 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free syn-dioxygenation of alkenes
Michael J.
Rawling
and
Nicholas C. O.
Tomkinson
*
WestCHEM, Department of Pure and Applied Chemistry, Thomas Graham Building, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: Nicholas.Tomkinson@strath.ac.uk; Fax: +44 (0) 141 5485743; Tel: +44 (0) 141 5482276
First published on 11th January 2013
Abstract
Reactions employing inexpensive reagents from sustainable sources and with low toxicity are becoming increasingly desirable from an academic and industrial perspective. A fascinating example of a synthetic transformation that requires development of alternative procedures is the osmium catalysed dihydroxylation. Recently there has been considerable interest in achieving this reaction through metal-free procedures. This review describes the methods available for metal-free syn-dioxygenation of alkenes.
 Michael J. Rawling | Mike Rawling was born in Plymouth, England, UK in 1987. He studied Chemistry at The University of Sheffield where he was awarded an EPSRC Summer Vacation Bursary to carry out research under the supervision of Dr Simon Jones. He then completed his final year research project with Professor Nicholas Williams, receiving his MChem in 2009. An interest in organocatalysis led him to undertake a PhD with Professor Nick Tomkinson at Cardiff University. This included a three-month placement at GlaxoSmithKline, Stevenage, UK, under the supervision of Matthew Campbell. He is now completing his PhD at The University of Strathclyde (2009–present). |
 Nicholas C. O. Tomkinson | Nick Tomkinson was born in St Andrews, Scotland in 1969. He studied Chemistry at The University of Sheffield and received his BSc in 1992 and PhD in 1996 under the supervision of Dr D. Neville Jones and Professor Jim Anderson. After postdoctoral studies with Dr Tim Willson at GlaxoSmithKline, Research Triangle Park, North Carolina (1996–1998), he was appointed to the staff at Cardiff University in 1999. In June 2011 he took up a position in the Department of Pure and Applied Chemistry at the University of Strathclyde. His research interests are centred on the development of practical synthetic methodology. |
1 Introduction
The Sharpless asymmetric dihydroxylation is one of the most celebrated, influential and inspiring synthetic methods developed.1 The broad substrate scope, high yields, exceptional levels of asymmetric induction, and convenient reaction conditions renders the process both robust and practical. Despite this success, the high cost and toxicity of osmium together with limited availability provide the impetus to develop new and alternative methods. In response to this requirement the community has delivered a number of transition-metal catalysed processes but none have yet reached the gold-standard required.2Along with transition-metal catalysed reactions there have been a number of recent successes in the development of metal-free methods. Although a general metal-free catalytic asymmetric alkene syn-dihydroxylation remains an elusive and attractive challenge a large body of work has been directed towards achieving this ultimate goal. This review outlines progress towards this target.
2 Peroxides
The use of organic peroxides for metal-free syn-dihydroxylation of alkenes has received much attention in recent years. The inherent high reactivity of peroxides together with their low cost and often non-toxic nature mean that reactions are frequently efficient and allow for effective procedures that do not require the use of transition-metal catalysts to be developed.2.1 Malonoyl peroxides
A promising area is the use of cyclic diacyl peroxides and in particular malonoyl peroxides.3 Cyclopropyl malonoyl peroxide 3 has been shown to be an effective reagent for the syn-dihydroxylation of a series of stabilised alkenes containing a wide range of functionality (22 examples) (Scheme 1).4 Treatment of an alkene with just 1.2 equivalents of peroxide 3 in the presence of 1 equivalent of water followed by basic hydrolysis leads to the corresponding syn-diol in excellent yield and very high levels of diastereoselectivity.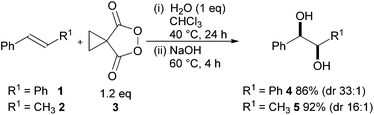 | ||
| Scheme 1 Cyclopropyl malonoyl peroxide 3 mediated dihydroxylation. | ||
The proposed mechanistic course for the reaction is outlined in Scheme 2. Reaction of alkene 1 and peroxide 3 leads to 6 which undergoes ring closure, forming dioxonium 7. Hydrolysis with the molecule of water necessary for reaction gives observed ester 9. Basic hydrolysis of 9 affords diol 4 (86%) and diacid 10, which can be recovered and converted into peroxide 3 in one simple synthetic step.
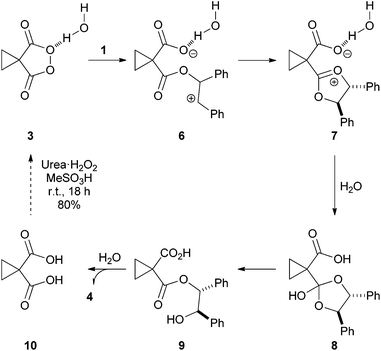 | ||
| Scheme 2 Proposed pathway for the cyclopropyl malonoyl peroxide mediated dihydroxylation of stilbene. | ||
In a related study it was shown that the overall rate of this reaction could be controlled through modification of the peroxide structure.5 Although these reactions proceed with lower yield and stereoselectivity than reaction with cyclopropyl malonoyl peroxide 3 the ability to control the overall rate of reaction could have important implications in the development of a catalytic procedure.6
2.2 Phthaloyl peroxides
In a series of largely neglected reports,7 Greene showed that trans-stilbene 1 reacted with phthaloyl peroxide 11 to give two dioxygenated products 12 and 13 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio. Alkaline hydrolysis of 12 and 13 leads to (±)-hydrobenzoin 4 (Scheme 3).8 Significantly, this syn-dihydroxylation was shown to be stereospecific for the reaction of both cis- and trans-stilbene providing a powerful piece of synthetic methodology.
:3 ratio. Alkaline hydrolysis of 12 and 13 leads to (±)-hydrobenzoin 4 (Scheme 3).8 Significantly, this syn-dihydroxylation was shown to be stereospecific for the reaction of both cis- and trans-stilbene providing a powerful piece of synthetic methodology.
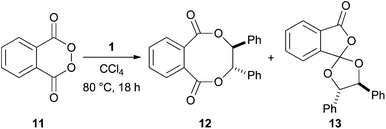 | ||
| Scheme 3 Phthaloyl peroxide dioxygenation of stilbene 1. | ||
Greene did not develop this methodology further, presumably because of the explosive nature of the phthaloyl peroxide 11. Siegel realised the potential of this method and recently reported on the improved reactivity of 3,4-dichlorophthaloyl peroxide 14 (20 examples) (Scheme 4).9
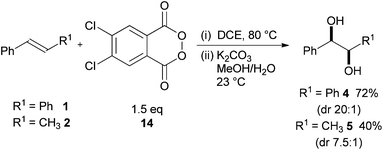 | ||
| Scheme 4 3,4-Dichlorophthaloyl peroxide 14 dihydroxylation of alkenes. | ||
The reaction was simple to perform although elevated temperatures of 80 °C were required and 14 showed similar instability to 11. 3,4-Dichlorophthaloyl peroxide 14 was shown to dihydroxylate a range of nucleophilic alkenes containing a variety of functionality with good diastereoselectivity. The reactions were frequently lower yielding and exhibited poorer diastereoselectivity when compared to cyclopropyl malonoyl peroxide 3 although the reaction of selected challenging aliphatic substrates was described.
2.3 Oxone
It is well established that Oxone is an effective reagent for the dihydroxylation of alkenes with anti-selectivity.10 De Kimpe and co-workers have described an unprecedented example of Oxone mediated syn-dioxygenation of mollugin 15 (Scheme 5).11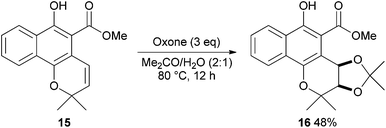 | ||
| Scheme 5 Oxone mediated syn-dioxygenation of mollugin 15. | ||
Under specific conditions, mollugin 15 reacts with Oxone to produce cis-3,4-dihydroxymollugin acetonide 16 (48%) preferentially over the trans isomer. Protection of the phenol group in 15 prior to reaction leads to formation of the expected trans-diol illustrating that this method is highly substrate dependent. However, application of this method as a more general procedure for syn-dioxygenation presents significant opportunity.
3 Hypervalent iodine
Iodine reagents are attractive because of their low toxicity, ready availability and ease of handling and there have been a number of reviews on their applications in synthesis.12 The use of hypervalent iodine compounds in the metal-free dioxygenation of alkenes has undergone a renaissance over recent years.3.1 Achiral hypervalent iodine reagents
The first reported use of hypervalent iodine in the dioxygenation of alkenes was in 1939 when Criegee and Beucker showed that (diacetoxyiodo)arenes 17, ArI(OAc)2, could be used in the syn-diacetoxylation of trans-anethole and cyclopentadiene.13 In the following 50 years, analogous syn-ditosyloxylation,14syn-trifluoroacetoxylation15 and syn-methoxylation/perchlorination16 reactions have been described using alternative hypervalent iodine reagents 18–20 (Scheme 6).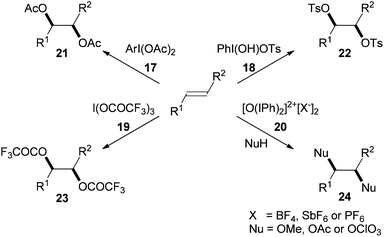 | ||
| Scheme 6 Early examples of hypervalent iodine reagents in the dioxygenation of alkenes. | ||
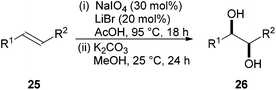
The Woodward17 and Prévost18 reactions represent established methods for the syn- and anti-dihydroxylation of alkenes respectively. A major drawback of these transformations is the requirement for a stoichiometric amount of silver salt. A synthetically useful alternative for syn-dihydroxylation was reported by Sudalai and co-workers in 200519 using NaIO4 as the oxidant (Scheme 7). Reaction of an alkene 25 with catalytic amounts of lithium bromide using sodium periodate or (diacetoxyiodo)benzene as the oxidant in wet acetic acid gives the syn-diol 26 in high yield after basic hydrolysis (21 examples).
 | ||
| Scheme 7 Hypervalent iodine mediated Woodward–Prévost dihydroxylation. | ||
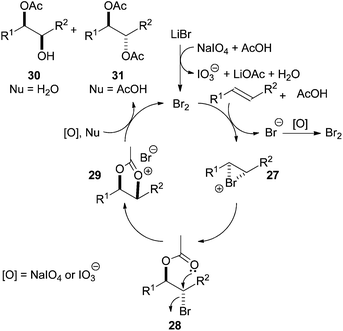
Mechanistically it was proposed that Br2, generated in situ by oxidation of lithium bromide, brings about the bromoacetoxylation of alkenes via bromonium ion 27 to give the trans-1,2-bromoacetate derivative 28. Intramolecular cyclisation gives 1,3-dioxolan-2-yl cation 29, which is hydrolysed (H2O) to give syn-hydroxy acetate 30 (Scheme 8). Interestingly, reaction in dry acetic acid leads to the trans-diacetate 31.
 | ||
| Scheme 8 Proposed catalytic cycle for hypervalent iodine mediated Woodward–Prévost dihydroxylation. | ||
The reaction works well for electron rich and electron deficient alkenes (21 examples), leading to the corresponding syn-diols in excellent yield and good diastereoselectivity. The reaction is simple to perform and all reagents are commercially available but the elevated reaction temperature (95 °C) and the solvent (acetic acid) mean the process is not suitable for more sensitive substrates.
Further improvement to this protocol was reported by Li and co-workers who addressed the issue of high temperature by employing PhI(OAc)2 in the presence of BF3·OEt220 resulting in a scalable, convenient and practical procedure for the diastereoselective syn-diacetoxylation of alkenes (19 examples). Interestingly, the reaction was also effective for electron deficient alkenes. For example, ethyl cinnamate 32 gave the syn-dioxygenation product 33 in 97% yield and >19:1 diastereoselectivity (Scheme 9).
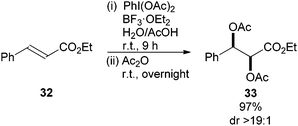 | ||
| Scheme 9 BF3·OEt2 catalysed diacetoxylation of alkenes with PhI(OAc)2. | ||
The reaction proved general for a range of alkenes producing the corresponding syn-diacetates in good to excellent yield and diastereoselectivity when wet acetic acid was used as the solvent. It was proposed that BF3·OEt2 activated PhI(OAc)2 through a Lewis acid coordination pathway. However, Gade21 demonstrated that protons can catalyse dioxygenations when using PhI(OAc)2 as the oxidant, so it is also possible that a strong Brønsted acid produced by the BF3·OEt2/AcOH could also be responsible for catalysis. The possibility of preparing the anti-diacetate by performing the reaction in a mixture of AcOH–Ac2O further enhances the appeal of this reaction sequence.
An interesting extension to the use of (diacetoxyiodo)benzene in the syn-dioxygenation of alkenes comes from the reaction of amines (12 examples).22 For example, PhI(OAc)2 mediated oxidation of N-phenylpiperidine 34 gives the corresponding enamine 35 which is dioxygenated in situ to give 36 in 42% yield (Scheme 10). Although 2.2 equivalents of oxidant are required, the potential to expand the scope of this transformation further is significant.
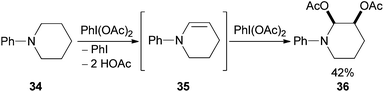 | ||
| Scheme 10 Selective functionalisation of amines with PhI(OAc)2. | ||
Çelik23 reported that alkene syn-dioxygenation can be achieved using the more reactive phenyliodine(III) bis(trifluoroacetate) in the absence of additive or catalyst (Scheme 11). Although the examples reported were limited and functional group tolerance was not explored the overall transformation clearly has potential for development, particularly for substrates not tolerant of the acidic Woodward–Prévost conditions described above.
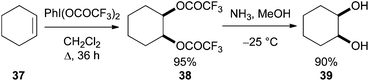 | ||
| Scheme 11 Alkene dihydroxylation using PhI(OCOCF3)2. | ||
3.2 Chiral hypervalent iodine reagents
Although there is significant precedent for the use of stoichiometric chiral hypervalent iodine reagents in synthesis, it is only recently that application of this knowledge to alkene dihydroxylation has been described. Fujita reported an asymmetric variant of the Woodward and Prévost reactions using optically active hypervalent iodine reagents 40–43 (Scheme 12).24,25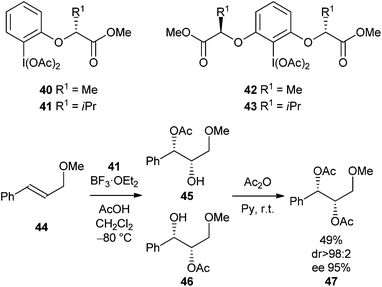 | ||
| Scheme 12 Enantioselective alkene dioxygenation. | ||
Reaction of alkene 44 with hypervalent iodine reagent 41 (1.2 equivalents) at −80 °C, quenching with water at −40 °C (Conditions A) gave a mixture of hydroxyacetate products 45 and 46 which were directly acetylated to give 47 (dr > 98:2, 95% ee) (Scheme 12). In an additional experiment it was shown that enantioselectivity could be switched by performing the reaction at −80 °C in the presence of TMSOAc, followed by quenching at room temperature (Conditions B) (Scheme 13).
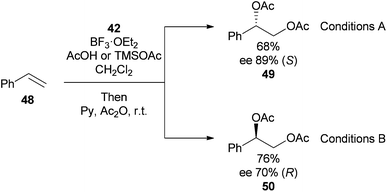 | ||
| Scheme 13 Switching enantioselectivity in the dihydroxylation of styrene. | ||
Although the ee's observed with these reagents are lower than would be expected by contemporary standards, the ability to alter both stereoselectivity and enantioselectivity simply by altering reaction conditions is particularly exciting and bodes well for further development in the area of alkene dioxygenation using hypervalent iodine reagents.
More recently Fujita has gone on to show that chiral hypervalent iodine reagent 42 can also be used in the enantioselective oxylactonisation of methyl ortho-alk-1-enylbenzoate 51 in 90% ee (Scheme 14).26
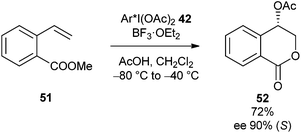 | ||
| Scheme 14 Enantioselective acetoxylactonisation. | ||
3.3 Catalytic hypervalent iodine reagents
Over recent years, a significant understanding of how to use sub-stoichiometric amounts of hypervalent iodine reagents has emerged.27 This knowledge has been applied by Li and co-workers to the alkene dihydroxylation described above (Scheme 9) providing an effective organocatalytic alkene dihydroxylation.28 Treatment of an alkene 25 with 0.2 equivalents of the aryl iodide 53 in the presence of either hydrogen peroxide or mCPBA as the stoichiometric oxidant followed by direct acetylation of the product leads to 54 (Scheme 15). Although functional group tolerance was not widely explored within this work, the 25 examples reported suggest the method has outstanding potential and the development of a catalytic asymmetric protocol should soon be forthcoming.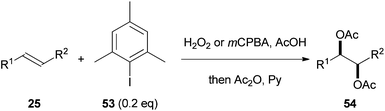 | ||
| Scheme 15 Organocatalytic diacetoxylation of alkenes. | ||
4 Selenium
4.1 Selenium dioxide
The use of selenium reagents in the syn-dioxygenation of alkenes was first reported by Tsutsumi and co-workers whilst they were investigating the acid-catalysed oxidation of alkenes.29 They described the stereoselective oxidation of cis-but-2-ene 55 and trans-but-2-ene 57 to give the diacetates 56 and 58 respectively as the major reaction products, along with small amounts of the corresponding syn-monoacetates (<5%) (Scheme 16). Although yields of the products were low, the mild conditions and high selectivity provide an excellent starting point for those wishing to develop this transformation further.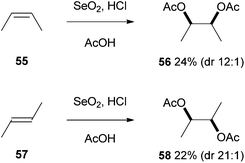 | ||
| Scheme 16 Selenium dioxide mediated syn-diacetoxylation. | ||
More recent reports by Nguyen and Lee examined selenium dioxide in the dioxygenation of diene substrates to prepare both 1,2- and 1,4-diols.30 Once again this methodology has not been significantly developed suggesting that oxidation reactions with selenium dioxide may have a high substrate dependence and lack of generality.
4.2 Diaryl diselenide
A more general selenium-mediated dihydroxylation method was published by Tiecco in 2008. This report contained the first example of a diphenyl diselenide catalysed dihydroxylation of alkenes using ammonium persulphate or hydrogen peroxide as the stoichiometric oxidant (Scheme 17).31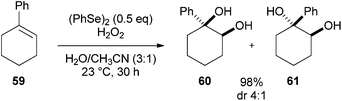 | ||
| Scheme 17 Diphenyl diselenide catalysed dihydroxylation. | ||
It was proposed that this one-pot procedure occurred by oxidation of diphenyl diselenide 62 to perseleninic acid 64, which epoxidised the alkene substrate giving 65. SN2 ring-opening of 65 by a molecule of water leads to the anti-diol 68 (Path B). SN1 ring-opening of 65 (Path A) provided both the syn (67) and anti (68) diols. It was rationalised that hydrogen bonding between the water molecule and the hydroxyl group of intermediate 66 was responsible for the preferential syn-addition of the water molecule with selected substrates (Scheme 18).
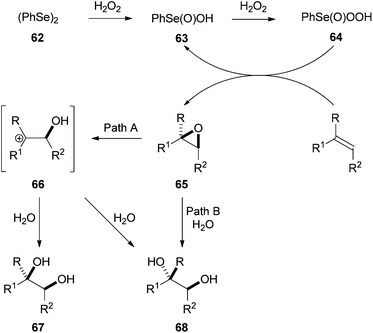 | ||
| Scheme 18 Proposed mechanism for the diphenyl diselenide catalysed dihydroxylation. | ||
Despite the reaction proceeding in good yield, none of the substrates reported contained functionality suggesting the reaction may have limited scope. Further drawbacks of this method include long reaction times (up to 12 days) and the poor stereoselectivities observed. Nevertheless this represents a novel catalytic process for the syn-dihydroxylation of alkenes.
4.3 Chiral diaryl diselenide
Significantly, Tiecco went on to show that use of chiral diselenide 69 in the reaction with 1-phenyl cyclohexene 59 gave the syn-diol 60 in an excellent 92% ee (Scheme 19). This is a particularly significant result in the field of dihydroxylation although substrate scope appears to be limited from the examples reported.32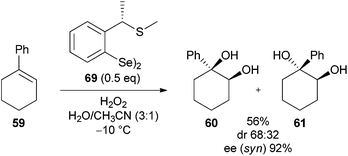 | ||
| Scheme 19 Catalytic asymmetric dihydroxylation using chiral diselenide. | ||
Organoselenium compounds clearly have great potential as convenient syn-dihydroxylation reagents but important mechanistic understanding and incorporation of functionality into substrates is required before a general protocol is established.
5 Sulphur
The use of sulphur in alkene syn-dihydroxylation is rare but an exciting report by Yoshida suggests that this area has potential for development. The method involves an oxidative syn-dihydroxylation mediated by electrochemically generated alkoxysulfonium ions (Scheme 20).33 Five stabilised alkenes were oxidised in the presence of DMSO to give bisalkoxysulfonium ions 71 which underwent rapid hydrolysis with aqueous sodium hydroxide to afford the corresponding diols in good yield (52–86%). It was proposed that preference for syn-addition arises from sulfonium ion directed attack of the second molecule of DMSO on 70 to give 71. Although electrochemical dioxygenation of alkenes has been known for some time,34 this represents the first example of a direct electrochemical dihydroxylation of alkenes. Expanding the substrate scope beyond stabilised alkenes and improving diastereoselectivity would increase the significance of this work.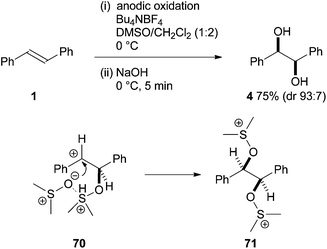 | ||
| Scheme 20 Electrochemical syn-dihydroxylation. | ||
6 Conclusions
Recent years have seen a substantial amount of activity in the area of metal-free syn-dioxygenation of alkenes. This review has highlighted advances using peroxides, hypervalent iodine, selenium and sulphur as vehicles to deliver the oxygen atoms to the alkene. Whilst significant strides have been made, the bench-mark for alkene dihydroxylation, the osmium catalysed Upjohn procedure35 and its asymmetric version developed by Sharpless1 are truly outstanding synthetic procedures that represent the gold-standard for synthetic transformations when considering scope, selectivity, yield and versatility. With such formidable precedent, it is an exciting time for the metal-free methods being developed where catalytic and asymmetric variants are starting to be described. Further understanding of mechanism, reactivity and scope will be central to advancement in this area to address the pressing needs for this pivotal and industrially relevant transformation.Notes and references
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS.
- For a recent review on osmium-free syn-dihydroxylation see: C. J. R. Bataille and T. J. Donohoe, Chem. Soc. Rev., 2011, 40, 114 RSC.
- M. Schwarz and O. Reiser, Angew. Chem., Int. Ed., 2011, 50, 10495 CrossRef CAS.
- J. C. Griffith, K. M. Jones, S. Picon, M. J. Rawling, B. M. Kariuki, M. Campbell and N. C. O. Tomkinson, J. Am. Chem. Soc., 2010, 132, 14409 CrossRef CAS.
- K. M. Jones and N. C. O. Tomkinson, J. Org. Chem., 2012, 77, 921 CrossRef CAS.
- S. Picon, M. Rawling, M. Campbell and N. C. O. Tomkinson, Org. Lett., 2012, 14, 6250 CrossRef CAS.
- F. D. Greene, J. Am. Chem. Soc., 1956, 78, 2246 CrossRef CAS; F. D. Greene, J. Am. Chem. Soc., 1956, 78, 2250 CrossRef; F. D. Greene and W. W. Rees, J. Am. Chem. Soc., 1958, 80, 3432 CrossRef; F. D. Greene, J. Am. Chem. Soc., 1959, 81, 1503 CrossRef; F. D. Greene and W. W. Rees, J. Am. Chem. Soc., 1960, 82, 890 CrossRef; F. D. Greene and W. W. Rees, J. Am. Chem. Soc., 1960, 82, 893 CrossRef.
- A. Tadokoro, T. Takata and T. Endo, Macromolecules, 1993, 26, 2388 CrossRef CAS.
- C. Yuan, A. Axelrod, M. Varela, L. Danysh and D. Siegel, Tetrahedron Lett., 2011, 52, 2540 CrossRef CAS.
- W. Zhu and W. T. Ford, J. Org. Chem., 1991, 56, 7022 CrossRef CAS; S. Rani and Y. D. Vankar, Tetrahedron Lett., 2003, 44, 907 CrossRef.
- N. V. S. Mudiganti, S. Claessens, P. Habonimana and N. De Kimpe, J. Org. Chem., 2008, 73, 3867 CrossRef CAS.
- T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656 CrossRef CAS; H. Liang and M. A. Ciufolini, Angew. Chem., Int. Ed., 2011, 50, 11849 CrossRef; L. F. Silva, Jr. and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722 RSC.
- R. Criegee and H. Beucker, Justus Liebigs Ann. Chem., 1939, 541, 218 CrossRef CAS.
- L. Rebrovic and G. F. Koser, J. Org. Chem., 1984, 49, 2462 CrossRef CAS.
- J. Buddrus, Angew. Chem., Int. Ed. Engl., 1973, 12, 163 CrossRef CAS; J. Buddrus and H. Plettenberg, Chem. Ber., 1980, 113, 1494 CrossRef.
- V. V. Zhdankin, R. Tykwinski, B. Berglund, M. Mullikin, R. Caple, N. S. Zefirov and A. S. Koz'min, J. Org. Chem., 1989, 54, 2609 CrossRef CAS.
- R. B. Woodward and F. V. Brutcher, Jr., J. Am. Chem. Soc., 1958, 80, 209 CrossRef CAS.
- C. Prevost, Compt. Rend., 1933, 196, 1129 CAS; C. Prevost, Compt. Rend., 1933, 197, 1661 Search PubMed; C. Prevost and J. Wiemann, Compt. Rend., 1937, 204, 700 Search PubMed.
- L. Emmanuvel, T. M. A. Shaikh and A. Sudalai, Org. Lett., 2005, 7, 5071 CrossRef CAS.
- W. Zhong, J. Yang, X. Meng and Z. Li, J. Org. Chem., 2011, 76, 9997 CrossRef CAS.
- Y.-B. Kang and L. H. Gade, J. Am. Chem. Soc., 2011, 133, 3658 CrossRef CAS.
- X.-Z. Shu, X.-F. Xia, Y.-F. Yang, K.-G. Ji, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2009, 74, 7464 CrossRef CAS.
- M. Çelik, C. Alp, B. Coşkun, M. S. Gültekin and M. Balci, Tetrahedron Lett., 2006, 47, 3659 CrossRef.
- M. Fujita, M. Wakita and T. Sugimura, Chem. Commun., 2011, 47, 3983 RSC . Diastereoselective Prévost and Woodward reactions of chiral alkene substrates have been reported, see: J. H. Kim, M. J. C. Long, J. Y. Kim and K. H. Park, Org. Lett., 2004, 6, 2273 CrossRef CAS; J. H. Kim, M. J. Curtis-Long, W. D. Seo, Y. B. Ryu, M. S. Yang and K. H. Park, J. Org. Chem., 2005, 70, 4082 CrossRef; A. D'Alfonso, M. Pasi, A. Porta, G. Zanoni and G. Vidari, Org. Lett., 2010, 12, 596 CrossRef.
- The use of chiral hypervalent iodine reagents in the dioxygenation of styrene was first reported by Wirth et al., but low enantioselectivity was observed. See: T. Wirth and U. H. Hirt, Tetrahedron: Asymmetry, 1997, 8, 23 CrossRef CAS; U. H. Hirt, B. Spingler and T. Wirth, J. Org. Chem., 1998, 63, 7674 CrossRef; U. H. Hirt, M. F. H. Schuster, A. N. French, O. G. Wiest and T. Wirth, Eur. J. Org. Chem., 2001, 1569 CrossRef.
- M. Fujita, Y. Yoshida, K. Miyata, A. Wakisaka and T. Sugimura, Angew. Chem., Int. Ed., 2010, 49, 7068 CrossRef CAS . For analogous asymmetric cycloetherification reactions, see: M. Fujita, S. Okuno, H. J. Lee, T. Sugimura and T. Okuyama, Tetrahedron Lett., 2007, 48, 8691 CrossRef; M. Fujita, Y. Ookubo and T. Sugimura, Tetrahedron Lett., 2009, 50, 1298 CrossRef.
- R. D. Richardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402 CrossRef CAS; T. Dohi and Y. Kita, Chem. Commun., 2009, 2073 RSC; F. C. Kuepper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther, Z. Lu, M. Jonsson and L. Kloo, Angew. Chem., Int. Ed., 2011, 50, 11598 CrossRef; M. S. Yusubov and V. V. Zhdankin, Curr. Org. Synth., 2012, 9, 247 CrossRef.
- W. Zhong, S. Liu, J. Yang, X. Meng and Z. Li, Org. Lett., 2012, 14, 3336 CrossRef CAS.
- K. A. Javaid, N. Sonoda and S. Tsutsumi, Tetrahedron Lett., 1969, 10, 4439 CrossRef CAS; K. A. Javaid, N. Sonoda and S. Tsutsumi, Bull. Chem. Soc. Jpn., 1970, 43, 3475 CrossRef; N. Sonoda, S. Furui, K. A. Javaid and S. Tsutsumi, Ann. N. Y. Acad. Sci., 1972, 192, 49 CrossRef.
- T. M. Nguyen and D. Lee, Org. Lett., 2001, 3, 3161 CrossRef CAS.
- C. Santi, M. Tiecco, L. Testaferri, C. Tomassini, S. Santoro and G. Bizzoca, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 956 CrossRef CAS; S. Santoro, C. Santi, M. Sabatini, L. Testaferri and M. Tiecco, Adv. Synth. Catal., 2008, 350, 2881 CrossRef.
- We are grateful to a reviewer who suggested that a limitation is oxidation of the sulphur atoms in the catalyst. Further developments using diselenide catalysts in an anti-dioxygenation have recently been reported, see: C. Santi, R. Di Lorenzo, C. Tidei, L. Bagnoli and T. Wirth, Tetrahedron, 2012, 68, 10530 CrossRef CAS.
- Y. Ashikari, T. Nokami and J.-I. Yoshida, Org. Lett., 2012, 14, 938 CrossRef CAS.
- K. Koyama, T. Ebara, T. Tani and S. Tsutsumi, Can. J. Chem., 1969, 47, 2484 CrossRef CAS.
- V. VanRheenen, R. C. Kelly and D. Y. Cha, Tetrahedron Lett., 1976, 17, 1973 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |