Tautomerism and metal complexation of 2-acylmethyl-2-oxazolines: a combined synthetic, spectroscopic, crystallographic and theoretical treatment†
Received
6th May 2012
, Accepted 3rd April 2013
First published on 3rd April 2013
Abstract
A synthetic, structural and theoretical investigation into the solid-state, solution and gas phase structure(s) of six 2-acylmethyl-4,4-dimethyl-2-oxazolines is reported. Four of these materials, viz. α-[(4,5-dihydro-4,4-dimethyl-2-oxazolyl)methylene]benzenemethanol (3a), α-[(4,5-dihydro-4,4-dimethyl-2-oxazolyl)methylene]-(4-nitrobenzene)methanol (3b), 1-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-3,3-dimethyl-1-buten-2-ol (3d) and (E)-1-phenyl-2-((3aR)-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-ylidene)ethanone (3f) have been characterised in the solid-state by single crystal X-ray diffraction studies. These data represent the first solid-state structural studies of this class of compounds and details the first synthesis and full characterisation of chiral derivative 3f. All four of these materials are shown to exist in the solid phase in the enamine tautomeric form (e.g., 3a is best described as 2-[4,4-dimethyl-2-oxazolidinylidene]-1-phenylethanone) and it is suggested (NMR, IR) that this isomeric form is likely also retained in solution (e.g., CDCl3) as the more stable isomer. An investigation of the relative gas phase stabilities of the three possible (i.e., the (Z)-enol, keto and enamine) isomers of all five compounds by DFT at the B3LYP/6-311G(d) level of theory confirms the latter as the most stable form. The energy differences between the enamine and keto tautomers have been calculated to be the lowest for derivative 3d. These results are compared and contrasted with the previously reported NMR studies of such compounds which have identified the keto form as being a minor (albeit solution) tautomer. Equilibrium solution tautomer distributions for 3d are found to be solvent dependent. The protonated form of 3a, isolated as the HSO4− salt (i.e.4a), has been further characterised in the solid state by single crystal X-ray diffraction. These data represent the first example of a protonated oxazoline to be structurally elucidated and confirms that upon protonation, the keto (oxazoline) tautomer is the energetically favoured form in the solid-state. This observation is further supported by DFT studies for the gas phase protonated forms of such materials. Further DFT (B3LYP/6-311G(d)) calculations employing the SM8 or SMD solvation models were then applied to address the observed solution isomeric distribution for 3d; these results corroborate the gas phase theoretical treatment and also yield values that predict the higher solution stability of the enamine form as observed, although they fail to account for the existence of the keto form as a minor solution state tautomer. To access the availability of an enol-form, via hypothetical de-protonation to the enolate, compound 3a was treated with hydrated Cu(NO3)2 in EtOH solution. The resulting isolated green-coloured product (5), the first metal derivative of this entire class of ligands, is best described (IR, X-ray diffraction) as a coordinated enolate complex, i.e., Cu(3a-H)2. Complex 5 crystallizes in the P21/c space group with four molecules in the unit cell. The coordination geometry around the formal Cu2+ metal centre is determined to be highly distorted square planar in nature (τ4 = 0.442). TD-DFT is used to give a reasonable explanation for the intensity of the absorbance band observed in the visible region for solutions of 5. These latter experiments strongly suggest that the title class of compounds may have considerable potential as ligands in coordination chemistry and/or metal-mediated catalysis.
Introduction
Azoles, specifically the 2-oxazoline (i.e., 4,5-dihydro-2-oxazole) sub-class of azoles, represent an important group of chiral auxiliaries and metal binding agents which are used extensively in both enantio-selective catalysis and materials science.1 In addition, this heterocycle has been employed for some time as a monomer in polymer chemistry2 and as a directing and/or protecting group in organic syntheses; azoles are also present in a wide variety of natural products and synthetic drugs.3,4 Our own interests lie in the areas of metal coordination chemistry and ligand design strategies of both chiral and achiral oxazolines.5 In this regard, we became interested in a class of oxazoline-derived organics first described by the late A. I. Meyers;6 simple protocols (Scheme 1) for a variety of other examples being revealed sometime later by Tohda and co-workers.7a–c These compounds, in one tautomeric form, contain an oxazoline linked to a ketone functionality via a carbon atom emanating from oxazoline ring position-2 (Scheme 1).6,7 A potential application of these materials involves the sequestering of the enol-form (Scheme 1: A) via κ2-N,O metal chelation either proceeding or subsequent to H+ loss (i.e., bidentate metal-binding via the resulting enolate).8 There are several reasonable resonance forms for this class of heterocycles including two oxazoline forms (Scheme 1: A [enol] and B [keto]) and a secondary amine of the enamine class (Scheme 1: C).
 |
| | Scheme 1 Synthetic scheme leading to 3-type derivatives (enamine isomeric product shown); conditions: (i) 2 × RC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)Cl/NEt3/MeCN/Δ; (ii) KOH/alcohol; (iii) +1/2 × Cu(NO3)2·2.5H2O/EtOH; –HNO3 −1.25 H2. O)Cl/NEt3/MeCN/Δ; (ii) KOH/alcohol; (iii) +1/2 × Cu(NO3)2·2.5H2O/EtOH; –HNO3 −1.25 H2. | |
The investigation reported herein examines several of these materials in both the solid-state (X-ray diffraction, IR), solution (NMR) and in a theoretical sense (DFT) to determine the relative stabilities and properties of such tautomeric possibilities.9 In this regard, reaction of enamine 3a with divalent copper nitrate results in the isolation and full characterisation of the corresponding bis-κ2-N,O-enolate complex. This confirms the accessibility, at least under these conditions, of a hypothetical enol-tautomer of this class of materials and thus establishes the groundwork for a coordination chemistry exploration of ligands such as 3a. A further consequence of this work is the establishment of the facile accessibility of chiral versions of this potential ligand class via example 3f. The forms depicted in Scheme 1 also serve to demonstrate that this class of heterocycles will allow for a rare opportunity10 to examine an enamine, keto and enol-trio, not only in terms of their stabilities, but also with respect to their general structural properties in terms of the resonance-assisted H-bonding (RAHB) theory.11
Results and discussion
As mentioned previously, compounds such as 3a–3e have found application as useful intermediates in the syntheses of other heterocyclic materials.6,7 Both Meyers6 and Tohda7a–c independently surmised that the three tautomers, shown in Scheme 2, could be accessible for these compounds and that this aspect might be a contributing factor to their observed reactivity. Indeed for alkyl derivatives such as 3e, both researchers reported a minor solution tautomer (∼10%) unequivocally identified as the keto-form12 in addition to a major species assigned to the enamine. This latter conclusion was supported by 13C NMR data.6,7 No theoretical studies or any solid-state characterisation (X-ray) data have been reported for any of these materials. An initial selection of three of these compounds was chosen for investigation by single crystal X-ray diffraction and these results are described below.
 |
| | Scheme 2 Schematic representations of the isomers and organic compounds studied herein. | |
Solid-state characterisation
Suitable single crystals of compounds 3a, 3c and 3d (Scheme 2) were obtained by re-crystallisation of the said materials from hexanes–toluene mixtures. Fig. 1–3 give graphical representations (ORTEP13) of a unit cell molecule of each of the respective compounds.
 |
| | Fig. 1 ORTEP representation of a unit cell molecule of 3a, thermal ellipsoids are at the 50% probability level. | |
 |
| | Fig. 2 ORTEP representation of a unit cell molecule of 3c, thermal ellipsoids are at the 50% probability level. | |
 |
| | Fig. 3 ORTEP representation of a unit cell molecule of 3d, thermal ellipsoids are at the 50% probability level. | |
Details of the crystal data and tables of bond lengths and angles can be found in the ESI.† The general crystal parameters reveal nothing unusual nor surprising.14 However, the identification of an N–H group and a formal CC initiating from ring position-2 on the heterocycle are observations of obvious relevance. These results unequivocally identify all three of these compounds as existing in the enamine isomeric form (Scheme 1: C) in the crystalline solid-state.15 This tautomeric situation has been previously noted in, for example, compounds such as (E)-N-(1,3-oxazolidin-2-ylidene)benzamide16a and other related species.10a,16b It is therefore suggested that this isomeric form is the likely thermodynamically more stable solid-state isomer for such compounds.
