 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic effects within a C2-symmetric organocatalyst: the potential formation of a chiral catalytic pocket†
Joshua P.
Delaney
a,
Hannah L.
Brozinski
a and
Luke C.
Henderson
*ab
aStrategic Research Centre, Faculty of Science and Technology, Deakin University, Geelong, Victoria 3216, Australia. E-mail: luke.henderson@deakin.edu.au; Fax: (+613) 52271045; Tel: (+613)52272767
bInstitute for Frontier Materials, Deakin University, Geelong, Victoria 3216, Australia
First published on 18th February 2013
Abstract
This study describes the synthesis of five novel C2-symmetric organocatalysts that facilitate the on-water asymmetric aldol reaction at low catalyst loading (1 mol%) without the use of additives. Each catalyst is composed of two diprolinamide units joined by a symmetric alkyl bridging group allowing for systematic modulation of catalytic site proximity. Typically, catalysts in this manuscript which bear the catalytic units in close proximity gave the best reaction outcomes in terms of conversion (up to >99%), diastereomeric ratio (4/96, syn/anti) and enantiomeric excess (up to 97%). This effect has been attributed to the assembly of a chiral pocket, facilitated by hydrogen bonding at the oil-in-water interface.
Introduction
Since the seminal work of List and Barbas, organocatalysis has experienced a dramatic increase in the intensity of investigation.1 Proline-derived organocatalysts have been the most explored scaffold having been applied to a vast range of chiral bond forming reactions in a broad selection of solvents and conditions. In order to improve the activity, versatility and chiral induction of proline based catalysts; the proline motif has been subjected to substantial modification and elaboration.2Rigorous modification of L-proline throughout the past decade has seen a myriad innovations to the catalyst structure. Fundamental elaborations such as functionalization at the 4 position, as demonstrated by Hayashi et al.,3 have resulted in vastly improved activity and chiral induction of proline catalysts. Similarly, elaboration via the carboxyl group of proline have seen great success.4 To this end, there has been an increasingly broad range of organic transformations carried out by highly derivatised, novel proline-based organocatalysts such as small peptides, proline-thioamides and ionic liquid tagged prolines are but a few.5 Despite the progress made towards catalyst generality, organocatalysed asymmetric transformations in water at low catalyst loadings remain a topic of great interest in the synthetic chemistry community.
Aside from the prominent use of steric influences, properties such as catalytic site proximity and catalyst pre-organisation are largely overlooked, thus spurring our interest in C2-symmetric organocatalysts. The use of C2-symmetric ligands is commonplace among transition metal catalysis but has been applied to organocatalysis only a few times.6 Embracing C2-symmetric architectures provides a means to control spatial properties within an oligomeric catalyst, an aspect that has yet to be embraced as a significant aspect of organocatalyst design.
Recently, we reported the synthesis and evaluation of a novel C2-symmetric organocatalyst which demonstrated high catalytic activity for the asymmetric aldol reaction in an ‘on-water’ reaction system (Fig. 1).7
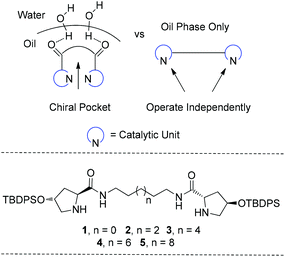 | ||
| Fig. 1 Proposed catalytic conformation adopted by C2-symmetric organocatalysts, on-water (left) and organic (right) reaction medium. Diprolinamide schematic is below. | ||
Based on this previous work, our hypothesis for this study was that the intramolecular distance of the prolinamide units within the organocatalyst influences their cooperation and therefore may lead to highly stereoselective aldol reactions. To test this hypothesis several organocatalysts were synthesised with sequentially increasing distance between the prolinamide units. The rationale being that the shorter spacer lengths, such as that present in catalysts 1 (4 methylene units) would encourage prolinamide cooperation, while the larger intramolecular distances present in catalysts 4 and 5 (10 and 12 methylene units, respectively) would minimize the catalytic site interaction and thus diminishing the cooperative potential of each diprolinamide. Herein we present the comparison of organocatalysts 1–5 using the asymmetric aldol reaction on-water. Based on this experimental evidence we propose the formation of a chiral catalytic pocket by this class of catalyst.
Results and discussion
The described organocatalysts are easily accessed via a four step synthesis utilizing affordable starting materials (Scheme 1). Incorporation of a tert-butyldiphenylsilyl moiety onto trans-4-hydroxyproline 6 was achieved in excellent yield (96%) following a protocol reported by McQuade et al.8 and the crude product was directly N-protected using a tert-butoxycarbamate (Boc) group furnishing 7 in a high yield (82%) over two steps. The protected proline unit was then tethered to each end of a series of linear diamines via an EDCI mediated peptide coupling to afford bis-prolinamides 8–12. Removal of the N-Boc protecting groups was obtained in moderate to excellent yield using a TFA–CH2Cl2 (10% v/v) protocol furnishing the desired organocatalysts 1–5.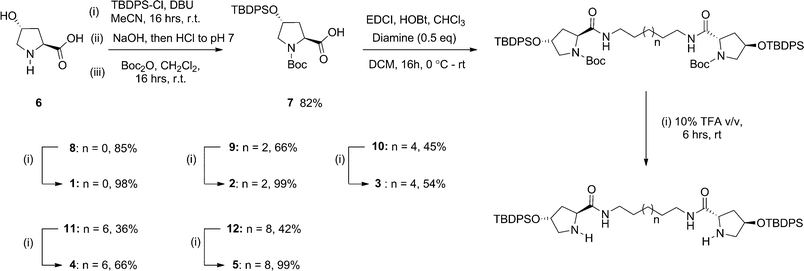 | ||
| Scheme 1 | ||
By utilizing the well understood aldol reaction as a model system, variations in reaction outcome can be attributed to catalyst structure with greater confidence. The clearer insight into potential transition states of each C2-symmetric diprolinamide can be achieved. Additionally the aldol reaction is one of the most important C–C bond forming reaction in organic chemistry.9 The on-water aldol reaction between cyclohexanone 13 and benzaldehyde 14 (Scheme 1) was chosen as the model reaction.
