Synthesis of nucleosides and dNTPs bearing oligopyridine ligands linked through an octadiyne tether, their incorporation into DNA and complexation with transition metal cations†
Lubica
Kalachova
a,
Radek
Pohl
a,
Lucie
Bednárová
a,
Jindřich
Fanfrlík
a and
Michal
Hocek
*ab
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Gilead & IOCB Research Center, Flemingovo nam. 2, CZ-16610 Prague 6, Czech Republic. E-mail: hocek@uochb.cas.cz; Fax: +420 220183559; Tel: +420 220183324
bDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, CZ-12843 Prague 2, Czech Republic
First published on 9th October 2012
Abstract
Modified nucleosides (dARs and dCRs) bearing bipyridine or terpyridine ligands attached through an octadiyne linker were prepared by single-step aqueous-phase Sonogashira cross-coupling of 7-iodo-7-deaza-2′-deoxyadenosine and 5-iodo-2′-deoxycytidine with the corresponding bipyridine- or terpyridine-octadiynes and were triphosphorylated to the corresponding nucleoside triphosphates (dARTPs and dCRTPs). The modified dNRTPs were successfully incorporated into the oligonucleotides by primer extension experiment (PEX) using different DNA polymerases and the PEX products were used for post-synthetic complexation with divalent metal cations. The complexation of these DNAs containing flexibly-tethered ligands was compared with the previously reported ones bearing rigid acetylene-linked ligands suggesting the possible formation of both inter- and intra-strand complexes with Ni2+ or Fe2+.
Introduction
DNA is a suitable template for construction of nanomolecular components, which assemble in a deliberately designed manner,1 due to its nanometric dimension, high stability, selective recognition of the complementary strand through the Watson–Crick base-pairing and facile synthesis. Incorporation of transition metals into these biomacromolecules allows DNA to form many other spatial arrangements2,3 due to the geometrical preference of metal ions in coordinating with different ligands. Moreover, incorporation of transition metals into DNA can lead to photoactive,4 electroactive5 or even catalytically active6 species and can significantly influence the stability.7 Transition metals can be placed within (metal–base-pairs8 or intercalators9) or outside (metal complex covalently attached to a nucleobase, sugar or phosphate)10 the DNA duplex. Considering metal-induced assembly of conjugates of nucleic acids and metal-chelating moieties, it is important to point out that most of the studies have been carried out with terminally modified oligonucleotides via long and flexible linkers. Recently, we,11 Wagenknecht12 and Stulz13 independently published enzymatic or chemical synthesis of oligonucleotides bearing terpyridine (tpy) ligands linked through a short and rigid acetylene tether and their post-synthetic side by side complexation with M2+ ions in order to prepare intermolecular DNA-complexes. However, presumably due to the rigidity of the linker and rather short distance from the DNA duplex, the complexation properties of these DNA constructs were rather limited. Using a longer and more flexible linker might enhance the complexation ability and even allow formation of cyclic metal–DNA chelates, which were reported very rarely.14 Therefore, we report here on the enzymatic synthesis of oligonucleotides bearing oligopyridine ligands attached through a long and flexible 1,7-octadiyne linker, which is often used15 for click-conjugations, and their post-synthetic complexation with diverse transition metals.Results and discussion
Synthesis of ligand building blocks
In order to prepare functionalized nucleosides bearing oligopyridine ligands attached via a long and flexible linker, suitable ligand–octadiyne building blocks had to be synthesized first. The Sonogashira cross-coupling reactions of an activated oligopyridine 1a–b16 with 3 equiv. of 1,7-octadiyne (2) in the presence of Pd(PPh3)2Cl2, CuI and Et3N (Scheme 1) were performed at 70 °C for 3 h. Desired products were obtained in good yields of 66% for 3a or 75% for 3b.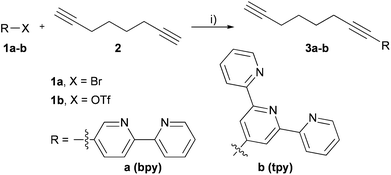 | ||
| Scheme 1 Synthesis of ligand building blocks. Reagents and conditions: (i) Pd(PPh3)2Cl2 (5 mol%), CuI (5 mol%), Et3N (10 equiv.), THF, 3 h, yields: 66% (3a); 75% (3b). | ||
Synthesis of modified nucleosides and dNTPs
The Sonogashira cross-coupling reaction was also successfully used for the attachment of oligopyridine–octadiyne building blocks to position 5 of 2′-deoxycytidine and to position 7 of 7-deaza-2′-deoxyadenosine. The Pd-catalyzed Cu-mediated cross-coupling reactions of 5-iodo-2′-deoxycytidine (dCI, 5) or 7-iodo-7-deaza-2′-deoxyadenosine (dAI, 4) with 3a or 3b were performed in the presence of 5 mol% Pd(OAc)2, water soluble tris(3-sulfonatophenyl)phosphine (TPPTS), CuI and Hünnig's base in DMF at 75 °C for 2 h to reach full conversion (Scheme 2, Table 1) in analogy to previously developed procedures.17 Products were purified by flash chromatography on reverse phase and after the crystallization were isolated as brownish solids in acceptable yields (taking into account that non-polar building blocks were attached to polar nucleoside in a single-step reaction without any use of protecting groups) (Table 1).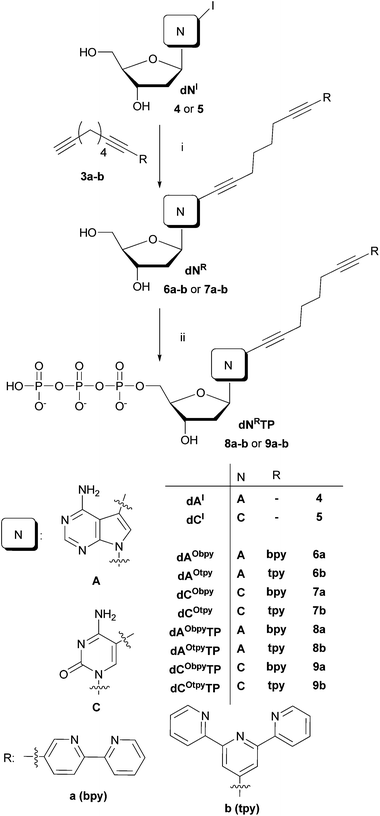 | ||
| Scheme 2 Reagents and conditions: (i) Pd(OAc)2 (5 mol%), TPPTS (2.5 equiv. to Pd), CuI (10 mol%), Et(i-Pr)2N (10 equiv.), DMF, 75 °C, 2 h; (ii) 1. PO(OMe)3, POCl3 (1.3 equiv.), 0 °C, 1 h; 2. (NHBu3)2H2P2O7 (5 equiv.), Bu3N (4.5 equiv), DMF, 0 °C, 1.5 h; 3. 2 M TEAB. | ||
| Entry | Nucleoside | Oligopyridine | Product | Yield |
|---|---|---|---|---|
| 1 | dAI (4) | 3a | dAObpy (6a) | 46% |
| 2 | dCI (5) | 3a | dCObpy (7a) | 38% |
| 3 | dAI (4) | 3b | dAOtpy (6b) | 45% |
| 4 | dCI (5) | 3b | dCOtpy (7b) | 72% |
A previously developed procedure of direct functionalization of halogenated dNTPs by aqueous-phase Sonogashira cross-coupling11,18 could not be used due to low solubility of ligand building blocks in the mixture water–acetonitrile (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) which resulted in very poor yields even after extensive optimizations. Therefore, the desired modified triphosphates dNRTPs (8a–b and 9a–b) were prepared by classical triphosphorylation19 of modified nucleosides (Scheme 2). The solution of dNR (6a–b and 7a–b) in PO(OMe)3 was treated with POCl3 at 0 °C for 1 h, followed by an addition of a pre-formed mixture of tributylammonium pyrophosphate in DMF with an addition of Bu3N. The reaction mixture was stirred at 0 °C for another 1.5 h and quenched by an addition of 2 M TEAB. Desired products were isolated in the yields shown in Table 2. The rather moderate yields are the consequence of difficult isolation of amphiphilic compounds 8a–b and 9a–b, involving 2 step purification using DEAE Sephadex and semi-preparative RP HPLC.
:1) which resulted in very poor yields even after extensive optimizations. Therefore, the desired modified triphosphates dNRTPs (8a–b and 9a–b) were prepared by classical triphosphorylation19 of modified nucleosides (Scheme 2). The solution of dNR (6a–b and 7a–b) in PO(OMe)3 was treated with POCl3 at 0 °C for 1 h, followed by an addition of a pre-formed mixture of tributylammonium pyrophosphate in DMF with an addition of Bu3N. The reaction mixture was stirred at 0 °C for another 1.5 h and quenched by an addition of 2 M TEAB. Desired products were isolated in the yields shown in Table 2. The rather moderate yields are the consequence of difficult isolation of amphiphilic compounds 8a–b and 9a–b, involving 2 step purification using DEAE Sephadex and semi-preparative RP HPLC.
| Entry | Nucleoside | dNTPs | Yield |
|---|---|---|---|
| 1 | dAObpy (6a) | dAObpyTP (8a) | 35% |
| 2 | dAOtpy (6b) | dAOtpyTP (8b) | 14% |
| 3 | dCObpy (7a) | dCObpyTP (9a) | 39% |
| 4 | dCOtpy (7b) | dCOtpyTP (9b) | 31% |
Incorporation of dNRTPs by DNA polymerase
The enzymatic incorporation of all four functionalized dARTPs (8a–b) and dCRTPs (9a–b) was tested by using thermostable DNA polymerases in primer extension experiment (PEX). Similarly to our previous work,11,18 each PEX experiment, analyzed by denaturing polyacrylamide gel electrophoresis (PAGE), was compared with positive (all four natural dNTPs) and negative control experiments (absence of one natural dNTP) in order to exclude any miss-incorporation. Single and multiple incorporations of oligopyridine-functionalized triphosphates were tested (for sequences of primer and templates see Table 3).| a In the template (temp) ON segments that form a duplex with the primer are printed in italics, the replicated segments are printed in bold. In synthesized DNAs (DNA1-4), the underlined letters indicate modifications. For magnetic separation of the extended primer strands, the templates were 5′-end biotinylated. The acronyms used in the text for primer products are analogues to those introduced for templates (e.g. the PEX product pexrnd16 was synthesized on the temprnd16 template). | |
|---|---|
| primrnd | 5′-CATGGGCGGCATGGG-3′ |
| primcomp | 5′-CATGGGCGGCATCTC-3′ |
| temprnd16 | 5′-CTAGCATGAGCTCAGTCCCATGCCGCCCATG-3′ |
| tempcomp3gA | 5′-CAGACCAGCCCTCCCGAGATGCCGCCCATG-3′ |
| tempA | 5′-CCCTCCCATGCCGCCCATG-3′ |
| tempC | 5′-CCCGCCCATGCCGCCCATG-3′ |
| tempA1 | 5′-TCCCATGCCGCCCATG-3′ |
| tempC1 | 5′-GCCCATGCCGCCCATG-3′ |
| tempcompA1 | 5′-TGAGATGCCGCCCATG-3′ |
| DNA1 | 5′-CCCTCCCATGCCGCCCATG-3′ |
3′-GGG![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) GGGTACGGCGGGTAC-5′ GGGTACGGCGGGTAC-5′ |
|
| DNA2 | 5′-CCCGCCCATGCCGCCCATG-3′ |
3′-GGG![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) GGGTACGGCGGGTAC-5′ GGGTACGGCGGGTAC-5′ |
|
| DNA3 | 5′-CTAGCATGAGCTCAGTCCCATGCCGCCCATG-3′ |
| 3′-GTCGTCTCGGTCGGGTACGGCGGGTAC-5′ |
|
| DNA4 | 5′-CTAGCATGAGCTCAGTCCCATGCCGCCCATG-3′ |
| 3′-GATGTATGAGTAGGGTACGGCGGGTAC-5′ |
|
Single nucleotide extension experiments were tested separately with all modified dNRTPs (8a–b and 9a–b) by using 19-mer templates tempA and tempC and four different DNA polymerases: Pwo, DyNAzyme II, KOD XL and Deep Vent. While experiments using Pwo polymerase (Fig. 1) were successful to give fully extended products for all of the dNTPs and the lack of extension in negative control (A- or C-) proving that no miss-incorporation occurred, experiments using other DNA polymerases (see ESI†) lead to the mixture of products with different lengths or to some miss-incorporations and therefore could not be used for direct functionalization of DNA.
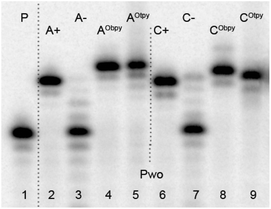 | ||
| Fig. 1 Denaturing PAGE analysis of PEX experiment synthesized on tempA (lanes 2–5) and tempC (lanes 6–9) with Pwo polymerases. 5′-32P-end labelled primer-template was incubated with different combinations of natural and functionalized dNTPs. P: primer; A+: natural dATP, dGTP; A−: dGTP; AObpy: dAObpyTP (8a), dGTP; AOtpy: dAOtpyTP (8b), dGTP; C+: natural dCTP, dGTP; C−: dGTP; CObpy: dCObpyTP (9a), dGTP; COtpy: dCOtpyTP (9b), dGTP. | ||
To compare the efficiency of incorporation in the natural and oligopyridine-modified dNTPs, we performed a simple kinetic study in single-nucleotide PEX-experiment. The experiments were performed using Pwo polymerase and a tempA1 template for experiments with natural and modified dATPs, whereas the tempC1 template was used for experiments with natural and modified dCTPs (Fig. 2). The PEX with natural dNTPs was finished within 1 min, whereas the PEX with dCRTP (9a–b) or dARTP (8a–b) took 2 or 5 min, respectively. Due to the slower incorporations of modified dNRTPs (8a–b and 9a–b) in comparison to the natural dNTPs, the reaction time for multiple incorporations must be prolonged to 30 min to ensure full length product formation.
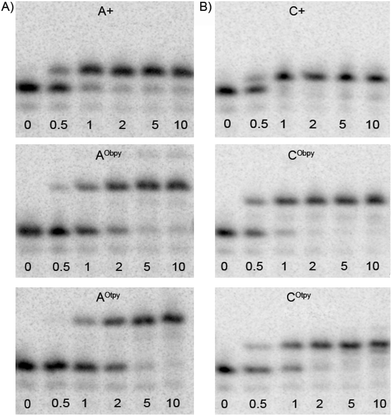 | ||
| Fig. 2 Comparison of the rate of the single-nucleotide PEX using Pwo polymerase: (A) with natural A+ (dATP) and modified dARTPs (dAObpyTP, 8a and dAOtpyTP, 8b) nucleotides using tempA1; (B) with natural C+ (dCTP) and modified dCRTPs (dCObpyTP, 9a and dCOtpyTP, 9b) nucleotides using tempC1. The reaction mixtures were incubated for time intervals indicated (in min), followed by stopping the reaction by addition of PAGE loading buffer and immediate heating. | ||
Multiple incorporations were tested on 31-mer template temprnd16 requiring incorporation of four modified dNTPs in separate positions. Modified dARTPs (8a–b) were successfully incorporated into the ONs by Pwo, KOD XL as well as Deep Vent polymerases. Using these polymerases gave fully extended ONs (for using Pwo polymerase see Fig. 3, lanes 5–6), while incorporation of dAtpyTP (8b) using DyNAzyme II polymerase was less feasible and resulted in early termination of PEX (lane 13). For incorporation of modified dCRTP (9a–b) only DyNAzyme II polymerase was suitable (lanes 14 and 15). Experiments using other polymerases lead to the mixture of ONs with different lengths (for incorporation using KOD XL or Deep Vent see ESI†).
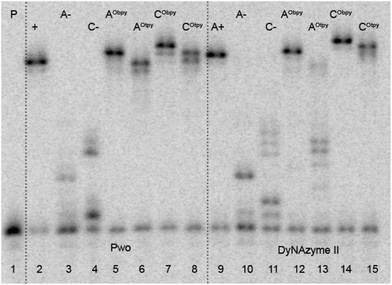 | ||
| Fig. 3 Denaturing PAGE analysis of PEX experiment synthesized on temprnd16 with Pwo (lanes 2–8) and DyNAzyme II (lanes 9–15) polymerases. 5′-32P-end labelled primer-template was incubated with different combinations of natural and functionalized dNTPs: P: primer; A+: unmodified DNA (dATP, dTTP, dCTP, dGTP); A−: unmodified DNA (dTTP, dCTP, dGTP); C−: unmodified DNA (dATP, dTTP, dGTP); AObpy: dAObpyTP (8a), dTTP, dCTP, dGTP; AOtpy: dAOtpyTP (8b), dTTP, dCTP, dGTP; CObpy: dATP, dTTP, dCObpyTP (9a), dGTP; COtpy: dATP, dTTP, dCOtpyTP (9b), dGTP. | ||
The products of PEX slightly differ in electrophoretic mobilities visible on gel, due to the higher molecular weight of modified ONs in combination with possible formation of different secondary structures. Therefore, the successful incorporations and full-length product formations were verified by measurement of MALDI mass spectra of PEX products. Single stranded DNA was prepared by PEX with biotinylated templates and then isolated by magnetoseparation.18b The correct masses were confirmed for all products (see ESI†).
Complexation studies
All oligopyridine-modified nucleosides dNR (6a–b and 7a–b) were tested as model compounds for further complexation studies proceeded on modified oligonucleotides. Methanolic solutions of dNR (6a–b and 7a–b) were mixed with 0.5 equiv. of divalent metal cations, i.e. Cu2+, Ni2+, Zn2+ and Fe2+. After incubation for 10 min at room temperature, the complex-formation was detected by UV/Vis spectroscopy. The spectra were recorded for non-metalated as well as for metalated nucleosides. Due to the fact, that an oligopyridine unit is not conjugated with the nucleobase (in contrast to the previous study when the oligopyridine unit was attached to the nucleobase via an acetylene linker11), the dominating absorbance is shifted to lower wavelengths and MLCT bands of all metal complexes of dNOtpy (6b and 7b) can be easily detected (Fig. 4). For UV/Vis spectra of dNObpy (6a and 7a) with metal cations see ESI.†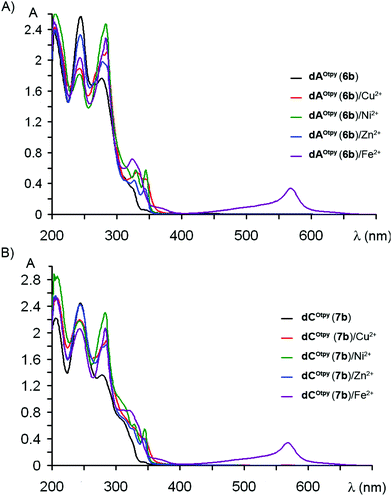 | ||
| Fig. 4 UV/Vis spectra of oligopyridine-modified nucleosides: (A) dAOtpy (6b), (B) dCOtpy (7b) with metal cations. | ||
After successful complexation of nucleosides, we proceeded to complexation of Otpy-modified oligonucleotides. For complexation studies with UV/Vis detection, pexrnd16 products were prepared in bigger scale by using Deep Vent (for incorporation of dAOtpyTP8b, synthesis of DNA3) or DyNAzyme II polymerases (for incorporation of dCOtpyTP9b, synthesis of DNA4). Oligonucleotides were well purified from unreacted dNOtpyTPs (8b or 9b) before complexation. After addition of 0.5 equiv. of M(BF4)2·nH2O per each modification to the aqueous solution of modified oligonucleotide, the mixture was incubated overnight at room temperature. The UV/Vis spectra were recorded for non-metalated and metalated DNA duplexes, either natural or modified ones (Fig. 5). The dominant absorbance at ca. 260 nm originated from the absorbance of natural nucleotides (grey line) while the small absorbance band at ca. 350 nm is due to the presence of Otpy-modification (black line). While MLCT bands of complexes formed by Otpy-modified ON with Cu2+ (red line), Ni2+ (green line), Zn2+ (blue line) are partially overlapped with the absorbance band of Otpy-modification, the complex formed by mixing Otpy-modified ON with Fe2+ can be easily detected due to the characteristic absorbance at 580 nm20 (magenta line). Similar MLCT bands were not observed for a natural DNA duplex mixed with the corresponding metals (see ESI†).
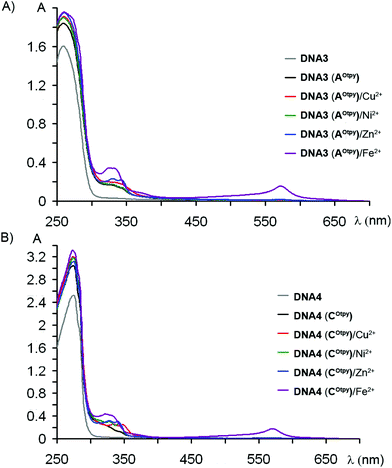 | ||
| Fig. 5 UV/Vis spectra of oligopyridine-modified DNA: (A) DNA3 (AOtpy), (B) DNA4 (COtpy) with metal cations. | ||
Complex formation was also detected by native polyacrylamide gel electrophoresis. The products of monoincorporations were prepared by PEX experiment using tempA and dATP (natural DNA) or dAOtpyTP, 8b (modified DNA, DNA1), or tempC and dCTP (natural DNA) or dCOtpyTP, 9b (modified DNA, DNA2). PEX products were directly, without previous purification, mixed with 1 equiv. of M(BF4)2·nH2O (calculated to the amount of modified dNOtpyTP in PEX experiment) and incubated at room temperature overnight. Despite longer time of incubation, compared to the time required for successful complexation of acetylene linked derivatives,11 none (for AOtpy) or only minor band (for COtpy) with slower mobility proving a successful complex formation was observed (see Fig. S9 in ESI†). In the case of pexrnd16 containing four Otpy-modifications successful complex formation with Ni2+ and Fe2+ ions, shown by bands with slower mobility, was observed (Fig. 6, lanes 8 and 10). A similar change in mobility was not observed for non-modified DNA mixed with these metal ions (lanes 3 and 5).
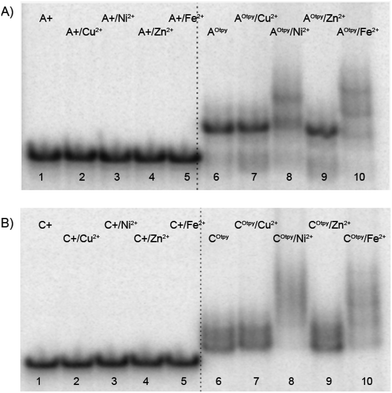 | ||
| Fig. 6 Non-denaturing gel electrophoresis (8% SB_PAGE) of DNA duplexes in the absence and in the presence of M2+ for pexrnd16. 5′-32P-end labelled primer-template was incubated with different combinations of natural and functionalized dNTPs: A+: unmodified DNA (dATP, dTTP, dCTP, dGTP); A+/M2+: unmodified DNA mixed with corresponding metal cations; AOtpy: Otpy-modified DNA (dAOtpyTP8b, dTTP, dCTP, dGTP); AOtpy/M2+: Otpy-modified DNA mixed with corresponding metal cations; C+: unmodified DNA (dATP, dTTP, dCTP, dGTP); C+/M2+: unmodified DNA mixed with corresponding divalent metals; COtpy: Otpy-modified DNA (dATP, dTTP, dCOtpyTP9b, dGTP); COtpy/M2+: Otpy-modified DNA mixed with corresponding metal cations. | ||
Intra-strand vs. inter-strand DNA complex
Since, the quantitative complex formation was observed only for DNAs containing four Otpy-modifications, the question arises whether intra-strand or inter-strand complexes are formed. A short and rigid acetylene linker predeterminated modified DNA to form inter-strand complexes (Fig. 7A), whereas the long and flexible octadiyne linker enables also metal-templated intra-strand cyclization of Otpy-modified oligonucleotides (Fig. 7B).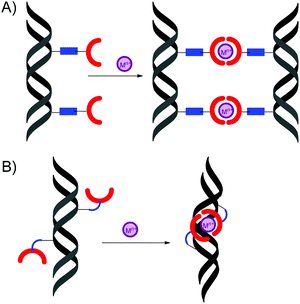 | ||
| Fig. 7 Schematic representation of: (A) inter-strand, (B) intra-strand DNA complexes. | ||
Therefore, we compared complexations of previously described DNA bearing tpy-modification attached via an acetylene linker (made from dNEtpyTP)11 with the complexation of DNA modified by an octadiyne linker (made from dNOtpyTP). Since the DNAs containing only one dNOtpy did not form metal complexes and dCEtpyTP was not a good substrate in multiple incorporations,11 we focused in our investigation on DNAs prepared by PEX experiment using temprnd16 and dATP (natural DNA), dAEtpyTP (modified DNA with acetylene linked oligopyridine units) and dAOtpyTP8b (modified DNA with octadiyne linked oligopyridine units).
First we compared the mobility of these complexes on native polyacrylamide gel (Fig. 8). The Fe2+ complex of DNA with acetylene linked oligopyridine tpy-units (lane 6) showed significantly slower mobility in comparison to non-metalated Etpy-modified DNA (lane 4) due to inter-duplex complexation. On the other hand, the Ni2+ or Fe2+ complexes (lanes 8 and 9) formed from Otpy-modified DNA moved only slightly more slowly compared to non-metalated Otpy-modified DNA (lane 7) suggesting possible formation of (less bulky) intra-strand complexes.
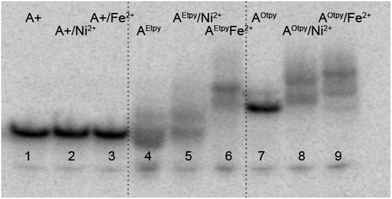 | ||
| Fig. 8 Non-denaturing gel electrophoresis (8% SB_PAGE) of DNA duplexes in the absence and in the presence of M2+ for pexrnd16. 5′-32P-end labelled primer-template was incubated with different combinations of natural and functionalized dNTPs: A+: unmodified DNA (dATP, dTTP, dCTP, dGTP); A+/M2+: unmodified DNA mixed with indicated metal cations; AEtpy: Etpy-modified DNA (dAEtpyTP, dTTP, dCTP, dGTP); AEtpy/M2+: Etpy-modified DNA mixed with indicated metal cations; AOtpy: Otpy-modified DNA (dAOtpyTP8b, dTTP, dCTP, dGTP); AOtpy/M2+: Otpy-modified DNA mixed with indicated metal cations. | ||
Metal-templated intra-strand cyclization of Otpy-modified oligonucleotide was also confirmed by measurement of MALDI spectra, where DNA3(AOtpy)·Fe2+ and DNA3(AOtpy)·2Fe2+ were successfully detected (Fig. 9), while no non-metalated modified DNA was observed.
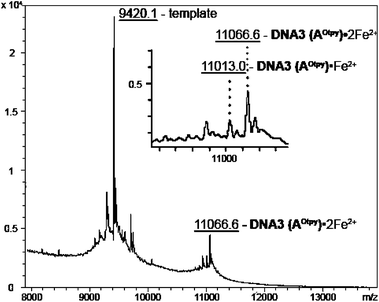 | ||
| Fig. 9 MALDI-TOF spectra of DNA3 (AOtpy) mixed with Fe2+ (calculated mass: 11010.7 Da for DNA3 (AOtpy)·1Fe2+ and 11060.9 Da for DNA3 (AOtpy)·2Fe2+). | ||
To further verify the hypothesis that the DNA containing Otpy-modification(s) allows formation of intra-strand complexes, we have prepared other DNA sequences (PEX-experiments using tempcompA1 and tempcomp3gA) bearing just one Otpy and tested their complexation. In these cases, very weak bands of slower mobility were also observed (Fig. 10). These must be due to inter-strand complexation since no intra-strand complexes could be formed with only one tpy-modification. Some other tested sequences are shown in ESI.† We were unable to find any rationale why some sequences did and some others did not show the formation of these inter-strand complexes. However, since the inter-strand complexes of DNAs bearing a single Otpy modification are either not formed or only very weak and the complexations of DNAs containing four Otpy units with Fe2+ and Ni2+ are quantitative, it seems that the intra-strand complexes were formed (or even preferred) in some sequences.
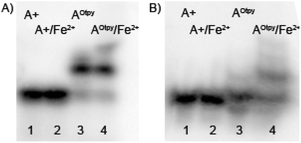 | ||
| Fig. 10 Non-denaturing gel electrophoresis (8% SB_PAGE) of DNA duplexes in the absence and in the presence of M2+ for tempcompA1(A) and tempcomp3gA (B). 5′-32P-end labelled primer-template was incubated with different combinations of natural and functionalized dNTPs: A+: unmodified DNA (dATP, dTTP, dCTP, dGTP); A+/Fe2+: unmodified DNA mixed with Fe2+; AOtpy: Otpy-modified DNA (dAOtpyTP8b, dTTP, dCTP, dGTP); AOtpy/Fe2+: Otpy-modified DNA mixed with Fe2+. | ||
The CD spectra of natural and tpy-modified DNA3 (either non-metalated or metalated) prepared by PEX using temprnd16 were measured in order to verify the possible intra-strand complex formation (Fig. 11). For natural, as well as for tpy-modified DNAs, classical B-structure was observed. While the negative minimum for natural (A+) and Etpy-modifed (AEtpy) DNAs was found at 245 nm, the negative minimum for Otpy-modified (AOtpy) DNA was shifted to 250 nm. A positive maximum was found at 270 nm with a shoulder at 290 nm for tpy-modified DNA (blue and magenta line). This shoulder is more significant after addition of Fe2+ ions, while for complexes formed from Otpy-modified DNA (AOtpy/Fe2+) (red line) it is even more obvious than for complexes formed from Etpy-modified DNA (AEtpy/Fe2+) (green line). Although the interpretation of CD-spectra of corresponding DNA complexes might be complicated by formation of diverse structures (inter-strand DNA complexes, products of inter- or intra-strand intercalation), small spectral changes in non-metalated tpy-modified DNA can be explained by diminishing the arrangement of the DNA double helix caused by the presence of the tpy-modification or by intercalation of tpy-units, while the formation of the band at 290 nm in the DNA complex formed from Otpy-modified DNA and Fe2+ ions (AOtpy/Fe2+) can be a result of partial distortion of the B-DNA double helix, required (according to the computational studies) for formation of intra-strand DNA complex DNA(AOtpy)·2Fe2+ (Fig. 11).
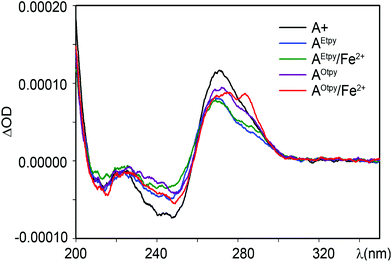 | ||
| Fig. 11 CD-spectra of natural and tpy-modified DNA duplexes prepared from DNA3 in the absence or presence of Fe2+ ions. | ||
Further, the possibility of intra-strand complex formation was studied by molecular modelling. In DNA3 containing four dAOtpy units, there are in principle four combinations of intramolecular complexes: 1–2 (separated by 4 base-pairs), 3–4 (separated by 3 bp), 1–3 (separated by 9 bp) and 2–4 (separated by 8 bp). The 1–2 and 3–4 complexes were built and the geometry was successfully minimized by using QM/MM methodology (Fig. 12). The 3–4 complex was by 3.1 kcal mol−1 more stable than the 1–2 complex due to better proximity and orientation of the modified nucleotides in the 3–4 complex. The formation of the complex 3–4 also induced smaller geometrical changes in the B-DNA duplex (RMSD 0.269 Å) compared to the 1–2 complex (RMSD 0.662 Å). We did not succeed in building the 1–3 and 2–4 complexes since the distances between modified nucleotides were too large for any complex formation. These calculations clearly confirm the possibility of intra-strand complex formation when the two tpy-modified nucleotides are separated by 3 or 4 bps in the DNA duplex.
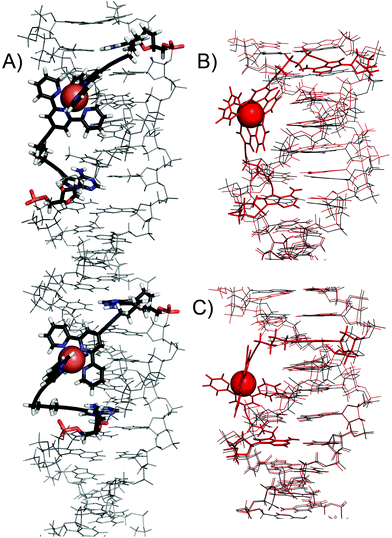 | ||
| Fig. 12 (A) Detailed view of the QM/MM optimized structure of DNA3 with 1–2 and 3–4 intra-strand DNA complexes. Comparison of (B) 1–2 and (C) 3–4 complexes (in red) with unmodified DNA (in black). | ||
Conclusions
Novel dNR bearing oligopyridine ligands attached via an octadiyne linker were prepared in a single-step Sonogashira cross-coupling reaction of iodinated nucleosides with the corresponding oligopyridine–octadiyne building blocks. Modified nucleosides dNR were used as starting materials for synthesis of functionalized dNRTPs by triphosphorylation. Modified dNRTPs were shown to be good substrates for DNA polymerases and were incorporated into the DNA by primer extension. Oligopyridine functionalized DNAs, containing either one or four modifications, were tested for post-synthetic complexation with M2+ ions. For DNAs containing only one tpy-unit, weak inter-strand complex formation was observed only for some sequences. On the other hand, DNAs containing four tpy-units (DNA3 and DNA4) form quantitatively complexes with Ni2+ and Fe2+ ions suggesting that intra-strand complexes might be involved. Unfortunately, no hard proof of the intra-strand complexes was found, probably due to reversibility of the complexation21 and complicated equilibrium of different types of complexes. However, we found some evidence for the possibility of intra-strand complexation from gel electrophoresis, MALDI, CD and QM/MM calculations. Therefore, we can conclude that the attachment of terpyridine ligands to DNA by a flexible octadiyne linker allows formation of intra-strand metal complexes when more tpy-units are present in the same DNA molecule separated by 3 or 4 bps. However, as the inter-strand complex formation is also possible, at least for some sequences, the reversible formation of inter- and intra-strand complexes may complicate future applications of tpy-modified ONs and DNA in self-assembly.Experimental
All reactions were performed under an argon atmosphere. POCl3 and PO(OMe)3 used for phosphorylation of nucleoside were distilled before using. Other chemicals were purchased from commercial suppliers and were used as received. Preparative flash chromatography on reverse phase was performed on a Biotage SP1 flash purification system. Semi-preparative HPLC separations were performed on a column packed with 10 μm C18 reversed phase (Phenomenex, Luna C18(2)). NMR spectra were measured on a Bruker Avance 500 (500.0 MHz for 1H, 125.7 MHz for 13C and 202.3 for 31P) or a Bruker Avance II 600 (600.1 MHz for 1H and 150.9 MHz for 13C) in CDCl3 (1H referenced to TMS as an internal standard (δ = 0 ppm); 13C referenced to the solvent signal (δ = 77.0 ppm)), in DMSO-d6 (1H referenced to the residual solvent signal (δ = 2.50 ppm); 13C referenced to the solvent signal (δ = 39.7 ppm)), or in CD3OD (1H referenced to the residual solvent signal (δ = 3.31 ppm); 13C referenced to the solvent signal (δ = 49.0 ppm); 31P referenced to H3PO4 (δ = 0 ppm) as an external standard). Chemical shifts are given in ppm (δ scale), coupling constants (J) in Hz. Complete assignment of all NMR signals was achieved by use of a combination of H,H-COSY, H,C-HSQC, and H,C-HMBC experiments. Mass spectra were measured on an LCQ classic (Thermo-Finnigan) spectrometer using ESI or Q-Tof Micro (Waters, ESI source, internal calibration with lockspray). Mass spectra of functionalized DNA were measured by Maldi-TOF, Reflex IV (Bruker) with nitrogen laser. UV/Vis spectra were measured on a Varian CARY 100 Bio spectrophotometer at room temperature. CD spectra were recorded on a Jasco 815 spectropolarimeter (Japan) at room temperature. The optical path length was 0.1 cm and the CD signal was monitored from 200 nm to 350 nm. For each experiment the data are average of 2 scans taken with the time constant of 32 s, a scanning speed of 5 nm min−1 with blank subtracted. The CD spectra are expressed in differential absorption (ΔOD = AL − AR).For a numbering scheme for NMR assignment see Fig. S1 in ESI.†
General procedure A: synthesis of ligand building blocks
To an argon-purged flask containing 5-bromo-2,2′-bipyridine (1a) or (2,2′:6′,2′′-terpyridine-4′-yl) trifluoromethanesulfonate (1b) (500 mg), Pd(PPh3)2Cl2 (5 mol%) and CuI (5 mol%) were added THF (10 mL), Et3N (10 equiv.) and 1,7-octadiyne (3 equiv.). The reaction mixture was heated at 75 °C for 3 h. After evaporation of the solvent under reduced pressure, the residue was extracted with three 100 mL portions of CHCl3. Organic phases were combined and dried over MgSO4. The residue was then purified by silica gel chromatography using hexane/ethyl acetate (0%–9%).4′-(Octa-1′′′,7′′′-diyn-1′′′-yl)-2,2′:6′,2′′-terpyridine (3b)
Product 3b was prepared according to general procedure A from (2,2′:6′,2′′-terpyridine-4′-yl) trifluoromethanesulfonate (1b). It was isolated as an orange oil in the yield of 75% (331.8 mg).Mp 69.0–69.5 °C; IR: 3207, 2940, 1581, 1564, 1467, 1391, 1263, 1113 cm−1; 1H NMR (500.0 MHz, CDCl3): 1.73 (m, 2H, HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CH2CH2CH2CH2–CC–tpy); 1.77 (m, 2H, HCC–CH2CH2CH2CH2–CC–tpy); 1.98 (t, 1H, 4J = 2.7, HCC–); 2.28 (td, 2H, Jvic = 6.8, 4J = 2.7, HCC–CH2CH2CH2CH2–CC–tpy); 2.51 (t, 2H, Jvic = 6.8, HCC–CH2CH2CH2CH2–CC–tpy); 7.43 (ddd, 2H, J5′,4′ = 7.5, J5′,6′ = 5.0, J5′,3′ = 1.2, H-5′); 7.97 (ddd, 2H, J4′,3′ = 8.0, J4′,5′ = 7.5, J4′,6′ = 1.8, H-4′); 8.48 (s, 2H, H-3,5); 8.69 (d, 1H, J3′,4′ = 8.0, H-3′); 8.78 (ddd, 1H, J6′,5′ = 5.0, J6′,4′ = 1.8, J6′,3′ = 0.9, H-6′); 13C NMR (125.7 MHz, CDCl3): 17.96 (HCC–CH2CH2CH2CH2–CC–tpy); 19.05 (HCC–CH2CH2CH2CH2–CC–tpy); 27.30, 27.47 (HCC–CH2CH2CH2CH2–CC–tpy); 68.67 (HCC); 79.04 (tpy–CC); 83.99 (HCC); 95.87 (tpy–CC); 121.93 (CH-3′); 123.83 (CH-3,5); 124.34 (CH-5′); 134.64 (C-4); 138.29 (CH-4′); 148.21 (CH-6′); 154.25 (C-2,6); 154.55 (C-2′); MS (ESI): m/z (%) = 3385 (100) [M+ + H]; HRMS-ESI: m/z [M + H]+ calcd for C23H23N3: 338.16517; found: 338.16516; Anal. Calcd for C23H19N3·1/5MeOH: C, 81.04; H, 5.80; N, 12.22. Found: C, 81.42; H, 5.58; N, 11.93.
C–CH2CH2CH2CH2–CC–tpy); 1.77 (m, 2H, HCC–CH2CH2CH2CH2–CC–tpy); 1.98 (t, 1H, 4J = 2.7, HCC–); 2.28 (td, 2H, Jvic = 6.8, 4J = 2.7, HCC–CH2CH2CH2CH2–CC–tpy); 2.51 (t, 2H, Jvic = 6.8, HCC–CH2CH2CH2CH2–CC–tpy); 7.43 (ddd, 2H, J5′,4′ = 7.5, J5′,6′ = 5.0, J5′,3′ = 1.2, H-5′); 7.97 (ddd, 2H, J4′,3′ = 8.0, J4′,5′ = 7.5, J4′,6′ = 1.8, H-4′); 8.48 (s, 2H, H-3,5); 8.69 (d, 1H, J3′,4′ = 8.0, H-3′); 8.78 (ddd, 1H, J6′,5′ = 5.0, J6′,4′ = 1.8, J6′,3′ = 0.9, H-6′); 13C NMR (125.7 MHz, CDCl3): 17.96 (HCC–CH2CH2CH2CH2–CC–tpy); 19.05 (HCC–CH2CH2CH2CH2–CC–tpy); 27.30, 27.47 (HCC–CH2CH2CH2CH2–CC–tpy); 68.67 (HCC); 79.04 (tpy–CC); 83.99 (HCC); 95.87 (tpy–CC); 121.93 (CH-3′); 123.83 (CH-3,5); 124.34 (CH-5′); 134.64 (C-4); 138.29 (CH-4′); 148.21 (CH-6′); 154.25 (C-2,6); 154.55 (C-2′); MS (ESI): m/z (%) = 3385 (100) [M+ + H]; HRMS-ESI: m/z [M + H]+ calcd for C23H23N3: 338.16517; found: 338.16516; Anal. Calcd for C23H19N3·1/5MeOH: C, 81.04; H, 5.80; N, 12.22. Found: C, 81.42; H, 5.58; N, 11.93.
General procedure B: Sonogashira cross-coupling reaction – synthesis of modified deoxynucleosides
DMF (1 mL) and Et(i-Pr)2N (10 equiv.) were added to an argon-purged flask containing nucleoside 5-iodo-2′-deoxycytidine (dCI, 5) or 7-iodo-7-deaza-2′-deoxyadenosine (dAI, 4) (50 mg), an octadiyne modified oligopyridine 3a–b (1.5 equiv.) and CuI (10 mol%). In a separate flask, Pd(OAc)2 (5 mol%) and P(Ph-SO3Na)3 (2.5 equiv. to Pd) were combined, evacuated and purged with argon followed by addition of DMF (0.5 mL). The mixture of catalyst was then injected into the reaction mixture and the reaction mixture was stirred at 75 °C for 2 h. The solvent was then evaporated in vacuo. Products were directly purified by flash chromatography on reverse phase using H2O/MeOH (0% to 100%) as an eluent. Products were recrystallized from the mixture MeOH–H2O.7-[8′′′′′-(2′′,2′′′:6′′′,2′′′′-Terpyridin-4′′′-yl)octa-1′′′′′,7′′′′′-diyn-l′′′′′-yl]-7-deaza-2′-deoxyadenosine (dAOtpy, 6b)
Product 6b was prepared according to the general procedure B from dAI (4) and 3b. It was isolated as a brownish powder in the yield of 45% (35.0 mg).Mp 109–113 °C; IR: 3444, 3185, 2928, 1584, 1566, 1468, 1393, 1305, 1046 cm−1; 1H NMR (600.1 MHz, DMSO-d6): 1.76 (m, 4H, dapur–CC–CH2CH2CH2CH2–CC–tpy); 2.15 (ddd, 1H, Jgem = 13.1, J2′b,1′ = 6.0, J2′b,3′ = 2.7, H-2′b); 2.46 (ddd, 1H, Jgem = 13.1, J2′a,1′ = 8.1, J2′a,3′ = 5.7, H-2′a); 2.58 (m, 2H, dapur–CC–CH2CH2CH2CH2–CC–tpy); 2.62 (m, 2H, dapur–CC–CH2CH2CH2CH2–CC–tpy); 3.49 (ddd, 1H, Jgem = 11.8, J5′b,OH = 6.0, J5′b,4′ = 4.4, H-5′b); 3.56 (ddd, 1H, Jgem = 11.8, J5′a,OH = 5.3, J5′a,4′ = 4.4, H-5′a); 3.81 (td, 1H, J4′,5′ = 4.4, J4′,3′ = 2.7, H-4′); 4.32 (m, 1H, J3′,2′ = 5.7, 2.7, J3′,OH = 4.0, J3′,4′ = 2.7, H-3′); 5.07 (dd, 1H, JOH,5′ = 6.0, 5.3, OH-5′); 5.26 (d, 1H, JOH,3′ = 4.0, OH-3′); 6.46 (dd, 1H, J1′,2′ = 8.1, 6.0, H-1′); 7.52 (ddd, 2H, J5′′′,4′′′ = 7.5, J5′′′,6′′′ = 4.7, J5′′′,3′′′ = 1.2, H-5′′′); 7.67 (s, 1H, H-6); 8.02 (ddd, 2H, J4′′′,3′′′ = 7.9, J4′′′,5′′′ = 7.5, J4′′′,6′′′ = 1.8, H-4′′′); 8.09 (s, 1H, H-2); 8.34 (s, 2H, H-3′′,5′′); 8.61 (ddd, 2H, J3′′′,4′′′ = 7.9, J3′′′,5′′′ = 1.2, J3′′′,6′′′ = 0.9, H-3′′′); 8.72 (ddd, 2H, J6′′′,5′′′ = 4.7, J6′′′,4′′′ = 1.8, J6′′′,3′′′ = 0.9, H-6′′′); 13C NMR (150.9 MHz, DMSO-d6): 18.52 (dapur–CC–CH2CH2CH2CH2–CC–tpy); 18.70 (dapur–CC–CH2CH2CH2CH2–CC–tpy); 27.34, 27.70 (dapur–CC–CH2CH2CH2CH2–CC–tpy); 39.93 (CH2-2′); 62.11 (CH2-5′); 71.18 (CH-3′); 74.07 (dapur–CC); 79.10 (tpy–CC); 83.28 (CH-1′); 87.67 (CH-4′); 92.45 (dapur–CC); 95.69 (C-5); 96.40 (tpy–CC); 102.52 (C-4a); 121.06 (CH-3′′′); 122.25 (CH-3′′,5′′); 124.96 (CH-5′′′); 125.75 (CH-6); 133.55 (C-4′′); 137.77 (CH-4′′′); 149.25 (C-7a); 149.64 (CH-6′′′); 152.78 (CH-2); 154.51 (C-2′′,6′′); 155.47 (C-2′′′); 157.77 (C-4); MS (ESI): m/z (%) = 586 (27) [M+ + H], 608 (100) [M+ + Na]; HRMS-ESI: m/z [M + H]+ calcd for C34H32O3N7: 586.25611; found: 586.25592.
General procedure C: phosphorylation of oligopyridine modified nucleosides (dNR) – synthesis of modified dNRTPs
Dry trimethyl phosphate (1 mL) was added to an argon-purged flask containing nucleoside analogue dNR (6a–b or 7a–b, 50 mg), cooled to 0 °C on ice followed by the addition of POCl3 (1.5 equiv.). A solution of (NHBu3)2H2P2O7 (5 equiv., 1 mL) in dry DMF with an addition of Bu3N (4.5 equiv.) was prepared in a separate flask and cooled down to 0 °C. Like this the prepared solution was then added to the reaction mixture and stirred for 1.5 h and quenched by 2 M TEAB buffer (2 mL). The product was isolated on a DEAE Sephadex column (150 mL) eluting with a gradient 0 to 1.2 M TEAB, evaporated, co-distilled with water (3 times) and re-purified by semi-preparative HPLC on a C18 column using a linear gradient of 0.1 M TEAB (triethylammonium bicarbonate) in H2O to 0.1 M TEAB in H2O–MeOH (1:1) as an eluent. Several co-distillations with water followed by freeze-drying from water gave the products as brownish powder.
7-[8′′′′′-(2′′,2′′′:6′′′,2′′′′-Terpyridin-4′′′-yl)octa-1′′′′′,7′′′′′-diyn-l′′′′′-yl]-7-deaza-2′-deoxyadenosine-5′-O-triphosphate (dAOtpyTP, 8b)
This compound was prepared according to the general procedure C from dAOtpy (6b) in the yield of 14% (13.2 mg).
1H NMR (600.1 MHz, CD3OD): 1.29 (t, 27H, Jvic = 7.3, CH3CH2N); 1.84 (m, 4H, dapur–CC–CH2CH2CH2CH2–CC–tpy); 2.30 (ddd, 1H, Jgem = 13.3, J2′b,1′ = 5.8, J2′b,3′ = 2.9, H-2′b); 2.46 (ddd, 1H, Jgem = 13.3, J2′a,1′ = 8.0, J2′a,3′ = 5.7, H-2′a); 2.58 (t, 2H, Jvic = 6.8, dapur–CC–CH2CH2CH2CH2–CC–tpy); 2.61 (t, 2H, Jvic = 6.8, dapur–CC–CH2CH2CH2CH2–CC–tpy); 3.18 (q, 18H, Jvic = 7.3, CH3CH2N); 4.14 (m, 1H, H-4′); 4.26 (m, 2H, H-5′); 4.60 (dt, 1H, J3′,2′ = 5.7, 2.9, J3′,4′ = 2.9, H-3′); 6.57 (dd, 1H, J1′,2′ = 8.0, 5.8, H-1′); 7.42 (ddd, 2H, J5′′′,4′′′ = 7.3, J5′′′,6′′′ = 4.8, J5′′′,3′′′ = 0.6, H-5′′′); 7.69 (s, 1H, H-6); 7.92 (ddd, 2H, J4′′′,3′′′ = 7.9, J4′′′,5′′′ = 7.3, J4′′′,6′′′ = 1.6, H-4′′′); 8.26 (s, 3H, H-2, H-3′′,5′′); 8.52 (bd, 2H, J3′′′,4′′′ = 7.9, H-3′′′); 8.62 (bd, 2H, J6′′′,5′′′ = 4.8, H-6′′′); 13C NMR (150.9 MHz, CD3OD): 9.12 (CH3CH2N); 19.75 (dapur–CC–CH2CH2CH2CH2–CC–tpy); 20.12 (dapur–CC–CH2CH2CH2CH2–CC–tpy); 28.87, 28.93 (dapur–CC–CH2CH2CH2CH2–CC–tpy); 41.85 (CH2-2′); 47.44 (CH3CH2N); 66.87 (d, JC,P = 5.5, CH2-5′); 72.56 (CH-3′); 72.74 (dapur–CC); 80.10 (tpy–CC); 84.92 (CH-1′); 87.62 (d, JC,P = 8.5, CH-4′); 95.16 (dapur–CC); 96.79 (tpy–CC); 99.97 (C-5); 102.80 (C-4a); 122.69 (CH-3′′′); 123.95 (CH-3′′,5′′); 125.55 (CH-5′′′); 128.22 (CH-6); 135.51 (C-4′′); 138.70 (CH-4′′′); 146.66 (CH-2); 148.17 (C-7a); 150.16 (CH-6′′′); 153.75 (C-4); 156.56 (C-2′′,6′′); 156.70 (C-2′′′); 31P{1H} NMR (202.3 MHz, CD3OD): −21.99 (bdd, J = 20.7, 19.3, Pβ); −9.56 (d, J = 20.7, Pα); −8.71 (d, J = 19.3, Pγ); MS (ES−): found m/z: 824.2 (M − 1), 744.3 (M − PO3H2 − 1); HRMS (ES): m/z calcd for C34H33O12N7P3: 824.14055; found: 824.13982.
Primer extension, purification and analysis of the PEX products
Synthetic ONs were purchased from Sigma Aldrich (USA). Primer: 5′-CAT GGG CGG CAT GGG-3′ (primrnd), 5′-CAT GGG CGG CAT CTC-3′ (primcomp); templates: 5′-CTA GCA TGA GCT CAG TCC CAT GCC GCC CAT G-3′ (temprnd16), 5′-CAG ACC AGC CCT CCCGAG ATG CCG CCC ATG-3′ (tempcomp3gA), 5′-CCC GCC CAT GCC GCC CAT G-3′ (tempC), 5′-CCC TCC CAT GCC GCC CAT G-3′ (tempA), TCC CAT GCC GCC CAT G-3′ (tempA1), GCC CAT GCC GCC CAT G-3′ (tempC1), 5′-TGA GAT GCC GCC CAT G-3′ (tempcompA1) (segments forming duplex with the primer are in italics, the replicated segments are in bold). Templates used in experiment involving the DBstv magnetoseparation procedure were biotinylated at their 5′ ends. Streptavidine magnetic beads MagPrep P-25 Streptavidine Particles were obtained from Novagen (EMD Chemicals, USA), Pwo DNA polymerase from PeqLab (Germany), DyNAzyme II DNA polymerases from Finnzymes (Finland), KOD XL DNA polymerase from Novagen (EMD Chemicals, USA), Deep Vent DNA polymerases as well as T4 polynukleotide kinase and natural nucleoside triphosphate (dATP, dCTP, dGTP and dCTP) from New England Biolabs (Great Britain) and γ-32P-ATP from Izotop, Institute of Isotopes Co, Ltd. (Hungary).For magnetoseparation were used unlabelled primers and biotinylated templates.
Acknowledgements
This work was supported by the Academy of Sciences of the Czech Republic (RVO: 61388963), the Czech Science Foundation (203/09/0317) and by Gilead Sciences, Inc. (Foster City, CA, U. S. A.)Notes and references
-
(a) N. C. Seeman, J. Theor. Biol., 1982, 99, 237 CrossRef CAS
; (b) P. W. K. Rothemund, Nature, 2006, 440, 297 CrossRef CAS
-
(a) F. D. Lewis, S. A. Helvoigt and R. L. Letsinger, Chem. Commun., 1999, 327 RSC
-
(a) I. Vargas-Baca, D. Mitra, H. J. Zulyniak, J. Banerjee and H. F. Sleiman, Angew. Chem., Int. Ed., 2001, 40, 4629 CrossRef CAS
-
(a) D. J. Hurley and Y. Tor, J. Am. Chem. Soc., 1998, 120, 2194 CrossRef CAS
- M. Vrábel, P. Horáková, H. Pivoňková, L. Kalachova, H. Černocká, H. Cahová, R. Pohl, P. Šebest, L. Havran, M. Hocek and M. Fojta, Chem.–Eur. J., 2009, 15, 1144 CrossRef
-
(a) G. Roelfes and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 3230 CrossRef CAS
-
(a) M. Kalek, A. S. Madsen and J. Wengel, J. Am. Soc. Chem., 2007, 129, 9392 CrossRef CAS
-
(a) G. H. Clever, C. Kaul and T. Carell, Angew. Chem., Int. Ed., 2001, 46, 6226 CrossRef
-
(a) A. A. Gorodetsky and J. K. Barton, Langmiur, 2006, 22, 7917 CrossRef CAS
- Review:
(a) K. Tanaka and M. Shionoya, Coord. Chem. Rev., 2007, 251, 2732 CrossRef CAS
- L. Kalachova, R. Pohl and M. Hocek, Org. Biomol. Chem., 2012, 10, 49 CAS
- T. Ehrenschwenderm, A. Barth, H. Puchta and H.-A. Wagenknecht, Org. Biomol. Chem., 2012, 10, 46 Search PubMed
- T. Ruhl and E. Stulz, Supramol. Chem., 2010, 22, 103 CrossRef CAS
- M. Göritz and R. Krämer, J. Am. Chem. Soc., 2005, 127, 18016 CrossRef
-
(a) J. Gierlich, K. Gutsmiedl, P. M. E. Gramlich, A. Schmidt, G. A. Burley and T. Carell, Chem.–Eur. J., 2007, 13, 9486 CrossRef CAS
- V. Grosshenny, F. M. Romero and R. Ziessel, J. Org. Chem., 1997, 62, 1491 CrossRef CAS
-
(a) M. Vrábel, R. Pohl, I. Votruba, M. Sajadi, A. S. Kovalenko, N. P. Ernsting and M. Hocek, Org. Biomol. Chem., 2008, 6, 2852 RSC
-
(a) P. Čapek, H. Cahová, R. Pohl, M. Hocek, C. Gloekner and A. Marx, Chem.–Eur. J., 2007, 13, 6196 CrossRef
- T. Kovacs and L. Ötvös, Tetrahedron Lett., 1988, 29, 4525 CrossRef CAS
- P. S. Braterman, J. I. Song and R. D. Peacock, Inorg. Chem., 1992, 31, 555 CrossRef CAS
- Very recently, the reversibility of tpy-Ni2+ formation in DNA was reported for DNA constructs containing tpy instead of some nucleobases: J. R. Morgan, D. V. X. Nguyen, A. R. Frohman, S. R. Rybka and J. A. Zebala, Bioconjugate Chem., 2012 DOI:10.1021/bc3003293
-
D. A. Case, T. A. Darden, T. E. Cheatham, III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossvai, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed
-
The PyMol Molecular Graphics System, Version 1.2, Schrodinger, LLC Search PubMed
- E. L. Stawart and J. P. Bowen, J. Comput. Chem., 1992, 13, 1125 CrossRef
- S. Dapprich, I. Komaromi, K. Suzie Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct. (Teochem), 1999, 461–462, 1 CrossRef CAS
- P. Jurecka, J. Cerny, P. Hobza and D. Salahub, J. Comput. Chem., 2007, 28, 555 CrossRef CAS
- J. Rezac, Cuby3, Prague.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental part and spectral data, additional PAGEs, MALDI and additional UV/Vis spectra. See DOI: 10.1039/c2ob26881g |
| This journal is © The Royal Society of Chemistry 2013 |