 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled functionalization of graphene oxide with sodium azide†
Siegfried
Eigler
*a,
Yichen
Hu
b,
Yoshitaka
Ishii
bc and
Andreas
Hirsch
a
aDepartment of Chemistry and Pharmacy, Institute of Advanced Materials and Processes (ZMP), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Dr-Mack Str. 81, 90762 Fürth, Germany. E-mail: siegfried.eigler@fau.de; Fax: +49 (0)911 6507865015; Tel: +49 (0)911 6507865005
bDepartment of Chemistry, University of Illinois at Chicago, 845 W Taylor St, Chicago, IL 60607, USA
cCenter for Structural Biology, University of Illinois at Chicago, 1100 South Ashland Street, Chicago, Illinois 60607, USA
First published on 14th October 2013
Abstract
We present the first example of azide functionalization on the surface of graphene oxide (GO), which preserves thermally unstable groups in GO through the mild reaction with sodium azide in solids. Experimental evidence, by 15N solid-state NMR and other spectroscopic methods, indicates the substitution of organosulfate with azide anions as the reaction mechanism.
Graphene oxide (GO) is a nanomaterial that has attracted interdisciplinary attention for its broad applications across the fields of physics, chemistry and medicine.1–4 It can be prepared from graphite in large amounts and processed from solution.5 The treatment of GO by thermal methods or by reducing agents yields an electrically conductive graphene-based material.6 Furthermore, single layers of graphene with tunable defect density and electronic properties can be obtained from GO.7,8
Despite its great promise, chemical functionalization of GO is still in its early stage, and novel chemical tools for the modification of GO are demanded.9 For example, new molecular architectures of graphene- or GO-based materials are highly desired in the field of sensors or to alter the electronic properties of graphene.10,11 Among a variety of functionalization methods for graphene-based materials, the use of nitrogen containing reagents has attracted great attention. Hydrazine has been utilized to reduce GO to restore sp2 conjugation.12 Production of high quality N-doped graphene was achieved in recent years by growing graphene using chemical vapor deposition using ammonia as the doping source.11,13 N-doping within the carbon framework of graphene highly improves probing of organic molecules by graphene-enhanced Raman spectroscopy.13 Approaches using GO as a precursor for N-doping lead to graphene that is rather N-doped due to functionalization at edges of defects than within the carbon framework.14–16 These materials perform well for rewritable non-volatile memories or n-type field effect transistors through reactions with a dimethylformamide or amino-benzene moiety.15,16 Nevertheless, one general drawback for making functionalized GO with these relatively reactive nitrogen containing reagents is its limited thermal stability and the dynamic change of GO's chemical structure in aqueous media.17–19 Therefore, GO decomposes in water, and permanent defects are introduced in an uncontrolled manner by the cleavage of the carbon framework.19 Thus, chemical functionalization, while preserving the carbon backbone, is one of the major challenges for preparing new GO-based molecular architectures with a controlled structure. Hence, we recently altered the synthetic protocol for the preparation of GO yielding a type of GO that exhibits an almost preserved carbon skeleton as a consequence of preventing the evolution of CO2 during synthesis.7 This material is highly reducible and the highest charge carrier mobility values exceeding 1000 cm2 V−1 s−1 could be measured. Further on, we realized that purified GO bears organosulfate groups on the basal plane as part of its chemical structure, in addition to epoxy and hydroxyl groups as described earlier.20–23
With this new type of GO in hand, here we introduce a novel synthetic path to functionalize GO in a controlled manner through selectively functionalizing the basal plane of GO with azide. This product azide-functionalized GO (GO-N3) provides a versatile reactive motif for further chemical reactions such as “click” reactions or heterocycle formation.24 We also give insight into the reaction mechanism that involves substitution of organosulfate and a small number of oxides by azide groups (Scheme 1).20 Our study is based on complementary analysis including thermogravimetry coupled with mass spectrometry (TG-MS), solid-state NMR (SSNMR) or Fourier transform infrared spectroscopy (FTIR). Importantly, we report that introducing azide at low temperature allows one to functionalize GO while largely preventing the decomposition of the basic structural frame of GO unlike previously proposed chemical or thermochemical reactions to modify GO. Furthermore, cleavage of N2 from azide followed by N-insertion within the carbon σ-bonds is expected to be an entry to N-doped graphene. Opening this field may result in new materials for charge storage devices, selective sensors or n-type transistors of high performance.
