-((2-methoxyethoxy)methyl)-2-(4-(methylsulfonyl)phenyl)-3H-imidazo[4,5-b]pyridine, 16i.
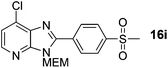
Prepared according to general procedure B from intermediate 8a (as 1.0 M solution in DMF) and 4-bromophenylmethyl sulfone, with added PivOH (15 mg, 0.15 mmol). Purification by flash column chromatography (0–50% EtOAc in CH2Cl2) yielded product as a yellow solid (275 mg, 69%); 1H NMR: (500 MHz, CDCl3) δH 8.44–8.39 (m, 2H), 8.35 (d, J = 5.2 Hz, 1H), 8.17–8.10 (m, 2H) 7.38 (d, J = 5.2 Hz, 1H), 5.77 (s, 2H), 3.99–3.94 (m, 2H), 3.63–3.58 (m, 2H) 3.39 (s, 3H), 3.13 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 153.7, 149.7, 145.2, 142.3, 134.9, 134.1, 132.8, 130.9, 127.9, 120.0, 72.4, 71.4, 69.3, 59.1, 44.5; LCMS tR = 2.55 min, m/z = 396 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C17H19ClN3O4S = 396.0785, found = 396.0772.
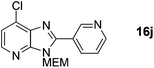
7-Chloro-3-((2-methoxyethoxy)methyl)-2-(pyridin-3-yl)-3H-imidazo[4,5-b]pyridine, 16j.
Prepared according to general procedure B from intermediate 8a (as 1.0 M solution in DMF) and 3-iodopyridine, with added PivOH (15 mg, 0.15 mmol). Purified by flash column chromatography (0–5% MeOH in CH2Cl2) affording product as a cream solid (110 mg, 69%). 1H NMR: (500 MHz, CDCl3) δH 9.38 (dd, J = 2.4, 0.8 Hz, 1H), 8.82 (dd, J = 4.8, 1.8 Hz, 1H), 8.50 (dt, J = 7.9, 1.8 Hz, 1H), 8.34 (d, J = 5.2 Hz, 1H), 7.51 (ddd, J = 7.9, 4.8, 0.8 Hz, 1H), 7.37 (d, J = 5.2 Hz, 1H), 5.77 (s, 2H), 3.99–3.90 (m, 2H), 3.64–3.56 (m, 2H), 3.39 (s, 3H); 13C NMR: (126 MHz, CDCl3) δC 153.2, 151.6, 150.6, 149.7, 144.9, 137.1, 134.6, 132.9, 125.4, 123.5, 119.8, 72.3, 71.4, 69.3, 59.08; LCMS tR = 2.45 min, m/z = 319 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C15H16ClN4O2 = 319.0956, found = 319.0956.
Synthesis of deprotected products 9 and 15b–l
General procedure D
The MEM-protected intermediate was taken in a 1:1 mixture of THF and 12 M HCl and stirred at rt for 16 h. The reaction mixture was neutralised with sat. aq. NaHCO3 and stirred for 1 h. Where product precipitated, this was collected by filtration and triturated with Et2O. Where product did not precipitate, the solution was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated in vacuo. The crude mixture was purified by column chromatography where required; conditions are given. Note: For all deprotected C2-aryl imidazo[4,5-b]pyridine products, carbon 7 does not appear in 13C NMR spectra, it can be visualised by taking the sample in DMSO-d6 and adding 1 drop of HCl.
2-(4-Methoxyphenyl)-3H-imidazo[4,5-b]pyridine, 9.
Prepared by general procedure D, product precipitated on neutralization and collected by filtration. Product was obtained as an off-white solid (312 mg, 98%). Lit m.p.9a 228–230 °C, observed m.p. 227–229 °C; 1H NMR: (500 MHz, CD3OD) δH 8.34 (d, J = 4.2, Hz, 1H), 8.16–8.11 (m, 2H), 7.98 (d, J = 7.9, Hz, 1H), 7.30 (dd, J = 8.0, 4.9 Hz, 1H), 7.17–7.07 (m, 2H), 3.91 (s, 3H); 13C NMR: (126 MHz, CD3OD) δC 162.2, 143.3, 128.5, 121.3, 118.1, 114.2, 54.6, C7 and four quaternary C's do not appear; LCMS tR = 1.88 min, m/z = 226 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C13H12N3O = 226.0975, found = 226.0975.