Resonance assisted H-bonding (RAHB) is a theory first introduced by Gilli et al.11 and is used to describe a special form of intramolecular H-bonding and its effect(s) on ground state molecular structure.11,17,18 This concept is used to evaluate the level of H-bonding and bond length distortions between groups D–H⋯A = where D and A are atoms which are connected through a π-conjugated system. A schematic representation of such a generalised system, with some selected bond labelling, appears in Scheme 3.18 Compounds such as 3a therefore provide an opportunity to examine the presence of the effects attributed to RAHB in enamine-containing molecules. If there is appreciable π-delocalisation present in a particular system, such as that depicted in Scheme 3, this will affect the various interatomic distances d1 through d4 in the following manner: (i) bond lengths d2 and d3 tend to become equal and (ii) d4 bond lengths shorten and d1 bond lengths increase vs. that of typical14 values. The common values for the relevant interatomic distances are shown in Table 1 (taken from the benchmark study of Allen et al.: ref. 14) as are the observed d1–d4 distances found in the solid-state for 3a, 3c and 3d. An examination of Table 1 reveals that in the case of the enamines examined here, the d1 (i.e., carbonyl CO) bond lengths do indeed elongate (∼2–3%) in all three cases and the d4 (i.e., C-NHR′′′) parameter is shorter than typical values by about 1–3%. Significant shortening of d2 (∼3%) and lengthening of d3 (∼3%) is also observed, but these latter two values are still significantly different from one another by an average of more than 0.03 Å. Although more examples will be necessary to make a more concrete conclusion, it can be suggested based on these simple observations that the H-bond resulting from the N–H⋯OC interaction appears to be relatively weak in these systems.17,18
 |
| | Scheme 3 Bond labelling of a hypothetical β-enaminone for discussions of RAHB. | |
Table 1 Bond lengths d1–d4 of β-enaminones with respect to discussions of RAHB.
| Parameter |
Typical valuesa |
Compound 3ab |
Compound 3cc |
Compound 3d |
|
Ref. 14.
Three molecules in the unit cell: average values shown.
Four molecules in the unit cell: average values shown.
Calculated (DFT) values in parentheses.
|
|
d
1 (Å)d |
1.221 for 3a and 3c; 1.222 for 3d |
1.25 |
1.25 |
1.26 |
| (1.25) |
(1.25) |
(1.24) |
|
d
2 (Å)d |
1.464 |
1.41 |
1.41 |
1.42 |
| (1.44) |
(1.43) |
(1.47) |
|
d
3 (Å)d |
1.340 |
1.37 |
1.38 |
1.37 |
| (1.37) |
(1.38) |
(1.37) |
|
d
4 (Å) |
1.355 |
1.32 |
1.32 |
1.34 |
| (1.35) |
(1.34) |
(1.35) |
In an attempt to observe and/or clarify the isomeric distributions in solution of the phenyl compounds such as 3a–c, an examination by VT-1H NMR spectroscopy was initiated. Unfortunately, under both low (−78 °C) and high (+40 °C) temperature conditions only a single (non-keto) isomer was observed in the case of 3a (CDCl3 solution) and indeed a similar result is noted for the RT spectrum of 3a recorded in acetone-d6. These observations are consistent with the room temperature (RT) data reported by Tohda.7a,c As mentioned previously, alkyl derivative 3e reveals the presence of a minor (unequivocally keto) species in addition to the major, enamine (NMR:6,7cvide supra) form. Our independent observations here confirm both Meyers6 and Tohda7c reports of the minor (∼10%) keto 3e tautomer being present at RT in chloroform-d. tert-Butyl compound 3d has also been examined previously via1H NMR spectroscopy. This material has been reported to exhibit a single set of resonances, again presumed to be representative of the enamine, when examined in CCl4 solution by NMR spectroscopy.7a In contrast, our observations confirm the presence of the minor keto form in CCl4 (external CDCl3 lock) being present in about 6.1%. Our further examination of this compound dissolved in CDCl3 also reveals the presence of this same minor species (8.1%: δH = 3.97 [s, 2H, O–CH2], 3.63 [s, 2H, –CH2–], 1.29 [s, 6H, –CH3], 1.18 [(s, 9H, –C(CH3)3]) in addition to the major enamine tautomer. These data suggest that solvent influences may be an important aspect of the isomer distributions in these materials17,19 and that such interactions could be energetically greater than any influences due to RAHB.6,7,17,19,20 Calculations (DFT calculations see ESI†) have revealed that upon protonation, the keto tautomers are considerably lower in energy than the respective protonated enamine or enol species (∼40 kJ mol−1). Therefore, the attempted syntheses of the protonated form of 3a and 3d was carried out (reaction of 3a/d and H2SO4) and the resulting products (e.g., 4a/d) isolated as a crystalline solids.† The nature of these materials was then confirmed by single crystal X-ray diffraction (Fig. 4) and these data represent, to our knowledge, the only crystallographically characterised protonated oxazoline in the literature.