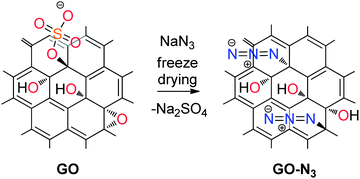 | ||
| Scheme 1 Illustration of the reaction of sodium azide with GO; substitution of organosulfate and assumedly epoxy groups by azide. | ||
GO was prepared by a modified Hummers' method that prevents the σ-framework of carbon atoms from rupture on the few nm scale.7 Moreover, organosulfate groups in addition to epoxy and hydroxyl groups remain stable in aqueous solution at least for months if stored at <10 °C.20 Thus, we find a sulfur content of about 5% by elemental analysis as part of the structure of GO. At first, we investigated the reaction of GO by FTIR spectroscopy on ZnSe windows. The amount of sodium azide added to a dispersion of GO was altered between 8% and 42%. As shown in Fig. S1A† we find two absorptions at 2123 cm−1 and 2065 cm−1 that originate from the stretching mode of chemically bound azide to GO and excess sodium azide, respectively. With increasing excess of sodium azide the absorption at 2065 cm−1 develops. With respect to these results we tried to isolate GO-N3 from solution by centrifugation. We purified the centrifuged material several times with water. Surprisingly, the final product did not show signals between 2000 and 2200 cm−1. From this experiment we concluded that the reaction does not proceed in solution but only in the solid state during drying of the dispersion on ZnSe used for FTIR spectroscopy. Because of that, a dispersion of GO and sodium azide was freeze-dried after mixing both using 0.8 mg sodium azide and 1 mg GO. After freeze-drying the material was purified by repeated centrifugation and redispersion in water and this time the reaction was successful. The final GO-N3 was freeze-dried again and the FTIR spectrum proves the azide functionalization as shown in Fig. 1A (black). Thus, we conclude that GO-N3 is hydrolytically stable at room-temperature. To further confirm the origin of the FTIR absorption at 2123 cm−1 we repeated the functionalization experiment using Na15N14N2 and found a 11 cm−1 shifted stretching vibration of azide at 2112 cm−1Fig. 1A (red).
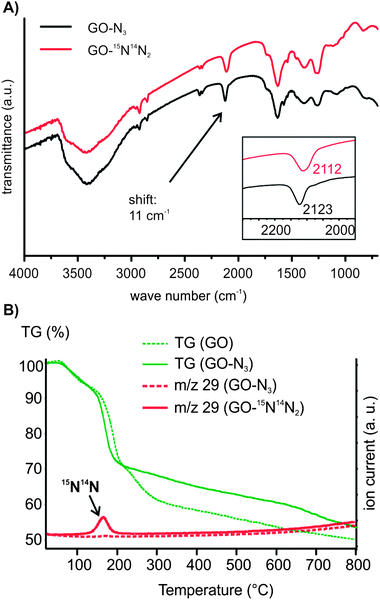 | ||
| Fig. 1 (A) FTIR spectra of GO-N3 and GO-15N14N2; (B) TGA curve of GO and GO-15N14N2, m/z 29 of GO-N3 and GO-15N14N2. | ||
To give further evidence of the controlled mild functionalization of GO with azide we used TG-MS analysis. As shown in Fig. 1B and S2† the main weight-loss step occurs between 150 °C and 200 °C. Water, CO, N2, and CO2 are the main decomposition products that originate from hydroxyl and epoxy groups for both materials. To differentiate between CO and N2 in GO-N3 we analyzed GO-15N14N2 as well and find a clear signal with m/z 29 for 15N14N formed between 150 and 200 °C. The total weigh-loss of GO-N3 and GO is about 30% up to 200 °C, for both materials. This is an indication for GO-N3 being highly functionalized with oxygen carrying groups in contrast to earlier reports.25 However, in contrast to GO-N3, the educt GO exhibits an additional weight-loss step above 200 °C that we recently assigned to the decomposition of organosulfate.20 This organosulfate can be identified in GO by the evolution of SO2 up to 300 °C during TG-MS analysis (m/z 64, Fig. S2†). TG-MS analysis for GO-N3 reveals no SO2 evolution.