2-(4-Tolyl)-3H-imidazo[4,5-b]pyridine, 15b.
Prepared by general procedure D, product precipitated on neutralization and collected by filtration. Product was obtained as a white solid (86 mg, 86%). Lit. m.p. 261–262 °C;29 observed m.p. 252–255 °C; 1H NMR: (500 MHz, CDCl3) δH 13.76 (s, 1H), 8.45 (dd, J = 1.1, 4.9 Hz, 1H), 8.19 (m, 3H), 7.45 (d, J = 7.9 Hz, 2H), 7.34 (dd, J = 4.9, 8.0 Hz, 1H), 2.51 (3H, s); 13C NMR: (126 MHz, CDCl3) δC 154.0, 149.5, 142.6, 141.3, 137.2, 129.9, 127.0, 127.2, 127.6, 118.4, 21.6, C7 does not appear; LCMS tR = 1.81 min, m/z = 210 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C13H12N3 = 210.1026, found = 210.1025.
2-(4-Fluorophenyl)-3H-imidazo[4,5-b]pyridine, 15c.
Prepared by general procedure D, product precipitated on neutralization and collected by filtration. Product was obtained as a cream solid (53 mg, 97%). Lit. m.p. 289–290 °C;30 observed m.p. 285–287 °C; 1H NMR: (500 MHz, CD3OD) δH 8.38 (dd, J = 4.9, 1.4 Hz, 1H), 8.25–8.17 (m, 2H), 8.03 (dd, J = 8.0, 1.4 Hz, 1H), 7.37–7.30 (m, 3H); 13C NMR: (126 MHz, CD3OD) δC 164.5 (d, J = 250.7), 143.8, 129.2 (d, J = 8.9 Hz), 125.6, 118.4, 115.8 (d, J = 22.4 Hz), four C's do not appear; 19F NMR: (500 MHz, CD3OD) δF −111.1; LCMS tR = 1.97 min, m/z = 214 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C12H9FN3 = 214.0775, found = 214.0784; CHN Microanalysis calculated for C12H8FN3 = C, 67.60; H, 3.78; N, 19.71%, observed = C, 67.54; H, 3.78; N, 19.67%.
2-(4-Chlorophenyl)-3H-imidazo[4,5-b]pyridine, 15d.
Prepared by general procedure D, product precipitated on neutralization and collected by filtration. Product was obtained as a cream solid (15 mg, 50%). Lit. m.p. 300 °C;9a Observed 301–304 °C; 1H NMR: (500 MHz, CD3OD) δH 8.38 (dd, J = 4.8, 1.5 Hz, 1H), 8.19–8.14 (m, 2H), 8.03 (dd, J = 8.0, 1.5 Hz, 1H), 7.63–7.58 (m, 2H), 7.32 (dd, J = 8.0, 4.8 Hz, 1H); 13C NMR: (126 MHz, CD3OD) δC 143.8, 129.0, 128.3, 118.3, 100.0, five C's do not appear; LCMS tR = 2.51 min, m/z = 230 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C12H9ClN3 = 230.0480, found = 230.0482.
2-(4-(Trifluoromethyl)phenyl)-3H-imidazo[4,5-b]pyridine, 15e.
Prepared by general procedure D, crude material was purified by flash column chromatography (0–50% EtOAc in CH2Cl2), to yield product as a white solid (14 mg, 53%); m.p. 289–293 °C; 1H NMR: (500 MHz, CD3OD) δH 8.43 (dd, J = 4.8, 1.4 Hz, 1H), 8.36 (d, J = 8.1 Hz, 2H), 8.08 (dd, J = 8.0, 1.4 Hz, 1H), 7.90 (d, J = 8.2 Hz, 2H), 7.37 (dd, J = 8.0, 4.8 Hz, 1H); 13C NMR: (126 MHz, CD3OD) δC 144.4, 127.4, 125.8, 125.7, 118.8, C7 and five quaternary C's do not appear; 19F NMR: (500 MHz, CD3OD) δF −64.4; LCMS tR = 2.79 min, m/z = 264; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C13H9N3F3 = 264.0743, found = 264.0752; CHN Microanalysis calculated for C13H8F3N3 = C, 59.32; H, 3.06; N, 15.96%, observed = C, 59.29; H, 3.13; N, 16.03%.