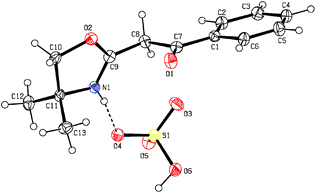 |
| | Fig. 4 ORTEP representation of a unit cell molecule of 4a, thermal ellipsoids are at the 50% probability level. | |
As expected, the data are wholly consistent with a keto-form for crystalline 4a; distinct NC (d = 1.29 Å) and clearly apparent sp3-hybridised C atom emanating from azole ring position-2 (∠C–CH2–C = 111.3°) are observed.† These data corroborate the DFT calculations on the relative stability of the keto following ring ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) -protonation. Unfortunately, samples of 3d treated in this way (to yield the desired 4d) resulted in the isolation of crystalline material of the oxazoline ring-opened ester product formed by the attack by water.¶ Therefore, only solution observations of 4d could be reliably reported. The 1H NMR chemical shift data for 4d (CDCl3: δH = 7.51 [br], 6.30 [br], 4.17 [s, 2H, O–CH2], 3.81 [s, 2H, –CH2–], 1.41 [s, 6H, –CH3], 1.15 [(s, 9H, –C(CH3)3]) do not correspond to the observed keto-form in samples of 3d in CDCl3 mentioned above, thus ruling out a possible assignment of the latter as 4d due to protonation of 3d by residual HCl present in chloroform.19f,g,20
-protonation. Unfortunately, samples of 3d treated in this way (to yield the desired 4d) resulted in the isolation of crystalline material of the oxazoline ring-opened ester product formed by the attack by water.¶ Therefore, only solution observations of 4d could be reliably reported. The 1H NMR chemical shift data for 4d (CDCl3: δH = 7.51 [br], 6.30 [br], 4.17 [s, 2H, O–CH2], 3.81 [s, 2H, –CH2–], 1.41 [s, 6H, –CH3], 1.15 [(s, 9H, –C(CH3)3]) do not correspond to the observed keto-form in samples of 3d in CDCl3 mentioned above, thus ruling out a possible assignment of the latter as 4d due to protonation of 3d by residual HCl present in chloroform.19f,g,20
To investigate this notion further, a sample of compound 3d was further examined (1H NMR) after dissolution in C6D6, acetone-d6, MeOH-d4 and DMSO-d6. The ratio of enamine to keto forms (KT) in these solvents (RT) were determined to be 24.3; 27.5; 6.4 and 40.9-to-one, respectively. Examination of these values does not lead to a logical correlation between KT and common solvent parameters such as donor number (DN), acceptor number (AN) or relative permittivity.19f,g,21 Such an investigation will be part of a separate treatment of these effects.
Theoretical gas phase and solution (DFT) treatments
The solid-state structures of compounds 3a, 3c and 3d determined above (X-ray) can be compared with calculated structures for the gas phase.† Using the solid-state structure of 3a, a study was made of the effect of the selected basis set on the results of the geometric optimisation. Calculations were carried out with the B3LYP/6-31G(d), 6-311G(d) and 6-311G(d,p) basis sets and the bond lengths, angles and torsion angles compared thereafter. Calculations with the 6-311G(d) basis set provided very good agreement with the experimental values, similar to but better than the results obtained from using the 6-31G(d) basis set. Subtle differences in the calculated structure of 3a when compared to the solid-state structure were consistent with the expected effects of crystal packing. The geometrically-optimised structure with the higher level 6-311G(d,p) basis set was not noticeably improved and not surprisingly required significantly more computation time to complete. Therefore, the 6-311G(d) basis set was considered the best compromise between accuracy and computational time requirements and was used accordingly for structure calculations of all the molecular isomers in this study. A comparison of the bond lengths, bond and torsion angles for the solid-state vs. DFT determined gas phase structures for 3a, 3c and 3d can be found in the ESI† and in Table 1. As noted in Table 2, in all five cases (3a–e), the enamine tautomer is calculated to be the most stable gas phase isomeric form, followed by the enol form. The keto-isomer is estimated to be the least stable ground state configuration. This result is in contrast to the solution (CDCl3) phase observations, notably for 3d and 3e in which both the keto (minor) and the enamine (major) isomer are present.6,7
Table 2 Calculated gas phase energies (DFT: B3LYP/cc-pVTZ) for the tautomers of 3a–e
| Tautomer |
Relative energy differences to enamine form (kJ mol−1) in gas phase |
|
3a-enamine |
0 |
|
3a-enol |
+6.81 |
|
3a-keto |
+29.88 |
|
|
|
3b-enamine |
0 |
|
3b-enol |
+9.02 |
|
3b-keto |
+26.36 |
|
|
|
3c-enamine |
0 |
|
3c-enol |
+11.30 |
|
3c-keto |
+40.55 |
|
|
|
3d-enamine |
0 |
|
3d-enol |
+4.06 |
|
3d-keto |
+30.89 |
|
|
|
3e-enamine |
0 |
|
3e-enol |
+4.47 |
|
3e-keto |
+30.23 |
In relation to the idea of RAHB, an examination of the calculated structure of the enol tautomer of 3a was carried out to evaluate the potential energetic contributions of RAHB in this particular isomeric form. Average14 bond length values for such enols are 1.333, 1.362, 1.455 and 1.314 Å respectively, for the d1 to d4 parameters (Scheme 3). The calculated values of enol-3a are in quite close agreement to these values for related known systems (1.34, 1.37, 1.44 and 1.29 Å respectively: ESI†).14 This result gives credence to the hypothesis that RAHB is also weak for this tautomer in a similar way as was found for the more stable enamine form (vide supra).
It appears from the observed data obtained by us and others that solvation may be an important contributor to the observed tautomeric distribution(s). Therefore, a theoretical study (DFT: B3LYP/6-311G(d)) was carried out on 3d which included the solvation modelling parameters SM8 or SMD.†22 These treatments of solution phenomena have been found to be useful22,23 in some cases for predicting solvation enthalpies and the relative stability of tautomeric isomers.23 Data for this investigation is tabulated in the ESI as Table S-3.† This data clearly parallel the gas phase studies, confirming the stability of the enamine tautomer in each case, but failing to account for the observation of the keto-tautomer. The results are, however, consistent with an energetically accessible enol-tautomer. In terms of applying such findings to metal–ligand complex syntheses, one might suggest that materials such as 3a may be amenable to conversion to a metal-bound enolate formed via proton loss. This concept is addressed below.
Chiral derivatives – proof-of-principle: the synthesis of 3f
The use of organic fragments as ligands in both coordination chemistry and catalysis has been a subject of growing importance in recent years as the interdisciplinary nature of synthesis continues to expand. A modular ligand design strategy, in which a similar set of protocols and/or starting materials can be used, facilitates the production of large libraries of ligands that can have pre-established steric and/or electronic properties. Our desire to use the Tohda inspired compounds as potential ligands is further enhanced by the realisation that many chiral 2-methyl-2-oxazoline precursors are available or readily synthesised. It has been noted that unsubstituted 2-methyl-2-oxazoline itself can be a troublesome reagent to employ under such methodology7g and hence this substrate provides a potential caveat to the realisation of such a ligand library. We therefore felt that the synthesis of a chiral analogue of 3a–3d would suggest that this potential class of ligands was worthy of further synthetic scrutiny. In this regard, the use of (3aS,8aR)-2-methyl-8,8a-dihydro-3aH-indeno[1,2-d]oxazole (1f: Scheme 2), readily available from (1S,2R)-cis-1-amino-2-indanol, was chosen as a proof-of-principle reactant. Treatment of 1f under identical conditions as those employed in the synthesis of 2a (and subsequently 3a) were found, to our delight, to yield chiral derivative 3f in reasonable overall yield (see Experimental) without employing column chromatography. As observed previously for aryl-derivatives such as 3a, no spectroscopic evidence for a second tautomer was observed via NMR spectroscopy. Isolation of 3f as single crystals and examination by X-ray crystallography unequivocally confirmed the nature of the structure of the desired material and again identifies the enamine form as the stable solid-state tautomer (Fig. 5). This promising result opens a gateway for an extensive examination of potential chiral ligands for applications in enantio-selective substrate modification.