These observations can be explained by the exchange of organosulfate with azide. To further prove this exchange we analyzed the supernatant after centrifugation of GO-N3 from the dispersed freeze dried GO–NaN3 reaction mixture. We allowed the supernatant to dry in air and dissolved the residue in 1 M HCl. To verify the existence of sulfate we added 0.1 M BaCl2, filtered the precipitate, washed with water and dried it at 160 °C. After that BaSO4 was identified by ATR-FTIR and the signature was compared to freshly precipitated BaSO4 prepared from Na2SO4 and BaCl2 (Fig. S3†). Elemental analyses of GO and GO-N3 give insight into the proceeded reaction. On the one hand, the nitrogen content increases from 0.0% to 4.1% and on the other hand the sulfur content is reduced from 3.6% for GO to 1.7% for GO-N3. The residual sulfur may originate from inorganic sulphate to some extent and is indicated by the weight-loss detected at about 700 °C (Fig. 2B). Using these data we estimate that there is about one azide group on 30 C-Atoms in GO-N3.
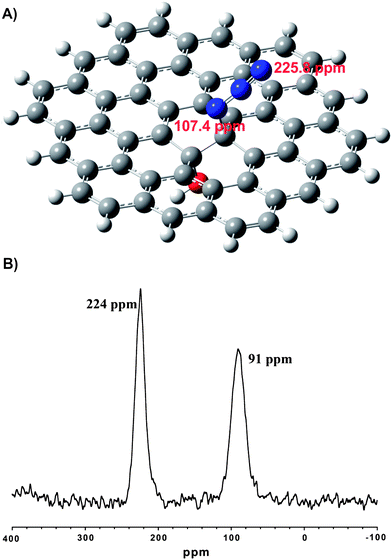 | ||
Fig. 2 (A) Simplified model of GO-N3 with an azide and a hydroxyl group connected to the carbon lattice in trans-configuration, calculated by ab initio methods to predict 15N NMR shifts (107.4 ppm and 225.8 ppm); (B) 15N SSNMR MAS spectrum of GO-15N14N2 with two peaks (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio). :1 ratio). | ||
To find further evidence for the azide functionalization of GO we conducted 15N SSNMR analysis. This technique was effectively used to evaluate the structural properties of reduced GO that was obtained by the reaction of 15N-labelled hydrazine and 13C-labeled GO.26Fig. 2 illustrates (B) a 15N MAS SSNMR spectrum for GO-N3 prepared from Na15N14N2 and GO with (A) a simplified structural model of GO-N3. The model shows, besides aromatic regions, a hydroxyl group adjacent to an azide group (trans-configuration). This model was obtained by geometry optimization using ab initio calculations and used to simulate the resonance frequency of 15N SSNMR signals obtained for GO-15N14N2. The ab initio geometry optimized structure showed no signs of instability. Since 15N in 15N14N2− can be bound to GO in two configurations (C–15N–14N14N or C–14N–14N–15N), two signals are expected if 15N14N2 is chemically bound to GO. Consequently, two signals with an integral intensity ratio of 1:1.05 are obtained in the 15N SSNMR spectrum (Fig. 2B) for the two possible bounding configurations. No other peaks were experimentally confirmed. Furthermore, the 15N chemical shifts at 224 ppm and 91 ppm are consistent with 15N shifts predicted by the ab initio calculations for the azide that is bound in end-on configuration (226 ppm) or with 15N being connected to the sp3 carbon atom (107 ppm). These results are an excellent indication for the controlled functionalization of GO with azide without formation of any other nitrogen containing species as a major impurity incorporated into N3-GO.
