 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-terminal dual protein functionalization by strain-promoted alkyne–nitrone cycloaddition†
Rinske P.
Temming
a,
Loek
Eggermont
a,
Mark B.
van Eldijk
b,
Jan C. M.
van Hest
b and
Floris L.
van Delft
*a
aSynthetic Organic Chemistry, Institute for Molecules and Materials, Radboud University Nijmegen, Nijmegen, The Netherlands. E-mail: f.vandelft@science.ru.nl; Fax: +31-(0)24-3653393; Tel: +31-(0)24-3652373
bBioorganic Chemistry, Institute for Molecules and Materials, Radboud University Nijmegen, Nijmegen, The Netherlands
First published on 20th February 2013
Abstract
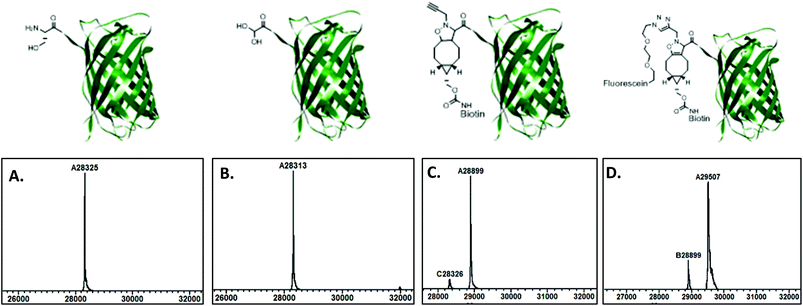
Strain-promoted alkyne–nitrone cycloadditon (SPANC) was optimized as a versatile strategy for dual functionalization of peptides and proteins. The usefulness of the dual labeling protocol is first exemplified by the simultaneous introduction of a chloroquine and a stearyl moiety, two endosomal escape-improving functional groups, into the cell-penetrating peptide hLF (human lactoferrin). Additionally, we demonstrate that dual labeling of proteins is feasible by combining metal-free and copper-catalyzed click chemistry. First, SPANC is applied to enhanced green fluorescent protein to introduce both biotin and a terminal alkyne. The terminal acetylene then serves as a convenient anchor point for the CuAAC reaction with azido-containing fluorescein, thereby demonstrating the potential of combined SPANC and CuAAC for the straightforward, dual functionalization of proteins.
Introduction
Chemical conjugation of a functional tag, e.g. a polyethylene glycol (PEG) tail, a small molecule toxin, or a reporter molecule, is a well-established technique to modulate behavior or study the biological function of proteins. As a further extension of the strategy, interest is growing in the dual conjugation of two orthogonal functionalities to a single protein. For example, multimodality imaging is offering increased accuracy for the diagnosis and surgical treatment of human disease. To this end, an antibody (or another targeting protein) with high affinity for a site of disease is labeled with two different imaging reporters to enable the simultaneous visualization of a single target with two diagnostic techniques, e.g. positron emission tomography (PET), computated tomography (CT), fluorescence imaging, or magnetic resonance imaging (MRI).1 Synthesis of the requisite dual-labeled targeting agents, however, is more challenging than attachment of a single reporter molecule, as schematically depicted for three different strategies in Fig. 1. Approach A, involving conjugation of a bifunctional linker to the protein, appears most attractive but requires the potentially complex synthesis of a branched structure before conjugation. Approach B is more straightforward in design and is based on combining two existing tools for protein conjugation, which traditionally rely on the selective targeting of canonical amino acids such as cysteine, lysine, tyrosine or tryptophan.2 However, such a process typically proceeds with low site-specificity and poor control over the number of reactions. As a consequence, protein function may be attenuated in case conjugation takes place in close proximity to for example the active site or a binding region. Some approaches for selective N-terminal labeling have been developed in recent years, however these require specific enzymes3,4 or careful fine-tuning of the reaction conditions to reach full conversion to a singly modified product.5 | ||
| Fig. 1 Different approaches for the introduction of two functional labels to a peptide or protein. A. Via a pre-synthesized complex containing the two labels; B. via two separate functional handles; C. via the N-terminal, one-pot SPANC reaction. | ||
To overcome the limitations associated with conjugation to natural amino acids, labeling techniques based on non-native functional handles have been developed, for example hydrazone6 or oxime ligation7 to an aldehyde. An aldehyde can be chemically introduced into a protein by oxidation of the N-terminal amino acid upon the action of sodium periodate8 or pyridoxal 5-phosphate,9 or at an internal site by genetically encoding a cysteine-containing pentapeptide tag for enzymatic oxidation.10 Another often applied non-natural functional handle is azide,11 for reaction with a phosphine probe by Staudinger ligation,12 with a terminal acetylene by the copper-catalyzed click reaction (CuAAC),13,14 or with a strained alkyne by strain-promoted azide–alkyne cycloaddition (SPAAC).15 Several methods have been developed for the site-selective introduction of an azide into a protein, such as post-translational diazotransfer,16,17 by protein expression in auxotrophic E. coli,18 or by genetic encoding of azide-containing amino acids in the presence of a heterologous suppressor tRNA/aminoacyl tRNA synthetase pair with altered specificity.19 The latter technique has also been found suitable for site-specific incorporation of cyclooctyne-charged lysine into proteins,20,21 thereby enabling the alternative SPAAC route for protein conjugation by reaction with an azide-containing probe.
Despite the existence of a multitude of conjugation methods, it requires great effort to introduce two functional handles into a protein, and to our knowledge only a few groups have successfully adapted this approach.22–24 In summary, there is great need for a straightforward and practical method for the simultaneous introduction of two imaging modalities or other functional labels into a peptide or protein.
In 2010, we reported that cycloaddition between cyclooctynes and nitrones, the so-called strain-promoted alkyne–nitrone cycloaddition (SPANC),25 is exceptionally suitable for the site-specific functionalization of peptides and proteins at the N-terminus. It was postulated by us, but not yet demonstrated, that the SPANC procedure would also enable dual protein functionalization. In addition, our earlier protocol required relatively high concentrations of the peptide or protein and a large excess of reagents.26 We now report dual functionalization of peptides and proteins using an optimized SPANC procedure (Fig. 1, approach C). The usefulness of the dual labeling protocol is exemplified by the simultaneous introduction of two endosomal escape-improving functional groups into the cell-penetrating peptide hLF (human lactoferrin). Additionally, we demonstrate that dual labeling of proteins is feasible by combining metal-free and copper-catalyzed click chemistry, enabling the introduction of both biotin and a fluorophore into enhanced green fluorescent protein (eGFP).
Results and discussion
Optimization of SPANC on S-hLF
Optimization of the one-pot three-step protocol for the introduction of two functional labels into a biologically relevant system was performed on hLF, a recently established cell-penetrating peptide (CPP). hLF is a 20-mer peptide derived from the N-terminal domain of human lactoferrin and it was recently found to transport cargo across cell membranes in a concentration-dependent manner.27 It was reported that at a concentration of 20 μM nearly all cells showed a homogeneous cytoplasmic and nuclear peptide distribution next to vesicular staining. However, at lower concentrations (up to 10 μM), the peptide was localized predominantly in vesicles, thus hampering cytosolic delivery. In order to facilitate escape from the acidic endosomal compartment and enabling cytoplasmic targeting, Andaloussi et al.28 introduced a chloroquine and a stearyl moiety into the cell-penetrating peptide TP10 by coupling of stearic acid to the N-terminus, and a chloroquinoline analog to a lysine residue of resin-bound TP10. We envisioned that the SPANC procedure could be employed to simultaneously introduce both the chloroquine analog and the stearyl group post-synthetically to hLF, providing a method for the improvement of the cell-penetrating properties of commercially available CPPs. To this end, screening of SPANC conditions was undertaken first, in order to determine the optimized stoichiometry of reagents, because relatively high concentrations of the peptide or protein (up to 8 mM) and/or a large excess of reagents (60–80 equiv.)26 were earlier required. As depicted in Scheme 1, the SPANC protocol involves the following sequence of events: first oxidative cleavage of an N-terminal serine (or threonine) with sodium periodate, followed by a benzenethiol quench to remove excess (per)iodate. Then, without work-up or intermediate purification, an alkylated hydroxylamine is added to the reaction mixture, preferably in the presence of p-anisidine, leading to in situ nitrone formation and cycloaddition with a strained alkyne. As the strained alkyne, we selected bicyclo[6.1.0]nonyne (BCN)26 over the more lipophilic cyclooctyne DIBAC,29 or DIBO.30 After a first round of optimization on a model hexapeptide (ESI, Table S1†), it was found that, at a peptide concentration of 1 mM, the SPANC procedure was best executed with 10 equivalents of both MeNHOH·HCl and BCN-CH2OH. In the next step, an hLF analog containing the requisite N-terminal serine residue (S-hLF, Scheme 1) was oxidized under carefully controlled conditions (0 °C, 1.0 equiv. NaIO4, 10 min) in order to avoid oxidation of the disulfide linkage. Under these conditions, mass spectrometric analysis (ESI, Fig. S1b†) showed the formation of the desired aldehyde 2 along with only a minor amount of a p-methoxybenzenethiol adduct, presumably by opening of the disulfide linkage. Then, the optimized SPANC conditions were applied to the resulting glyoxyl-hLF derivative, by addition of N-methyl hydroxylamine and BCN-CH2OH 5, leading to full conversion of the desired isoxazoline product 4.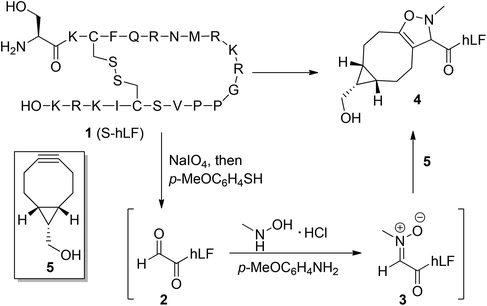
Dual functionalization of S-hLF
Now, the stage was set for the introduction of two functional labels onto hLF via the SPANC procedure, i.e. a chloroquine analog and a stearyl moiety for endosomal escape. Thus, we synthesized the required BCN-trifluoromethylquinoline analog 8 (BCN-QN, Scheme 2) from commercially available BCN succinimidyl carbonate and amine-functionalized trifluoromethylquinoline 7.28 Similarly straightforward, N-stearylhydroxylamine 10 was synthesized by nucleophilic substitution of stearyl bromide with N,O-diBoc-protected hydroxylamine31 followed by Boc deprotection (TFA in DCM) to give the desired product 10 in 80% overall yield. These reagents were then applied in the SPANC reaction on glyoxyl-hLF 2 in the stoichiometry that was also applied before (10 equiv. of N-stearylhydroxylamine 10, 10 equiv. of BCN-QN 8). However, overnight incubation gave low conversion (20%) to the desired isoxazoline product, probably caused by the increased steric hindrance of both BCN-QN and stearylhydroxylamine compared to their simpler analogs. Much to our satisfaction, by partially increasing the stoichiometry of BCN-QN (40 equiv.) and by prolonging the reaction time (3 days), full conversion to the desired dual functionalized product was observed (Fig. 2C).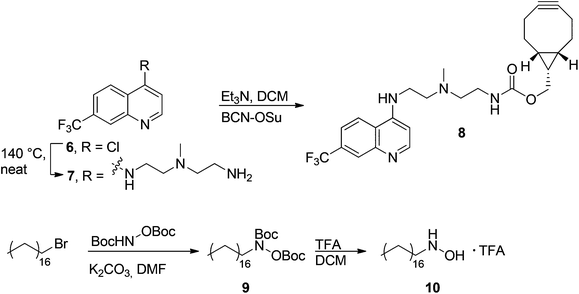 | ||
| Scheme 2 Synthesis of BCN-trifluoromethylquinoline 8 and N-stearylhydroxylamine 10. | ||
 | ||
| Fig. 2 Deconvoluted mass spectra of S-hLF. A. Native peptide; B. after oxidation with NaIO4; C. after dual SPANC labeling with BCN-trifluoromethylquinoline and N-stearylhydroxylamine. | ||
Optimization of SPANC on the protein HspB2
The next stage of SPANC labeling of a biomolecule involved the application to a model medium-sized protein (20 kDa). To this end, we selected heat shock protein HspB2, a protein with an N-terminal serine that was conveniently expressed in-house (along with 25% of HspB3).32 Not unexpectedly, it was found that periodate cleavage of the N-terminal aminoalcohol of HspB2 was accompanied by significant concomitant oxidation of the free cysteine. Gratifyingly, alkylation of the free cysteine with iodoacetamide33 prior to periodate oxidation led to clean conversion of the alkylated HspB2 to the desired glyoxyl product (ESI†). Given the compromised introduction of the sterically hindered stearyl hydroxylamine, we now opted to introduce propargyl hydroxylamine instead. Propargyl hydroxylamine is small, readily synthesized and may serve as a convenient handle for the subsequent further introduction of azide-substituted functional handles by means of copper-catalyzed click chemistry.13,14 Thus, varying amounts of reagents were applied to HspB2 (ESI, Table S2†) for p-anisidine (10 equiv.–100 mM), BCN-CH2OH (10–100 equiv.) and N-propargylhydroxylamine (10–100 equiv.), while opting to keep the BCN-CH2OH stoichiometry to a minimum. From these experiments it was concluded that optimal conditions involve 50 equivalents of N-propargylhydroxylamine and 20 equivalents of BCN-CH2OH, leading to efficient formation of the desired propargyl isoxazoline derivative, as indicated by mass spectrometric analysis (ESI, Fig. S2†).Dual functionalization of S-eGFP
Finally, the 28 kDa protein enhanced green fluorescent protein (eGFP)34 was selected to explore the generality of the optimized SPANC reaction conditions, as well as to investigate the envisioned further functionalization of the protein by a second, copper-catalyzed derivatization. To this end, eGFP with an N-terminal serine residue was expressed in E. coli. The resulting S-eGFP was subjected to oxidation with sodium periodate (1.5 equiv., MW = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 313, Fig. 3B), followed by quenching with p-methoxybenzenethiol (10 equiv.), leading to the near quantitative formation of the aldehyde derivative of eGFP as expected. To our delight, in this particular case no competitive oxidation of the cysteine residues in eGFP was observed. Then, p-anisidine (100 mM final concentration), N-propargylhydroxylamine (50 equiv.) and BCN-biotin (20 equiv.) were added. After 4 h, mass spectrometric analysis showed full conversion of glyoxyl-eGFP to the dual labeled product (MW = 28898 Da, Fig. 3C). The minor peak in the MS spectrum belongs to residual (unoxidized) native S-eGFP (MW = 28326 Da).
313, Fig. 3B), followed by quenching with p-methoxybenzenethiol (10 equiv.), leading to the near quantitative formation of the aldehyde derivative of eGFP as expected. To our delight, in this particular case no competitive oxidation of the cysteine residues in eGFP was observed. Then, p-anisidine (100 mM final concentration), N-propargylhydroxylamine (50 equiv.) and BCN-biotin (20 equiv.) were added. After 4 h, mass spectrometric analysis showed full conversion of glyoxyl-eGFP to the dual labeled product (MW = 28898 Da, Fig. 3C). The minor peak in the MS spectrum belongs to residual (unoxidized) native S-eGFP (MW = 28326 Da).
 | ||
| Fig. 3 Deconvoluted mass spectra of S-eGFP. A. Native protein; B. after NaIO4 oxidation (hydrate form); C. after dual SPANC labeling with BCN-biotin and N-propargylhydroxylamine; D. after CuAAC reaction with azido-fluorescein 12. | ||
CuAAC on alkyne-functionalized eGFP
To show the full extent of this technology for dual functional labeling, a CuAAC reaction was next performed between the N-terminal propargyl function and an azido-containing fluorescein derivative 12 (Scheme 3). To this end, isoxazoline 11 was dissolved in 20 mM phosphate buffer (pH 7.6) and subjected to typical CuAAC conditions (CuSO4, TBTA, sodium ascorbate and azido-fluorescein 12). After overnight incubation at room temperature, mass spectrometry analysis showed the near complete conversion of the starting alkyne to the desired triazole product 13 (MW = 29506, Fig. 3D), thereby demonstrating the potential of combined SPANC and CuAAC for the dual functionalization of proteins.

Experimental procedures
General procedures
1H NMR spectra were recorded in CDCl3, DMSO or D2O on Bruker DMX 300 or Varian Inova-400 spectrometers at 300 K. TMS (δH 0.00), DMSO (δH 2.50) or D2O (δH 4.79) was used as the internal reference. 13C NMR spectra were recorded in CDCl3, DMSO or MeOD at 75 MHz on a Bruker DMX 300 spectrometer, using the central resonance of CDCl3 (δC 77.2), DMSO (δC 39.5) or MeOD (δC 49.0) as the reference. Mass spectra were obtained on a JEOL AccuToF or a Thermo LCQ Fleet. Deconvoluted mass spectra were obtained with MagTran 1.03b2. Chemicals were purchased from Sigma-Aldrich or Acros and used without further purification, unless otherwise indicated. TBTA was synthesized according to the literature procedure,35 azido fluorescein 1236 was synthesized according to the literature procedure with DCM as the solvent. All reactions were monitored by TLC on Kieselgel F254 (Merck). Detection was by examination under UV light (254 nm) and by charring with aqueous KMnO4. Silica gel (Acros 0.035–0.070 mm) was used for chromatography.Dual labeling of S-hLF with BCN-CH2OH 5 and N-methylhydroxylamine
S-hLF-K-Fluo (EMC microcollections, SKCFQWQRNMRKVRGPPVSCIKRK(ε-carboxyfluorescein)-NH21, 1.6 mg, 486 nmol) was dissolved in 1.1 mL of 0.1 M NH4OAc buffer (adjusted to pH 8.34 using NaOH) and shaken (300 rpm) at 40 °C for 2 h to form the disulphide bridge. 0.1 M HCl (5.5 μL) and 0.1 M NH4OAc buffer pH 6.8 (1.1 mL) were added to adjust to pH 6.8. The reaction mixture was cooled to 0 °C and NaIO4 (0.093 μg, 486 nmol in water) was added. The reaction mixture was incubated for 10 min at 0 °C and immediately quenched with p-methoxybenzenethiol (1.22 mL, 9.8 μmol dissolved in THF). After incubation at 0 °C for 1 h, the reaction mixture was warmed to rt and diluted with THF (2.2 mL) to get a mixture of THF–water 1:1. Then, p-anisidine (1.20 mg, 9.8 μmol dissolved in THF), N-methylhydroxylamine (0.82 mg, 9.8 μmol in water) and BCN-CH2OH 5 (1.5 mg, 9.8 μmol in THF–water 1:1) were added. The reaction was incubated at rt for 16 h, until mass spectrometry showed full conversion to isoxazoline 4. ESI: calculated 3440.1 Da, found 3438.2 Da.
Synthesis of N1-methyl-N1-(2-((7-trifluoromethyl)quinolin-4-yl)aminoethyl)propane-1,3-diamine (QN, 7)
4-Chloro-7-(trifluoromethyl)quinoline (ABCR, 1.02 g, 4.4 mmol) and 2,2′-diamino-N-methyldiethylamine (TCI, 6.6 mL, 50.9 mmol) were heated neat to 140 °C for 2 hours to give a red solution. After cooling to rt, DCM (20 mL) was added and the solution was washed with a 5% aqueous NaHCO3 solution (2 × 20 mL) and water (2 × 20 mL). The organic phase was dried (Na2SO4), filtered and the solvent was removed under reduced pressure to give QN 7 (1.14 g, 83%). RF = 0.10 (MeOH–DCM 1:2); 1H NMR (300 MHz, CDCl3) δ 8.61 (d, J = 5.3 Hz, 1H), 8.25 (s, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.57 (dd, J = 8.8, 1.9 Hz, 1H), 6.46 (d, J = 5.4 Hz, 1H), 6.25 (bs, 1H), 3.33 (m, 2H), 2.90 (t, J = 6.1 Hz, 2H), 2.81 (dd, J = 6.4, 5.1 Hz, 2H), 2.57 (t, J = 6.1 Hz, 2H), 2.33 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 152.4, 149.9, 147.7, 131.0, 130.6, 127.5, 125.9, 121.6, 120.0, 100.2, 59.7, 55.1, 41.9, 40.2, 39.5; FT-IR νmax (cm−1): 3250–2794, 1584, 1537, 1324, 1161, 1122, 811; HRMS (ESI+) m/z calcd for C15H20F3N4 (M + H)+: 313.1640, found: 313.1631.
Synthesis of (1R,8S,9S)-bicyclo[6.1.0]non-4-yn-9-ylmethyl 2-(methyl(2-((7-(trifluoromethyl)quinolin-4-yl)amino)ethylamino)ethyl)carbamate (BCN-QN, 8)
BCN succinimidyl carbamate (Synaffix B.V., 50.6 mg, 0.17 mmol) was dissolved in DCM (2 mL), QN 7 (82 mg, 0.26 mmol) and triethylamine (75 μL, 0.54 mmol) were added and the reaction mixture was stirred at rt for 90 min. Saturated NH4Cl solution (15 mL) was added and extracted with dichloromethane (2 × 15 mL). The DCM layer was dried using Na2SO4, filtered and solvents were removed in vacuo. Products were separated by silica gel column chromatography (DCM–MeOH 19:1) to give BCN-QN 8 (79 mg, 93%). RF = 0.20 (DCM–MeOH 19:1); 1H NMR (300 MHz, CDCl3) δ 8.56 (d, J = 5.4 Hz, 1H), 8.26 (s, 1H), 8.07 (d, J = 8.9 Hz, 1H), 7.57 (d, J = 9.2 Hz, 1H), 6.44 (d, J = 5.4 Hz, 1H), 5.13 (bs, 1H), 4.07 (d, J = 7.9 Hz, 2H), 3.36–3.33 (m, 4H), 3.13 (bs, 1H), 2.83 (t, J = 5.7 Hz, 2H), 2.61 (t, J = 5.9 Hz, 2H), 2.33 (s, 3H), 2.29–2.08 (m, 6H), 1.55–1.40 (m, 2H), 1.29–1.16 (m, 1H), 0.92–0.81 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 157.2, 151.5, 150.2, 146.8, 131.4, 131.0, 126.7, 125.8, 121.9, 120.6, 100.1, 98.8, 63.1, 57.3, 55.1, 42.1, 40.2, 38.7, 29.0, 21.5, 20.2, 17.8; FT-IR νmax (cm−1): 3334, 2924, 2846, 1701, 1588, 1571, 1537, 1372, 1325, 1268, 1156, 1126, 1070; HRMS (ESI+) m/z calcd for C26H32N4O2F3 (M + H)+: 313.1640, found: 313.1631.
Synthesis of N,O-diBoc-N-stearylhydroxylamine (9)
Stearyl bromide (1.7 g, 5.1 mmol) was dissolved in DMF (20 mL) before addition of N,O-diBoc hydroxylamine (1.2 g, 5.1 mmol) and K2CO3 (861 mg, 6.2 mmol). The reaction mixture was stirred for 3 days at room temperature. Water (40 mL) was added and the product was extracted using EtOAc (3 × 30 mL). The organic layer was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The product was purified by silica gel column chromatography (heptane–EtOAc 15:1) to give N,O-diBoc-N-stearylhydroxylamine 9 (1.98 g, 80%). RF = 0.58 (heptane–EtOAc 9:1); 1H NMR (400 MHz, CDCl3) δ 3.56 (bs, 2H), 1.62–1.56 (m, 2H), 1.53 (s, 9H), 1.48 (s, 9H), 1.25 (s, 30H), 0.90–0.86 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 155.1, 152.5, 84.7, 82.2, 50.4, 32.1, 29.9, 29.5, 29.4, 28.4, 28.3, 27.9, 27.7, 26.7, 22.8, 14.3; FT-IR νmax (cm−1): 2920, 2855, 1783, 1718, 1476, 1394, 1372, 1247, 1130, 841; HRMS (ESI+) m/z calcd for C28H56NO5 (M + H)+: 486.4159, found: 486.4181.
Synthesis of N-stearylhydroxylamine TFA salt (10)
N,O-diBoc-N-stearylhydroxylamine 9 (84.3 mg, 0.17 mmol) was dissolved in DCM (1 mL) and TFA (1 mL) was added dropwise. After stirring at rt for 1 h, solvents were removed in vacuo and the product was co-evaporated with toluene (3 × 60 mL) to give the TFA salt of N-stearylhydroxylamine 10 (67 mg, quant.). RF = not determined; 1H NMR (300 MHz, DMSO) δ 3.10–3.05 (m, 2H), 1.60–1.52 (m, 2H), 1.24 (s, 30H), 0.89–0.82 (m, 3H); 13C NMR (75 MHz, DMSO) δ = 50.2, 31.3, 30.7, 29.0, 28.9, 28.8, 28.7, 28.5, 25.7, 23.0, 22.1, 13.9; FT-IR νmax (cm−1): 3386, 2906, 2859, 1653, 1467, 1200, 1018, 616; HRMS (ESI+) m/z calcd for C18H40NO (M + H)+: 286.3110, found: 286.3102.Dual labeling of S-hLF with BCN-QN 8 and N-stearylhydroxylamine 10
S-hLF-K-Fluo (SKCFQWQRNMRKVRGPPVSCIKRK(carboxyfluorescein)-NH2, 1.6 mg, 486 nmol) was dissolved in 1.1 mL of 0.1 M NH4OAc buffer (adjusted to pH 8.34 using NaOH) and shaken (300 rpm) at 40 °C for 2 h to form the disulphide bridge. 0.1 M HCl (5.5 μL) and 0.1 M NH4OAc buffer pH 6.8 (1.1 mL) were added to adjust to pH 6.8. The reaction mixture was cooled to 0 °C and NaIO4 (0.093 μg, 486 nmol in water) was added. The reaction mixture was incubated for 10 min at 0 °C and immediately quenched with p-methoxybenzenethiol (1.22 mL, 9.8 μmol dissolved in THF). After incubation at 0 °C for 1 h, the reaction mixture was warmed to rt and diluted with THF (2.2 mL) to get a mixture of THF–water 1:1. Then, p-anisidine (1.20 mg, 9.8 μmol dissolved in THF), N-stearylhydroxylamine 10 (3.92 mg, 9.8 μmol in water) and BCN-QN 8 (4.8 mg, 9.8 μmol in THF–water 1:1) were added. The reaction was incubated at rt for 3 days, until mass spectrometry showed full conversion to the product. ESI: calculated 4016.9, found 4015.4 Da.
Synthesis of N,O-diBoc-N-propargylhydroxylamine
N,O-diBoc hydroxylamine (435 mg, 1.86 mmol) was dissolved in dry DMF (15 mL) under an argon atmosphere and K2CO3 (343 mg, 2.48 mmol) was added. Propargyl bromide (80% solution in toluene, 230 μL, 2.07 mmol) was added dropwise, and the reaction mixture was stirred at rt for 20 h. Water (150 mL) was added and extracted with DCM (2 × 200 mL). The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. The product was purified by silica gel column chromatography (heptane–EtOAc 5:1) to give the title compound as a colourless oil (488 mg, 93%). RF = 0.29 (heptane–EtOAc 5:1); 1H NMR (400 MHz, CDCl3) δ 4.31 (bs, 2H), 2.25 (t, J = 2.4 Hz, 1H), 1.51 (s, 9H), 1.47 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 154.7, 152.1, 85.1, 83.5, 77.1, 72.7, 40.6, 28.2, 28.1, 27.7, 27.7; FT-IR νmax (cm−1): 3296, 2980, 1792, 1718, 1372, 1230, 1148, 1091, 832; HRMS (CI+) m/z calcd for C13H22NO5 (M + H)+: 272.1498, found: 272.1487.
Synthesis of N-propargylhydroxylamine HCl salt
N,O-diBoc-N-propargylhydroxylamine (563 mg, 2.075 mmol) was dissolved in EtOAc (10 mL) and H2O (5 mL) was added. To this mixture was added conc. HCl (5 mL) and the reaction was stirred at rt for 2 h. The solvents were evaporated in vacuo to give N-propargylhydroxylamine HCl salt as an off-white solid (223 mg, quant.). RF = not determined; 1H NMR (400 MHz, D2O) δ 4.14 (d, J = 2.6 Hz, 2H), 2.98 (t, J = 2.6 Hz, 1H); 13C NMR (75 MHz, MeOD) δ 79.4, 72.8, 42.1; FT-IR νmax (cm−1): 3378, 3291, 2915, 2747, 2613, 1632, 664; HRMS (ESI+) m/z calcd for C3H6NO (M + H)+: 72.0449, found: 72.0436.Dual labeling of HspB2/3 with N-propargylhydroxylamine and BCN-CH2OH 5
A 245 μM stock solution of rHspB2/3 complex 3:1 in PBS buffer (150 μL) was diluted with 0.1 M NH4OAc buffer pH 6.8 (600 μL) to give a 50 μM solution. An equal amount of a 10 mM stock solution of iodoacetamide in water (750 μM) was added, and the mixture was incubated for 3.5 h at rt. The reaction mixture was purified with a Millipore Amicon ultra centrifugal filter (10 kDa cut-off). After each spin cycle the spinfilter was refilled with 0.1 M NH4OAc buffer pH = 6.8, finally resulting in a 100 μM solution. 1.1 equivalent NaIO4 was added (40.6 nmol, 7.8 μg in water) and the reaction mixture was incubated for 1 h at rt. 10 Equivalents of p-methoxybenzenethiol (370 nmol, 53 μg in MeCN) was added and allowed to react for 16 h at rt. The reaction mixture was divided in 7 and to each tube was added the required amount of p-anisidine (0 equiv.–100 mM in MeCN), N-propargylhydroxylamine (10–100 equiv. in water) and BCN-CH2OH 5 (10–100 equiv. in MeCN–water 1:1). After 16 h at rt, the conversion was analysed with mass spectrometry. ESI-TOF: calculated 20444, found 20448.
Dual labeling of S-eGFP with N-propargylhydroxylamine and BCN-biotin
A 92 μM solution of S-eGFP in 0.1 M NH4OAc buffer pH 6.8 (65 μL, 6.0 nmol S-eGFP) was diluted with the same buffer (130 μL), to give a 30 μM solution. NaIO4 was added (14.3 nmol, 2.7 μg in water) and the reaction mixture was incubated for 65 min at rt. p-Methoxybenzenethiol (60 nmol, 8.4 μg in MeCN) was added and allowed to react for 16 h at rt. p-Anisidine was added to give a final concentration of 100 mM (2.9 μmol, 0.36 mg in MeCN) and N-propargylhydroxylamine HCl salt (285 nmol, 30.6 μg in water) and BCN-biotin (Synaffix B.V., 114 nmol, 62.8 μg in MeCN–water 1:1) were added. After incubation for 4 h at rt, mass spectrometry analysis showed full conversion to isoxazoline product 11, along with a minor amount of unoxidized, native S-eGFP. The product was purified using a Millipore Amicon Ultra-0.5 mL centrifugal filter (10 kDa cut-off). After each spin cycle the spinfilter was refilled with 20 mM phosphate buffer (pH 7.6) to finally give 187 μL of a 31 μM solution. ESI-TOF: calculated 28898 Da, found 28899 Da.
CuAAC on propargyl-isoxazoline 11 with azido-fluorescein 12
A 20 mM solution of CuSO4·5H2O in water was prepared and mixed in a 1:1 ratio with a 20 mM solution of sodium ascorbate in water. A 10 mM solution of the Cu(I) ligand Tris-(benzyltriazolylmethyl)amine (TBTA) in tBuOH–DMSO 4:1 was prepared, and this was mixed in a 1:1 ratio with the Cu/ascorbate mixture to give a 5 mM solution of all reagents. 20 μL of the previously prepared 31 μM solution of 11 in 20 mM phosphate buffer (pH 7.6) was taken (0.6 nmol protein) and to this mixture was added 30 equivalents of Cu/ascorbate/ligand and 20 equivalents of azido-fluorescein 12 (12 nmol, 7.3 μg in MeCN–water 1:1). After incubation at rt for 19 h, mass spectrometry analysis showed approximately 90% conversion to triazole product 13. ESI-TOF: calculated 29505 Da, found 29507 Da.
Conclusions and outlook
For the first time, the SPANC protocol was employed for the site-selective one-pot introduction of two functional groups to a peptide and a protein. First, a chloroquine analog and a stearyl moiety were introduced into the CPP hLF, thus potentially providing a method for the improvement of the cell-penetrating properties of commercially available CPPs. Currently, investigations on the effectiveness of the dual labeled hLF derivatives are ongoing. Further optimization of the SPANC protocol for proteins led to conditions highly suitable for introduction of N-propargyl hydroxylamine and BCN-biotin into S-eGFP within a few hours. The terminal alkyne served as a convenient anchor point for a CuAAC reaction with azido-containing fluorescein, thereby demonstrating the potential of combined SPANC and CuAAC for the dual functionalization of proteins. In this respect, it is of relevance to note that the applicability of combined SPANC and CuAAC is particularly facilitated by the commercial availability of a wide range of functionalized azides and cyclooctynes, in contrast to for example N-functionalized hydroxylamines. We are currently investigating various alternative synthetic routes towards such functionalized N-hydroxylamines. We anticipate that the SPANC protocol will find in particular application in the simultaneous visualization of a single target with two diagnostic techniques, i.e. multimodality imaging. However, numerous other (combinations of) functional groups can be envisioned, e.g. polyethylene glycol (PEG) chains, imaging modalities, drugs, targeting elements, nanoparticles and solid surfaces, thereby further enhancing the prospect of SPANC for dual labeling of peptides and proteins.Acknowledgements
Marjoke Debets is kindly acknowledged for the synthesis of azido-fluorescein. This research was supported financially by the Council for Chemical Sciences of The Netherlands Organization for Scientific Research (NMW-CW, F.L.v.D.)Notes and references
- H. Xu, K. Baidoo, A. J. Gunn, C. A. Boswell, D. E. Milenic, P. L. Choyke and M. W. Brechbiel, J. Med. Chem., 2007, 50, 4759–4765 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- R. E. Connor, K. Piatkov, A. Varshavsky and D. A. Tirrell, ChemBioChem, 2008, 9, 366–369 CrossRef CAS.
- W. P. Heal, M. H. Wright, E. Thinon and E. W. Tate, Nat. Protocols, 2012, 7, 105–117 CAS.
- A. O.-Y. Chan, C.-M. Ho, H.-C. Chong, Y.-C. Leung, J.-S. Huang, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2012, 134, 2589–2598 CrossRef CAS.
- H. F. Gaertner, K. Rose, R. Cotton, D. Timms, R. Camble and R. E. Offord, Bioconjugate Chem., 1992, 3, 262–268 CrossRef CAS.
- K. Rose, J. Am. Chem. Soc., 1994, 116, 30–33 CrossRef CAS.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., 2006, 118, 5433–5437 CrossRef.
- T. Dierks, A. Dickmanns, A. Preusser-Kunze, B. Schmidt, M. Mariappan, K. von Figura, R. Ficner and M. G. Rudolph, Cell, 2005, 121, 541–552 CrossRef CAS.
- M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168–1184 CrossRef CAS.
- S. S. Van Berkel, M. B. van Eldijk and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 8806–8827 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- S. F. M. van Dongen, R. L. M. Teeuwen, M. Nallani, S. S. van Berkel, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Bioconjugate Chem., 2008, 20, 20–23 CrossRef.
- S. Schoffelen, M. B. van Eldijk, B. Rooijakkers, R. Raijmakers, A. J. R. Heck and J. C. M. van Hest, Chem. Sci., 2011, 2, 701–705 RSC.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS.
- A. Borrmann, S. Milles, T. Plass, J. Dommerholt, J. M. M. Verkade, M. Wießler, C. Schultz, J. C. M. van Hest, F. L. van Delft and E. A. Lemke, ChemBioChem, 2012, 13, 2094–2099 CrossRef CAS.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS.
- E. M. Brustad, E. A. Lemke, P. G. Schultz and A. A. Deniz, J. Am. Chem. Soc., 2008, 130, 17664–17665 CrossRef CAS.
- M.-H. Seo, T.-S. Lee, E. Kim, Y. L. Cho, H.-S. Park, T.-Y. Yoon and H.-S. Kim, Anal. Chem., 2011, 83, 8849–8854 CrossRef CAS.
- M. Simon, U. Zangemeister-Wittke and A. Plückthun, Bioconjugate Chem., 2011, 23, 279–286 CrossRef.
- X. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G.-J. Boons and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 3065–3068 CrossRef CAS.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS.
- F. Duchardt, I. R. Ruttekolk, W. P. R. Verdurmen, H. Lortat-Jacob, J. Bürck, H. Hufnagel, R. Fischer, M. van den Heuvel, D. W. P. M. Löwik, G. W. Vuister, A. Ulrich, M. de Waard and R. Brock, J. Biol. Chem., 2009, 284, 36099–36108 CrossRef CAS.
- S. E. L. Andaloussi, T. Lehto, I. Mäger, K. Rosenthal-Aizman, I. I. Oprea, O. E. Simonson, H. Sork, K. Ezzat, D. M. Copolovici, K. Kurrikoff, J. R. Viola, E. M. Zaghloul, R. Sillard, H. J. Johansson, F. Said Hassane, P. Guterstam, J. Suhorutšenko, P. M. D. Moreno, N. Oskolkov, J. Hälldin, U. Tedebark, A. Metspalu, B. Lebleu, J. Lehtiö, C. I. E. Smith and Ü. Langel, Nucleic Acids Res., 2011, 39, 3972–3987 CrossRef.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- Y. Zeng, B. T. Smith, J. Hershberger and J. Aubé, J. Org. Chem., 2003, 68, 8065–8067 CrossRef CAS.
- J. den Engelsman, S. Boros, P. Y. W. Dankers, B. Kamps, W. T. Vree Egberts, C. S. Böde, L. A. Lane, J. A. Aquilina, J. L. P. Benesch, C. V. Robinson, W. W. de Jong and W. C. Boelens, J. Mol. Biol., 2009, 393, 1022–1032 CrossRef CAS.
- R. van Geel, G. J. M. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392–398 CrossRef CAS.
- R. Heim, A. B. Cubitt and R. Y. Tsien, Nature, 1995, 373, 663–664 CrossRef CAS.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS.
- C. M. Santos, A. Kumar, W. Zhang and C. Cai, Chem. Commun., 2009, 2854–2856 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Results from SPANC condition optimization on SKYRAG and HspB2, mass spectra of S-hLF after functionalization with BCN-CH2OH and N-methyl hydroxylamine, mass spectra of HspB2 after functionalization with BCN-CH2OH and N-propargyl hydroxylamine, synthesis of SKYRAG, expression and purification of S-eGFP and 1H and 13C NMR spectra of described compounds. See DOI: 10.1039/c3ob00043e |
| This journal is © The Royal Society of Chemistry 2013 |