 |
| | Fig. 5 ORTEP representation of a unit cell molecule of 3f, thermal ellipsoids are at the 50% probability level. | |
Sequestering an enolate – metal complex formation via3a
The solid-state and solution examination of the materials described herein has clearly indicated the preference of the enamine form with only alkyl derivatives displaying the spectroscopic presence of the keto tautomer. Despite our calculations to the contrary, unequivocal observation or isolation of the hypothetical enol form has not been observed. The sequestering of an enol form, as a precursor to metal-bound formally anionic enolates, would make these materials even more attractive from both a coordination chemistry (e.g., 3a) and enantio-selective catalysis (3f) point of view. Thus, in our opinion a further fundamentally attractive aspect of these compounds is their potential as ligands. We therefore tested 3a in reactions with hydrated Cu(NO3)2 in an attempt to replace the nitrate and aqua ligands with two equivalents of a hypothetical enolate derived via proton loss from 3a. This chemistry is prompted by the known propensity of Cu2+ ions to form stable and isolable metal-enolates24 and hence this reaction provides a good starting point for a coordination chemistry study. The connection between the structure of a deprotonated enolate such as [3a-H]– and the highly successful β-ketoamine class of ligands, is also of obvious relevance.25 The treatment of light yellow-coloured solutions of 3a with a one-half equivalent of Cu(NO3)2·2.5(H2O) leads to an immediate colour change to deep green. Isolation of the resulting green powder (5: Experimental), followed by characterisation and crystallisation of this material, reveals elemental analyses data consistent with the formation of a complex of stoichiometry Cu(3a-H)2. To unequivocally confirm the structural nature of 5, a single crystal X-ray diffraction study was again undertaken. Complex 5 crystallises in the P21/c space group (Table S-1†); a schematic representation of a unit cell molecule of 5 is shown in Fig. 6. These data confirm the coordination of two units of the predicted enolate anion resulting from the de-protonation of 3a. The resulting product 5 is a mononuclear formally Cu2+ species with trans-spanning N- and O-donor atoms (i.e., κ2-N,O-chelating) from the two ligands obviously arriving via formal loss of water and HNO3. The IR and solution UV/Vis. data are likewise consistent with this formulation. The coordination sphere around the metal centre is best described as highly distorted square planar in nature with a calculated Houser τ4 value of 0.441.26 Bond lengths and angles† of this complex are similar to related Cu-oxazoline and related compounds with a trans-N2O2 ligand atom donor set and are likewise unsurprising27,28 although strictly square planar is a more common geometry within similar species.27a–d,o–q The main visible absorption band (λmax = 443 nm) cannot be adequately explained by a simple d–d transition model due to the relatively large ε value (>103). We therefore examined this complex by time dependent DFT (TD-DFT) employing the B3LYP-LANDZD basis set. These results (ESI†) clearly indicate the interaction of two, primarily ligand-based, MOs and a predominately metal-based LUMO and hence this observation can be best described as ligand–metal charge transfer in nature.
 |
| | Fig. 6 ORTEP representation of a unit cell molecule of 5, thermal ellipsoids are at the 50% probability level. | |
Conclusions
An examination of the solid-state, solution and gas phase tautomers for a number of 2-acylmethyl-2-oxazolines has been carried out and the first solid-state structural studies of this class of compounds has been revealed. The stability of the possible isomers of these materials seems to be phase dependent with solely the enamine isomer being found in the (crystalline) solid-state and dominating the solution phase. Keto isomers are observed in solution for the alkyl derivatives 3d and 3e; this appears to be a solvent dependent observation. The enol form has not been unequivocally observed but is calculated (DFT) to be a more stable gas phase isomer than the keto-forms. An examination of the potential of RAHB as an energetic contribution to the stability of the enamine and enol forms of a number of these materials was also examined and it is surmised that such contributions are likely to be fairly weak in these systems. The B3LYP/6-311G(d) basis set was found to be a suitable level of theory to emulate the solid-state molecular properties of compounds such as 3a and no clear advantage to employing the B3LYP/6-311G(d,p) basis set was found. A further result of these investigations has been the first isolation and solid-state characterisation of any protonated oxazoline (4a) and, under these conditions, the keto isomer is clearly established by crystallography and theoretical calculations to be the more stable form. This series of organic derivatives has been expanded to include a chiral analogue (i.e., 3f) based on the utilisation of the known 2-methyl-2-oxazoline derivative (3aS,8aR)-2-methyl-8,8a-dihydro-3aH-indeno[1,2-d]oxazole. The new compound 3f is formed under similar reaction protocols to those disclosed by Tohda and co-workers. One of the title materials (3a) has also been tested for the first time in the synthesis of a transition metal coordination complex. The resulting compound formed between Cu2+ ions in the presence of excess 3a is shown to be derived from deprotonation of the organic fragment and coordination, in a κ2-N,O-chelating sense, of a resulting enolate. An expanded exploration into the use of this class of potential anionic ligands is currently underway.
Experimental
General remarks
All chemical reagents were purchased commercially and used as received. Melting points were determined on a Fisher Scientific Model 12-144 melting point apparatus and are uncorrected. IR spectra were recorded as KBr pellets using a Perkin Elmer Spectrum One IR spectrometer and near-IR spectra were recorded from CCl4 solutions on a Varian Carey 5 UV/Vis/near-IR spectrophotometer. 1H and 13C{1H} NMR spectra were recorded in CDCl3 solution or other solvents as indicated at RT by a Bruker Avance 70 AC-400 NMR spectrometer. Spectra are referenced to external TMS (δ = 0.00 ppm) which is in turn assigned by the residual non-deuterated solvent as internal standard. The syntheses of 3a, 3c and 3d were carried out as described previously using 2,4,4-trimethyl-2-oxazoline (1) which is first converted to the corresponding di-acylated products (2: Scheme 1).7a–c Toluene was used in lieu of benzene7a for the purposes of re-crystallization for 3a, 3c and 3d. Compound 3e was synthesised as reported by Meyers et al.6 The purity of all of the products was confirmed by a combination of mp and IR and NMR spectroscopies. The synthesis of 4a and attempted synthesis of 4b was carried out by the addition of excess conc. H2SO4 to a methanol solution of the organic starting material; crystals were obtained by slow evaporation of the resulting mixture. These materials were only examined by NMR spectroscopy and/or X-ray diffraction and hence were not characterised further nor purified. The examination of compounds 3a, 3c, 3d, 3f, 4a and 5via single crystal X-ray diffraction was carried out as described5a,d,e previously.†
Synthesis of 3b7,29.