All the analytical results indicate that a large amount of organosulfate becomes accessible for azide to be substituted (Scheme 1). Since azide is a good anionic nucleophile and anionic organosulfate is present on both sides of GO's carbon backbone, a reaction in solution is hindered by steric effects and especially due to electrostatic repulsion. Thus, this is an explanation why the reaction of sodium azide and GO was conducted at high temperature and why long reaction times had been necessary causing significant decomposition as reported by Salvio et al.25 Furthermore, Salvio et al. proposed “click” reactions, which however proceed rather at edges than on the surface of GO. Such chemistry is not effective to develop GO based materials with minimal defects. Whereas, in the solid state the situation is different and since azide is adsorbed on the surface of GO the local azide concentration is very high and therefore, organosulfate can now react as a leaving group assuming a nucleophilic reaction. Besides, we assume that few epoxy groups can act as reaction partners as well, as indicated in Scheme 1. Finally, we tested the thermal stability of GO-N3 both in solids and in aqueous dispersion (Fig. S4†). In solids we find that degradation of GO-N3 starts at about 80 °C. The degradation is indicated by CO2 formation and the CO2 is trapped between layers of the GO-N3 film. This trapped CO2 is analytically accessible for FTIR analysis (Fig. S4A†).18 At higher temperature azide bound to GO degrades in agreement with TG-MS analysis (Fig. 1B). Next, GO-N3 was treated for 30 min at a certain temperature with water. We find that azide in GO-N3 is cleaved in aqueous dispersion starting at 80 °C and especially at 100 °C, as indicated by the FTIR vibration at 2058 cm−1 (Fig. S4B†). After 16 h of treatment little degradation is indicated for the 60 °C sample, and for the 80 °C sample decomposition is detected (Fig. S5†). These results reveal that GO-N3 is hydrolytically stable in a range between room temperature and 60 °C, which has been utilized in azide-based chemistry such as “click” reactions. Thus, thermal activation can be used when adopting GO-N3 as a precursor for other reactions as long as attention is paid to the compound stability.
Conclusions
We successfully established the direct functionalization of GO with N3 preserving functional groups in GO for the first time. Our analysis is based on FTIR, TG-MS, EA and 15N SSNMR to prove the successful functionalization of GO using reactions at low temperature in solids. This new method preserves thermally unstable groups present in GO as indicated by TG-MS analysis. Moreover, azide is predominately located on the surface of GO and not at edges. Although edge structures of GO involving chemical species such as carbonyl and carboxyl groups have been reported,27 the possibility of the reactions of such edge species with azide will be examined in our future studies. Based on our findings we estimate approximately one azide group on 30 carbon atoms. Further on, the azide group in GO-N3 is hydrolytically stable up to 60 °C and therefore we believe that GO-N3 is a suitable precursor for other reactions, opening an avenue for the field of azide chemistry for graphene-based materials. Further development will lead to n-doped graphene, amine functionalized graphene after reduction or positively charged graphene after quarterisation. Besides, “click” reactions and Staudinger reactions become feasible to generate molecular architectures while the basic structure of GO is preserved. Further development of mild reactions with GO is highly demanded to find an access to highly conductive and functionalized graphene. Such new structures may e.g. make new sensor materials possible based on GO-N3 that selectively bind substrates and thus potentially enhance the field of medical applications. Another possible research field may be the development of n-type transistors with enhanced charge carrier mobility values.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG-SFB 953, Project A1 “Synthetic Carbon Allotropes”), the European Research Council (ERC; grant 246622 – GRAPHENOCHEM), and the Cluster of Excellence ‘Engineering of Advanced Materials (EAM)’ for financial support. The solid-state NMR analysis was supported by funding from the U.S. National Science Foundation (NSF; CHE 957793, CHE 1310363) for YI. The purchase of the NMR instrument used in this study was, in part, supported by the NSF (CHE 957793), the U.S. Department of Energy Basic Sciences (DE-SC001951), and the U.S. National Institutes of Health (GM098033).Notes and references
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- C. Chung, Y. K. Kim, D. Shin, S. R. Ryoo, B. H. Hong and D. H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- H. Y. Mao, S. Laurent, W. Chen, O. Akhavan, M. Imani, A. A. Ashkarran and M. Mahmoudi, Chem. Rev., 2013, 113, 3407–3424 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- S. Eigler, M. Enzelberger-Heim, S. Grimm, P. Hofmann, W. Kroener, A. Geworski, C. Dotzer, M. Rockert, J. Xiao, C. Papp, O. Lytken, H. P. Steinruck, P. Muller and A. Hirsch, Adv. Mater., 2013, 25, 3583–3587 CrossRef CAS PubMed.
- S. Eigler, S. Grimm, M. Enzelberger-Heim, P. Muller and A. Hirsch, Chem. Commun., 2013, 49, 7391–7393 RSC.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Biosens. Bioelectron., 2011, 26, 4637–4648 CrossRef CAS PubMed.
- L. Zhao, R. He, K. T. Rim, T. Schiros, K. S. Kim, H. Zhou, C. Gutierrez, S. P. Chockalingam, C. J. Arguello, L. Palova, D. Nordlund, M. S. Hybertsen, D. R. Reichman, T. F. Heinz, P. Kim, A. Pinczuk, G. W. Flynn and A. N. Pasupathy, Science, 2011, 333, 999–1003 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- R. Lv, Q. Li, A. R. Botello-Mendez, T. Hayashi, B. Wang, A. Berkdemir, Q. Hao, A. L. Elias, R. Cruz-Silva, H. R. Gutierrez, Y. A. Kim, H. Muramatsu, J. Zhu, M. Endo, H. Terrones, J. C. Charlier, M. Pan and M. Terrones, Sci. Rep., 2012, 2, 586 Search PubMed.
- T. Palaniselvam, H. B. Aiyappa and S. Kurungot, J. Mater. Chem., 2012, 22, 23799–23805 RSC.
- S. Seo, Y. Yoon, J. Lee, Y. Park and H. Lee, ACS Nano, 2013, 7, 3607–3615 CrossRef CAS PubMed.
- D. W. Chang, E. K. Lee, E. Y. Park, H. Yu, H. J. Choi, I. Y. Jeon, G. J. Sohn, D. Shin, N. Park, J. H. Oh, L. Dai and J. B. Baek, J. Am. Chem. Soc., 2013, 135, 8981–8988 CrossRef CAS PubMed.
- S. Kim, S. Zhou, Y. Hu, M. Acik, Y. J. Chabal, C. Berger, W. de Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed.
- S. Eigler, C. Dotzer, A. Hirsch, M. Enzelberger and P. Müller, Chem. Mater., 2012, 24, 1276–1282 CrossRef CAS.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- S. Eigler, C. Dotzer, F. Hof, W. Bauer and A. Hirsch, Chem.–Eur. J., 2013, 19, 9490–9496 CrossRef CAS PubMed.
- A. Lerf, H. Heb, T. Riedl, M. Forster and J. Klinowskib, Solid State Ionics, 1997, 101–103, 857–862 CrossRef CAS.
- W. Cai, R. D. Pine, F. Stadermann, S. Park, M. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. M. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1818 CrossRef CAS PubMed.
- L. B. Casabianca, M. Shaibat, W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- R. Salvio, S. Krabbenborg, W. J. Naber, A. H. Velders, D. N. Reinhoudt and W. G. van der Wiel, Chem.–Eur. J., 2009, 15, 8235–8240 CrossRef CAS PubMed.
- S. Park, Y. Hu, J. O. Hwang, E.-S. Lee, L. B. Casabianca, W. Cai, J. R. Potts, H.-W. Ha, S. Chen, J. Oh, S. O. Kim, Y.-H. Kim, Y. Ishii and R. S. Ruoff, Nat. Commun., 2012, 3, 638 CrossRef PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General methods, solid-state NMR analysis, ab initio calculations, preparation procedures, FTIR analysis of GO films on ZnSe, and detailed TG-MS analysis. See DOI: 10.1039/c3nr04332k |
| This journal is © The Royal Society of Chemistry 2013 |