Employing diprolinamide 1 (Table 1, entry 1) at a catalyst loading of 1 mol%, we were pleased to see that the aldol product 15a was obtained in good yield and high stereopurity, dr of 10/90 (syn-/anti-) and ee of 86%. Continuing the comparison with the remaining catalysts (Table 1, entries 2–5) afforded inferior ee's in all cases compared to 1, and similar sacrifices were observed regarding dr. The general decline in stereochemical purity of the aldol products as prolinamide tether length increases supports our original hypothesis of potential catalytic site cooperation.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conversiona (%) | dra (syn/anti) | eeb (%) |
a Determined by integration of key signals in the 1H NMR spectrum.
b Determined by Chiral HPLC, Chiralpak AD-H, 1 mL min−1, 2-propanol–n-hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.
c Results obtained from a previous study but provided here for clarity.7a :9.
c Results obtained from a previous study but provided here for clarity.7a
|
||||
| 1 | 1 | 68 | 10/90 | 86 |
| 2c | 2 | 45 | 8/92 | 66 |
| 3 | 3 | 70 | 16/84 | 55 |
| 4 | 4 | 87 | 20/80 | 70 |
| 5 | 5 | 32 | 15/85 | 68 |

Encouraged by these preliminary results our attention turned to investigating each catalyst in combination with other commonly employed benzaldehydes and a second ketone (cyclopentanone) to see if catalyst 1 outperforming catalysts 2–5 was indeed a general trend.
Employing cyclopentanone 16 as the donor ketone (Table 2, entries 1–5) demonstrated the same general trend with respect to catalyst 1 as was observed in Table 1 whereby the smallest prolinamide linker furnishes the superior reaction outcomes.
|
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst→ | 1 | 2 | 3 | 4 | 5 | |||||||||||||
| Entry | n | R | 15 | Con.a (%) | dr (syn/anti)a | eeb (%) | Con.a (%) | dra (syn/anti) | eeb (%) | Con.a (%) | dra (syn/anti) | eeb (%) | Con.a (%) | dra (syn/anti) | eeb (%) | Con.a (%) | dra (syn/anti) | eeb (%) |
|
a Determined by integration of key signals in the 1H NMR spectrum, Chromatography was restricted to reaction mixtures showing conversions <20%, similar to such protocols employed by groups such as Penhoat,11a Giacalone11b and Guellena11c in order to accurately quantify catalyst activity.
b Determined by Chiral HPLC, Chiralpak AD-H, 1 mL min−1, IPA–Hexane, 1:9.
c Results obtained from a previous study but provided here for clarity of comparison.7a Results in bold were determined to be the best of the series by the authors. |
||||||||||||||||||
| 1 | 0 | H | 15b | 92 | 25/72 | 74 | 90 | 64/36 | 50 | 99 | 61/39 | 64 | 92 | 60/40 | 68 | 56 | 60/40 | 70 |
| 2 | 0 | 4-Br | 15c | 96 | 50/50 | 72 | 99 | 59/41 | 52 | 98 | 47/53 | 68 | 99 | 43/57 | 54 | 99 | 61/39 | 56 |
| 3 | 0 | 4-F | 15d | 97 | 27/73 | 80 | 85 | 64/36 | 42 | 99 | 43/57 | 65 | 99 | 42/58 | 68 | 99 | 62/38 | 66 |
| 4 | 0 | 4-NO2 | 15e | 97 | 39/61 | 8 | 61 | 24/76 | 14 | 99 | 73/27 | 8 | 72 | 49/51 | 14 | 40 | 66/34 | 12 |
| 5 | 0 | 3-NO2 | 15f | 55 | 88/12 | 96 | 99 | 54/46 | 52 | 99 | 52/48 | 56 | 98 | 53/47 | 46 | 99 | 51/49 | 50 |
| 6 | 0 | 2-NO2 | 15g | 74 | 91/9 | 99 | 37 | 34/66 | 78 | 99 | 40/60 | 82 | 92 | 30/70 | 66 | 36 | 35/65 | 69 |
| 5 | 0 | 4-Ph | 15h | 78 | 38/62 | 80 | 70 | 59/41 | 46 | 78 | 58/42 | 50 | 76 | 63/37 | 40 | 38 | 57/43 | 44 |
| 6 | 1 | 4-Br | 15i | 99 | 9/91 | 88 | 97 | 14/86 | 86 | 96 | 14/86 | 80 | 37 | 19/81 | 56 | 77 | 16/84 | 82 |
| 7 | 1 | 4-F | 15j | 71 | 6/94 | 86 | 88 | 9/91 | 92 | 86 | 21/79 | 82 | 99 | 19/81 | 84 | 76 | 17/83 | 86 |
| 8 | 1 | 4-NO2 | 15k | 99 | 4/96 | 97 | 91 | 4/96 | 99 | 62 | 23/77 | 80 | 50 | 25/75 | 52 | 65 | 20/80 | 64 |
| 9 | 1 | 3-NO2 | 15l | 85 | 7/93 | 99 | 57 | 16/84 | 88 | 73 | 19/81 | 86 | 36 | 17/83 | 84 | 61 | 17/83 | 86 |
| 10 | 1 | 2-NO2 | 15m | 73 | 3/97 | 92 | 71 | 15/85 | 72 | 65 | 16/84 | 74 | 92 | 16/84 | 64 | 61 | 13/87 | 46 |
| 11 | 1 | 4-Ph | 15n | 67 | 12/88 | 94 | 37 | 15/85 | 80 | 44 | 18/82 | 80 | 44 | 21/79 | 89 | 19 | 15/85 | 80 |

In all cases the catalyst bearing the smallest intramolecular distance between proline moieties exhibited the best optical purity of aldol products obtained. In general, the reaction conversions were good to excellent, though no discernible trend was obvious when comparing conversion values to catalyst structure. When comparing the diastereomeric ratio of the products obtained from each of the catalysts examined, the predominant diastereomer is syn, a stark contrast to those in Table 1. Cyclopentanone has been reported to exhibit moderate diastereoisomeric ratio in the aldol reaction and indeed this was the case with catalysts 1–5.10 However, catalyst 1 displays behaviour which is countermand whereby in most cases the anti diastereoisomer is preferred, while the aldol products furnished by 2–5 were primarily composed of the syn diastereomer. This aberration in results suggests that there is an important role being played by the intramolecular distance between these prolinamide units that contributes to the diastereomeric purity of the generated aldol products. We were further pleased as examination of the ees furnished by catalyst 1 were superior to those in all cases to those shown by catalysts 2–5.