Using similar reaction protocols to those described by Tohda et al. (Scheme 1),7a a sample of 2,4,4-trimethyl-2-oxazoline (1: 5.65 g: 50.0 mmol) was placed in a 500 mL round-bottomed flask and heated to reflux temperature for 3 h in the presence of 4-methoxybenzoyl chloride (17.05 g: 100 mmol), NEt3 (17.4 mL: 125 mmol) and acetonitrile (100 mL). The mixture was then cooled, filtered and all volatile components of the reaction mixture were then removed in vacuo; the residue was then treated with CHCl3 (50 mL) and water (50 mL). The organic layer was washed with 5% aq. NaHCO3 (15 mL × 3), dried (Na2SO4), filtered and the solvents removed by rotary evaporation. The crude solid material was then recrystallised (hot hexanes) and then re-dissolved in EtOAc (100 mL), extracted with dil. KOH, dried (Na2SO4), and then evaporated. This procedure yielded the di-acyl product 2b (9.77 g: 51%) in the form of a pale yellow powder (mp: 175–176 °C).30 To a sample of 2b isolated in this way (3.50 g: 9.18 mmol), a solution of 100 mL of 1.5 M methanolic KOH was added. The mixture was stirred at RT for 16 h, filtered, and then the volatile components were removed in vacuo. The residue was suspended in water (50 mL) and then extracted with CHCl3 (20 mL). The organic layer was then washed with water (thrice: 15 mL), dried (Na2SO4), filtered again and then evaporated to dryness. The crude solid product was then recrystallised (hot hexanes) to give the colourless product3b (1.65 g: 74%) having a mp of 108–109 °C (lit.29 mp: 105–106 °C). 1H NMR (400 MHz, RT, CDCl3): δH = 10.10 (br, 1H, OH), 7.84–7.82 (d, 2H, J = 9.2 Hz, ArH), 6.89–6.87 (d, 2H, J = 8.8 Hz, ArH), 5.53 (s, 1H, CH), 4.10 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 1.39 (s, 6H, CH3); these data are consistent with that reported29 previously. 13C{1H} NMR (75 MHz, RT, CDCl3): δC = 27.1, 55.3, 58.4, 73.3, 78.8, 113.3, 128.6, 130.9, 132.6, 161.6, 169.4, 186.7.
Synthesis of 3f.
Stage 1: a sample of (3aS,8aR)-2-methyl-8,8a-dihydro-3aH-indeno[1,2-d]oxazole (1f; 0.42 g: 2.43 mmol; synthesised and characterised as previously reported31) was dissolved in a mixture of MeCN (20 mL) and NEt3 (0.67 mL: 4.86 mmol). Benzoyl chloride (0.56 mL: 4.86 mmol) was then added by syringe and the mixture slowly heated to reflux temperature. A gradual colour change from colourless to yellow to orange was noted. After 4 h, the reaction flask was cooled to RT. All volatile components were then removed by rotary evaporation using a bath temperature of between 50–60 °C. The resulting brown-coloured solid was then extracted with EtOAc (2 × 30 mL) and water (50 mL). The residual solids and aqueous layer was again extracted (CHCl3: 30 mL) and the organics combined, washed with 5% aq. NaHCO3, and dried (Na2SO4). This mixture was then filtered, and evaporated (rotovap) to give the crude, dark yellow solid of (Z)-2-((3aR)-3-benzoyl-3,3a,8,8a-tetrahydro-2H-indeno[1,2-d]oxazol-2-ylidene)-1-phenylethanone (2f). Recrystallisation of this material (toluene–hexanes) gave the pale yellow product (mp: 206–206.5 °C. Elemental analysis (%); calc'd. (found) for C25H19NO3: C 78.72 (78.29); H 5.02 (5.00); N 3.67 (3.69). 1H NMR (400 MHz, RT, CDCl3): δH = 7.60–7.21 (m, 14H, ArH), 6.12 (d, 1H, J = 6.0 Hz, CHN), 5.56 (m, 1H, CHHCHO), 5.37 (s, 1H, CCH), 3.68–3.63 (dd, 1H, J = 18.0 Hz, CHHCH), 3.45–3.39 (dd, 1H, J = 5.6, 18.0 Hz, CHHCH); 13C{1H} NMR (75 MHz, RT, CDCl3): δC = 37.8, 65.8, 83.6, 85.4, 125.5, 126.8, 127.2, 128.0, 128.1, 128.2, 129.0, 129.9, 131.3, 131.9, 134.3, 138.4, 139.8, 140.0, 158.9, 168.5, 186.8) in 50% yield (0.54 g). This material was then used directly in stage 2. Stage 2: A 0.38 g sample of 2f (1.0 mmol), isolated as above, was treated with 50 mL of 1.5 M KOH in MeOH and the resulting deep yellow coloured mixture stirred at RT for 6 h. The (now dark orange coloured) solution was then filtered and the mixture extracted with CHCl3 (30 mL) and water (3 × 50 mL). The organic fraction was isolated, dried (Na2SO4), filtered and evaporated to give the crude brown coloured solid. This material was then recrystallised from a toluene–hexanes mixture; yield: 0.14 g (50%). mp: 211.5–212 °C. Elemental analysis (%); calc'd. (found) for C18H15NO2: C 77.96 (77.07); H 5.45 (5.57); N 5.05 (4.93). 1H NMR (400 MHz, RT, CDCl3): δH = 10.63 (s, 1H, NH), 7.84–7.30 (m, 9H, ArH), 5.57 (s, 1H, CCH), 5.55–5.51 (m, 1H, J = 1.6, 5.6, 7.2 Hz, CHHCH), 5.50–5.48 (dd, 1H, J = 7.2 Hz, CHN), 4.02–3.98 (dd, 1H, J = 16 Hz), 3.91–3.87 (dd, 1H, J = 16 Hz), 3.52–3.46 (dd, 1H, J = 5.6, 18.0 Hz, CHHCH), 3.44–3.39 (dd, 1H, J = 1.2, 18.0 Hz, CHHCH); 13C{1H} NMR (75 MHz, RT, CDCl3): δC = 38.5, 64.5, 74.1, 83.8, 125.0, 125.5, 126.8, 128.0, 128.1, 128.6, 129.4, 130.5, 139.3, 139.7, 169.7, 187.0.
Synthesis of 5.
To a 50 mL round-bottomed flask was added 3a (0.50 g: 2.3 mmol) and 95% aq. EtOH (10 mL). The mixture was then stirred until all contents had dissolved. A sample of Cu(NO3)2·2.5H2O (0.27 g: 1.2 mmol) was added to the solution; an immediate colour change from yellow to green was observed. After stirring for 6 h, the solvent was reduced (vacuo) to a volume of about 2 mL and the flask then placed in a freezer (−4 °C) for 3 h to complete precipitation of the resulting powder. These green coloured solids were filtered off and then dried in vacuo. Recrystallisation was carried out using CHCl3 as solvent which was then layered with an equal volume of EtOH. This yielded material suitable for X-ray diffraction and analytical purposes after a period of 2–5 d. Yield: 0.23 g (41%). mp: 165 °C (decomp.). IR (KBr, ν, cm−1): 3100–3000 (vw), 2962 (m), 1595 (vs), 1574 (s), 1536 (s), 1489 (s), 1308 (s), 1194 (br, m), 1107 (m), 756 (m), 696 (m). λmax (acetone: 10−4 M) = 443 nm (ε = 1048 L mol−1 cm−1). Elemental analysis (%); calc'd. (found) for C36H28N2O4Cu: C 62.95 (63.13); H 5.69 (5.76); N 5.65 (5.66).
Theoretical calculations
Full geometry optimisations were carried out with the use of the B3LYP density functional level of theory32 using the 6-31G(d), 6-311G(d) and 6-311G(d,p) basis sets33 on all atoms (see ESI†) for all materials studied except Cu complex 5; in this latter case the 6-31G(d) level only was employed. For the optimised geometries, harmonic vibrational frequencies were calculated at the B3LYP level. Single-point energies for the 6-311G(d) optimised geometries were calculated at the B3LYP/cc-pVTZ level of theory (except 5: 6-31G(d)).32,33 Solvation modelling was carried out using the SM8 or SMD solvation parameters at the 6-311G(d) level of DFT.33 All calculations were carried out with using the Spartan 8.0, Spartan 10.0 and the Gaussian 3.0 suites of software.34,35
Acknowledgements
The authors thank the support of the Dean's Research Fund (Ryerson University) and NSERC (Canada) in the form of a Discovery Grant (RAG) and an USRA (TM), the ARC and our respective universities (Acadia, Ryerson, Saskatchewan, Toronto and Tasmania) for further support of this research. RAG is also indebted to the Royal Society of Chemistry for provision of a J. W. T. Jones Travelling Fellowship which provided key additional funding for this work.
References
- For example:
(a) M. Gómez, G. Muller and M. Rocamora, Coord. Chem. Rev., 1999, 193–195, 769–835 CrossRef;
(b) A. Pfaltz, Acta Chem. Scand., 1996, 50, 189–194 CrossRef CAS;
(c) G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505–2550 CrossRef CAS;
(d) H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151–4202 CrossRef CAS;
(e) A. I. Meyers, J. Org. Chem., 2005, 70, 6137–6151 CrossRef CAS;
(f) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS;
(g) V. A. Chebanov, K. A. Gura and S. M. Desenko, Top. Heterocycl. Chem., 2010, 23, 41–84 CrossRef CAS.
- B. M. Culbertson, Prog. Polym. Sci., 2002, 27, 579–626 CrossRef CAS and references therein.
- For example:
(a) R. H. Wiley and L. L. Bennett Jr., Chem. Rev., 1949, 44, 447–476 CrossRef CAS;
(b) J. A. Frump, Chem. Rev., 1971, 71, 483–505 CrossRef CAS;
(c) J. L. C. Wright, Proc. Nova Scotian Inst. Sci., 1984, 34, 133–162 Search PubMed;
(d) A. I. Meyers, J. Heterocycl. Chem., 1998, 35, 991–1002 CrossRef CAS;
(e) C. Lamberth, J. Prakt. Chem., 2000, 342, 518–522 CrossRef CAS;
(f) V. I. Akhmedzhanova, D. Batsrunén and R. Sh. Shakirov, Khim. Prir. Soedin., 1993, 29, 873–876 Search PubMed;
(g) N. Okujo, M. Saito, S. Yamamoto, T. Yoshida, S. Miyoshi and S. Shinoda, BioMetals, 1994, 7, 109–116 CAS;
(h) W. E. Houssen and M. Jaspars, ChemBioChem, 2010, 11, 1803–1815 CrossRef CAS.
- For example: W. W. Christie, Lipids, 1998, 33, 343–353 CrossRef CAS.
- For example:
(a) R. A. Gossage, P. N. Yadav, T. D. MacInnis, J. W. Quail and A. Decken, Can. J. Chem., 2009, 87, 368–379 CrossRef CAS;
(b) R. A. Gossage, H. A. Jenkins, N. D. Jones, R. C. Jones and B. F. Yates, Dalton Trans., 2008, 3115–3122 RSC;
(c) F. Baerlocher, R. Bucur, A. Decken, C. R. Eisnor, R. A. Gossage, S. M. Jackson, L. Jolly, S. L. Wheaton and R. S. Wylie, Aust. J. Chem., 2010, 63, 47–55 CrossRef CAS;
(d) A. A. Deshpande, R. A. Gossage, S. M. Jackson, J. W. Quail, A. L. Sadowy and P. N. Yadav, Z. Naturforsch., B: Chem. Sci., 2009, 64, 1046–1052 CAS;
(e) M. W. Chojnacka, A. J. Lough, R. S. Wylie and R. A. Gossage, J. Mol. Struct., 2011, 991, 158–161 CrossRef CAS.
- A. I. Meyers, D. L. Temple, R. L. Nolen and E. D. Mihelich, J. Org. Chem., 1974, 39, 2778–2783 CrossRef CAS.
-
(a) Y. Tohda, T. Kawashima, M. Ariga, R. Akiyama, H. Shudoh and Y. Mori, Bull. Chem. Soc. Jpn., 1984, 57, 2329–2330 CrossRef CAS;
(b) Y. Tohda, M. Morikawa, T. Kawashima, M. Ariga and Y. Mori, Chem. Lett., 1986, 273–274 CrossRef CAS;
(c) Y. Tohda, T. Yanagidani, S. Hiramatsu, N. Nishiwaki, K. Tani, K. Imagawa and M. Ariga, Bull. Chem. Soc. Jpn., 1997, 70, 2781–2790 CrossRef CAS . Also see: ;
(d) F. Castan, F. Denonne and D. C. H. Bigg, Synthesis, 1993, 1081–1083 CrossRef CAS;
(e) S. Chatterjee, G. Ye, Y. Song, B. L. Barker and C. U. Pittman Jr., Synthesis, 2010, 3384–3394 CAS;
(f) A. Zhou and C. U. Pittman Jr., Tetrahedron Lett., 2005, 46, 2045–2048 CrossRef CAS;
(g) Y. Song, H. I. De Silva, W. P. Henry, G. Ye, S. Chatterjee and C. U. Pittman Jr., Tetrahedron Lett., 2011, 52, 4507–4511 CrossRef CAS.
-
(a) A. D. Garnovskii and I. S. Vasil'chenko, Usp. Khim., 2005, 74, 211–234 (
Russ. Chem. Rev.
, 2005
, 74
, 193–215
) CrossRef;
(b) A. D. Garnovskii, A. P. Sadimenko, I. S. Vasilchenko, D. A. Garnovskii, E. V. Sennikova and V. I. Einkin, Adv. Heterocycl. Chem., 2009, 97, 291–392 CrossRef CAS;
(c) F. Dénès, A. Pérez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366–2447 CrossRef;
(d) C. Spino, Org. Prep. Proced. Int., 2003, 35, 1–140 CrossRef CAS.
-
(a)
J. Elguero, C. Marzin, A. R. Katritzky and P. Linda, The Tautomerism of Heterocycles (Adv. Heterocyclic Chem. Supplement 1), Academic Press, New York, 1976, ch. 4, pp. 266–501 Search PubMed;
(b) V. I. Minkin, A. D. Garnovskii, J. Elguero, A. R. Katritzky and O. V. Denisko, Adv. Heterocycl. Chem., 2000, 76, 157–323 CrossRef CAS.
- For example:
(a) Y. J. Park, N. S. Sickerman, J. W. Ziller and A. S. Borovik, Chem. Commun., 2010, 46, 2584–2586 RSC;
(b) K. Pyta, P. Przybylski, A. Huczyński, A. Hoser, K. Woźniak, W. Schilf, B. Kamieński, E. Grech and B. Brzezinski, J. Mol. Struct., 2010, 970, 147–154 CrossRef CAS;
(c) A. N. Dixit, K. V. Reddy, A. R. A. S. Deshmukh, S. Rajappa, B. Ganguly and J. Chandrasekhar, Tetrahedron, 1995, 51, 1437–1448 CrossRef CAS;
(d) M. Juhász, L. Lázár and F. Fülöp, J. Heterocycl. Chem., 2007, 44, 1465–1473 CrossRef;
(e) M. M. Venter and V. Zaharia, Stud. Univ. Babes-Bolyai, Chem., 2007, 52, 103–109 CAS;
(f) A. Amar, H. Meghezzi, A. Boucekkine, R. Kaoua and B. Kolli, C. R. Chim., 2010, 13, 553–560 CrossRef CAS;
(g) V. Zaharia, A. Silvestru, P. Verite, M. Vlassa, S. Imre and C. Silvestru, Rev. Chim., 2008, 59, 1249–1254 CAS;
(h) G. Ye, S. Chatterjee, M. Li, A. Zhou, Y. Song, B. Lloyd Barker, C. Chen, D. J. Beard, W. P. Henry and C. U. Pittman Jr., Tetrahedron, 2010, 66, 2919–2927 CrossRef CAS.
- G. Gilli and P. Gilli, J. Mol. Struct., 2000, 552, 1–15 CrossRef CAS and references therein.
-
N.b. The keto-isomer can be distinguished from both the (Z)-enol and enamine forms due to the presence of a –CH2 group appending the C-atom emanating from oxazoline ring position-2. This functionality therefore provides, via integration of the 1H NMR signals, a method to distinguish this isomer versus that of the other two CH– containing forms.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
-
N.b., These materials give solid-state IR data (Nujol®) which does not yield clear evidence for NC nor N–H functionalities due to considerable signal overlap in both of these regions.7 We attempted to observe the near-IR overtone vibrations associated with the N–H unit (3a: CCl4 solution) but did not observe the characteristic overtone absorption around 6500–6900 cm−1 typical of heterocyclic secondary amines (e.g., pyrrole). A weak absorption observed at 5944 cm−1 cannot be distinguished from an aromatic C–H overtone vibration; see, for example:
(a) Z. Shen and J. R. Schlup, J. Appl. Polym. Sci., 1998, 67, 267–276 CrossRef CAS;
(b) D. L. Snavely, F. R. Blackburn, Y. Ranasinghe, V. A. Walters and M. Gonzalez del Riego, J. Phys. Chem., 1992, 96, 3599–3605 CrossRef CAS;
(c) E. W. Crandall, J. Chem. Educ., 1987, 64, 466–467 CrossRef CAS;
(d) R. F. Goddu and D. A. Delker, Anal. Chem., 1960, 32, 140–141 CAS;
(e) K. B. Whetsel, W. E. Roberson and M. W. Krell, Anal. Chem., 1958, 30, 1598–1604 CrossRef CAS;
(f) R. A. Russell and H. W. Thompson, Proc. R. Soc. London, Ser. A, 1956, 234, 318–326 CrossRef CAS.
-
(a) H.-G. An, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3357 Search PubMed;
(b) J. Wang, J. Bai and Y. Pan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o106 Search PubMed.
-
(a) V. Bertolasi, V. Ferretti, P. Gilli, X. Yao and C.-J. Li, New J. Chem., 2008, 32, 694–704 RSC;
(b) R. I. Zubatyuk, O. V. Shishkin, L. Gorb and J. Leszczynski, J. Phys. Chem. A, 2009, 113, 2943–2952 CrossRef CAS;
(c) T. Steiner, Chem. Commun., 1998, 411–412 RSC;
(d) A. Mohajeri, J. Mol. Struct. (THEOCHEM), 2004, 678, 201–205 CrossRef CAS;
(e) P. Lenain, M. Mandado, R. A. Mosquera and P. Bultinck, J. Phys. Chem. A, 2008, 112, 7898–7904 CrossRef CAS;
(f) R. I. Zubatyuk, Y. M. Volovenko, O. V. Shishkin, L. Gorb and J. Leszczynski, J. Org. Chem., 2007, 72, 725–735 CrossRef CAS;
(g) S. Abdalla and M. Springborg, J. Phys. Chem. A, 2010, 114, 5823–5829 CrossRef CAS;
(h) V. Bertolasi, P. Gilli, V. Ferretti, G. Gilli and K. Vaughan, New J. Chem., 1999, 23, 1261–1267 RSC;
(i) P. Sanz, O. Mó, M. Yáñez and J. Elguero, J. Phys. Chem. A, 2007, 111, 3585–3591 CrossRef CAS;
(j) W. M. F. Fabian, L. Antonov, D. Nedeltcheva, F. S. Kamounah and P. J. Taylor, J. Phys. Chem. A, 2004, 108, 7603–7612 CrossRef CAS.
- M. Małecka, Struct. Chem., 2010, 21, 175–184 CrossRef.
-
(a) A. R. E. Carey, R. A. More O'Ferrall and B. A. Murray, J. Chem. Soc., Perkin Trans. 2, 1993, 2297–2302 RSC;
(b) R. A. More O'Ferrall and B. A. Murray, J. Chem. Soc., Perkin Trans. 2, 1994, 2461–2470 RSC;
(c) A. R. E. Carey, S. Eustance, R. A. More O'Ferrall and B. A. Murray, J. Chem. Soc., Perkin Trans. 2, 1993, 2285–2296 RSC;
(d) A. R. E. Carey, R. A. More O'Ferrall, M. G. Murphy and B. A. Murray, J. Chem. Soc.,
Perkin Trans. 2, 1994, 2471–2479 CAS;
(e) A. R. Katritzky, I. Ghiviriga, D. C. Oniciu, R. A. More O'Ferrall and S. M. Walsh, J. Chem. Soc., Perkin Trans. 2, 1997, 2605–2608 RSC;
(f) V. I. Minkin, A. V. Tsukanov, A. D. Dubonosov and V. A. Bren, J. Mol. Struct., 2011, 998, 179–191 CrossRef CAS;
(g) L. Antonov, W. M. F. Fabian, D. Nedeltcheva and F. S. Kamounah, J. Chem. Soc., Perkin Trans. 2, 2000, 1173–1179 RSC;
(h) M. Flores-Leonar, N. Esturau-Escofet, J. M. Méndez-Stivalet, A. Marín-Becerra and C. Amador-Bedolla, J. Mol. Struct., 2011, 1006, 600–605 CrossRef CAS;
(i) R. Dobosz, B. Ośmiałowski and R. Gawinecki, Struct. Chem., 2010, 21, 1037–1041 CrossRef CAS.
-
(a) R. P. Bell, J. Chem. Soc., 1931, 1371–1382 RSC;
(b) J. J. Howland Jr., D. R. Miller and J. E. Willard, J. Am. Chem. Soc., 1941, 63, 2807–2811 CrossRef;
(c) R. Battino and H. L. Clever, Chem. Rev., 1966, 66, 395–463 CrossRef CAS.
-
(a) V. Gutmann, Electrochim. Acta, 1976, 21, 661–670 CrossRef CAS;
(b) Y. Marcus, J. Solution Chem., 1984, 13, 599–624 CrossRef CAS;
(c) F. J. Luque, J. M. López-Bes, J. Cemeli, M. Aroztegui and M. Orozco, Theor. Chem. Acc., 1997, 96, 105–113 CrossRef CAS.
- A. V. Marenich, R. M. Olsen, C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2007, 3, 2011–2033 CrossRef CAS.
-
(a) R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Comput. Aided Mol. Des., 2010, 24, 317–333 CrossRef CAS;
(b) H. Fang and Y. Kim, J. Phys. Chem. B, 2011, 115, 15048–15058 CrossRef CAS;
(c) S. Ito, T. Hirano, A. Sugimoto, H. Kagechika, S. Takechi and T. Yamaguchi, Chem. Pharm. Bull., 2010, 58, 922–927 CrossRef CAS.
-
(a)
J. Vicente, in The Chemistry of Metal Enolates: Part 1, ed. J. Zabricky, Wiley, 2009, ch. 6, pp. 313–353 Search PubMed;
(b)
H. Lang and R. Buschbeck, in The Chemistry of Metal Enolates: Part 2, ed. J. Zabricky, Wiley, 2009, ch. 17, pp. 929–1017 Search PubMed.
- For example:
(a) W. N. Wallis and S. C. Cummings, Inorg. Chem., 1974, 13, 988–991 CrossRef CAS;
(b) F. Bao, R. Ma, R. Ma and Y. Jiao, J. Coord. Chem., 2007, 60, 557–560 CrossRef CAS;
(c) X.-Q. Lü, F. Bao, B.-S. Kang, Q. Wu, H.-Q. Liu and F.-M. Zhu, J. Organomet. Chem., 2006, 691, 821–828 CrossRef;
(d) D. M. Granum, P. J. Riedel, J. A. Crawford, T. K. Mahle, C. M. Wyss, A. K. Begej, N. Arulsamy, B. S. Pierce and M. P. Mehn, Dalton Trans., 2011, 40, 5881–5890 RSC;
(e)
I.-M. Lee, in Focus on Organometallic Chemistry Research, ed. M. A. Cato, Nova Science Publishers, Hauppauge, 2005, ch. 5, pp. 133–145 Search PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
-
(a) P. Sténson, Acta Chem. Scand., 1969, 23, 1514–1524 CrossRef;
(b) W.-K. Dong, X.-Y. Dong, Y.-X. Sun, J.-C. Wu and S.-J. Xing, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m1626 Search PubMed;
(c) W.-J. Tai, C.-H. Li, C.-Y. Li and B.-T. Ko, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m1315 Search PubMed;
(d) W.-K. Dong, Y. Wang, J.-C. Wu and S.-T. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m91 Search PubMed;
(e) E. Wehman, G. van Koten, J. T. B. H. Jastrzebski, M. A. Rotteveel and C. H. Stam, Organometallics, 1988, 7, 1477–1485 CrossRef CAS;
(f) J. Thorhauge, M. Roberson, R. G. Hazell and K. A. Jørgensen, Chem.–Eur. J., 2002, 8, 1888–1898 CrossRef CAS;
(g) S. Cabaleiro, P. Pérez-Lourido, J. Castro, J. Romero and J. A. García-Vázquez-Sousa, Transition Met. Chem., 2001, 26, 709–716 CrossRef CAS;
(h) P. Segĺa, M. Jamnický, M. Koman and T. Glowiak, Polyhedron, 1998, 17, 2765–2772 CrossRef;
(i) G. K. Patra, I. Goldberg, A. Sarkar, S. Chowdbury and D. Datta, Inorg. Chim. Acta, 2003, 344, 7–14 CrossRef CAS;
(j) Z.-L. Lü, Z.-L. Liu and D.-Q. Zhang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m147–m150 Search PubMed;
(k) M. Koman, P. Segĺa, M. Jamnický and T. Glowiak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 554–555 Search PubMed;
(l) P. Segĺa, M. Koman and T. Glowiak, J. Coord. Chem., 2000, 50, 105–117 CrossRef;
(m) Y.-Z. Zhu, Z.-P. Li, J.-A. Ma, F.-Y. Tang, L. Kang, Q.-L. Zhou and A. S. C. Chan, Tetrahedron: Asymmetry, 2002, 13, 161–165 CrossRef CAS;
(n) D. A. Evans, J. S. Johnson and E. J. Olhava, J. Am. Chem. Soc., 2000, 122, 1635–1649 CrossRef CAS;
(o) J. Jiang, C. Duan, J. Bai and Y. Pan, Anal. Sci., 2006, 22, x153–x154 CrossRef CAS;
(p) Y.-W. Chang and J.-J. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2126 Search PubMed;
(q) Y.-M. Zhang, K. Xian, T.-B. Wei and K.-B. Yu, J. Chem. Res., 2003, 27(S), 798–799 CrossRef;
(r) M. Corsini, E. Grigiotti, F. Laschi, P. Zanello, J. Burgess, J. Fawcett and S. R. Gilani, Coll. Czech. Chem. Commun., 2003, 68, 1449–1460 CrossRef CAS;
(s) P. N. Yadav, T. M. Barclay and R. A. Gossage, J. Nepal Chem. Soc., 2011, 28, 54–58 Search PubMed;
(t) P. S. E. Bungu, M. Schutte and G. Steyl, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m1373 Search PubMed.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard and D. G. Watson, J. Chem. Soc., Dalton Trans., 1989, S1–S83 RSC.
- X. Chen, X.-L. Wang, H.-Z. Lian, J.-J. Chen, Y. Pan and Y.-Z. Shi, Chin. J. Chem., 1999, 17, 80–83 CrossRef CAS.
- The intermediate compound 2b has been further characterised in the solid-state by single crystal X-ray diffraction and these data confirm the surmised formulation as depicted in Scheme 1; see: A. Petrov, R. C. Jones, D. G. Vaughan, A. J. Lough and R. A. Gossage, Crystals, 2011, 1, 229–235 CrossRef CAS.
-
(a) K. Watanabe, T. Hirasawa and K. Hiroi, Chem. Pharm. Bull., 1992, 50, 372–379 CrossRef;
(b) A. I. Meyers, K. Kamata and I. Agata, J. Org. Chem., 1998, 63, 3113–3116 CrossRef.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 295–300 Search PubMed.
-
(a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS;
(b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS;
(c) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
-
Spartan 8.0 and Spartan 10.0, Wavefunction, Inc., Irvine, CA, 2008 and 2011 Search PubMed.
-
GAUSSIAN 3.0 (Revision E.01), Gaussian Inc., CT, 2008 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Oxazoles XXIX. DFT calculations, general crystal data, cif files for X-ray data, spectroscopic data for 2f, 3f and 5. CCDC 799949–799951 (3a, 3c–d), 915339 (3f), 860691 (4a), 915340 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob25867j |
| ‡ Undergraduate research participants; these authors contributed equally to this research. |
| § Authors to whom correspondence should be directed concerning the crystallographic work; compounds 3a and 3c (E-mail: E-mail: wilson.quail@usask.ca; ); 3d, 3f, 4a and 5 (E-mail: alough@chem.utoronto.ca). |
| ¶ Crystallographic characterisation of the products resulting from the ring-opening of protonated 3d will be reported separately. For related examples, see: (a) A. M. Korolev, L. T. Eremenko and L. V. Meshikhina, Izv. Akad. Nauk. Ser. Khim., 1999, 812–814 (Russ. Chem. Bull., 1999, 48, 808–809); (b) R. A. Gossage, H. A. Jenkins and J. W. Quail, J. Chem. Crystallogr., 2010, 40, 272–277; (c) P. Deslongchamps, S. Dubé, C. Lebreux, D. R. Patterson and R. J. Taillefer, Can. J. Chem., 1975, 53, 2791–2807; (d) P. Deslongchamps, C. Lebreux and R. Taillefer, Can. J. Chem., 1973, 51, 1665–1669. |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence