 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-organizing surface-initiated polymerization, templated self-sorting and templated stack exchange: synthetic methods to build complex systems
Marco
Lista
,
Edvinas
Orentas
,
Jetsuda
Areephong
,
Pierre
Charbonnaz
,
Adam
Wilson
,
Yingjie
Zhao
,
Altan
Bolag
,
Giuseppe
Sforazzini
,
Raluca
Turdean
,
Hironobu
Hayashi
,
Yuya
Domoto
,
Adam
Sobczuk
,
Naomi
Sakai
and
Stefan
Matile
*
Department of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/ Fax: +41 22 379 5123; Tel: +41 22 379 6523
First published on 29th January 2013
Abstract
In nature, spectacular function is achieved by highly sophisticated supramolecular architectures. Little is known what we would obtain if we could create complexity with similar precision, because the synthetic methods to do so are not available. This account summarizes recent approaches conceived to improve on this situation. With self-organizing surface-initiated polymerization (SOSIP), charge-transporting stacks can be grown directly on solid substrates with molecular-level precision. The extension to templated self-sorting (SOSIP-TSS) offers a supramolecular approach to multicomponent architectures. A solid theoretical framework for the transcription of information by templated self-sorting has been introduced, intrinsic templation efficiencies up to 97% have been achieved, and the existence of self-repair has been shown. The extension to templated stack exchange (SOSIP-TSE) offers the complementary covalent approach. Compatibility of this robust method with the creation of double-channel architectures with antiparallel two-component gradients has been demonstrated.
Introduction
Supramolecular architectures of highest sophistication account for the excellent functionality of biological systems.1,2 Photosystem II from the cyanobacterium Thermosynechococcus elongatus, for example, a 650 kD dimeric multisubunit complex, uses 35 transmembrane α-helices as scaffolds to precisely position the involved chromophores, 72 chlorophylls and 14 carotenoids, in a directional manner.2 The active site of biological photosystems is characterized by a molecular pathway with a four-component gradient for the directional transport of electrons after their generation with light. The hole left behind in the special pair of chlorophylls is guided along a similar gradient in another direction to separate the charges before they can recombine and everything is lost. One of the big open questions is what we would get if we could build functional supramolecular materials with the same level of sophistication. Today, this question cannot be answered because the synthetic organic chemistry to build complex architectures with high precision on the molecular level is largely missing, despite much effort worldwide.3–8 Highest standards of molecular precision in organic synthesis are known from fields such as chemical biology or medicinal chemistry. To contribute to the development of synthetic methods to build large functional systems with similar precision, we first focused on the creation of multifunctional photosystems that work, like their biological counterparts, in lipid bilayer membranes.9,10 However, we soon decided to move on to solid surfaces for several reasons. Early on, we recognized that we would have to learn how to grow multicomponent architectures directly on solid substrates to preserve the directionality needed for the creation of oriented gradients. It would be very difficult to achieve such architectures by solution processing. The first technique for constructing gradients directly on surfaces was zipper assembly, a sticky-end layer-by-layer method.11 It provides access to multicomponent architectures on gold.Zipper assembly was successfully applied to the construction of photosystems with co-axial channels for the transport of holes and electrons (i.e., supramolecular n/p-heterojunctions, SHJs).12 Moreover, two-component redox gradients could be engineered into both channels to drive electrons and holes in opposite directions, affording oriented multicomponent antiparallel redox gradients (OMARGs).13 As in biological photosystems,1,2 these quite sophisticated architectures are of interest to prevent the fast recombination of charges after their generation with light and their separation along the two channels of SHJ photosystems. Synthetic methods to construct OMARG SHJs did not exist prior to zipper assembly. The obtained systems were able to separate holes and electrons over very long distances. However, the organic synthesis required for zipper assembly was too demanding to progress with reasonable speed. We thus tried to accomplish the impossible and find synthetic methods that would provide access to complex surface architectures without overwhelming synthetic effort. The result was the discovery of self-organizing surface-initiated polymerization, that is, SOSIP.14
SOSIP
SOSIP was conceived as a synthetic method for providing facile access to complex architectures on solid substrates (Fig. 1).14 Polymer brushes15 appeared ideal to tackle this challenge, but all our attempts to grow brushes with established methods failed as soon as more demanding chemistry was involved. Since this advanced chemistry is unavoidable for the creation of function, we decided to search for new methods for surface-initiated polymerization that work well for this purpose.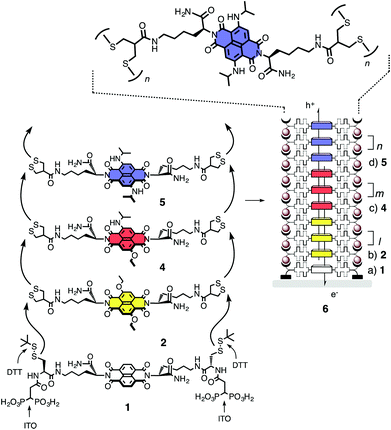 | ||
| Fig. 1 Schematic outline of the SOSIP concept with structures of initiators (1), propagators (2, 4, 5), and photosystem 6. | ||
Lessons from nature were applied with regard to two central problems. Firstly, we sought to preorganize the monomers for ordered polymerization by molecular recognition between the propagator and the growing polymer. With preceding molecular recognition, polymerization was expected to become a covalent capture process16 that would occur favorably and with a minimum of defects. This process is now referred to as SOSIP. Secondly, lessons from nature were applied to optimize the chemistry of the polymerization itself. Screening of promising processes suggested that disulfide exchange polymerization17 would be ideal. The reversible formation of disulfide bridges is the process that controls protein folding and stabilizes their structure. Applied to surface-initiated polymerization, we found that the ring-opening disulfide exchange polymerization of strained disulfides in asparagusic acid was the most suitable reaction for surface-initiated polymerization (Fig. 1). Asparagusic acid is the natural product whose metabolites are responsible for the smell in our urine after having eaten asparagus.
SOSIP was first realized with initiator 1 and propagator 2 (Fig. 1). They both contain a central aromatic unit, here a naphthalenediimide (NDI).4 These NDIs were expected to form π-stacks that could serve as electron- or hole-transporting channels in the final SOSIP architectures 3 (Fig. 1 and 2). These stacks are embedded in hydrogen-bonded networks in the peptide-based self-organizing subunits (Fig. 2). Initiator 1 further contains two diphosphonate “feet” for covalent anchoring on oxide surfaces (e.g., ITO, indium tin oxide). Moreover, initiator 1 contains two protected thiols. For SOSIP, these thiols are deprotected directly on the ITO surface by a brief incubation with DTT (dithiothreitol, Fig. 1). SOSIP is then initiated by incubation with propagator 2 under mildly basic conditions. Molecular recognition of the propagator by the initiator on the surface is designed to place the strained disulfides right on top of the thiolate nucleophiles. Ring-opening disulfide exchange covalently captures the propagator on top of the initiator and regenerates two thiolates on the new surface for continuing polymerization (Fig. 1).
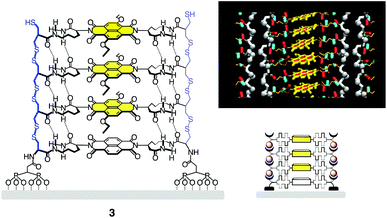 | ||
| Fig. 2 Full structure, molecular model and schematic structure of SOSIP architecture 3. Adapted from ref. 14 with permission. © 2011 American Chemical Society. | ||
The obtained surface architecture 3 is a poly(disulfide) ladderphane18 with a central π-stack (Fig. 2). The structures in Fig. 2 are idealized structures shown only to describe the concept. Defects will naturally exist in reality. So far, it has not been possible to directly quantify their abundance. However, several indirect measurements suggest that self-organization coupled with reversible polymerization really helps to minimize defects. For instance, activities of SOSIP architectures are better than those of controls, surfaces are smoother, and processes that require high order to work work well (e.g., templation, self-sorting). Intrinsic templation efficiencies have been introduced to indirectly quantify the abundance of defects. Efficiencies up to 97% have been measured, which indicates that the occurrence of errors in self-sorting during co-SOSIP is as low as 3% (see below). The abundance of defects in simple single-component SOSIP should thus be far below 3%. Moreover, experiments have been developed to directly follow the removal of artificially added defects during co-SOSIP, i.e., to secure experimental evidence for the occurrence of self-repair (see below).
The key question concerning the existence of SOSIP was whether or not the polymerization really occurs on the surface. This question was addressed with dose response curves and confirmed with microcontact printing (Fig. 3). In dose response curves, the dependence of SOSIP on the concentration of the propagator is measured. To record dose response curves, all experiments have to be conducted in parallel with ITO plates that contain activated initiators and ITO plates that do not contain activated initiators. The pairs of plates are then incubated in a deaerated solution of, for example (Fig. 3), 5–30 mM propagator 2 in CHCl3–MeOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, at ambient temperature, for 12 h. The number of NDIs on the ITO surface is then assessed from the absorbance of the chromophore. Comparison of the two dose response curves – with and without initiator – reveals the existence of an ideal concentration for SOSIP. For the example in Fig. 3, this SOSIP concentration was around cSOSIP ∼ 12 mM. At lower concentrations, SOSIP did not occur. At c > 15 mM, NDI absorption was also found on initiator-free ITO (Fig. 3, ○). This absorption demonstrated the onset of random polymerization in solution above cSOSIP, with insoluble polymers precipitating from the solution onto the ITO surface. The SOSIP concentration for a given propagator depends strongly on solvent, temperature and the nature of the propagator. For successful SOSIP, it is thus essential to optimize the solvent mixture used by recording dose response curves and identifying SOSIP concentrations for the chosen compound.
:1, at ambient temperature, for 12 h. The number of NDIs on the ITO surface is then assessed from the absorbance of the chromophore. Comparison of the two dose response curves – with and without initiator – reveals the existence of an ideal concentration for SOSIP. For the example in Fig. 3, this SOSIP concentration was around cSOSIP ∼ 12 mM. At lower concentrations, SOSIP did not occur. At c > 15 mM, NDI absorption was also found on initiator-free ITO (Fig. 3, ○). This absorption demonstrated the onset of random polymerization in solution above cSOSIP, with insoluble polymers precipitating from the solution onto the ITO surface. The SOSIP concentration for a given propagator depends strongly on solvent, temperature and the nature of the propagator. For successful SOSIP, it is thus essential to optimize the solvent mixture used by recording dose response curves and identifying SOSIP concentrations for the chosen compound.
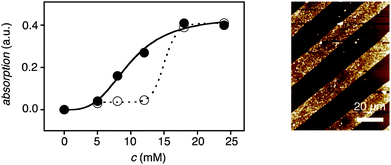 | ||
| Fig. 3 Evidence that SOSIP architectures grow directly on the surface. Left side: Absorption of ITO electrodes with (●) and without (○) initiators 1 after incubation with propagator 2. cSOSIP is the propagator concentration high enough for SOSIP but low enough to avoid random polymerization in solution (here ∼12 mM). Right side: AFM height image of SOSIP architectures obtained by μCP of initiator 1 on ITO followed by incubation with 2 at cSOSIP. Adapted from ref. 14 with permission. © 2011 American Chemical Society. | ||
Microcontact printing (μCP) was used to probe the validity of these interpretations. Initiators were printed on the ITO surface and activated with DTT. AFM height images demonstrated that, at cSOSIP, polymers were found to grow only on areas where the initiators have been placed (Fig. 3). AFM analysis further revealed that the surfaces of SOSIP architectures were at least as smooth as that of the bare ITO plates used, whereas control systems showed significantly increased surface roughness. Phase images evidenced the long-range, low-defect self-organization of SOSIP architectures. Compared to “grafting-to” controls, SOSIP photosystems also generated much more photocurrent.
SOSIP architectures can be readily dissolved by reductive depolymerization with DTT, and the components obtained can be characterized in solution. However, brief incubation with dilute DTT can reactivate the surface of SOSIP architectures for continuing SOSIP. Surface reactivation could be repeated up to 23 times without any deviation from linear growth to give an absorbance of ∼0.9. The thickness reached at this point was ∼250 nm, that is, formal stacks of ∼750 NDIs.
With SOSIP, the construction of π-stacks with oriented redox gradients for directional charge transfer was very straightforward.14 All that was needed was to continue incubation with the yellow NDI propagator 2 with the more electron-rich red and blue NDI propagators 4 and 5 (Fig. 1).
In the obtained multicomponent photosystem 6, the yellow NDI generated much more photocurrent than in the single-component system 3, presumably because the holes are driven into the blue domain while the electrons are prevented from going there.
After these initial studies with NDIs 1–6, the scope and limitations of SOSIP were explored with an increasingly rich collection of initiators and propagators 7–29 (Fig. 4). Increasingly long alkyl groups were introduced in the self-organizing subunit of 7–17 for studies devoted to self-sorting (below).19–21 Pyrrolidines and sulfides were introduced into the core of NDIs 18–20 for the same purpose.19–21 Dynamic hydrazone bridges were placed in the self-organizing subunits of NDIs 21–23 to prepare for stack exchange (below).22 All new initiators and propagators were found to be compatible with SOSIP.
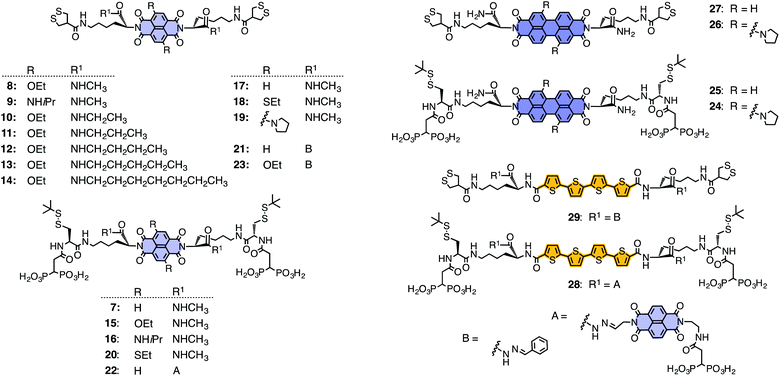 | ||
| Fig. 4 Initiators and propagators used so far for SOSIP. | ||
More important modifications concerned the replacement of the central NDIs by perylenediimides (PDIs) in 24–27 and oligothiophenes 28 and 29.23,24 Both PDIs5 and oligothiophenes6 are among the most common components in electronic, photonic, and optoelectronic materials, including organic photovoltaics. SOSIP with the π-acidic PDIs 27 was particularly attractive to us because their absorption spectra report on the nature of the produced π-stacks.23 The identified stacks with a face-to-face arrangement and a characteristic helical offset were in agreement with the design of SOSIP. SOSIP with the electron-rich oligothiophenes 29 was interesting because their π-stacking should be less favorable than in electron-poor stacks obtained from NDIs and PDIs.24 More demanding SOSIP with more electron-rich NDIs 5 or PDIs 26 gave reasons for concern.23 However, unproblematic SOSIP with oligothiophenes demonstrated the compatibility of SOSIP with the creation of not only electron- but also hole-transporting channels.24 SOSIP thus emerges as a general, robust and user-friendly method to grow oriented charge-transporting pathways directly on solid substrates with molecular-level precision. This important conclusion stimulated research toward more sophisticated multicomponent architectures. Templated self-sorting (TSS) and templated stack exchange (TSE) have been invented for this purpose. The two methods will be summarized in the following.
SOSIP-TSS
For the organic chemist, SOSIP became really interesting as soon as the polymerization of two or more propagators at the same time was considered (Fig. 5).19 Consider the yellow NDI propagator 8 and the blue NDI propagator 9, for example. Their co-SOSIP on activated initiators 7 (Fig. 5, 30) can occur randomly and produce total disorder. Alternatively, their co-SOSIP can occur with self-sorting.7,19 With alternate or “social” self-sorting, blue propagators prefer to surround themselves in stacks with yellow propagators, and vice versa. Alternate self-sorting in both axial and lateral directions affords chessboard-like photosystems 31. Uniform or “narcissistic” axial self-sorting leads to single-component π-stacks. Alternate self-sorting of these stacks in the lateral dimension affords SHJ photosystems 32 with yellow stacks surrounded by blue ones. Uniform self-sorting in lateral and axial dimension affords microdomains 33. The observation of self-sorting during co-SOSIP is nearly impossible on the structural level. On the functional level, donor–acceptor photosystems 31 could be expected to show good charge separation but poor charge mobility after the generation of charges with light. Despite providing good charge mobility, microdomains 33 are also not expected to generate much photocurrent because the donor–acceptor contact area should be insufficient for good charge separation. Only SHJ photosystems 32 should achieve good charge separation between and good charge mobility within their co-axial hole- and electron-transporting channels.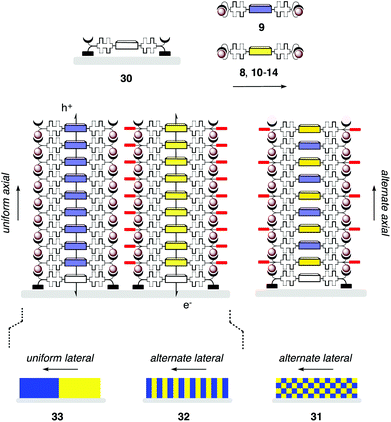 | ||
| Fig. 5 Co-SOSIP on uniform initiators can occur randomly or with alternate (31) as well as uniform axial self-sorting. Alternate lateral self-sorting with the latter results in SHJ architectures (32), uniform lateral and axial self-sorting in microdomains (33). | ||
These predictions were tested with yellow propagators 8 and 10–14 with increasingly long alkyl chains in their self-organizing subunit.19 Photocurrent generation of single-component SOSIP photosystems was independent of these alkyl chains. SOSIP of the blue NDI 9 alone gave practically inactive photosystems. SOSIP with yellow NDIs 8 and 10–14 gave strongly increasing photocurrent with decreasing alkyl chain length in the yellow NDIs. This was consistent with a transition from microdomains 33 to SHJ photosystems 32 as the structural similarity of the propagators increased. Poor photocurrent for co-SOSIP with hexyl NDI 14 and methyl NDI 9 was consistent with poor charge separation at the small contact areas in microdomains 33 (directly visible with the optical microscope). Strongly increased photocurrent generation for co-SOSIP with methyl NDI 8 and methyl NDI 9 – also compared to single-component SOSIP with 8 – demonstrated the formation of the ideal SHJ photosystems 32 at high structural similarity of the propagators.
Considering the often favorable formation of donor–acceptor complexes in solution, we were actually puzzled not to observe alternate self-sorting into chessboard photosystem 31. Propagators 8 and 9 have an identical self-organizing subunit and differ only in their substituents in the NDI core. To further increase similarity with the yellow NDI 8 with two ethoxy substituents in the core, the hydrogen-bond donors in the blue NDI 9 were replaced by hydrogen-bond acceptors in the similarly blue pyrrolidino NDI 19 (Fig. 4). Co-SOSIP with yellow propagators 8 and 10–14 gave the highest activity at intermediate structural similarity between propyl NDI 11 and methyl NDI 19. From there, decreasing activity with increasing structural similarity toward methyl NDI 8 and methyl NDI 19 implied the emergence of alternate self-sorting into the less active chessboard architectures 31 at maximized structural similarity of the propagators (Fig. 5).
In summary, self-sorting studies by co-SOSIP gave reliable and consistent results. Uniform axial self-sorting is clearly favorable over alternate axial self-sorting because topological matching in confined space is naturally ideal. When the propagators are structurally highly similar, lateral alternate self-sorting of the uniform stacks provides access to double-channel photosystems with high activity. Very high structural similarity can enforce alternate axial self-sorting into less active chessboard architectures.
These results were very encouraging. They also provided a system to explore one of the most challenging questions concerning SOSIP, i.e., that of self-repair.19 This question is interesting because polymer brushes with ladderphane structure are expected to be error prone, whereas self-organization by molecular recognition preceding covalent capture should minimize defects (see above). To probe for self-repair, strategies for the design of experiments were adapted from studies on gene repair.25 First, highly active SHJ photosystems 32 were prepared by co-SOSIP of the yellow NDI 8 and the blue NDI 9 (Fig. 6). Then, a defect was introduced by short incubation with only blue propagator 9. Single-component photosystems with NDI 9 only generate very little photocurrent. The defect placed in photosystem 34 inhibits photocurrent generation and thus causes increasing inactivation with increasing thickness of the barrier. Then the photosystem is exposed again to co-SOSIP with NDIs 8 and 9, and self-repair is measured as recovery of activity.
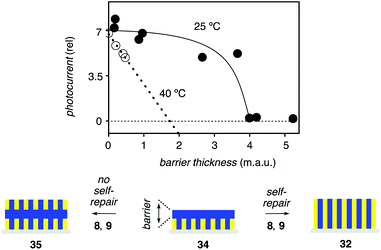 | ||
| Fig. 6 Self-repair during co-SOSIP of 8 and 9 is assessed from the removal of a blue barrier in 34 at 25 °C (●) and 40 °C (○) to restore the most active SHJ architecture 32. Adapted from ref. 19 with permission. © 2011 American Chemical Society. | ||
Little self-repair occurred at 40 °C (Fig. 6, ○). Rapidly decreasing photocurrent generation with increasing barrier thickness was consistent with the formation of the inactive photosystem 35 with a blue barrier in the middle. Poor self-repair on thermal denaturation was meaningful because without operational molecular recognition, there is little reason to repair, and polymerization is faster. At ambient temperature, in clear contrast, increasing thickness of the barrier placed in photosystem 34 did not significantly reduce the activity of the final photosystem until a critical thickness was reached and all self-repair vanishes (Fig. 6, ●). This non-linear response is similar to the one known from genetics and consistent with operational self-repair to give the active photosystem 32. This compelling experimental evidence for the occurrence of self-repair underscores the significance of SOSIP as a powerful method to synthesize complex systems. However, the occurrence of self-repair does not imply that SOSIP is error free, as the following studies on templated self-sorting nicely demonstrate.
The question whether or not self-sorting during co-SOSIP could be templated from the surface was an important one from both a theoretical and a practical point of view. Templated synthesis has been explored extensively with small molecules and is increasingly considered as a promising synthetic approach toward larger architectures.8 Examples reach from giant porphyrin macrocycles to many elegant processes achieved with DNA nanotechnology.8 The central dogma of biology stands out as a powerful example from nature that general synthetic routes to complex systems will ultimately have to work with the transfer of information.
From a practical point of view, templated self-sorting was of interest to allow the composition of photosystems obtained by co-SOSIP to be freely varied.20 One limitation of co-SOSIP is that the composition of the resulting photosystem is determined by the invariable mole fraction xP of the two propagators used, defined by their respective cSOSIP. With the red propagator 18 and the colorless propagator 17, for example, this is xP = 0.75 for the red propagator 18 (Fig. 4 and 7). However, the mole fraction xT of the red initiator 20 in the mixed monolayer 36 is freely variable. This suggested that, if red stacks could be grown on red initiators and colorless stacks on colorless initiators, the composition of the 3D photosystem 37 could be freely chosen with the composition of the 2D template 36. With perfect transcription of information from the 2D template 36 to the 3D architecture 37, the mole fraction of the red component in the photosystem xPS would equal xT. Without templation, the composition of the photosystem 38 would be identical with that of the feeding solution, that is xPS = xP. The most likely situation to occur was partial templation into photosystem 39.
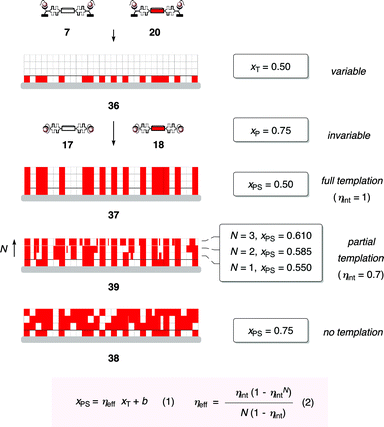 | ||
| Fig. 7 The non-empirical eqns (1) and (2) provide a quantitative theoretical framework for the synthesis of complex architectures by information transfer. The transcription of the variable 2D information in monolayer 36 occurs by templated self-sorting during co-SOSIP with propagators 17 and 18 at the invariable concentration ratio xP. The mole fraction xT is the 2D input in monolayer 36, that is the mole fraction of initiators on the ITO surface. xPS is the 3D output in photosystems 37–39, that is the arrangement and constitution of the SOSIP architecture. ηeff is the effective templation efficiency and b an error. The intrinsic templation efficiency ηint, reporting the fidelity of templation per layer N, is the key parameter to describe all possible systems, together with intrinsic errors α and β (not shown). | ||
When we first entered the field, we could not find a theoretical framework to quantitatively describe templated self-sorting, that is the transcription of 2D input into 3D output. This was surprising considering the significance of the process. So we decided to make one and derived eqns (1) and (2).20 Eqn (1) describes the transcription plot, with the 2D input xT on the x-axis and the 3D output xPS on the y-axis (Fig. 8a). The slope gives the effective templation efficiency ηeff, the intercept the error b. Analysis of the thickness dependence of ηeff with eqn (2) gives the intrinsic templation efficiency ηint, a thickness-independent value that describes the fidelity of templation per layer N (Fig. 8b).
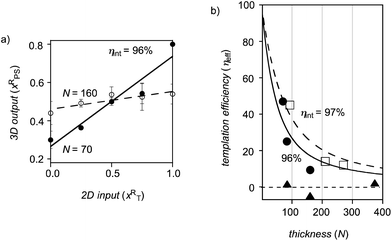 | ||
| Fig. 8 (a) Transcription plot for templated self-sorting of initiator 7 and propagator 17 against initiator 20 and propagator 18. (b) Thickness dependence for templated self-sorting of 7/17 against the red 20/18 (●), 7/17 against the yellow 15/8 (□), and 20/18 against the isosteres 15/8 (▲). Reproduced from ref. 20 with permission. © 2012 Nature Publishing Group. | ||
Theoretical simulations revealed that exceedingly high intrinsic templation efficiencies ηint will be required to observe any templated self-sorting. The situation is reminiscent of peptide or DNA synthesis, where the yield of each step has to be near perfection to obtain reasonable amounts of the desired oligomer. Already with ηint = 90%, templated self-sorting will be barely visible at a thickness of 100 layers N (ca. 30 nm), ηint < 85% will essentially pass unnoticed. Gratifyingly, these exceedingly high values of ηint were readily accessible with templated self-sorting of the red pair 20/18 against the colorless pair 7/17 (Fig. 8). The transcription plot gave an impressive ηeff = 47% at N = 70 (Fig. 8a, ●). Moreover, templated self-sorting remained clearly detectable at N = 160 (Fig. 8a, ○). Analysis of the thickness dependence of ηeff revealed an outstanding ηint = 96% for templated self-sorting of 20/18 against 7/17 (Fig. 8b, ●).
 | ||
| Fig. 9 Alternate templated self-sorting of pseudo-racemic NDIs ent-8 and 18 into layered photosystems 40 around xT = 1 or xT = 0 and chessboard photosystems 41 around xT = 0.5 is indistinguishable from disordered photosystems 42 by spectroscopic means because their composition is always xPS = 0.5. Their activity differs, however, with 41 > 42 > 40. | ||
Just as outstanding ηint = 97% was obtained for templated self-sorting of the yellow NDIs 15/8 against the colorless NDIs 7/17 (Fig. 8b, □). Moreover, templated self-sorting had a positive influence on function. Compared to the best single-component photosystem, photocurrent generation by photosystems obtained by templated self-sorting was up to 13 times better.
Templated self-sorting of the yellow NDIs 15/8 against the red NDIs 20/18 failed (Fig. 8b, ▲). This result was understandable because the two partners are isosteres. The only difference between them is an oxygen compared to a sulfur atom in their core substituents. The irresponsiveness of NDI isosteres was interesting to probe for other means to induce templated self-sorting. To look at stereochemistry, for example, the enantiomer of the yellow NDI propagator 8 and a diastereoisomer of yellow NDI initiator 15 were prepared.21 According to spectroscopic data, the transcription of 2D information into 3D architectures was as ineffective with the pseudo-racemic propagators ent-8 and 18 as it was with the pseudo-homochiral isosteres 8 and 18. However, spectroscopic analysis would report on uniform axial self-sorting (above) but fail to tell the difference between alternate photosystems such as 40 and 41 and disordered photosystem 42 (Fig. 9). With enantiomers, uniform axial self-sorting corresponds to the formation of conglomerates. This process worked well for Louis Pasteur and a few others, but the majority of enantiomers crystallize as racemic crystals. Dimerization studies of face-protected NDI enantiomers confirmed a clear preference for heterodimerization in solution and in the crystal.26 It was thus reasonable to consider that pseudo-enantiomeric isosteres would prefer alternate over uniform axial self-sorting. However, the photosystems produced by alternate axial self-sorting, e.g., 40 and 41, have all identical overall composition (Fig. 9). The only conceivable method to detect alternate self-sorting of pseudo-racemic isosteres was thus functional analysis. Photocurrent generation by pseudo-racemic photosystems obtained from templated self-sorting of isosteres ent-8/18 was indeed sensitive to structural differences. Compared to disorganized pseudo-homochiral photosystems obtained from 8/18, pseudo-racemic photosystems obtained from ent-8/18 were slightly less active around xT = 1 and xT = 0 and slightly more active around xT = 0.5. This suggested that both layered photosystem 40 and chessboard photosystem 41 exist with pseudo-racemic isosteres and are a bit more and a bit less active than the disorganized photosystem 42 with pseudo-homochiral isosteres, respectively.
In summary, to explore templated self-sorting, it was first necessary to develop a theoretical framework for the transcription of 2D information into 3D architectures.20 Our non-empirical model describes the transfer of information with the thickness-independent intrinsic templation efficiency ηint, that is the fidelity of templation per layer. For co-SOSIP with structurally similar partners, templation efficiencies up to ηint = 97% confirmed the compatibility of the synthetic method with information transfer. Lessons from nature, including the central dogma of biology, suggest that this might ultimately be the only way to synthesize sophisticated multicomponent architectures.
Templated self-sorting during co-SOSIP of pseudo-racemic isosteres was found to result in alternate templated self-sorting, a process corresponding to racemic crystallization.21 Enantiopure and homochiral partners are thus required to achieve the more desirable uniform axial self-sorting. With self-sorting during co-SOSIP, it became also possible to probe for self-repair during SOSIP. The results show non-linear behavior as with gene repair.
SOSIP-TSE
Templated stack exchange (TSE) was developed as a covalent alternative to templated self-sorting (TSS) approaches to double-channel architectures (Fig. 10).22 Both approaches can in principle lead to the same architectures. The covalent chemistry in TSE might be easier to control, at least in the short term, whereas the non-covalent chemistry in TSS might perhaps be more rewarding in the long term. For TSE, initiators and propagators were equipped with dynamic hydrazone bridges in the self-organizing subunits. In initiator 22, hydrazone exchange is used to install two additional NDIs as templates for stack exchange. In total, initiator 22 stands with four feet on the ITO surface (compare 43, Fig. 10). In propagator 21, the same hydrazone bridges connect to benzaldehydes, meant to serve as templates for stack exchange. Standard SOSIP with propagator 21 on initiator 43 yields the SOSIP photosystem 44 with the benzaldehyde templates along the oriented NDI stacks. Their removal with hydroxylamine drills large pores with reactive hydrazides along the NDI stacks. These pores in photosystem 45 are then filled with aldehydes of variable structure. The red NDI 46 was selected to build hole-transporting channels next to the original electron-transporting stacks of unsubstituted NDIs in SHJ photosystem 47.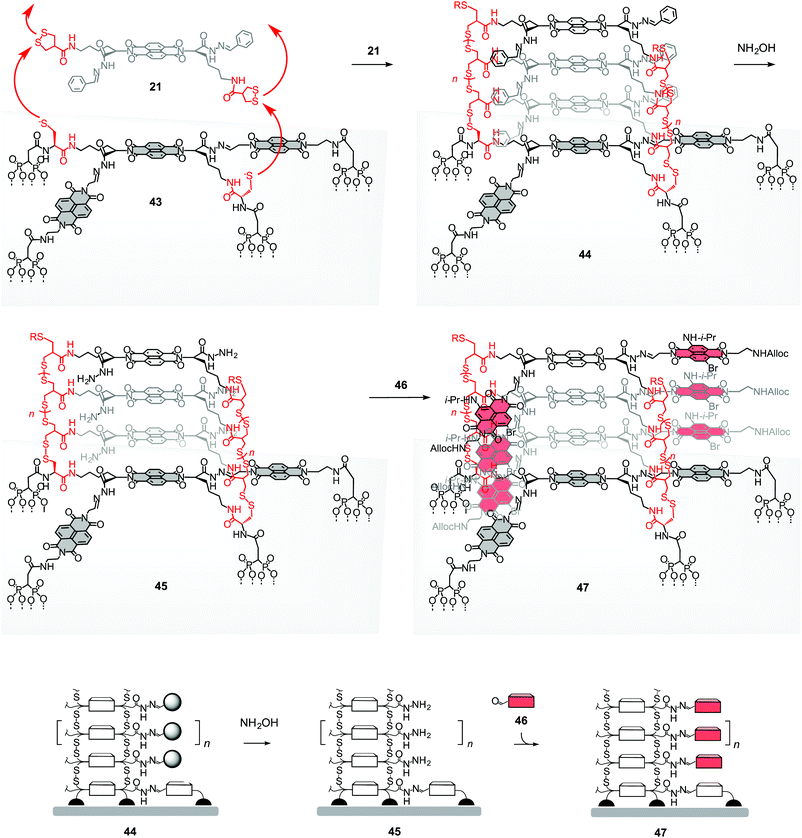 | ||
| Fig. 10 Concept and representative full structures for templated stack exchange. SOSIP with propagator 21 on initiator 22 in 43 is followed by removal of the benzaldehyde templates in 44 with hydroxylamine and filling of the holes drilled into photosystem 45 with aldehydes 46 to give the double-channel photosystem 47. R = H or oxidized form of thiol. | ||
Stack exchange was easily detectable in the absorption spectra of the corresponding photosystems. To quantify with monomer absorptions in solution, the SOSIP-TSE photosystems were routinely disassembled with DTT after use. At high enough concentrations of the stack exchangers, SOSIP-TSE turned out to work quantitatively, independent of the thickness of the photosystem. The two NDI templates in initiator 22 are essential for success. With template-free initiators 1, stack exchange gave very poor yield, decreasing with increasing thickness of the photosystem. Other obvious controls were all positive. For instance, stack exchangers without reactive aldehydes were not bound to the photosystems.
Templated stack exchange has been realized so far with stack exchangers 46 and 48–53 (Fig. 11).22,24 Electron-transporting channels obtained from NDI 50 were clearly more active than those from smaller exchangers such as aminophenyl 48 or the popular triphenylamine 49. Preliminary results indicate that TSE is also compatible with stack exchangers as large as phthalocyanines and porphyrins.
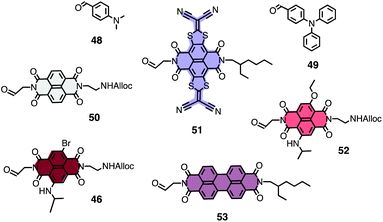 | ||
| Fig. 11 Structure of stack exchangers used so far for SOSIP-TSE. | ||
Most interesting was stack exchange along hole-transporting stacks 54 obtained from SOSIP with oligothiophenes (Fig. 12).24 TSE occurred with >90% yield with NDI exchangers 46 (Fig. 12a) and 50, and with PDI 53. After TSE, the PDI absorption showed the classical signature of twisted face-to-face stacks. Only the core-expanded NDI 51, a very promising compound with low bandgap and high charge mobility from the group of Daoben Zhu,4 gave low yield with TSE. This failure was, however, attributed to insufficient concentration of exchanger 51 at saturation despite solubilizing swallowtails.
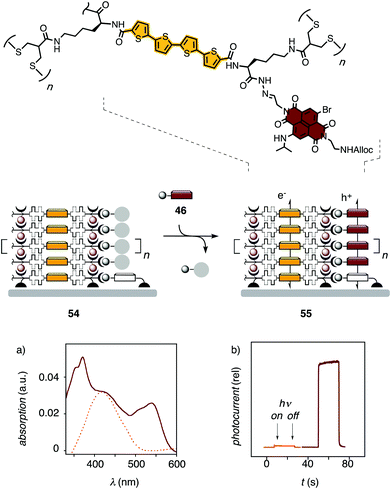 | ||
| Fig. 12 Absorption spectra (a) and relative photocurrent (b) of pre-TSE photosystem 54 (dotted, gold) and photosystem 55 after TSE with aldehyde 46 (solid, grenat). Adapted from ref. 24, reproduced by permission of The Royal Society of Chemistry. | ||
Before stack exchange, oligothiophene photosystems 54 were essentially inactive (Fig. 12b). This poor photocurrent generation was attributed to the absence of electron-transporting channels. With the introduction of these channels by directional TSE along the oligothiophene channels, photocurrent generation increased significantly in all cases. The post-TSE photosystem 55 generated 67 times more photocurrent than pre-TSE photosystem 54, a direct consequence of TSE with NDI 46 (Fig. 12b).
With SOSIP-TSE, double-channel photosystems with antiparallel gradients became accessible without overly demanding synthetic efforts (i.e., OMARG-SHJs).22 For this purpose, SOSIP was executed first with the colorless NDI propagator 21 on initiator 22 as described (Fig. 13a and b). Then, SOSIP was continued by incubation with the yellow NDI propagator 23 (Fig. 13c). This created a two-component gradient in the electron-transporting channel (Fig. 13f). As a consequence of the oriented growth of SOSIP architectures directly on the solid surface, the electrons transported in this channel would thus be directed toward the ITO electrode.
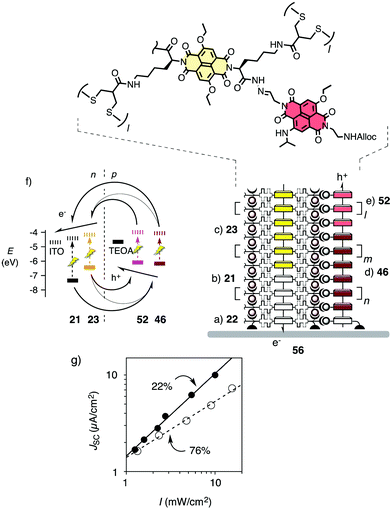 | ||
| Fig. 13 Structure (a–e) and HOMO/LUMO levels (f) of photosystem 56 with oriented antiparallel gradients in co-axial hole- and electron-transporting channels (half arrows). (g) Dependence of the short-circuit current density JSC on the irradiation intensity I for photosystem 56 with constructive gradients (●) compared to a control with destructive gradients (○). Bimolecular charge recombination efficiencies ηBR are indicated. Adapted from ref. 22 with permission. © 2011 American Chemical Society. | ||
For TSE, the obtained electrodes were incubated with hydroxylamine to remove the benzaldehyde templates. The obtained pores were first filled completely with the red NDI 46 because partial stack addition is difficult to control. However, partial stack removal is unproblematic. To install a gradient in the hole-transporting channel of photosystem 56, the stacks from 46 were thus partially removed with hydroxylamine, and the more shallow pores were filled with NDI 52.
With stack exchangers 52 placed on top of stack exchangers 46 by directional TSE, the holes are guided away from the ITO surface. This is opposite to the directionality of the gradient engineered into the electron-transporting SOSIP stacks. Photosystem 56 thus contains formal antiparallel redox gradients. The minimalist gradients are composed of only two components each, plus additional hole barriers from the TSE template at the bottom of the hole-transporting channels. Preliminary results imply high functional relevance of the hole barriers in photosystem 56 and call for electron barriers in the SOSIP channels, e.g., blue NDIs as in photosystem 6 (Fig. 1). As in biological photosystems, the antiparallel two-component gradients in photosystem 56 could be expected to drive photo-generated holes and electrons in opposite directions before they can recombine and everything is lost.
The functional relevance of antiparallel gradients was assessed from the dependence of photocurrent generation on the intensity of irradiation. From the slope, the bimolecular charge recombination efficiency ηBR could be calculated. An ηBR = 22% was obtained for photosystem 56 with an antiparallel two-component gradient (Fig. 13g, ●). For controls without gradients, ηBR = 50% was obtained. In photosystems with gradients that point in the wrong direction, charge recombination became dominant (ηBR = 76%, Fig. 13g, ○). These results support the idea that charge recombination can indeed be minimized with oriented antiparallel gradients in double-channel photosystems such as 56 (i.e., OMARG SHJs). They encourage ongoing efforts to build antiparallel gradients with more than two components.
Taken together, SOSIP-TSE emerges as a solid covalent approach to build multicomponent surface architectures. Yields for stack exchange are high and thickness independent as long as the suitable templates are in place. Structural tolerance is remarkable with regard to both SOSIP scaffolds as well as stack exchangers; preliminary results suggest compatibility also with porphyrins, phthalocyanines or fullerenes. Moreover, SOSIP-TSE has been validated as the current method of choice to build double-channel architectures with antiparallel redox gradients.
Conclusions
In summary, this account describes recent methods conceived to synthesize complex surface architectures with high precision. SOSIP allows us to grow charge-transporting pathways directly on solid substrates. SOSIP-TSS offers a supramolecular approach to multicomponent architectures. A solid theoretical framework for the transcription of information by template self-sorting has been introduced, and high efficiencies have been obtained. SOSIP-TSE offers the complementary covalent approach. Compatibility of this robust method with the creation of double-channel architectures with antiparallel two-component gradients has been demonstrated. Whereas SOSIP-TSS provides a privileged platform to explore fundamental processes such as the transcription and translation of information in organic synthesis, SOSIP-TSE is ready for practical applications. Current emphasis is on the creation of multichannel systems with multicomponent gradients in the broadest sense.Acknowledgements
We thank all past and present coworkers and collaborators for their contributions, particularly the groups of Eric Vauthey, Jiri Mareda and Michal Borkovec (all at the University of Geneva), and the University of Geneva, the European Research Council (ERC Advanced Investigator), the National Centre of Competence in Research (NCCR) Chemical Biology and the Swiss NSF for financial support.References
- (a) J. Deisenhofer and H. Michel, Science, 1989, 245, 1463–1473 CAS; (b) N. Nelson and A. Ben-Shem, Nat. Rev. Mol. Cell Biol., 2004, 5, 971–982 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS.
- (a) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS; (b) F. Würthner and K. Meerholz, Chem.–Eur. J., 2010, 16, 9366–9373 CrossRef; (c) D. M. Bassani, L. Jonusauskaite, A. Lavie-Cambot, N. D. McClenaghan, J.-L. Pozzo, D. Ray and G. Vives, Coord. Chem. Rev., 2010, 254, 2429–2445 CrossRef CAS; (d) J. L. Delgado, P.-A. Bouit, S. Filippone, M. A. Herranz and N. Martín, Chem. Commun., 2010, 46, 4853–4865 RSC; (e) M. V. Martínez-Díaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC; (f) R. Bhosale, J. Míšek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC; (g) A. Kira, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, J. K. Park, D. Kim and H. Imahori, J. Am. Chem. Soc., 2009, 131, 3198–3200 CrossRef; (h) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177–195 CrossRef CAS; (i) M. Morisue, S. Yamatsu, N. Haruta and Y. Kobuke, Chem.–Eur. J., 2005, 11, 5563–5574 CrossRef CAS; (j) D. M. Guldi, J. Phys. Chem. B, 2005, 109, 11432–11441 CrossRef CAS; (k) F. Yang, M. Shtein and S. Forrest, Nat. Mater., 2005, 4, 37–41 CrossRef CAS; (l) D. M. Guldi, I. Zilbermann, G. Anderson, A. Li, D. Balbinot, N. Jux, M. Hatzimarinaki, A. Hirsch and M. Prato, Chem. Commun., 2004, 726–727 RSC; (m) V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2003 Search PubMed.
- NDIs: (a) S. V. Bhosale, S. V. Bhosale and S. K. Bhargava, Org. Biomol. Chem., 2012, 10, 6455–6468 RSC; (b) Y. Hu, X. Gao, C. Di, X. Yang, F. Zhang, Y. Liu, H. Li and D. Zhu, Chem. Mater., 2011, 23, 1204–1215 CrossRef CAS; (c) S. V. Bhosale, C. H. Jani, C. H. Lalander, S. J. Langford, I. Nerush, J. G. Shapter, D. Villamaina and E. Vauthey, Chem. Commun., 2011, 47, 8226–8228 RSC; (d) N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225–4237 RSC; (e) B. J. H. Oh, S. L. Suraru, W. Y. Lee, M. Könemann, H. W. Höffken, C. Röger, R. Schmidt, Y. Chung, W. C. Chen, F. Würthner and Z. Bao, Adv. Funct. Mater., 2010, 20, 2148–2156 CrossRef; (f) C. W. Marquardt, S. Grunder, A. Błaszczyk, S. Dehm, F. Hennrich, H. V. Löhneysen, M. Mayor and R. Krupke, Nat. Nanotechnol., 2010, 5, 863–867 CrossRef CAS; (g) P. M. Alvey, J. J. Reczek, V. Lynch and B. L. Iverson, J. Org. Chem., 2010, 75, 7682–7690 CrossRef CAS.
- PDIs: (a) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef; (b) W. Li, A. Saeki, Y. Yamamoto, T. Fukushima, S. Seki, N. Ishii, K. Kato, M. Takata and T. Aida, Chem.–Asian J., 2010, 5, 1566–1572 CrossRef CAS; (c) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS; (d) F. Würthner, Chem. Commun., 2004, 1564–1579 RSC; (e) S. Foster, C. E. Finlayson, P. E. Keivanidis, Y.-S. Huang, I. Hwang, R. H. Friend, M. B. J. Otten, L.-P. Lu, E. Schwartz, R. J. M. Nolte and A. E. Rowan, Macromolecules, 2009, 42, 2023–2030 CrossRef CAS; (f) L. Bu, X. Guo, B. Yu, Y. Qu, Z. Xie, D. Yan, Y. Geng and F. Wang, J. Am. Chem. Soc., 2009, 131, 13242–13243 CrossRef CAS; (g) K. Sugiyasu, S. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS; (h) P. Jonkheijm, N. Stutzmann, Z. Chen, D. M. de Leeuw, E. W. Meijer, A. P. H. J. Schenning and F. Würthner, J. Am. Chem. Soc., 2006, 128, 9535–9540 CrossRef CAS; (i) A. B. F. Martinson, A. M. Massari, S. J. Lee, R. W. Gurney, K. E. Splan, J. T. Hupp and S. T. Nguyen, J. Electrochem. Soc., 2006, 153, A527–A532 CrossRef CAS.
- Oligothiophenes: (a) H. Goto, Y. Yokochi and E. Yashima, Chem. Commun., 2012, 48, 3291–3293 RSC; (b) R. Fitzner, E. Reinold, A. Mishra, E. Mena-Osteritz, H. Ziehlke, C. Körner, K. Leo, M. Riede, M. Weil, O. Tsaryova, A. Weiß, C. Uhrich, M. Pfeiffer and P. Bäuerle, Adv. Funct. Mater., 2011, 21, 897–910 CrossRef CAS; (c) T. W. Holcombe, J. E. Norton, J. Rivnay, C. H. Woo, L. Goris, C. Piliego, G. Griffini, A. Sellinger, J. L. Brédas, A. Salleo and J. M. Fréchet, J. Am. Chem. Soc., 2011, 133, 12106–12114 CrossRef CAS; (d) R. J. Kumar, J. M. MacDonald, T. B. Singh, L. J. Waddington and A. B. Holmes, J. Am. Chem. Soc., 2011, 133, 8564–8573 CrossRef CAS; (e) J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS; (f) A. Mishra, C. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS; (g) Y. Ie, A. Han, T. Otsubo and Y. Aso, Chem. Commun., 2009, 3020–3022 RSC; (h) I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS; (i) M. Zambianchi, F. Di Maria, A. Cazzato, G. Gigli, M. Piacenza, F. Della Sala and G. Barbarella, J. Am. Chem. Soc., 2009, 131, 10892–10900 CrossRef CAS; (j) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2002, 100, 2537–2574 CrossRef.
- Self-sorting: (a) M. Lal Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651–4684 RSC; (b) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS; (c) M. M. Safont-Sempere, P. Osswald, M. Grüne, M. Renz, M. Kaupp, K. Radacki, H. Braunschweig and F. Würthner, J. Am. Chem. Soc., 2011, 133, 9580–9591 CrossRef CAS; (d) M. R. Molla, A. Das and S. Ghosh, Chem.–Eur. J., 2010, 16, 10084–10093 CrossRef CAS; (e) W. Jiang and C. A. Schalley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS; (f) D. Ajami, J. L. Hou, T. J. Dale, E. Barrett and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10430–10434 CrossRef CAS; (g) N. E. Botterhuis, S. Karthikeyan, A. J. H. Spiering and R. P. Sijbesma, Macromolecules, 2010, 43, 745–751 CrossRef CAS; (h) A. D. Shaller, W. Wang, H. Gan and A. D. Q. Li, Angew. Chem., Int. Ed., 2008, 47, 7705–7709 CrossRef CAS; (i) T. Baumgart, G. Hunt, E. R. Farkas, W. W. Webb and G. W. Feigenson, Biochim. Biophys. Acta, 2007, 1768, 2182–2194 CrossRef CAS; (j) J. Zhang, B. Jing, V. Janout and S. L. Regen, Langmuir, 2007, 23, 8709–8712 CrossRef CAS; (k) N. C. Yoder and K. Kumar, J. Am. Chem. Soc., 2006, 128, 188–191 CrossRef CAS; (l) A. X. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS; (m) N. A. Schnarr and A. J. Kennan, J. Am. Chem. Soc., 2003, 125, 6364–6365 CrossRef CAS; (n) P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef CAS; (o) C. D. Bain and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 7164–7175 CrossRef CAS.
- Templated synthesis: (a) M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef CAS; (b) M. Röthlingshöfer, K. Gorska and N. Winssinger, J. Am. Chem. Soc., 2011, 133, 18110–18113 CrossRef; (c) F. B. L. Cougnon, H. Y. Au-Yeung, G. D. Pantos and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3198–3207 CrossRef CAS; (d) M. von Delius, E. M. Geertsema and D. A. Leigh, Nat. Chem., 2010, 2, 96–101 CrossRef CAS; (e) J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS; (f) K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–210 CrossRef CAS; (g) M. F. Jacobsen, J. B. Ravnsbæk and K. V. Gothelf, Org. Biomol. Chem., 2010, 8, 50–52 RSC; (h) X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS; (i) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS; (j) Templated Organic Synthesis, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1999 Search PubMed.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- A. Perez-Velasco, V. Gorteau and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 921–923 CrossRef CAS.
- N. Sakai, A. L. Sisson, T. Bürgi and S. Matile, J. Am. Chem. Soc., 2007, 129, 15758–15759 CrossRef CAS.
- (a) R. Bhosale, R. S. K. Kishore, V. Ravikumar, O. Kel, E. Vauthey, N. Sakai and S. Matile, Chem. Sci., 2010, 1, 357–368 RSC; (b) S. Maity, R. Bhosale, N. Banerji, E. Vauthey, N. Sakai and S. Matile, Org. Biomol. Chem., 2010, 8, 1052–1057 RSC; (c) R. Bhosale, A. Perez-Velasco, V. Ravikumar, R. S. K. Kishore, O. Kel, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, T. Sawada, E. Vauthey, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2009, 48, 6461–6464 CrossRef CAS; (d) R. S. K. Kishore, O. Kel, N. Banerji, D. Emery, G. Bollot, J. Mareda, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, E. Vauthey, N. Sakai and S. Matile, J. Am. Chem. Soc., 2009, 131, 11106–11116 CrossRef CAS; (e) M. Lista, N. Sakai and S. Matile, Supramol. Chem., 2009, 21, 238–244 CrossRef CAS; (f) N. Sakai, R. S. K. Kishore and S. Matile, Org. Biomol. Chem., 2008, 6, 3970–3976 RSC; (g) A. L. Sisson, N. Sakai, N. Banerji, A. Fürstenberg, E. Vauthey and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 3727–3729 CrossRef CAS.
- N. Sakai, R. Bhosale, D. Emery, J. Mareda and S. Matile, J. Am. Chem. Soc., 2010, 132, 6923–6925 CrossRef CAS.
- N. Sakai, M. Lista, O. Kel, S. Sakurai, D. Emery, J. Mareda, E. Vauthey and S. Matile, J. Am. Chem. Soc., 2011, 133, 15224–15227 CrossRef CAS.
- (a) J. Bünsow, T. S. Kelby and W. T. Huck, Acc. Chem. Res., 2010, 43, 466–474 CrossRef; (b) R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H. A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS; (c) J. Pyun and K. Matyjaszewski, Chem. Mater., 2001, 13, 3436–3448 CrossRef CAS; (d) M. Weck, J. J. Jackiw, R. R. Rossi, P. S. Weiss and R. H. Grubbs, J. Am. Chem. Soc., 1999, 121, 4088–4089 CrossRef CAS.
- L. J. Prins and P. Scrimin, Angew. Chem., Int. Ed., 2009, 48, 2288–2306 CrossRef CAS.
- E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- T.-Y. Luh, Acc. Chem. Res., 2013 DOI:10.1021/ar300170b.
- M. Lista, J. Areephong, N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 15228–15230 CrossRef CAS.
- E. Orentas, M. Lista, N.-T. Lin, N. Sakai and S. Matile, Nat. Chem., 2012, 4, 746–750 CrossRef CAS.
- E. Orentas, N. Sakai and S. Matile, Chirality, 2013 DOI:10.1002/chir.22118.
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 18542–18545 CrossRef CAS.
- P. Charbonnaz, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1492–1496 RSC.
- J. Areephong, E. Orentas, N. Sakai and S. Matile, Chem. Commun., 2012, 48, 10618–10620 RSC.
- K. Negishi, D. Loakes and R. M. Schaaper, Genetics, 2002, 161, 1363–1371 CAS.
- N.-T. Lin, A. Vargas Jentzsch, L. Guénée, N.-M. Neudörfl, S. Aziz, A. Berkessel, E. Orentas, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1121–1127 RSC.
| This journal is © The Royal Society of Chemistry 2013 |