 Open Access Article
Open Access ArticleProtein destabilisation by ruthenium(II) tris-bipyridine based protein-surface mimetics†
Andrew J. Wilson*ab, James R. Aultbc, Maria H. Filbyab, Hazel I. A. Philipsd, Alison E. Ashcroftbc and Nicholas C. Fletcherd
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, United Kingdom. E-mail: A.J.Wilson@leeds.ac.uk; Fax: +44 (0) 113 3436565; Tel: +44 (0) 113 3431409
bAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, United Kingdom
cInstitute of Molecular and Cellular Biology, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, United Kingdom
dSchool of Chemistry and Chemical Engineering, Queen's University, Belfast, BT9 5AG, UK
First published on 30th January 2013
Abstract
Highly functionalised ruthenium(II) tris-bipyridine receptor 1 which acts as a selective sensor for equine cytochrome c (cyt c) is shown to destabilise the native protein conformation by around 25 °C. Receptors 2 and 3 do not exert this effect confirming the behaviour is a specific effect of molecular recognition between 1 and cyt c, whilst the absence of a destabilising effect on 60% acetylated cyt c demonstrates the behaviour of 1 to be protein specific. Molecular recognition also modifies the conformational properties of the target protein at room temperature as evidenced by ion-mobility spectrometry (IMS) and accelerated trypsin proteolysis.
Introduction
The central role of protein–protein interactions1 in all biological processes, and in particular defective pathways, renders them as attractive, but until recently, intractable targets for molecular intervention.2,3 Achieving high affinity recognition of protein surfaces3,4 represents a starting point for the development of competitive inhibitors of protein–protein interactions (PPIs), hence the design and synthesis of receptors that bind to the solvent exposed surface of proteins is an area of important current investigation.3 Whilst a significant number of receptors for protein surfaces have been described,5–15 including ligands capable of selective recognition6,7,14,16,17 and inhibition of target protein–protein interactions,8,10 structural studies18,19 and the structural consequences11–13,20–22 of binding are less well-documented. Proteins are dynamic, continuously undergoing folding and refolding;23 ligand binding changes this equilibrium as evidenced by natural biological systems which exploit this feature in the form of chaperones that can “fold” a given protein following synthesis and also “unfold” a protein prior to degradation.24 Thus, the potential for protein-surface receptors to stabilise a native conformation or interact with non-native conformations25 to effect protein function represents a fundamentally different approach (than for competitive PPI inhibition) to modulate biological function.9,26,27We6,14 and others28–30 previously reported a series of ruthenium-based receptors for protein-surface recognition. Our studies have illustrated that the sensing of proteins can be performed by following the emission of the ruthenium tris-bipyridine core and that selective recognition of equine cytochrome c (cyt c) can be achieved in comparison to a series of other proteins.14 Furthermore, we have shown that the affinity towards protein targets is dependent upon the geometrical arrangement of ligands around the ruthenium core (fac/mer).6 The current paper describes our studies on the effect that binding of these receptors has on the conformational stability of cyt c. We illustrate that receptor 1 specifically destabilizes cyt c and does so in a potent and dose dependent manner, recreating some of the essential features of chaperones.
Results and discussion
Binding properties of ruthenium based receptors
The synthesis and binding affinities of ruthenium(II) tris bipyridine receptors 1–3 have been previously reported (Fig. 1).14 Briefly, receptor 1 was shown to bind selectively to cyt c with nanomolar affinity, Kd ∼ 2 nM (pH 7.4, 5 mM sodium phosphate) as evidenced by luminescence quenching upon titration of the ruthenium-based receptor with cyt c. The interaction was shown to be specific and selective as evidenced by the absence of binding towards 60% acetylated cyt c (Kd > 30 μM) and much weaker binding towards lysozyme – a protein of comparable pI/surface composition (Kd ∼ 294 nM) under the conditions of the experiment. Compound 2 was shown to bind cyt c with much lower affinity (Kd ∼ 23 nM) whereas compound 3, which is charge mismatched with the target protein, did not bind (Kd > 30 μM) as predicted. Experimental data point to an interaction of 1 with the haem exposed edge of cyt c; notably, a large reduction in the rate of ascorbate oxidation is observed consistent with blocking the approach of this small reducing agent. This is additionally supported by the aforementioned titration with 60% acetylated cyt c; this randomly acetylated protein preparation has its surface exposed basic residues capped by the acetyl modification and is therefore incapable of participating in strong electrostatic interactions. The majority of lysine residues in cyt c surround the haem exposed edge (Fig. 1b).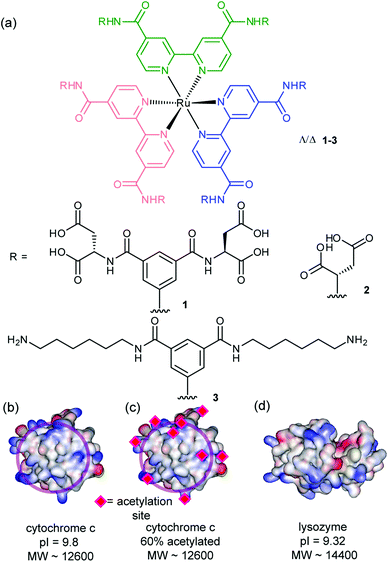 | ||
| Fig. 1 Structures of receptors and proteins (together with relevant properties) described in this work (a) highly functionalized ruthenium tris-bipyridine receptors 1–3 (b) cytochrome c (PDB ID 1HRC),31 (c) 60% acetylated cytochrome c (d) lysozyme (PDB ID 2LYM).32 | ||
Structural consequences of binding
To probe the structural consequences of binding the ruthenium receptor to cyt c, we performed circular dichroism (CD) experiments. CD is a useful technique for studying the secondary structural composition and stability of biomacromolecules, in particular, for giving a diagnostic signature for secondary structural components of peptides and proteins. At 25 °C, cyt c exhibits minima at 222 nm and 210 nm consistent with its high α-helical content (Fig. 2a). Addition of an excess of receptor 1 does not change the circular dichroism spectrum in the 300–200 nm region (Fig. 2a) indicating that supramolecular interaction between receptor 1 and protein does not result in observable conformational changes. In contrast the thermal melting profile of cyt c is dramatically affected by the presence of receptor 1 (Fig. 2b); normally a very thermally stable protein (Tm ∼ 87 °C), the melting temperature of the protein is reduced by 25 °C (Tm ∼ 62 °C) as a consequence of receptor binding. To determine if this effect was dependent on molecular recognition between protein and receptor we performed a titration of cyt c with 1 at 70 °C (Fig. 2c). This temperature is below the point at which thermal unfolding of cyt c is observed in the absence of 1 and hence represents an ideal temperature at which to ascertain if the reduction in Tm observed for the thermal melt derives from molecular recognition. As expected, titration of cyt c with 1 at 70 °C results in a dose dependent decrease in the circular dichroism spectrum. Curiously when the change in signal at 222 nm is plotted against the concentration of 1, the response saturates at 0.5 equiv. of 1, i.e. 2 unfolded proteins bind to 1 at 70 °C. An accurate estimation of binding affinity is not possible on the basis of this titration as Kd is much smaller than the concentration at which it is possible to perform CD experiments. The behaviour is distinct from that seen with porphyrins which bind with increasing stoichiometry to cyt c as it unfolds,11 although ruthenium complexes of this nature have been shown to bind with different stoichiometries to different proteins.30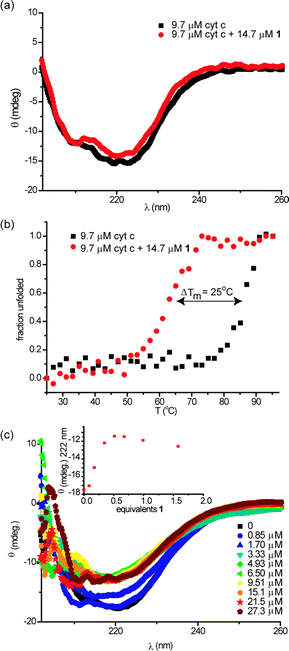 | ||