Although, compound 15e (Table 2, entry 4) was deemed an outlier due to a consistently poor compatibility among the catalysts series. In this instance we considered the superior conversion demonstrated by catalyst 1 to be the optimal outcome.
Repeating this series with cyclohexanone (Table 2, entries 6–9) gave a similar trend where the smallest organocatalyst 1 gave the best reaction outcomes. In this series of reaction employing cyclohexanone 13, albeit the superiority of catalyst 1 to catalysts 2–5 was not as pronounced as it was when employing cyclopentanone. It should be noted that in two instances the results obtained for catalyst 1 and 2 were comparable in both conversion and stereochemical aspects. When using 4-fluorobenzaldehyde as the reaction partner (Table 2, entry 7) catalyst 2 was deemed superior to catalyst 1, though only by a marginal amount (17% higher yield and 6% improved ee).
These catalysts were also employed to catalyse the aldol reaction using various ketones and electron rich aldehydes. Unfortunately, under these conditions the catalysts proved ineffective at facilitating high levels of conversion or optical purity. These data are presented in the ESI.†
A prominent trend becomes apparent when considering the dr of each aldol product, with diastereochemical control becoming markedly reduced among the larger catalysts 3–5. This may be a result of a more flexible, and thus less stereochemically defined, transition state allowing for multiple approaches of the incoming aldehyde.
Based on the experimental evidence, it is apparent that the transition state which is formed is significantly influenced by catalytic site proximity. It was reasoned that each diprolinamide must adopt a transition state that allows for potential catalytic site cooperation, be that via hydrogen bonding effects or purely in a steric fashion i.e. the second prolinamide serving as a directing group for the first, and its degree of cooperation that results in the observed discrepancies in catalytic ability. To address the trends observed within each reaction outcome across the catalysts 1–5, the formation of a chiral pocket has been proposed (Fig. 2).
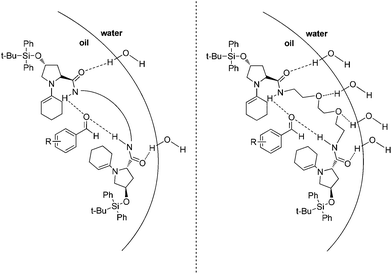 | ||
| Fig. 2 Proposed conformation taken by catalysts 1–5 (left) and a proposed catalyst 16 which is H-bond stabilized (right). | ||
It has been noted that on-water organocatalysis allows for particular advantages derived from abundant H-bonding interactions at the oil–water interace.9,12 This participation of water has led to proposed transition states such as that by Pedrosa et al.13 who describe an on water organocatalysed aldol reaction by a prolinamide bearing multiple amide groups. This transition state presents each amide as both a H-bond acceptor, leading to organization at the oil–water interface, and H-bond donors which facilitate a controlled means of aldehyde approach. Additionally, the concept of multiple hydrogen bonding donors to facilitate the asymmetric aldol reaction with high stereochemical outcome has been used previously by Gong et al.14 and Singh et al.15 who employed a prolinamide based amino alcohol to assist in direct the approach of the incoming aldehyde.
Through hydrogen bonding involving the amide groups at the oil–water interphase, each diprolinamide could potentially align along the surface of the oil droplet, a confirmation supported by lipophilic TBDPSO groups pointing into the oil phase. The impact of this potential formation has dramatic implications regarding both the catalytic activity and chiral induction furnished by each catalyst.
Due to the symmetric nature of the proposed transition state, each catalytic unit is situated equidistant to the approaching aldehyde, and so the activity would be bolstered due to a high relative concentration of the activated chiral enamine. From a chiral standpoint, this symmetry ensures that each enamine reacts with the appropriate Re or Si face of the incoming aldehyde, furnishing the observed enantioenriched aldol product. This proximal advantage would diminish as each catalytic unit is moved apart, reducing the likelihood of cooperation between diprolinamide units and resulting in the poor stereochemical outcomes which were observed for the larger diprolinamides.
This alignment at the interface also presents an opportunity for the linking group to act as a sterically directing moiety, restricting the approach of the aldehyde through the formation of a chiral catalytic pocket – a stereochemically defined environment. This further restricts the configuration by which the reacting aldehyde may approach, significantly improving the optical purity of the obtained products.
Considering the proposed catalytic pocket in Fig. 2 (left), the alignment of the hydrocarbon linker along the oil–water interface was considered to be an unfavourable interaction. This effect would be minimal for catalysts bearing the smaller hydrocarbon chains (such as catalysts 1 and 2). Though for the larger catalysts, such as 3–5, this unfavourable interaction may be responsible for the observed reductions in catalyst performance for catalysts bearing the larger, and thus more hydrophobic, alkyl chains.
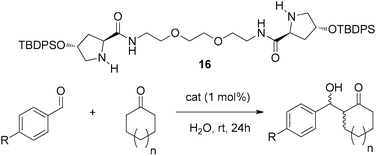
To address this unfavourable interaction, an oxygen rich catalyst 16 (analogous in size to catalyst 3) was synthesised and evaluated in the on-water asymmetric aldol system. It was reasoned that the oxygenated linker between prolinamide units would reinforce the chiral assembly, as proposed in Fig. 2 (right), via H-bonding at the oil–water interface while retaining the hydrophobic nature of the TBDPS groups on the prolinamide units.
It is important to note that the intramolecular distance between prolinamide units in catalyst 16 is approximately the same as that of catalyst 3. Thus a comparison could be drawn between each catalysts and the effect of this ethereal linker.
To probe how the absence of H-bonding may impact the activity of the oxygenated dimer, catalyst 16 was initially evaluated in the absence of water by conducting the reaction in neat cyclohexanone 13.
These experiments (Table 3, entries 1 and 2) show the similarly sized catalysts 3 and 16 affect near identical reaction outcomes, suggesting that without the presence of water, the oxygenated linking chain has no bearing on catalytic ability.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | n | Catalyst | R | Solvent | Conv.a (%) | dra (syn/anti) | eeb |
|
a Determined by integration of key signals in the 1H NMR spectrum.
b Determined by Chiral HPLC, Chiralpak AD-H, 1 mL min−1, IPA–n-Hexane, 1:9.
|
|||||||
| 1 | 1 | 3 | H | Neat | 88 | 13/87 | 64 |
| 2 | 1 | 16 | H | Neat | 90 | 25/75 | 66 |
| 3 | 1 | 16 | H | H2O | 98 | 15/85 | 76 |
| 4 | 1 | 16 | NO2 | H2O | 92 | 14/86 | 87 |
| 5 | 1 | 16 | F | H2O | 96 | 11/88 | 76 |
| 6 | 0 | 16 | H | H2O | 99 | 61/39 | 62 |
| 7 | 0 | 16 | NO2 | H2O | 56 | 63/37 | 20 |
| 8 | 0 | 16 | F | H2O | 99 | 61/39 | 70 |
Significant improvements to reaction outcome were observed when water was added to the reaction vessel. With respect to the cyclohexanone containing examples (Table 3, entries 3–5), 16 shows a marked improvement in both conversion, dr and ee compared to the purely alkyl bridged organocatalyst 3. Thus suggesting that there is some effect elicited by the combination of oxygenated linker group and water at the emulsion interface. In a similar trend observed throughout this study, the use of cyclopentanone as the donor ketone resulted in decreased enaniomeric excesses and only moderate control over diastereomeric ratio.
Although not definitive, the experimental results presented in this manuscript can be rationalised by the formation of a chiral assembly at the oil-in-water interface. We believe that full elucidation of this catalytic mechanism is beyond the scope of this comparative study. Never-the-less the data presented herein will assist in the design of organocatalysts bearing multiple proline groups.
Conclusions
In conclusion, this manuscript describes the synthesis of a range of C2-symmetric organocatalysts 1–5 and 16 consisting of multiple trans-TBDPS-prolinamides bearing systematically increasing linker lengths. Each of these organocatalysts were evaluated for their ability to facilitate the asymmetric aldol reaction on-water at a low catalyst loading of 1 mol% without the use of additives or co-solvents. It was observed that as the distance between the prolinamide units increased, both the conversion to the desired product and the optical purity (dr and ee) decreased among each aldol reactions. Also described is a new intramolecular organocatalytic interaction between prolinamide moieties that allows for a cooperative catalytic process. This effect was attributed to the formation of a hypothesized chiral ‘catalytic pocket’ providing conformational influences giving rise to aldol products of high optical purity at low loadings.The results obtained from this study will aid the rational design of organocatalysts and introduces the concept of catalytic site proximity within an oligomeric prolinamide system as being a key property that contributes to the activity and chiral induction of future novel catalysts.
Experimental section
General procedure for organocatalysed aldol reactions
Water (1.6 mL) was added to a round-bottom flask charge with organocatalyst (0.002 g, 0.01 eq.) dissolved in ketone (0.127 mL, 1.22 mmol, 5 eq.). Benzaldehyde (0.244 mmol, 37 mg, 1 equiv.) was introduced to the emulsion and the reaction mixture was then stirred vigorously at room temperature for 24 h. The emulsion was then extracted with CHCl3 (2 × 10 mL) and the combined organic phase was washed with 10% citric acid (10 mL). The organic phase was then dried over MgSO4 and the solvent was removed under vacuum to give the crude product as a yellow solid. Analysis by 1H NMR spectroscopy was used to determine the desired product, reaction conversion and the diastereomeric ratio. In cases where conversion was determined to be <20%, the crude product was purified by silica gel column chromatography (1/3 EtOAc:Pet. Spirits). The pure products were analysed by chiral HPLC (Chiral Pak AD-H, hexane:2-propanol, 90:10, 1 mL min−1) to determine the enantiomeric excess. All reactions were performed a minimum of three times and the data presented was considered representative of these results.
:1 ratio of Et2O was added and the biphasic mixture was allowed to stand for 24 h, resulting in crystals forming in the organic phase. The solid was filtered and washed with cold Et2O to give the silylated intermediate as white crystals (2.71 g, 0.733 mol, 96%). 1H NMR (400 MHz, CD3OD) δ 7.67–7.64 (m, 4H), 7.45–7.42 (m, 6H), 4.59 (s, J = 0.27, 1H), 4.23 (1H, m), 3.30 (dd, J = 10.8, 5.4 Hz, 1H), 3.29 (dt, J = 13.5, 2.7 Hz, 1H), 2.31 (ddt, J = 13.5, 7.56, 1.88 Hz, 1H), 1.93 (ddd, J = 13.5, 9.99, 4.05 Hz, 1H), 1.08 (s, 9H). Compound was identified by 1H NMR and was consistent with literature values.8 Without further purification, TBDPSO-proline (2.71 g, 0.734 mmol) was dissolved in a 1:1 ratio of THF–H2O (20 mL:20 mL). To this mixture was added NaOH (0.733 g, 0.018 mol) along with Boc2O (2.08 g, 0.953 mmol) and the solution was stirred for 16 h at room temperature. The resulting solution was then acidified with 2 M HCl and extracted into diethyl ether (3 × 20 mL). The combined organic phase was dried over MgSO4 and the solvent was removed in vacuo to afford clear viscous oil. The crude mixture was purified via flash chromatography (1/4 EtOAc:Pet spirits) to give the pure N-Boc protected monomer as a colourless oil (2.82 g, 0.6 mol, 86%). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.64 (m, 4H), 7.39 (m, 6H), 4.56 (t, J = 8 Hz, 1H), 4.43 (m, 1H), 3.59–3.29 (m, 2H), 2.28–2.04 (m, 2H), 1.47 (m, 9H), 1.07 (m, 9H). Compound was identified by 1H NMR and was consistent with literature values.8
:Pet spirits → 1/1 EtOAc:Pet spirits) to give the pure dimer as a viscous colourless oil (0.292 g, 0.254 mmol, 85%). Rf = 8/33 (1:1 EtOAc:Pet. Spirits); 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.62 (m, 8H), 7.40 (m, 12H), 4.40 (br, 4H), 3.72 (br, 1H), 3.43 (br, 1H), 3.23 (br, 4H), 2.22 (br, 2H), 1.88–1.76 (br. m, 2H), 1.43 (br, 18H), 1.25 (br, 4H), 1.03 (br, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) = 172.0, 155.9, 135.7, 133.6, 129.9, 127.9, 71.0, 60.5, 59.1, 55.2, 40.2, 38.9, 37.7, 28., 26.9, 19.2; [α]22.2D = −30.8° (0.00089, CHCl3); IRmaxλ = 2930 (m), 1660 (s), 1105 (s), 700 (s); HRMS calculated for [C56H79N4O8Si2+] M = 991.5431, found 991.5401.
:9 MeOH:EtOAc); Mp = 205.5–207 °C; 1H NMR (400 MHz, CDCl3) d (ppm) = 7.61 (m, 8H), 7.36 (m, 12H), 4.35 (br m, 2H), 4.00 (t, J = 8.4 Hz, 2H), 3.14 (m, 4H), 2.90 (d, J = 12.1, 2H), 2.58 (d, J = 2.94, 12.1, 2H), 2.26 (m, 2H), 1.70 (m, 2H), 1.45 (br, 4H), 1.04 (br, 18H). 13C NMR (100 MHz, CDCl3) δ (ppm) = 173.7, 135.8, 133.2, 129.9, 127.9, 71.8, 65.9, 59.4, 51.9, 38.9, 26.7, 19, 15.4; [α]23.3D = −37.6° (c = 0.0025, CHCl3); IR νmax = 2925 (m), 1624 (s) 698 (s); HRMS calculated for [C46H63N4O4Si2+] M = 791.4382, found 791.4395.
:Pet spirits) to give the pure dimer as a white amorphous solid (0.484 g, 0.475 mmol, 66%). 1H NMR (270 MHz, CDCl3) δ (ppm) = 7.59–7.25 (m, 20H), 4.39 (m, 4H), 3.69–3.42 (m, 4H), 3.17 (br s, 4 H), 2.39–1.97 (br m, 4H), 1.43 (s, 18H), 1.40 (br, 4H), 1.24 (br, 4H), 1.02 (s, 18H). The compound was confirmed by correlation to published spectra.7
:Pet spirits) to give the pure dimer 10 as a viscous colourless oil (0.507 g, 0.544 mmol, 45%). Rf = 11/31 (1/3 EtOAc:Pet. Spirits); 1H NMR (270 MHz, CDCl3) δ (ppm) = 7.69–7.36 (m, 20H), 4.52–4.36 (m, 4H), 3.67–3.65 (m, 4H), 3.52–3.37 (m, 4H), 2.29–1.79 (m, 4H), 1.57 (s, 4H), 1.43 (s, 18H), 1.24 (s, 8H), 1.03 (s, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) = 173.7, 153.9, 135.7, 133.6, 123.0, 127.7, 71.6, 70.8, 58.3, 57.8, 54.9, 54.6, 52.2, 52.0, 39.7, 38.9, 28.5, 28.4, 26.9, 19.4. [α]21.2D = −89.2° (c = 0.001, CHCl3); IR νmax = 2928 (s), 1664 (s), 1162 (s), 702 (s); HRMS calculated for [C60H87N4O8Si2+] M = 1047.6057, found 1047.6075.
:9 MeOH–EtOAc); M.p. = 88.9–90 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.6 (m, 8H), 7.38 (m, 12H), 4.36 (br, 2H), 4.08 (t, J = 8 Hz, 2H), 3.15 (q, J = 4 Hz, 4H), 2.95–2.91 (dt, J = 12, 1.6 Hz, 2H), 2.62 (dd, J = 12, 3.2 Hz, 2H), 2.29 (m, 2H), 1.74 (ddd, J = 13.2, 8.4, 4.4 Hz, 2H), 1.42 (m, 8H), 1.23 (m, 4H), 1.04 (m, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) 174.37, 135.74, 135.69, 134.03, 133.73, 129.90, 129.86, 127.84, 127.82, 77.49, 77.17, 76.86, 74.81, 59.93, 55.62, 40.03, 38.86, 29.61, 29.15, 27.00, 26.82, 19.17; [α]22.5D = −16.0° (c = 0.001, CHCl3); IR νmax = 2929 (s), 1657 (s), 702 (s); HRMS calculated for [C50H71N2O4Si2+] M = 847.5008, found 847.5016.
:Pet spirits) to give the pure dimer 11 as a viscous colourless oil (0.246 g, 0.23 mmol, 36%). Rf = 18/31 (1/3 EtOAc:Pet. Spirits); 1H NMR (400 MHz, CDCl3) δ (ppm) 7.59 (m, 8H), 7.37 (m, 12H), 4.39 (s, 4H), 3.64–3.44 (m, 4H), 3.17 (br, 4H), 2.60 (dd, J = 12, 4 Hz, 2H), 2.28 (m, 2H), 1.73 (m, 2H), 1.43 (m, 4H), 1.28–1.90 (m, 4H), 1.44 (br, 18H), 1.4 (br, 4H), 1.2 (br, 12H), 1.02 (s, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) = 173.66, 153.91, 135.69, 133.62, 129.99, 127.87, 70.85, 58.28, 57.78, 54.90, 54.56, 52.21, 52.01, 39.66, 38.88, 28.48, 28.35, 26.90, 19.14; [α]23.6D = −29.4° (c = 0.001, CHCl3); IR νmax = 2929 (s), 1700 (s), 1162 (s), 702 (s); HRMS calculated for [C62H91N4O8Si2] M+ = 1075.6370, found 1075.6345.
:9 MeOH–EtOAc); 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.62 (m, 8H), 2.38 (m, 12H), 4.36 (s, 2H), 4.03 (t, J = 8.4 Hz, 2H), 3.15 (m, 4H), 2.92 (d, J = 12 Hz, 2H), 2.60 (dd, J = 12, 3.2 Hz, 2H), 2.32–2.26 (m, 2H), 1.73 (ddd, J = 12.8, 8, 4.4 Hz, 2H), 4.13 (br m, 4H), 1.22 (br s, 12H), 1.04 (s, 18H); 13C NMR (125 MHz, CDCl3) δ (ppm) = 174.3, 135.7, 134.0, 133.6, 129.8, 121.8, 59.9, 55.5, 47.9, 40.0, 38.8, 29.6, 29.4, 29.2, 26.9, 26.85, 19.0; [α]24.1D = −49.4° (c = 0.001, CHCl3); IR νmax = 2928 (s), 1662 (s), 702 (s); HRMS calculated for [C52H75N4O4Si2+] M = 875.5321, found 875.5337.
:Pet spirits) to give the pure dimer 12 as a viscous colourless oil (0.14 g, 0.127 mmol, 42%). Rf = 1/3 (1:1 EtOAc:Pet. Spirits); 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.62 (m, 8H), 7.38 (m, 12H), 4.41–4.32 (br m, 4H), 3.71 (br, 2H), 3.44 (br, 2H), 3.16 (m, 4H), 2.31 (br, 2H), 2.06–1.94 (br m, 2H), 1.42 (br, 18H), 1.25 (br, 20H), 1.04 (br, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) = 171.4, 156.5, 135.7, 133.6, 129.9, 127.9, 71.0, 60.6, 58.9, 55.2, 40.2, 39.4, 36.9, 32.0, 29.8, 29.6, 29.34, 28.4, 26.9, 19.2; [α]24.6D = −46.7° (0.00033, CHCl3); IR νmax = 2927 (s), 11656 (s), 700 (s); HRMS calculated for [C64H95N4O8Si2+] M = 1103.6683, found 1103.6698.
:9 MeOH:EtOAc); 1H NMR (270 MHz, CDCl3) δ (ppm) = 7.59 (br m, 8H), 7.38 (m, 12H), 4.36 (br, 1H), 4.09 (t, J = 8.4 Hz, 1H), 3.14 (m, 4H), 2.94 (d, J = 11.9 Hz, 2H), 2.65 (dd, J = 11.9, 3.32 Hz, 2H), 2.27 (m, 2H), 1.76 (m, 2H), 1.41 (br, 4H), 1.21 (br, 16H), 1.03 (br, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) = 173.9, 135.7, 133.9, 133.6, 129.1, 127.8, 74.6, 65.9, 59.8, 55.5, 39.7, 39.03, 29.78, 29.62, 29.52, 29.24, 26.7, 19.1, 15.4; [α]24.3D = −12.8° (0.00113, CHCl3); IR νmax = 2927 (s), 1658 (s), 1105 (s), 700 (s); HRMS calculated for [C54H79N4O4Si2+] M = 903.5634, found 903.56196.
:EtOH) to give the pure dimer as a viscous colourless oil (0.165 g, 0.157 mmol, 29%). Rf = 20/37 (99/1 EtOAc:EtOH); 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.62–7.36 (m, 20H), 4.39 (m, 4H), 3.66 (m, 4H), 3.51 (m, 6H), 3.36 (m, 6H), 2.29–1.90 (m, 4H), 1.47 (s, 18H), 1.08 (s, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) = 135.73, 135.66, 133.56, 129.95, 127.86, 70.29, 55.23, 53.73, 28.42, 26.89, 19.15; [α]21.1D = −1.70° (c = 0.002, CHCl3); IR νmax = 2930 (s), 1701 (s), 1162 (s), 703 (s); HRMS calculated for [C58H83N4O10Si2+] M = 1051.5642, found 1051.5623.
:9 MeOH–EtOAc); M.p. = 61–62.5 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.6 (m, 8H), 7.37 (m, 12H), 4.38 (br s, 2H), 4.14 (t, J = 8 Hz, 2H), 3.47 (m, 12H), 2.97 (d, J = 12 Hz), 2.71 (dd, J = 12, 4 Hz), 2.26 (m, 2H), 1.79 (m, 2H), 1.02 (m, 18H); 13C NMR (100 MHz, CDCl3) δ (ppm) = 135.75, 135.73, 135.72, 135.69, 133.84, 133.61, 129.93, 129.91, 127.88, 127.86, 127.84, 127.83, 74.38, 70.27, 69.94, 65.94, 59.83, 55.34, 39.80, 38.88, 26.96, 19.15, 19.14, 19.13, 19.08, 15.36; [α]22.5D = −0.17° (c = 0.01, CHCl3); IR νmax = 2929 (s), 1662 (s), 1111 (s), 703 (s); HRMS calculated for [C48H67N4O4Si2+] M = 851.4594, found 851.4569.
Representative syntheses for aldol product racemates (benzaldehyde)
Procedure A: A stirred solution of water (15 mL) was charged with 10 M NaOH (1 mL). Ketone (0.28 mL, 2.7 mol, 5 equiv.) was added and the solution was stirred for 1 min. To the mixture was added aryl aldehyde (0.6 mL, 0.54 mmol, 1 equiv.) and the solution was stirred for 3 h. The mixture was extracted into CHCl3 (3 × 20 mL) and the combined organic phase was dried with MgSO4. The racemic mixture was isolated via flash chromatography (1/3 EtOAc/Petroleum spirits).Procedure B: Pyrrolidine (0.44 mL, 0.53 mmol, 1 equiv.) was added to a stirred solution of CHCl3. To the stirred mixture was added ketone (0.223 mL, 2.65 mmol, 5 equiv.) and aldehyde (80 mg, 0.53 mmol, 1 equiv.). Benzoic acid (20 mg, 0.16 mmol, 0.3 equiv.) was added to the mixture and the reaction was stirred at room temperature for 16 h. The reaction mixture was taken up in additional CHCl3 and washed with 2 M HCl (2 × 20 mL). The organic phase was dried with MgSO4. The racemic mixture was analysed by chiral HPLC without purification.
Acknowledgements
We would like to thank the Deakin University Central Research Grants Scheme. The Institute for Frontier Materials and the Strategic Research Centre for Chemistry and biotechnology for Funding. We would also like to thank Simren Khosa for manuscript preparation and the Australian Government for an Australian Postgraduate Award for JD.Notes and references
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- For selected examples: (a) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37(8), 580–591 CrossRef CAS; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS; (c) J. Mlynarski and B. Gut, Chem. Soc. Rev., 2012, 41, 587–596 RSC; (d) G. Guillena, C. Nájera and D. J. Ramón, Tetrahedron: Asymmetry, 2007, 18, 2249–2293 CrossRef CAS; (e) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (f) J. Mlynarski and J. Paradowska, Chem. Soc. Rev., 2008, 37, 1502–1511 RSC; (g) E. Marqués-López, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138–1167 RSC; (h) H.-F. Jiang, J.-H. Li and Z.-W. Chen, Tetrahedron, 2010, 66, 9721–9728 CrossRef CAS; (i) S. Guizzetti, M. Benaglia, L. Pignataro and A. Puglisi, Tetrahedron: Asymmetry, 2006, 17, 2754–2760 CrossRef CAS; (j) D.-F. Yu, Y. Wang and P.-F. Xu, Tetrahedron, 2011, 67, 3273–3277 CrossRef CAS; (k) L. H. Choudhury and T. Parvin, Tetrahedron, 2011, 67, 8213–8228 CrossRef CAS; (l) D. Enders and S. Chow, Eur. J. Org. Chem., 2006, 4578–4584 CrossRef CAS; (m) D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058–11076 CrossRef CAS; (n) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS; (o) B. List, Chem. Rev., 2007, 107(12), 5413–5415 CrossRef CAS; (p) V. Bisai, A. Bisai and V. K. Singh, Tetrahedron, 2012, 68, 4541–4580 CrossRef CAS.
- (a) S. Aratake, T. Itoh, T. Okano, N. Nagae, T. Sumiya, M. Shoji and Y. Hayashi, Chem.–Eur. J., 2007, 13, 10246–10256 CrossRef CAS; (b) Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., 2006, 6, 972–975 CrossRef; (c) Y. Hayashi, J. Yamaguchi, K. Hibino, T. Sumiya, T. Urushima, M. Shoji, D. Hashizumi and H. Koshino, Adv. Synth. Catal., 2004, 12, 1435–1439 CrossRef.
- Selected examples include: (a) L. Albrecht, F. Cruz Acosta, A. Fraile, A. Albrecht, J. Christensen and K. Jørgensen, Angew. Chem., Int. Ed., 2012, 36, 9088–9092 CrossRef; (b) K. L. Jensen, G. Dickmeiss, H. Jang, L. Albrecht and K. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS; (c) G. Lelais and D. W. C. Macmillan, Aldrichim. Acta, 2006, 39, 79–87 CAS.
- Selected examples include: (a) E. Emer, P. Galletti and D. Giacomini, Tetrahedron, 2008, 64, 11205–11208 CrossRef CAS; (b) Y. H. Chen, P. H. Sung and K. Sung, Amino Acids, 2009, 38, 839–845 CrossRef; (c) S. Aratake, T. Itoh, T. Okano, T. Usui, M. Shoji and Y. Hayashi, Chem. Commun., 2007, 2524 RSC; (d) S. Singh Chimni, S. Singh and D. Mahajan, Tetrahedron: Asymmetry, 2008, 19, 2276–2284 CrossRef; (e) M. Raj and V. K. Singh, Chem. Commun., 2009, 6687–6703 RSC; (f) Y. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 8103–8104 CrossRef CAS; (g) Z.-H. Tzeng, H.-Y. Chen, C.-T. Huang and K. Chen, Tetrahedron Lett., 2008, 49, 4134–4137 CrossRef CAS; (h) D. Font, C. Jimeno and M. A. Pericás, Org. Lett., 2006, 8, 4653–4655 CrossRef CAS; (i) F. Giacalone, M. Gruttadauria, A. M. Marculescu and R. Noto, Tetrahedron Lett., 2007, 48, 255–259 CrossRef CAS; (j) D. Font, S. Sayalero, A. Bastero, C. Jimeno and M. A. Pericás, Org. Lett., 2008, 10, 337–340 CrossRef CAS; (k) Z. Jiang, Z. Liang, X. Wu and Y. Lu, Chem. Commun., 2006, 2801–28036 RSC; (l) Y. C. Teo, G.-L. Chua, C.-Y. Ong and C. Y. Poh, Tetrahedron Lett., 2009, 50, 4854–4856 CrossRef CAS; (m) D. E. Siyutkin, A. S. Kucherenko and S. G. Zlotin, Tetrahedron, 2009, 65, 1366–1372 CrossRef CAS; (n) T. Payagala and D. W. Armstrong, Chirality, 2012, 24, 17–53 CrossRef CAS; (o) M. Lombardo, S. Easwar, F. Pasi and C. Trombini, Adv. Synth. Catal., 2009, 351, 276–282 CrossRef CAS; (p) R. Pedrosa, J. M. Andrés, R. Manzano and P. Rodríguez, Eur. J. Org. Chem., 2010, 5310–5319 CrossRef CAS; (q) F. Chen, S. Huang, H. Zhang, F. Liu and Y. Peng, Tetrahedron, 2008, 64, 9585–9591 CrossRef CAS; (r) X. Liu, L. Lin and X. Feng, Chem. Commun., 2009, 6145–6158 RSC.
- (a) G. Guillena, M. del Carmen Hita and C. Nájera, Tetrahedron: Asymmetry, 2006, 17, 729–733 CrossRef CAS; (b) G. Guillena, M. d. C. Hita and C. Nájera, Tetrahedron: Asymmetry, 2006, 17, 1027–1031 CrossRef CAS; (c) S. Samanta, J. Liu, R. Dodda and C.-G. Zhao, Org. Lett., 2005, 7, 5321–5323 CrossRef CAS; (d) Y. Ma, Y. J. Zhang, S. Jin, Q. Li, C. Li, J. Lee and W. Zhang, Tetrahedron Lett., 2009, 50, 7388–7391 CrossRef CAS.
- (a) J. P. Delaney and L. C. Henderson, Adv. Synth. Catal., 2012, 354, 197–204 CrossRef CAS; (b) J. P. Delaney and L. C. Henderson, Int. J. Mol. Sci., 2011, 12, 9083–9094 CrossRef CAS.
- S. M. Opalka, J. L. Steinbacher, B. A. Lambiris and D. T. McQuade, J. Org. Chem., 2011, 76, 6503–6517 CrossRef CAS.
- (a) N. Mase and C. F. Barbas III, Org. Biomol. Chem., 2010, 8, 4043–4050 RSC; (b) J. K. Beattie, C. S. P. McErlean and C. B. W. Phippen, Chem.–Eur. J., 2010, 16, 8972–8974 CrossRef CAS; (c) M. R. Vishnumaya and V. K. Singh, J. Org. Chem., 2009, 4289–4297 CrossRef CAS; (d) G. Casiraghi and F. Zanardi, Chem. Rev., 2000, 100, 1929–1972 CrossRef CAS; (e) R. Mahrwald, in Modern Aldol Reactions Vol. 1, Wiley-VCH, 2004 Search PubMed; (f) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC.
- (a) C. Wu, X. Fu and S. Li, Eur. J. Org. Chem., 2011, 1291–1299 CrossRef CAS; (b) S. S. Chimni, S. Singh and D. Mahajan, Tetrahedron: Asymmetry, 2008, 19, 2276–2284 CrossRef CAS; (c) C. Wu, X. Fu, X. Ma and S. Li, Tetrahedron: Asymmetry, 2010, 21, 2465–2470 CrossRef CAS.
- (a) M. l. Penhoat, D. Barbry and C. Rolando, Tetrahedron Lett., 2011, 52, 159–162 CrossRef CAS; (b) F. Giacalone, M. Gruttadauria, P. L. Meo, S. Riela and R. Noto, Adv. Synth. Catal., 2008, 350, 2747–2760 CrossRef CAS; (c) G. Guillena, M. Hita, C. Najera and S. F. VioÌzquez, J. Org. Chem., 2008, 73, 5933–5943 CrossRef CAS.
- (a) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS; (b) A. Lombardo, A. Quintavalla, M. Chiarucci and C. Trombini, Synlett, 2010, 1746–1765 CrossRef; (c) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110(10), 6302–6337 CrossRef CAS.
- R. Pedrosa, J. M. Andres, R. Manzano, D. Roman and S. Tellez, Org. Biomol. Chem., 2011, 9, 935–940 CAS.
- Z. Tang, Z.-H. Yang, X.-H. Chen, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, J. Am. Chem. Soc., 2005, 127, 9285–9289 CrossRef CAS.
- (a) M. Raj, V. Vishnumaya, S. K. Ginotra and V. K. Singh, Org. Lett., 2006, 8, 4097–4099 CrossRef CAS; (b) V. Maya, M. Raj and V. K. Singh, Org. Lett., 2007, 9, 2593–2595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR of all novel compounds and chiral HPLC traces and their corresponding racemates is provided. See DOI: 10.1039/c3ob27250h |
| This journal is © The Royal Society of Chemistry 2013 |