2-(4-(Methylsulfonyl)phenyl)-3H-imidazo[4,5-b]pyridine, 15f.
Prepared by general procedure D, product precipitated on neutralization and collected by filtration. Product was obtained as a light orange solid (27 mg, 67%). Lit. m.p.31 286 °C, observed 268–270 °C; IR νmax/cm−1 1283s and 1266s (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR: (500 MHz, CD3OD) δH 8.47–8.42 (m, 2H), 8.39 (dd, J = 4.8, 1.5 Hz, 1H), 8.17–8.12 (m, 2H), 8.07 (dd, J = 8.0, 1.5 Hz, 1H), 7.32 (dd, J = 8.0, 4.8 Hz, 1H), 3.21 (s, 3H); 13C NMR: (126 MHz, CD3OD) δC 143.9, 127.8, 127.6, 118.2, 42.8, C7 and five quaternary C's do not appear; LCMS tR = 1.83 min, m/z = 274 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C13H12N3O2S = 274.0645, found = 274.0641.
O); 1H NMR: (500 MHz, CD3OD) δH 8.47–8.42 (m, 2H), 8.39 (dd, J = 4.8, 1.5 Hz, 1H), 8.17–8.12 (m, 2H), 8.07 (dd, J = 8.0, 1.5 Hz, 1H), 7.32 (dd, J = 8.0, 4.8 Hz, 1H), 3.21 (s, 3H); 13C NMR: (126 MHz, CD3OD) δC 143.9, 127.8, 127.6, 118.2, 42.8, C7 and five quaternary C's do not appear; LCMS tR = 1.83 min, m/z = 274 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C13H12N3O2S = 274.0645, found = 274.0641.
2-Phenyl-3H-imidazo[4,5-b]pyridine, 15g.
Prepared by general procedure D. Purified by flash column chromatography (20% EtOAc in CH2Cl2) to yield product as an off-white solid (136 mg, 81%). Lit. m.p. 288–290 °C;9a observed m.p. 283–285 °C; 1H NMR: (500 MHz, CD3OD) δH 8.39 (dd, J = 4.8, 1.0 Hz, 1H), 8.21–8.14 (m, 2H), 8.03 (dd, J = 8.0, 1.0 Hz, 1H), 7.64–7.55 (m, 3H), 7.33 (dd, J = 8.0, 4.9 Hz, 1H); 13C NMR: (126 MHz, CD3OD) δC 143.8, 130.8, 128.9, 126.8, 118.4, C7 and four quaternary C's do not appear; LCMS tR = 1.81, m/z = 196 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C12H10N3 = 196.0869, found = 196.0864.
2-(3-Fluorophenyl)-3H-imidazo[4,5-b]pyridine, 15h.
Prepared by general procedure D, product precipitated on neutralization and collected by filtration. Product was obtained as a white solid (54 mg, 90%). Lit. m.p. >300 °C,9a observed 271–274 °C; 1H NMR: (500 MHz, CD3OD) δH 8.40 (dd, J = 4.8, 1.4 Hz, 1H), 8.05 (dd, J = 8.0, 1.4 Hz, 1H), 8.01 (ddd, J = 0.9, 1.7, 7.8 Hz, 1H), 7.93 (ddd, J = 1.7, 2.4, 9.9 Hz, 1H), 7.61 (td, 5.3, 8.0 Hz, 1H) 7.38–7.29 (m, 2H); 13C NMR: (126 MHz, CD3OD) δC 164.5 (d, J = 250.7 Hz), 143.8, 129.2 (d, J = 8.9 Hz), 125.6, 118.4, 115.8 (d, J = 22.4 Hz), C7 and four quaternary C's do not appear; 19F NMR: (500 MHz, CD3OD) δF −113.9; LCMS tR = 2.16 min, m/z = 214.0 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C12H9FN3 = 214.0775, found = 214.0779.
2-(o-Tolyl)-3H-imidazo[4,5-b]pyridine, 15i.
Prepared by general procedure D, product precipitated on neutralization and collected by filtration. Product was obtained as a cream solid (56 mg, 76%). Observed m.p. 205–207 °C; 1H NMR: (500 MHz, CD3OD) δH 8.40 (d, J = 4.3, 1H), 8.06 (d, J = 7.8, Hz, 1H), 7.68 (d, J = 7.7 Hz, 1H), 7.51–7.32 (m, 4H) 1.02 (s, 3H); 13C NMR: (126 MHz, CD3OD) δC 143.5 (C5), 137.5, 136.8, 130.9, 130.1 129.5, 125.8, 118.2, 19.2, C7 and three quaternary C's do not appear; LCMS tR = 1.85 min, m/z = 210 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C13H12N3 = 210.1026, found = 210.1024.