| Fig. 2 Perturbations to secondary structure of cyt c in the presence of 1 (5 mM sodium phosphate, pH 7.4), (a) circular dichroism spectra of cyt c (9.7 μM) in the absence and presence of 1 (14.7 μM) at 25 °C, (b) thermal melting profiles of samples from (a) and (c) circular dichroism spectrum of 9.6 μM cyt c upon titration with 1 at 70 °C (inset: illustrates titration curve). | ||
To study the specificity of the denaturing abilities of 1 we performed melting experiments with 2 and 3 alongside different proteins. Compound 3 binds with no appreciable affinity to cyt c and had no effect on the CD spectrum of cyt c at 25 °C (see ESI†) or its melting profile (Fig. 3a). Similarly, compound 2 did not affect the CD spectrum of cyt c at 25 °C, but in contrast to 3, it did affect the thermal melting profile (Tm = 80 °C) (Fig. 3a). This is consistent with the weaker binding affinity to the native state; the melting temperature of cyt c is diminished to a lesser extent (ΔTm = 7 °C) in the presence of 2 than in the presence of compound 1 (ΔTm = 25 °C). These data point to a specific interaction between 1 and cyt c as the origin of the conformational changes observed in the protein. For acetylated cyt c (Tm = 65 °C), compound 1 had no effect on the CD spectra at 25 °C (see ESI†) or on the melting profile of the protein (Fig. 3b). This suggests the destabilizing effect of 1 is specific for cyt c. The melting behaviour of lysozyme gave inconsistent and irreproducible results and is currently under further investigation.
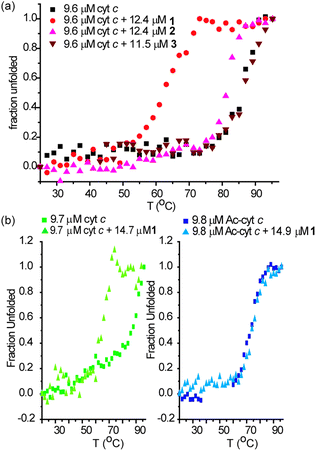 | ||
| Fig. 3 Perturbations to secondary structure of cyt c in the presence of 1–3 and of cyt c and 60% acetylated cyt c in the presence of 1 (5 mM sodium phosphate, pH 7.4), (a) thermal melting profiles of 9.6 μM cyt c in the presence of an excess of 1–3 (b) thermal melting profiles of 9.7 μM cyt c and 9.8 μM 60% acetylated cyt c in the presence of an excess of 1. | ||
In rationalising the effects observed above, a number of additional points are worthy of note. At 25 °C where the affinity of 1 for cyt c is Kd = 2 nM,14 a binding efficiency of 2 kJ mol−1 per carboxylate may be estimated, whereas for 2 with a Kd = 23 nM, a binding efficiency of 4 kJ mol−1 per carboxylate may be estimated. This indicates that a significant proportion of 1 is redundant in terms of maximising non-covalent contacts with cyt c and is not surprising given the symmetrical nature of the receptor i.e. both faces of 1 cannot make contacts with a single cyt c molecule at room temperature. Comparison of 60% acetylated cyt c (Tm = 73 °C) with cyt c (Tm = 87 °C), reveals a difference in Tm between the two proteins of ΔTm = 14 °C33 suggesting that the lysine residues play an essential role in maintaining the stability of cyt c.33 It is noteworthy that P. aeruginosa cyt c which lacks lysine residues on its haem exposed edge is also less stable than equine cyt c.9 Finally, the decrease in Tm for cyt c in the presence of 1 (to ∼62 °C) and the Tm for 60% acetylated cyt c are both significantly lowered relative to cyt c. A hypothesis whereby interaction between the carboxylates of receptor 1 and the surface exposed lysines results in structural loosening34 emerges (Fig. 4a). Such an effect would be driven by entropy and thus manifested more prominently at higher temperatures. Similarly the different stoichiometries can be accounted for with this hypothesis – at ambient temperature the stoichiometry is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 whereas at higher temperatures more of the carboxylates in 1 can engage in multivalent interactions with two proteins both of which possess greater chain entropy in a (partially) unfolded state (Fig. 4b).
:1 whereas at higher temperatures more of the carboxylates in 1 can engage in multivalent interactions with two proteins both of which possess greater chain entropy in a (partially) unfolded state (Fig. 4b).
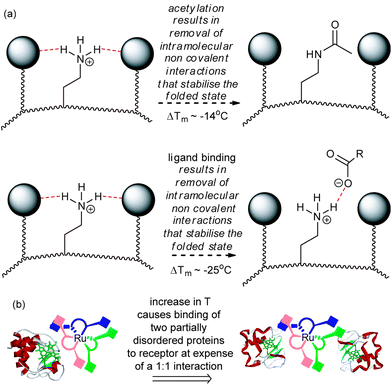 | ||
| Fig. 4 Schematic depicting the basis of reduced Tm in cyt c (a) post covalent modification and binding to surface receptors results in structural loosening in order to maximise non-covalent contacts (b) a change in stoichiometry results on increasing the temperature and is promoted by binding of more disordered proteins. | ||
Ion mobility spectrometry-mass spectrometry
To study the effects of binding upon the structural properties of cyt c we sought additional techniques complementary to the spectroscopic methods described above. Electrospray ionisation-mass spectrometry (ESI-MS) gives rise to a series of multiply charged ions from which the molecular mass of a protein can be calculated. In general, a higher number of charges on an ion indicates the more unfolded nature of the protein in solution, and vice versa. Ion mobility spectrometry-mass spectrometry (IMS-MS) is a technique that has been used to measure the molecular mass and also to study the collision cross-sectional area (Ω) of proteins in a single, rapid experiment.35–38 Notably, measurements on cyt c have shown that it adopts a range of conformational states under different conditions and that different charge states can adopt a number of conformational states.37–39 Binding of the receptor 1 to cyt c does not result in any change to the charge state distribution in the mass spectrum indicating that folding of cyt c in solution is not perturbed which is consistent with the room temperature CD experiments, however, changes to the cross-sectional areas of the protein in the gas phase indicate that the complex can remain bound to and access multiple conformational states (Fig. 5). This is most pronounced for binding of the complex to the +7 charge state of cyt c; in the absence of receptor, two conformational states are adopted (1565 and 1716 Å2) with the latter more prominent, whereas in the presence of receptor 1, two lower cross sectional areas are observed (1344 and 1523 Å2) with the former now the more prominent. The cross-sectional area appears to be reduced, at least in the gas phase, as a consequence of protein surface recognition. This may be considered surprising, however in prior studies of cyt c, the +7 and +8 charge states exhibit a range of cross-sections consistent with a transition to a less compact protein conformation with increasing charge.37 A plausible hypothesis here is that binding of the ruthenium receptor retards this gas phase property by mitigating the effects of charge repulsion. Overall the IMS-MS data support the CD data that indicate that the cyt c remains folded at room temperature in the presence of receptor 1.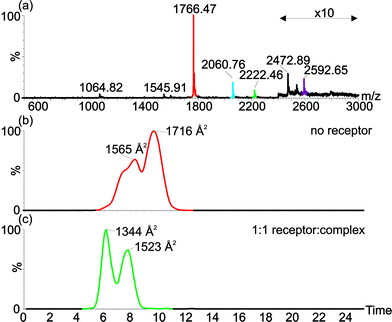 | ||
| Fig. 5 Electrospray ionisation-ion mobility spectrometry-mass spectrometry shows changes in average collision cross-sectional area of cyt c in presence and absence of the receptor indicating that the receptor can bind to multiple conformations of cyt c. (a) ESI-MS of cyt c in presence of 1 equivalent of receptor, shows receptor binding to the +7 (red-unbound; green-receptor bound) and +6 (blue-unbound; purple-receptor bound) charge states. Extracted ion mobility chromatograms show collisional cross-sectional areas for (b) the +7 charge state signals for unbound (red) and receptor bound (green) cyt c. | ||
Trypsin proteolysis
Having (a) shown binding of the receptor (1) to cyt c lowers the melting temperature of the protein and (b) observed receptor binding to multiple conformational states in IMS-MS, we postulated that binding of the ruthenium receptor 1 to the surface of cyt c might affect the rate of proteolysis. Trypsin itself is unfolded and therefore inactive at the temperature where unfolding of cyt c is promoted by 1, however, although no effect on secondary structure is evident from the CD studies at ambient temperature, accelerated proteolysis might still be observed under these conditions. As CD spectroscopy reports on gross secondary structure, the experiments described above do not necessarily report on the accessibility of residues within loops etc. whilst if the rate of folding transitions is affected, proteolysis could be accelerated because it represents a thermodynamic sink. Indeed, proteolysis of cyt c is accelerated in the presence of stoichiometric and sub-stoichiometric quantities of 1 (Fig. 6 and ESI†). Fig. 6 illustrates that the proportion of digested peptide fragments after 1200 min is greater in the presence of 1. There is no evidence of a change in the preferred cleavage sites for cyt c in the presence of 1 which is consistent with a general increase in accessibility of cleavable residues. The difference in rate of proteolysis is evident after 120 min (see ESI†).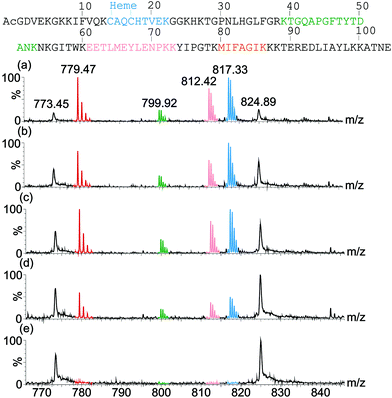 | ||
| Fig. 6 ESI-MS spectra of proteolysis samples after 1200 min digestion showing different rates of digestion of cyt c in the presence of (a) 2 equivalents of receptor, (b) 1 equivalent of receptor, (c) 0.2 equivalents of receptor and (d) no receptor. A control containing no trypsin and no receptor was also analysed (e). Tryptic peptide signals in the MS are colour coded corresponding to their sequence in the protein (top). | ||
Conclusions
In conclusion we have reported that the ruthenium tris-chelate 1 perturbs the stability of its target protein–cytochrome c. 1:1 Binding between 1 and the native state of cyt c at room temperature is strong (Kd ∼ 2 nM), but at higher temperatures specific dose responsive binding to an unfolded form of the protein is promoted, accompanied by a change in stoichiometry to 1:2 1:cyt c. The effect is also specific for cyt c. In addition, receptor 1 can remain bound to more than one conformational state and can change the distribution of states of cyt c at room temperature in the gas phase as evidenced by IMS-MS. Finally, trypsin proteolysis of cyt c is accelerated in the presence of receptor 1. Overall, the results illustrate that solvent exposed protein surface recognition of cyt c by 1 causes changes to the conformational properties of the protein. Our own future work will focus on probing the generality of these observations with other protein–ligand interactions.Experimental
General considerations
All reagents were purchased from Sigma-Aldrich or Alfa-Aesar and used without further purification unless otherwise stated. The synthesis and characterisation of receptors 1–3 was reported previously.14 Stock solutions of receptors 1–3 were prepared to a concentration of ∼1 mM in 5 mM phosphate buffer. Once the receptors had been dissolved, the pH was adjusted to 7.4 via the addition of 1 N sodium hydroxide or 1 N HCl. Likewise, stock solutions of acetylated horse heart or equine cytochrome c (all obtained from Sigma and used without further purification) was prepared to a concentration of ∼1 mM in 5 mM sodium phosphate buffer and the pH adjusted to 7.4 via the addition of 1 N sodium hydroxide or 1 N HCl and diluted appropriately. The concentrations of horse heart cyt c and acetylated cyt c were determined using the molar extinction coefficient at 550 nM of 2.95 × 104 after reduction using dithionite.Circular dichroism
CD measurements were performed on a JASCO J-715 or JASCO J-815 instrument using quartz 0.1 cm cuvettes with temperature controlled via a peltier device. The machine settings were as follows:J-715 scan settings; 600 nm–190 nm scan range, 0.2 nm pitch, continuous scanning at 100 nm min−1, 100 mdeg sensitivity, 4 s response time, 1.0 nm band width, single scan accumulation.
J-715 temperature ramp settings; λ = 222 nm, 20–97 °C scan range, 2.0 °C pitch, 10 s delay, 2 °C min−1, 100 mdeg sensitivity, 4 s response time, 1.0 nm band width.
J-815 scan settings; 600 nm–190 nm scan range, 0.2 nm pitch, continuous scanning at 100 nm min−1, high sensitivity, DIT 0.25 s, 1.0 nm band width, single scan accumulation.
J-815 temperature ramp settings; λ = 222 nm, 20–97 °C scan range, 2.0 °C pitch, 10 s delay, 2 °C min−1, 0.2 nm pitch, continuous scanning at 100 nm min−1, high sensitivity, DIT 0.25 s, 1.0 nm band width.
Non-covalent mass spectrometry and ion mobility spectrometry-mass spectrometry
Samples in 10 mM ammonium acetate, pH 7.4 and standards in acetonitrile/water/formic acid (50/49/1; v/v/v) were analysed by Z-spray nano-electrospray ionisation (ESI)-MS using a quadrupole-IMS-orthogonal time-of-flight MS (Synapt HDMS, Waters UK Ltd, Manchester, U.K.) using gold/palladium coated nanospray capillaries prepared in-house. The MS was operated in positive ion mobility-TOF mode using an ESI capillary voltage of 1.5 kV, and cone voltage of 150 V. The source and desolvation temperatures were set at 80 °C and 150 °C, respectively. The nanoESI gas pressure was 0.1 bar, source backing pressure was 4.81 mbar. Trap collision energy was 21.8 V, transfer collision energy was 4.0 V and a trap bias of 20 V was used. For MS only measurements, trap and transfer argon gas pressure was 2.58 × 10−3 mbar and the IMS cell nitrogen gas pressure was 2.79 × 10−4 mbar. For IMS measurements trap and transfer argon pressure was 1.79 × 10−2 mbar and IMS nitrogen gas pressure was 4.86 × 10−1 mbar The IMS travelling wave speed was 250 m s−1 and the wave height was 6.2 V. Mass calibration was performed by a separate injection of aqueous sodium iodide solution at a concentration of 2 μg μL−1. IMS drift cell calibration was performed by injection of denatured standard proteins horse heart myoglobin, equine cytochrome c and bovine ubiquitin at 10 μM concentration in acetonitrile/water/formic acid (50/49/1; v/v/v). Reduced cross-sections (Ω′) were calculated from published cross-sections determined using conventional ion mobility measurements40,41 and were plotted against measured drift times (tD). An allometric (y = AxB) fit was applied to the data.38 Experimental cross-sections were determined after separate infusion of the analytes and measurement of the drift time centroid for each charge state signal. Data processing was performed using MassLynx v4.1.Proteolysis experiments
10 μM solutions of cyt c in 10 mM ammonium acetate, pH 7.4 were prepared containing either 0, 0.2 and 2 equivalents of the ruthenium based receptor complex. Trypsin was added to a final concentration of 0.02 μg μL−1. A control sample of 10 μM cytochrome c with no trypsin was also prepared. The samples were incubated at 21 °C with shaking for the duration of the experiment. 10 μL aliquots of solution were removed from each sample at 15 s, 60 s, 120 s, 5 min, 10 min, 15 min, 30 min, 45 min, 60 min, 90 min, 120 min and 1200 min. The aliquots were diluted with 10 μL acetonitrile/water/formic acid (75/24/1 v/v/v) to quench the trypsin reaction ready for MS analysis.Mass spectrometry of proteolysis samples
10 μL of each cyt c sample (5 μM) were analysed by Z-spray nano-ESI-MS using an orthogonal time-of-flight MS (LCT Premier, Waters UK Ltd, Manchester, U.K.) using a Nanomate autosampler infusion device (Advion Biosciences, Ithaca, NY, USA). The mass spectrometer was operated in positive TOF mode using an nanoESI capillary voltage of 1.5 kV, cone voltage of 20 V and nanoESI gas pressure of 0.4 psi. The source temperature was 80 °C. Mass calibration was performed by a separate injection of aqueous sodium iodide solution at a concentration of 2 μg μL−1. Data processing was performed using MassLynx v4.1.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [EP/F039069 and EP/F038712] and by the European Research Council [ERC-StG-240324]. The Synapt HDMS mass spectrometer was purchased with funds from the BBSRC (BB/E012558/1) and the LCT Premier was purchased with funds from the Wellcome Trust (075099/Z/04/Z). We would also like to thank G. Nasir Khan for assistance with additional CD experiments.Notes and references
- O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Chem. Rev., 2008, 108, 1225–1244 CrossRef CAS.
- J. A. Wells and C. L. McLendon, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- A. J. Wilson, Chem. Soc. Rev., 2009, 38, 3289–3300 RSC.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2493 CrossRef CAS.
- M. De, S. Rana, H. Akpinar, O. R. Miranda, R. R. Arvizo, U. H. F. Bunz and V. M. Rotello, Nat. Chem., 2009, 1, 461–465 CrossRef CAS.
- M. H. Filby, J. Muldoon, S. Dabb, N. C. Fletcher, A. E. Ashcroft and A. J. Wilson, Chem. Commun., 2011, 47, 559–561 RSC.
- A. J. Wilson, J. Hong, S. Fletcher and A. D. Hamilton, Org. Biomol. Chem., 2007, 5, 276–285 RSC.
- H. Bayraktar, P. S. Ghosh, V. M. Rotello and M. J. Knapp, Chem. Commun., 2006, 1390–1392 RSC.
- R. K. Jain and A. D. Hamilton, Angew. Chem., Int. Ed., 2002, 41, 641–643 CrossRef CAS.
- M. A. Blaskovich, Q. Lin, F. L. Delarue, J. Sun, H. S. Park, D. Coppola, A. D. Hamilton and S. M. Sebti, Nat. Biotechnol., 2000, 18, 1065–1070 CrossRef CAS.
- K. Groves, A. J. Wilson and A. D. Hamilton, J. Am. Chem. Soc., 2004, 126, 12833–12842 CrossRef CAS.
- A. J. Wilson, K. Groves, R. K. Jain, H. S. Park and A. D. Hamilton, J. Am. Chem. Soc., 2003, 125, 4420–4421 CrossRef CAS.
- D. M. Tragore, K. I. Sprinz, S. Fletcher, J. Jayawickramarajah and A. D. Hamilton, Angew. Chem., Int. Ed., 2006, 46, 223–225.
- J. Muldoon, A. E. Ashcroft and A. J. Wilson, Chem.–Eur. J., 2010, 16, 100–103 CrossRef CAS.
- R. Zadmard and T. Schrader, J. Am. Chem. Soc., 2005, 127, 904–915 CrossRef CAS.
- H. Zhou, L. Baldini, J. Hong, A. J. Wilson and A. D. Hamilton, J. Am. Chem. Soc., 2006, 128, 2421–2425 CrossRef CAS.
- L. Baldini, A. J. Wilson, J. Hong and A. D. Hamilton, J. Am. Chem. Soc., 2004, 126, 5656–5657 CrossRef CAS.
- P. B. Crowley, P. Ganji and H. Ibrahim, ChemBioChem, 2008, 9, 1029–1033 CrossRef CAS.
- R. I. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527–533 Search PubMed.
- S. Fletcher and A. D. Hamilton, New J. Chem., 2007, 31, 623–627 RSC.
- F. Chiba, G. Mann and L. J. Twyman, Org. Biomol. Chem., 2010, 8, 5056–5058 RSC.
- R. Hong, N. O. Fischer, A. Verma, C. M. Goodman, T. Emrick and V. M. Rotello, J. Am. Chem. Soc., 2004, 126, 739–743 CrossRef CAS.
- A. I. Bartlett and S. E. Radford, Nat. Struct. Mol. Biol., 2009, 16, 582–588 CrossRef CAS.
- S. Walter and J. Buchner, Angew. Chem., Int. Ed., 2002, 41, 1098–1099 CrossRef CAS.
- P. Thiel, M. Kaiser and C. Ottmann, Angew. Chem., Int. Ed., 2012, 51, 2012–2018 CrossRef CAS.
- B. A. Foster, H. A. Cofeey, M. J. Morin and F. Rastinejad, Science, 1999, 286, 2507–2510 CrossRef CAS.
- J. R. Peterson, R. S. Lokey, T. J. Mitchison and M. W. Kirschner, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10624–10629 CrossRef CAS.
- J. Ohkanda, R. Satoh and N. Kato, Chem. Commun., 2009, 6949–6951 RSC.
- H. Takashima, S. Shinkai and I. Hamachi, Chem. Commun., 1999, 2345–2346 RSC.
- Y. Yamaguchi, N. Kato, H. Azuma, T. Nagasaki and J. Ohkanda, Bioorg. Med. Chem. Lett., 2012, 22, 2354–2358 Search PubMed.
- G. W. Bushnell, G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 585–595 CAS.
- C. E. Kundrot and F. M. Richards, J. Mol. Biol., 1987, 193, 157–170 CrossRef CAS.
- Y. Hagihara, Y. Tan and Y. Goto, J. Mol. Biol., 1994, 237, 336–348 Search PubMed.
- D. H. Williams, E. Stephens, D. P. O'Brien and M. Zhou, Angew. Chem., Int. Ed., 2004, 43, 6596–6616 CrossRef CAS.
- D. Smith, K. Giles, R. Bateman, S. Radford and A. Ashcroft, J. Am. Soc. Mass Spectrom., 2007, 18, 2180–2190 CrossRef CAS.
- S. D. Pringle, K. Giles, J. L. Wildgoose, J. P. Williams, S. E. Slade, K. Thalassinos, R. H. Bateman, M. T. Bowers and J. H. Scrivens, Int. J. Mass Spectrom., 2007, 261, 1–12 Search PubMed.
- C. A. Scarff, K. Thalassinos, G. R. Hilton and J. H. Scrivens, Rapid Commun. Mass Spectrom., 2008, 22, 3297–3304 CrossRef CAS.
- D. P. Smith, T. W. Knapman, I. Campuzano, R. W. Malham, J. T. Berryman, S. E. Radford and A. E. Ashcroft, Eur. J. Mass Spectrom., 2009, 15, 113–130 CrossRef CAS.
- D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1995, 117, 10141–10142 CrossRef CAS.
- S. J. Valentine, A. E. Counterman and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 1999, 10, 1188–1211 CrossRef CAS.
- D. E. Clemmer, http://www.indiana.edu/~clemmer/Research/research.htm.
Footnote |
| † Electronic supplementary information (ESI) available: Additional CD curves and proteolysis data. See DOI: 10.1039/c3ob26251k |
| This journal is © The Royal Society of Chemistry 2013 |