2-(Pyridin-4-yl)-3H-imidazo[4,5-b]pyridine, 15j.
Prepared by general procedure D. Purification by flash column chromatography (0–8% MeOH in CH2Cl2) yielded product as a white solid (18 mg, 46%); Lit. m.p. 297 °C,32 observed m.p. 295–296 °C; 1H NMR: (500 MHz, CD3OD) δH 7.77 (dd, J = 4.6, 1.6 Hz, 2H), 8.46 (d, J = 4.8 Hz, 1H), 8.16 (dd, J = 4.6, 1.7 Hz, 2H), 8.11 (d, J = 7.8 Hz, 1H), 7.39 (dd, J = 8.1, 4.8 Hz, 1H); 13C NMR: (126 MHz, CD3OD) δC 151.3, 146.4, 138.9, 122.5, 120.6, C7 and three quaternary C's do not appear; LCMS tR = 1.28 min, m/z = 197 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C11H9N4 = 197.0822, found = 197.0819.
2-(Pyridin-3-yl)-3H-imidazo[4,5-b]pyridine, 15k.
Prepared by general procedure D. Purification by flash column chromatography (0–8% MeOH in CH2Cl2) yielded product as a white solid (20 mg, 38%); Lit m.p. 284 °C,30 observed m.p. 283–285 °C; 1H NMR: (500 MHz, DMSO-d6) δH 13.66 (bs, 1H), 9.34 (d, J = 1.8 Hz, 1H), 8.72 (dd, J = 4.9, 1.5 Hz, 1H), 8.61–8.53 (m, 1H), 8.46–8.38 (m, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.66 (dd, J = 8.0, 4.9 Hz, 1H), 7.36 (dd, J = 8.1, 4.9 Hz, 1H); 13C NMR: (126 MHz, CD3OD) δC 156.9, 152.1, 148.8, 145.8, 136.4, 127.5, 125.7, 120.2, C7 and two quaternary C's do not appear; LCMS (Method B) tR = 1.42 min, m/z = 197 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C11H9N4 = 197.0822, found = 197.0819; CHN Microanalysis calculated for C11H8N4 = C, 67.34; H, 4.11; N, 28.55%, observed = C, 67.29; H, 4.13; N, 28.49%.
2-(1-Methyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine, 15l.
Prepared by general procedure D. Purification by flash column chromatography (0–10% MeOH in CH2Cl2) yielded product as an off-white solid (19 mg, 48%); Observed m.p. 274–278 °C; 1H NMR: (500 MHz, CD3OD) δH 8.32 (bs, 1H), 8.29 (s, 1H), 8.13 (s, 1H), 7.95 (dd, J = 8.0, 1.2 Hz, 1H), 7.29 (dd, J = 8.0, 4.9 Hz, 1H), 4.01 (s, 3H); 13C NMR: (126 MHz, CD3OD) δC 151.6, 149.1, 142.8, 137.9, 130.9, 122.4, 118.1, 112.5, two quaternary C's do not appear; LCMS tR = 1.14 min, m/z = 200 (M + H)+; purity (AUC) ≥ 95%; HRMS (M + H)+calculated for C10H10N5 = 200.0931, found = 200.0929; CHN Microanalysis calculated for C10H9N5 = C, 60.29; H, 4.55; N, 35.16%, observed = C, 60.23; H, 4.59; N, 35.02%.
Acknowledgements
We thank Cancer Research UK (grants C309/A8274 (JM/VO), C309/A11566 (JB/VB)) for funding. We also thank Dr Maggie Liu, Dr Amin Mirza and Mr Meirion Richards assistance with NMR and LCMS analysis.
References
-
(a) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC;
(b) R. H. Crabtree, Chem. Rev., 2010, 110, 575–575 CrossRef CAS;
(c) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC;
(d) I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036–1045 RSC.
-
(a) D. Lapointe, T. Markiewicz, C. J. Whipp, A. Toderian and K. Fagnou, J. Org. Chem., 2011, 76, 749–759 CrossRef CAS;
(b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS;
(c) B. T. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826–1834 CrossRef.
-
(a) S. De Ornellas, T. E. Storr, T. J. Williams, C. G. Baumann and I. J. S. Fairlamb, Curr. Org. Synth., 2011, 8, 79–101 CrossRef CAS;
(b) O. Daugulis, Top. Curr. Chem., 2010, 292, 57–84 CrossRef CAS;
(c) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS.
-
(a) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS;
(b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2009, 110, 624–655 CrossRef.
- T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC.
-
(a) V. Bavetsias, S. Crumpler, C. Sun, S. Avery, B. Atrash, A. Faisal, A. S. Moore, M. Kosmopoulou, N. Brown, P. W. Sheldrake, K. Bush, A. Henley, G. Box, M. Valenti, A. de Haven Brandon, F. I. Raynaud, P. Workman, S. A. Eccles, R. Bayliss, S. Linardopoulos and J. Blagg, J. Med. Chem., 2012, 55, 8721–8734 CrossRef CAS;
(b) T. Wang, M. A. Block, S. Cowen, A. M. Davies, E. Devereaux, L. Gingipalli, J. Johannes, N. A. Larsen, Q. Su, J. A. Tucker, D. Whitston, J. Wu, H.-J. Zhang, M. Zinda and C. Chuaqui, Bioorg. Med. Chem. Lett., 2012, 22, 2063–2069 CrossRef CAS;
(c) Y. S. Cho, H. Angove, C. Brain, C. H.-T. Chen, H. Cheng, R. Cheng, R. Chopra, K. Chung, M. Congreve, C. Dagostin, D. J. Davis, R. Feltell, J. Giraldes, S. D. Hiscock, S. Kim, S. Kovats, B. Lagu, K. Lewry, A. Loo, Y. Lu, M. Luzzio, W. Maniara, R. McMenamin, P. N. Mortenson, R. Benning, M. O'Reilly, D. C. Rees, J. Shen, T. Smith, Y. Wang, G. Williams, A. J. A. Woolford, W. Wrona, M. Xu, F. Yang and S. Howard, ACS Med. Chem. Lett., 2012, 3, 445–449 CrossRef CAS;
(d) D. Chen, Y. Wang, Y. Ma, B. Xiong, J. Ai, Y. Chen, M. Geng and J. Shen, ChemMedChem, 2012, 7, 1057–1070 CrossRef CAS;
(e) V. Bavetsias, J. M. Large, C. Sun, N. Bouloc, M. Kosmopoulou, M. Matteucci, N. E. Wilsher, V. Martins, J. h. Reynisson, B. Atrash, A. Faisal, F. Urban, M. Valenti, A. de Haven Brandon, G. Box, F. I. Raynaud, P. Workman, S. A. Eccles, R. Bayliss, J. Blagg, S. Linardopoulos and E. McDonald, J. Med. Chem., 2010, 53, 5213–5228 CrossRef CAS.
- M. Mader, A. de Dios, C. Shih, R. Bonjouklian, T. Li, W. White, B. L. de Uralde, C. Sanchez-Martinez, M. del Prado, C. Jaramillo, E. de Diego, L. M. Martin Cabrejas, C. Dominguez, C. Montero, T. Shepherd, R. Dally, J. E. Toth, A. Chatterjee, S. Pleite, J. Blanco-Urgoiti, L. Perez, M. Barberis, M. J. Lorite, E. Jambrina, C. R. Nevill Jr., P. A. Lee, R. C. Schultz, J. A. Wolos, L. C. Li, R. M. Campbell and B. D. Anderson, Bioorg. Med. Chem. Lett., 2008, 18, 179–183 CrossRef CAS.
- S.-C. Lee, H. T. Kim, C.-H. Park, D. Y. Lee, H.-J. Chang, S. Park, J. M. Cho, S. Ro and Y.-G. Suh, Bioorg. Med. Chem. Lett., 2012, 22, 4221–4224 CrossRef CAS.
-
(a) R. P. Kale, M. U. Shaikh, G. R. Jadhav and C. H. Gill, Tetrahedron Lett., 2009, 50, 1780–1782 CrossRef CAS;
(b) V. Bavetsias, C. Sun, N. Bouloc, J. Reynisson, P. Workman, S. Linardopoulos and E. McDonald, Bioorg. Med. Chem. Lett., 2007, 17, 6567–6571 CrossRef CAS;
(c) S.-Y. Lin, Y. Isome, E. Stewart, J.-F. Liu, D. Yohannes and L. Yu, Tetrahedron Lett., 2006, 47, 2883–2886 CrossRef CAS.
- A. J. Rosenberg, J. Zhao and D. A. Clark, Org. Lett., 2012, 14, 1764–1767 CrossRef CAS.
- A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 1036–1041 CrossRef CAS.
-
(a) T. E. Storr, C. G. Baumann, R. J. Thatcher, S. De Ornellas, A. C. Whitwood and I. J. S. Fairlamb, J. Org. Chem., 2009, 74, 5810–5821 CrossRef CAS;
(b) I. Čerňa, R. Pohl, B. Klepetářová and M. Hocek, J. Org. Chem., 2008, 73, 9048–9054 CrossRef;
(c) I. Čerňa, R. Pohl and M. Hocek, Chem. Commun., 2007, 4729–4730 RSC;
(d) F. Bellina, C. Calandri, S. Cauteruccio and R. Rossi, Tetrahedron, 2007, 63, 1970–1980 CrossRef CAS.
-
(a) V. O. Iaroshenko, D. Ostrovskyi, M. Miliutina, A. Maalik, A. Villinger, A. Tolmachev, D. M. Volochnyuk and P. Langer, Adv. Synth. Catal., 2012, 354, 2495–2503 CrossRef CAS;
(b) N. Barbero, R. SanMartin and E. Dominguez, Org. Biomol. Chem., 2010, 8, 841–845 RSC.
- V. O. Iaroshenko, I. Ali, S. Mkrtchyan, V. Semeniachenko, D. Ostrovski and P. Langer, Synlett, 2012, 2603–2608 CrossRef CAS.
- M. P. Singh, Y. Bathini and J. W. Lown, Heterocycles, 1993, 36, 971–985 CrossRef CAS.
- I. Čerňa, R. Pohl, B. Klepetářová and M. Hocek, Org. Lett., 2006, 8, 5389–5392 CrossRef.
- F. Bellina, S. Cauteruccio and R. Rossi, Eur. J. Org. Chem., 2006, 1379–1382 CrossRef CAS.
-
(a) B. Sezen and D. Sames, J. Am. Chem. Soc., 2006, 128, 8364–8364 Search PubMed;
(b) B. Sezen and D. Sames, J. Am. Chem. Soc., 2003, 125, 5274–5275 CrossRef CAS.
- S. Pivsa-Art, T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1998, 71, 467–473 CrossRef CAS.
- X.-M. Yan, X.-R. Mao and Z.-Z. Huang, Heterocycles, 2011, 83, 1371–1376 CAS.
- N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926–2927 CrossRef CAS.
- L.-C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581–590 CrossRef CAS.
- F. Bellina, S. Cauteruccio, L. Mannina, R. Rossi and S. Viel, Eur. J. Org. Chem., 2006, 693–703 CrossRef CAS.
- T. E. Storr, A. G. Firth, K. Wilson, K. Darley, C. G. Baumann and I. J. S. Fairlamb, Tetrahedron, 2008, 64, 6125–6137 CrossRef CAS.
-
(a) H.-Y. Sun, S. I. Gorelsky, D. R. Stuart, L.-C. Campeau and K. Fagnou, J. Org. Chem., 2010, 75, 8180–8189 CrossRef CAS;
(b) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef;
(c) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS;
(d) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS.
- S. I. Gorelsky, Organometallics, 2012, 31, 794–797 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51(13), 3066–3072 CrossRef CAS.
- D. L. Garmaise and J. Komlossy, J. Org. Chem., 1964, 29, 3403–3405 CrossRef CAS.
- Y. M. Yutilov and L. I. Shcherbina, Chem. Heterocycl. Compd., 1987, 23, 529–535 CrossRef.
-
E. Kutter, V. Austel and W. Diederen, US Patent3985891, 1976 Search PubMed.
- J. J. Baldwin, P. K. Lumma, F. C. Novello, G. S. Ponticello, J. M. Sprague and D. E. Duggan, J. Med. Chem., 1977, 20, 1189–1193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic schemes to key intermediates, supplementary tables, data from kinetic isotope experiments and copies of spectroscopic data for new compounds is available free of charge. See DOI: 10.1039/c3ob27477b |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.