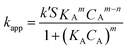DOI:
10.1039/C3NR03521B
(Paper)
Nanoscale, 2013,
5, 11919-11927
Revisiting catalytic model reaction p-nitrophenol/NaBH4 using metallic nanoparticles coated on polymeric spheres
Received
9th July 2013
, Accepted 17th August 2013
First published on 20th August 2013
Abstract
The early reported pseudo-first-order reaction kinetics of the polymer-supported metallic nanocatalysts for the model reaction of p-nitrophenol (p-NP)/NaBH4 were probably oversimplified. Here a detailed study of p-NP reduction by NaBH4 in the presence of the raspberry-like poly(allylamine hydrochloride)-modified polymer poly(glycidyl methacrylate) composite sub-microspheres with tunable gold nanoparticles (PGMA@PAH@AuNPs) was presented. Effects of polyelectrolyte concentration, the ratio of polymer spheres to gold nanoparticles, and the solution pH value for composite synthesis on the induction period, reaction time, average reaction rate and average turnover frequency were systematically investigated. Experimental results in all cases of our study revealed an nth order (n > 1) of the p-NP/NaBH4 catalytic reaction by the prepared polymer composite particles. The apparent order of reaction, n, is dependent on the total surface area of the coated gold nanoparticles on the polymer spheres, which can be closely correlated with the tunable gold nanoparticle surface coverage. The mechanism of the observed catalytic activity enhancement was proposed based on active epoxy groups of the polymer spheres and a large adsorption of p-nitrophenolate anions onto the positively-charged spheres.
1. Introduction
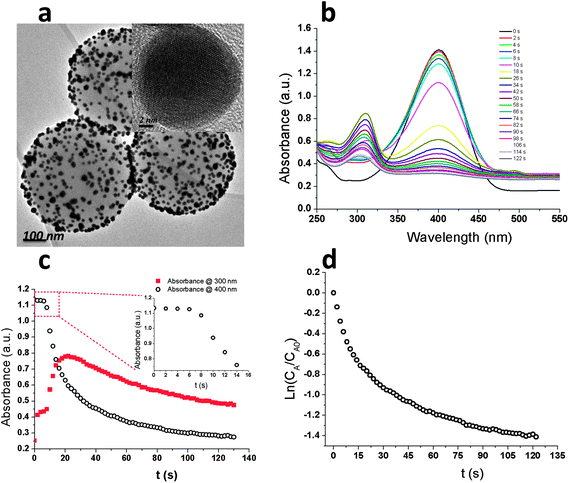
Catalytic reduction of p-nitrophenol by borohydride ions (BH4−) in the presence of a metal catalyst has become one of the model reactions for evaluating the catalytic activity of noble metallic nanoparticles.1–12 Although the reaction is thermodynamically feasible, it is kinetically restricted in the absence of a catalyst because the kinetic barrier between the mutually repelling negative ions p-NP and BH4− is very high.13 According to the traditional theory about the catalytic reduction of p-NP by metallic nanoparticles, electron transfer takes place from BH4− to p-NP through adsorption of reactant molecules onto the metallic nanoparticle surface. Metallic nanoparticles relay electrons to complete the redox reaction. The adsorption of reactant ions onto the particle surface contributes to overcoming the kinetic barrier of the reaction. Previous work demonstrated this model reaction is (pseudo-) first order in the concentration of p-NP (CA) when an excess of sodium borohydride is used.1,5,7,8,11,12 The rate of consumption of p-NP, rt, was often defined as, rt = −dCA/dt = kappCA = kSCA, where kapp refers to the apparent rate constant, k is the normalized rate constant with respect to the surface area (S) available on the nanoparticles responsible for catalytic activity. The correlation of Ln(CA/CA0) versus the reduction time t was estimated to be linear. However a deviation from the (pseudo-) first-order kinetics can be found if the published data are carefully studied.7–10 Some research work has already pointed out that the first-order kinetics could be changed by the metallic nanoparticle's supporting materials.14–16
Metal nanoparticles are important catalysts for oil refining and petrochemical industries, energy conversion and vehicle exhaust gas removal, since they can catalyze many important transformations including oxidation of hydrocarbons, C–C cross coupling, electron-transfer reaction, and hydrogenation–dehydrogenation.17–21 Research into noble metal nanoparticles as the catalyst has remained an active topic. However, their high surface energy due to the large surface-to-volume ratio and high collision frequency associated with their greater mobility cause serious stability problems, such as a tendency to aggregate, changes in shape and damage to their surface states during catalytic reactions, and eventual loss of their initial activity and selectivity. Therefore, it is necessary to load the noble metal nanoparticles onto the surface of supporting materials since such formed catalysts possess the advantages offered by the characteristics of heterogeneous catalysis (recovery and recyclability), and those of homogeneous catalysis (relatively low catalyst loadings and good selectivity). Recently in our research group, a new type of gold nanoparticle-coated composite sphere, based on chemically reactive poly(glycidyl methacrylate) (PGMA) colloids at sub-micrometer scale was successfully prepared in a raspberry-like fashion by an easy and facile controlled assembly method.22 These composites exhibited excellent catalytic activity, stability and recyclability. The catalytic activity of the composites was evaluated based on the catalytic reduction of p-NP with an excess amount of sodium borohydride (NaBH4). In the kinetic studies of the as-prepared composite catalyst, we found that the catalytic reduction of p-nitrophenol by metallic nanoparticles coated on PGMA spheres does not obey the first-order reaction law, in which the reaction rate does not depend linearly on the concentration of p-NP. The redox reaction kinetics of the model system p-NP/NaBH4 was changed probably by the supporting substrates, PGMA polymer spheres, which is worthwhile to be further investigated.
Here systematic kinetic studies were performed by reduction of p-NP with NaBH4 in the presence of raspberry-like PGMA@PAH@AuNP composites with different gold nanoparticle surface coverage, which could be tuned by the polyelectrolyte poly(allylamine hydrochloride) (PAH) concentration, the ratio of PGMA spheres to gold nanoparticles, and the solution pH value for the composite synthesis. The acquired kinetic data revealed an excellent catalytic activity of the synthesized composite catalysts. The mechanism for the enhancement in the catalytic activity was proposed based on active epoxy groups and positive electric charge of the modified PGMA spheres. The surface-controlled reaction was modeled following a monomolecular mechanism and Langmuir–Freundlich isotherm. The nth-order (n > 1) kinetic equation was obtained which was used to quantitatively fit the curves of time-dependent p-NP concentrations. The apparent reaction order, n, was correlated with the surface coverage of gold nanoparticles coated on the PGMA spheres.
2. Experimental section
2.1. Preparation of PAH-modified PGMA sub-microspheres (PGMA@PAH)
The PGMA@PAH sub-microspheres were synthesized in a way similar to our previous work.22 Briefly, 15 mL of inhibitor-free glycidyl methacrylate (GMA) (≥97.0%, Sigma-Aldrich, St Louis, MO, USA) was mixed in 150 mL of Nanopure water purged with N2 by stirring vigorously at 1200 rpm for 30 min at room temperature. The mixture was refluxed and heated to 90 °C following the dropwise addition of 10mL of (5% w/v) of potassium persulfate (KPS) (99+%, Sigma-Aldrich, St Louis, MO, USA). To avoid stripping of the monomer from the reaction mixture, the flow of nitrogen can be decreased through the nitrogen tank pressure regulator. The stirring rate of 1200 rpm and the temperature of 90 °C were kept constant until the end of the reaction. After 2 h of polymerization, the reaction mixture was stopped by bubbling oxygen for 30 min. Upon the completion of the reaction, the product was allowed to cool and dialyzed in a Cellulose Ester Dialysis membrane (MWCO 3500–5000 D, Spectrum Laboratory, Inc, Rancho Dominguez, CA) for 24 h and centrifuged at 5000 rpm (Eppendorf centrifuge 5804, VWR International, Radnor, PA, USA) for 30 min. After three successive centrifugation/wash cycles with Nanopure water, a certain amount of purified PGMA was obtained before 10 mL of it was mixed with 1 mL poly(allylamine hydrochloride) at different concentrations (Mw 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich, St Louis, MO, USA). The mixture was further dialyzed in a Cellulose Ester Dialysis membrane (MWCO 20000 D) for 24 h to remove the non-adsorbed PAH. The PAH-modified PGMA cores were separated from the supernatant by centrifugation for 30 min at 5000 rpm followed by cleaning with 11 mL Nanopure water. The PGMA@PAH mixture was obtained after three centrifugation/cleaning cycles and stored in a refrigerator prior to use.
000, Sigma-Aldrich, St Louis, MO, USA). The mixture was further dialyzed in a Cellulose Ester Dialysis membrane (MWCO 20000 D) for 24 h to remove the non-adsorbed PAH. The PAH-modified PGMA cores were separated from the supernatant by centrifugation for 30 min at 5000 rpm followed by cleaning with 11 mL Nanopure water. The PGMA@PAH mixture was obtained after three centrifugation/cleaning cycles and stored in a refrigerator prior to use.
2.2. Formation of raspberry-like PGMA@PAH@AuNP composites
Gold nanoparticles (AuNPs) (12 ± 3 nm) were prepared by reduction of HAuCl4·3H2O using trisodium citrate that acts as a reductant according to the Turkevich method.23 All glasswares were cleaned in aqua regia solution for 30 min prior to use. In a typical procedure, 250 mL of 1 mM HAuCl4·3H2O was brought to boiling on a IKA® RCT DIGITAL Basic Magnetic Hot Plate Stirrer (Chemglass Life Sciences, Vineland, NJ, USA). Under 500 rpm stirring, 25 mL of 38.3 mM of trisodium citrate was added. The reaction was allowed to run until the solution reached a ruby/red color and cooled to room temperature. A certain required amount of PGMA@PAH dispersion solution was suspended in 3 mL pristine as-synthesized AuNP solution for saturated adsorption of AuNPs through interactions between AuNPs and PGMA@PAH with opposite charges to form PGMA@PAH@AuNP composites in a raspberry-like fashion. To study the pH effect on the gold nanoparticle coverage, the PGMA@PAH dispersion solution was prepared by combining 10 mL of purified PGMA spheres with 1 mL of 0.5% (w/v) PAH solution followed by three centrifugation/wash cycles. The centrifuged PGMA@PAH sphere precipitates were then dispersed in 11 mL Nanopure water with different pHs (pH = 3, 7 and 11). The raspberry-like PGMA@PAH@AuNP composites were formed by adding 60 μL of PGMA@PAH dispersion to 3 mL of the as-synthesized AuNP solution. All samples were mixed by gentle shaking at around 1000 rpm for 30 min on a SH 2000 Finemixer shaker (Daigger, Vernon Hills, IL, USA). The PGMA@PAH@AuNP composites were collected after centrifugation and re-dispersed in Nanopure water.
2.3. Catalytic activity
The catalytic reactions were conducted by mixing 1.25 mL p-nitrophenol aqueous solution (0.1 mM) with 0.5 mL freshly prepared NaBH4 aqueous solution (0.1 M) in a 3 mL quartz cuvette under continuous stirring at 295 K. Subsequently, 0.4 mL of PGMA@PAH@AuNP composites were injected to the above reaction mixture to start the reduction. The reduction in the optical absorption peak of p-NP was determined from the UV-vis spectrum using a UV-vis spectrophotometer. The kinetics of the reaction was studied by following the optical absorption peak of p-NP at 400 nm with 2 second intervals using Agilent UV-vis Biochemical Analysis Software. To study the effect of PAH, at which the composite was prepared, on the catalytic activity of the composite particles, the composites were prepared at pH 7 with the fixed ratio of PGMA@PAH to AuNPs of 1:75 (v/v) at different PAH concentrations. In addition, to determine the effect of pH for the systems of PGMA@PAH@AuNPs on the catalytic activity of composites, the composite particles were prepared with a fixed PAH-modified (0.5% w/v) ratio of PGMA@PAH to AuNPs of 1:200 (v/v), and to investigate the effect of the volume ratio of PGMA to AuNPs on the catalytic activity of composites, the catalysts were prepared at pH 7 with 0.1% PAH (w/v) by varying the amount of PGMA@PAH while keeping the volume of AuNPs constant.
2.4. Characterization
A JEM-1400 transmission electron microscope (TEM) (JEOL, Peabody, MA, USA) at an accelerating voltage of 120 kV and a JEOL JEM2100F high-resolution analytical transmission electron microscope (HRTEM) (JEOL, Peabody, MA, USA) at an accelerating voltage of 200 kV were used to characterize the size and morphology of the raspberry-like particles. All samples were prepared by placing 50 μL of the sample solution on a carbon film coated copper grid (Electron Microscope Sciences, Hatfield, PA, USA) upon drying at room temperature. All UV-vis spectra were recorded using an Agilent 8453 UV-visible diode array spectrophotometer (Agilent Technologies, Santa Clara, CA, USA).
3. Results and discussion
3.1. Catalytic activity test
The catalytic reduction of 0.125 μmol p-nitrophenol to p-aminophenol (p-AP) in the presence of an excess amount of NaBH4 was chosen to investigate the catalytic activity of PGMA@PAH@AuNP composites with mild stirring at ambient conditions. As shown in Fig. 1a, PGMA spheres with a diameter of 484 nm were coated with gold nanoparticles of 12 ± 3 nm in diameter in a raspberry-like fashion. The crystallinity of the gold nanoparticles was checked by high-resolution TEM (the inset in Fig. 1a). The lattice plane spacing was 0.204 nm, which was indexed to the (200) plane of the face-centered-cubic (fcc) crystal structure of gold. The progress of the reaction was monitored by UV-Vis spectroscopy with 2 second interval. Fig. 1b shows the typical UV-vis spectra of absorbance change of p-nitrophenol in the presence of NaBH4 and PGMA@PAH@AuNP composites. Without addition of PGMA@PAH@AuNP composites, it was observed that the plasmon absorption band of p-nitrophenol (λmax = 317 nm) is red shifted to 400 nm, which is ascribed to the formation of p-nitrophenolate ions under alkaline conditions by the introduction of NaBH4, as is indicated by the color change from light yellow to dark yellow after addition of NaBH4. The intensity of the peak remained unaltered at 400 nm with time, which signified that no reduction occurred. However, in the presence of PGMA@PAH@AuNP composites, the dark yellow color of p-nitrophenol solution vanished quickly, indicated by the fast intensity decrease in the absorbance of 400 nm. Concomitantly, a new peak appeared at 300 nm, and increased with reduction time, which is assigned to p-aminophenol, confirming the reduction p-NP to p-AP. It is worth mentioning that the loss of isosbestic points in Fig. 1b is due to gas bubbles of H2 formed during the reaction. The black line shows that the reaction did not happen when no PGMA@PAH@AuNP composites were added. After introduction of PGMA@PAH@AuNP composites, the presence of gas bubbles shifted the UV-vis spectra to approximately 0.17 a.u. in absorbance as shown in Fig. 1b.24Fig. 1c shows the real-time changes in the absorbance of both the reactant p-NP and the product p-AP in the case of composite particles containing 4.4357 × 10−12 mol of AuNPs and 0.1494 mg of PGMA. There is no change in the absorbance of p-NP within the first 6 s, which we define as the induction period (t0). During this period, the composites do not seem to participate in the reduction reaction, and the catalysis reaction is paralyzed until the induction period is finished. After induction period, the reduction starts and the absorbance of p-nitrophenol decreases at a fast speed. After 20 seconds, the reduction of p-nitrophenol goes through a maximum, then the extinction of absorbance of p-NP becomes slower. Finally the absorbance levels off, which indicates the end of the reduction reaction. The overall reaction proceeds within 128 s. Fig. 1d shows the plot of Ln(CA/CA0) (CA: the concentration of p-NP at the reaction time t, which does not include t0, CA0: the original concentration of p-NP at 0 s) as a function of reduction time t for p-nitrophenol. A non-linear relationship between Ln(CA/CA0) and reduction time t clearly shows that the reduction of p-NP by our raspberry-like PGMA@PAH@AuNP composites does not follow a first-order reaction mechanism. In earlier reports, p-NP reduction using noble metal nanoparticles was shown to be pseudo-first-order in the presence of homogeneous25,26 and heterogeneous catalysts.27–33 However, in all cases of our study, a deviation from the pseudo-first-order kinetics was observed. Therefore, we propose that the reaction follows the nth-order-reaction mechanism (n > 1). The kinetic parameters for p-NP reduction were obtained by best fitting the curve CAvs. t, which will be discussed below.
 |
| | Fig. 1 Catalytic reduction of p-nitrophenol to p-aminophenol by an excess amount of NaBH4 in the presence of PGMA@PAH@AuNP composites prepared at a fixed ratio of PAH (0.5% w/v)-modified PGMA to AuNPs of 1:75 (v/v) and pH of 7. (a) TEM image of the as-prepared composite particles, the inset graph shows the HRTEM image of a gold nanoparticle. (b) Time-dependent UV-vis absorption spectra for the reduction of p-nitrophenol; (c) Absorbance vs. time plot of reduction of p-nitrophenol at 400 nm to p-aminophenol at 300 nm, the inset graph depicts absorbance of p-nitrophenol vs. induction period (t0); (d) Plot of Ln(CA/CA0) as a function of time for the reduction of p-nitrophenol. Reaction conditions: p-nitrophenol = 0.125 μmol, NaBH4 = 50 μmol, AuNPs = 4.4357 × 10−12 mol, T = 295 K. | |
3.2. Kinetic modeling
With an excess of BH4−, the catalytic reduction reaction occurred at the surface of catalytic nanoparticles following a monomolecular mechanism.
The reactant A (p-NP) interacted with the active sites on the catalyst surface C (the PGMA@PAH@AuNP composite) to form the adsorbed species AC, which underwent reaction to form the final product P (p-AP). The reaction rate rt is dependent on the fraction of catalyst surfaces covered by the substrate, θA, as described by the Langmuir–Freundlich isotherm.
| | 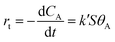 | (2) |
| | 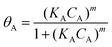 | (3) |
K
A is the adsorption constant of the p-NP, m is the exponent related to the heterogeneity of the sorbent.34 Combining eqn (2) and (3), the reaction rate can be expressed as follows:
| | 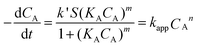 | (4) |
where
n refers to the apparent order of reaction with respect to the
p-NP, and
kapp can be defined as:
When n ≠ 1, the definite integration of the differential equation (eqn (4)) yields eqn (5), where CA0 is the initial concentration of the reactant p-NP.
| | | C1−nA = (n − 1)kappt + C1−nA0 | (5) |
Importantly, eqn (5) can be used to quantitatively fit the curve CAvs. t data, then the apparent reaction order, n, and the apparent rate constant, kapp, can be obtained.
3.3. Effect of PAH on the composite catalytic activity
Our previous studies have demonstrated that the amount of gold nanoparticle coverage on polymer spheres is related to the morphology of polyelectrolyte chains on the PGMA spheres.22 At low concentration of PAH polyelectrolyte, the negatively charged PGMA spheres compensate the positively charged PAH polyelectrolyte, leaving PGMA@PAH more neutralized, therefore, fewer negatively charged AuNPs are captured upon mixing PGMA@PAH with AuNPs. With the increase of PAH concentration, more AuNPs will be adsorbed on PGMA@PAH spheres. However, with a further increase of PAH concentration, the surface coverage of gold nanoparticles is decreased due to the pearl necklace structure of PAH chains with low surface charge density. Table 1 shows a similar result that the amount of AuNP coverage increases first in the range of 0.1–1% (w/v) PAH concentrations with a maximum gold nanoparticle coating at 0.75% (w/v) PAH concentration, and then decreases. The rate of reduction is directly proportional to the available surface area responsible for catalysis and the amount of catalyst.35 This assumption could be explained that a more vacant surface area of the catalyst available for electron transfer to take place, and more chances of diffusion of reactant molecules onto the AuNP surface lead to the higher catalytic efficiency. As shown in Table 1, the overall p-NP reduction reaction completed in 82 s in the case of 0.75% (w/v) PAH with the highest average reaction rate of 1.52 × 10−9 mol s−1 and the highest average TOF of 281 mol mol−1 s−1. However, the reduction time to full conversion of 0.125 μmol p-NP into p-AP for 0.1% (w/v) PAH and 0.5% (w/v) PAH is 126 s and 122 s, respectively, which leads to 9.92 × 10−10 and 1.02 × 10−9 mol s−1 for the average reaction rate, and 230.6 and 231 mol mol−1 s−1 for the average turnover efficiency (TOF) respectively. The catalytic activity can also be confirmed by the apparent order of reaction, n and the apparent rate constant, kapp, for the reduction reaction. By fitting the time dependence of the p-NP concentration curves using eqn (5) derived from the kinetic modeling, the apparent order of reaction, n, and the apparent rate constant, kapp, were obtained (Fig. 2b). The apparent reaction order of 5.16 was obtained for composites with the highest gold nanoparticle coverage at 0.75% (w/v) PAH concentration. But for composites with the lowest gold nanoparticle coverage at 1% (w/v) PAH concentration, the apparent reaction order was only 3.27.
Table 1 Kinetic parameters for composite particles at different PAH concentrations with a volume ratio of PGMA:AuNPs = 1:75 and pH = 7
| Composite particles |
AuNP coverage amount (moles) |
Reaction Time (s) |
R
ave
(mol s−1) |
TOFaveb (mol mol−1 s−1) |
t
0 (s) |
n
|
k
app
|
Correlation coefficient (R) |
|
R
ave is defined by the initial moles of p-NP over the overall reaction time.
TOFave is the average turnover frequency defined by the ratio of mole of p-NP per mole of AuNPs per second when p-NP reached full (100%) conversion.
n is defined by the apparent order of reaction.
k
app is defined by the apparent rate constant.
|
| 0.1% PAH |
4.3021 × 10−12 |
126 |
9.92 × 10−10 |
230.6 |
6 |
3.8 |
60.68 |
0.99227 |
| 0.5% PAH |
4.4357 × 10−12 |
122 |
1.02 × 10−9 |
231 |
6 |
3.71 |
49.87 |
0.99497 |
| 0.75% PAH |
5.4241 × 10−12 |
82 |
1.52 × 10−9 |
281 |
5 |
5.16 |
14100 |
0.99156 |
| 1% PAH |
3.3595 × 10−12 |
146 |
8.56 × 10−10 |
244.7 |
24 |
3.27 |
10.77 |
0.99224 |
 |
| | Fig. 2 (a) Plots of the concentration of p-NP versus time for the reduction of p-NP by NaBH4 in the presence of PGMA@PAH@AuNP composites at different PAH concentrations with a volume ratio of PGMA:AuNPs = 1:75. PAH concentration = 0.1% (w/v) ( ); 0.5% (w/v) ( ); 0.5% (w/v) ( ); 0.75% (w/v) ( ); 0.75% (w/v) ( ); 1% (w/v) ( ); 1% (w/v) ( ). The inset graph shows the plots of the concentration of p-NP versus induction period (t0). (b) Plots of the concentration of p-NP versus time (t0 is not included) for the reduction of p-NP by NaBH4 in the presence of PGMA@PAH@AuNP composites at different PAH concentrations with fitting curves. PAH concentration = 0.1% (w/v) ( ). The inset graph shows the plots of the concentration of p-NP versus induction period (t0). (b) Plots of the concentration of p-NP versus time (t0 is not included) for the reduction of p-NP by NaBH4 in the presence of PGMA@PAH@AuNP composites at different PAH concentrations with fitting curves. PAH concentration = 0.1% (w/v) ( ); 0.5% (w/v) ( ); 0.5% (w/v) ( ); 0.75% (w/v) ( ); 0.75% (w/v) ( ); 1% (w/v) ( ); 1% (w/v) ( ). ). | |
The rate constant for composites at 0.75% (w/v) PAH concentration is tremendously higher with a value of 1.41 × 104 M−4.16 s−1 compared to 10.77 M−2.27 s−1 for 1% (w/v) PAH concentration. When composites have comparable gold nanoparticle coverage in the case of 0.1% and 0.5% (w/v) PAH, the apparent reaction orders and the rate constants are close to each other as well, which are 3.8 vs. 3.71, and 60.68 M−2.8 s−1vs. 49.87 M−2.71 s−1 respectively (seen in Table 1). Another observation is that the induction period, t0, is also changed with the PAH concentration (the inset in Fig. 2a). The timescale on the order of seconds for t0 (5 to 24 s) may be caused by processes related to a substrate-induced surface reconstruction that is necessary to render the particles an active catalyst.24
3.4. Effect of the volume ratio of polymer spheres to AuNPs on the composite catalytic activity
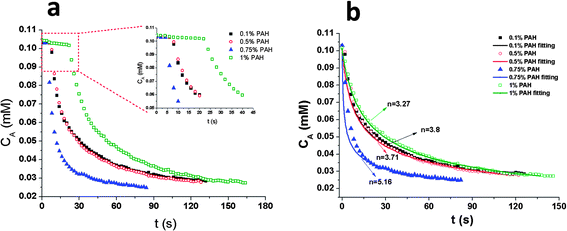
To investigate the effect of the volume ratio of polymer spheres to AuNPs on the composites' catalytic activity, we varied the volume ratio of polymer spheres to gold nanoparticles from 0.5:200 to 2:200, and kept the volume of gold nanoparticles constant but in an excess amount to ensure saturated adsorption of PAH-modified PGMA spheres with gold nanoparticles. The results are summarized in Table 2. An increase in the volume ratio of PGMA spheres to gold nanoparticles from 0.5:200 to 2:200 leads to an increase in the gold nanoparticle adsorption amount from 1.4772 × 10−12 to 2.4373 × 10−12 moles, a decrease in reaction time from 604 to 76 s, and an increase in the average reaction rate from 2.07 × 10−10 to 1.64 × 10−9 mol s−1. However, the average TOF is almost quadrupled from 140.1 to 674.8 mol mol−1 s−1 when the volume ratio of PGMA spheres to AuNPs is increased from 0.5:200 to 2:200. Reactants are vying for the available reactive sites on the surface of gold nanoparticles to form the surface-bound species until the reduction starts. If more reactive sites are vacant, more reactants are free to attach, the reaction will be expedited greatly.
Table 2 Kinetic parameters for composite particles at 0.1% PAH (w/v) concentration with different volume ratios of PGMA:AuNPs at pH 7
| Composite particles |
AuNP coverage amount (moles) |
Reaction time (s) |
R
ave (mol s−1) |
TOFave (mol mol−1 s−1) |
T
0 (s) |
n
|
k
app
|
Correlation coefficient (R) |
| 0.5:200 |
1.4772 × 10−12 |
604 |
2.07 × 10−10 |
140.1 |
22 |
1.60 |
1.97 × 10−2 |
0.99702 |
| 1:200 |
2.2782 × 10−12 |
174 |
7.18 × 10−10 |
315.2 |
8 |
2.47 |
1.38 |
0.99721 |
| 2:200 |
2.4373 × 10−12 |
76 |
1.64 × 10−9 |
674.8 |
2 |
4.31 |
2.46 × 103 |
0.98811 |
As discussed above, the reduction occurred only after the induction period t0. The t0 is increased from 2 s to 22 s when the volume ratio of PGMA to AuNPs decreased from 2:200 to 0.5:200 (the inset in Fig. 3a), which can be related to the catalysis-induced surface restructuring.36 The enhancement in catalytic activity is further proved by the apparent order of reaction, n and the apparent reaction rate, kapp. Both n and kapp increase with increasing the volume ratio of PGMA to AuNPs. The n for the composites with the volume ratio of PGMA to gold nanoparticles of 2:200 is 4.3, while the n is 1.6 for the composites with the volume ratio of PGMA to gold nanoparticles of 0.5:200. The apparent reaction constant is increasing exponentially as the volume ratio of PGMA to gold nanoparticles is increased. The kapp for the composites with the volume ratio of PGMA to gold nanoparticles 2:200 is 2.46 × 103 M−3.31 s−1, which is 1783 times larger than that with the volume ratio of PGMA to gold nanoparticles of 1:200, and more than 100000 times larger than that with the volume ratio of PGMA to gold nanoparticles of 0.5:200 (Table 2).
 |
| | Fig. 3 (a) Plots of the concentration of p-NP versus time for the reduction of p-NP by NaBH4 in the presence of 0.1% PAH-modified (w/v) PGMA@AuNP composites at pH 7. The ratio of PGMA spheres to AuNPs is 2:200 (v/v) ( ); 1:200 (v/v) ( ); 1:200 (v/v) ( ); 0.5:200 (v/v) ( ); 0.5:200 (v/v) ( ). The inset graph shows the plots of the concentration of p-NP versus induction period (t0). (b) Plots of the concentration of p-NP versus time (t0 is not included) for the reduction of p-NP by NaBH4 in the presence of 0.1% PAH-modified (w/v) PGMA@AuNP composites at pH 7 with fitting curves. The ratio of PGMA spheres to AuNPs is 2:200 (v/v) ( ). The inset graph shows the plots of the concentration of p-NP versus induction period (t0). (b) Plots of the concentration of p-NP versus time (t0 is not included) for the reduction of p-NP by NaBH4 in the presence of 0.1% PAH-modified (w/v) PGMA@AuNP composites at pH 7 with fitting curves. The ratio of PGMA spheres to AuNPs is 2:200 (v/v) ( ); 1:200 (v/v) ( ); 1:200 (v/v) ( ); 0.5:200 (v/v) ( ); 0.5:200 (v/v) ( ). ). | |
3.5. Effect of pH value on the composite catalytic activity
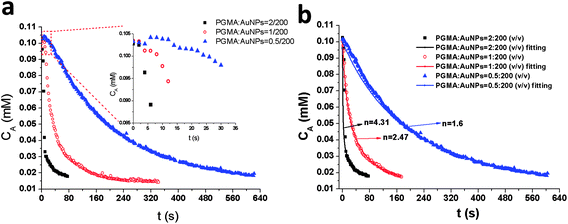
For the model reaction p-nitrophenol/NaBH4 reduction, the reaction pH should be maintained at alkaline conditions to ensure the formation of p-nitrophenolate ions with the plasmon absorption band at 400 nm. Therefore it is not meaningful to investigate the pH effect of the reduction reaction system on the composite catalytic activity. However, the AuNP surface coverage is dependent on the solution pH value when synthesizing the PGMA@PAH@AuNP composites, which can further change composite catalytic activity, so it is necessary to investigate the effect of pH for the synthesis of composite particles, not for the p-nitrophenol reduction reaction environment, on the composite catalytic activity. Here surface conformation transition from coil to globule through pearl-necklace structures can result in reduced interaction of gold nanoparticles with PAH-modified PGMA spheres when the solution pH is increased.22 The results are summarized in Table 3 which suggest the following: the reduction time increases with increasing pH value in composite solution. The average reaction rates and average TOFs decrease with increasing the solution pH value, indicating lower catalytic activity for composites at high pH values. The inset graph in Fig. 4a presents the analysis of induction time for composites obtained in different pH solutions. The results suggest that induction time is relying on the AuNP coverage amount of composites. By measuring the catalytic activity of composites prepared at different pHs, the apparent order of reaction, and apparent rate constant were determined by fitting the time dependence of concentration of the p-NP curve with eqn (5) from kinetic modeling (Fig. 4b). The apparent order of reaction, n and kapp, both decrease with an increasing pH in the range of 3–11. At the pH of 3, the model reaction p-NP/NaBH4 catalyzed by the as-prepared composite particles follows the 7th-order with the kapp of 1.19 × 105 M−5.97 s-1. On the other hand, the model reduction is the 5th-order reaction with the kapp of 7.59 × 102 M−3.99 s-1 at the pH of 11. The reduction reaction takes longer time as pH increases. The reduction time for composites in the cases of pH 3, 7 and 11 are 102, 216 and 288 s respectively.
Table 3 Kinetic parameters for composite particles at 0.5% PAH (w/v) concentration with a volume ratio of PGMA:AuNPs = 1:200 at pH 3, 7 and 11
| Composite particles |
AuNP coverage amount (moles) |
Reaction time (s) |
R
ave (mol s−1) |
TOFave (mol mol−1 s−1) |
t
0 (s) |
n
|
k
app
|
Correlation coefficient (R) |
| pH = 3 |
9.7619 × 10−12 |
102 |
1.23 × 10−9 |
125.5 |
6 |
6.97 |
1.19 × 105 |
0.99003 |
| pH = 7 |
8.0375 × 10−12 |
216 |
5.79 × 10−10 |
72 |
6 |
6.58 |
6.03 × 104 |
0.98926 |
| pH = 11 |
7.6197 × 10−12 |
288 |
4.36 × 10−10 |
57.2 |
14 |
4.99 |
7.59 × 102 |
0.99207 |
 |
| | Fig. 4 (a) Plots of the concentration of p-NP versus time for the reduction of p-NP by NaBH4 in the presence of 0.5% PAH-modified (w/v) PGMA@AuNP composites with a volume ratio of PGMA spheres to AuNPs of 1:200 at pH = 3 ( ); pH = 7 ( ); pH = 7 ( ); pH = 11 ( ); pH = 11 ( ), the inset graph is the plots of the concentration of p-NP versus induction period (t0). (b) Plots of the concentration of p-NP versus time (t0 is not included) for the reduction of p-NP by NaBH4 in the presence of 0.5% PAH-modified (w/v) PGMA@AuNP composites with a volume ratio of PGMA spheres to AuNPs of 1:200 with fitting curves. pH = 3 ( ), the inset graph is the plots of the concentration of p-NP versus induction period (t0). (b) Plots of the concentration of p-NP versus time (t0 is not included) for the reduction of p-NP by NaBH4 in the presence of 0.5% PAH-modified (w/v) PGMA@AuNP composites with a volume ratio of PGMA spheres to AuNPs of 1:200 with fitting curves. pH = 3 ( ); pH = 7 ( ); pH = 7 ( ); pH = 11 ( ); pH = 11 ( ). ). | |
3.6. Relationship between n and coated AuNP coverage
Previous analysis suggested that the reaction rate is related to the apparent reaction order, n. The activity of the coated gold nanoparticles as a catalyst is determined by the availability of surface on gold atoms with low coordination numbers and an associated electron density.37 Increasing the specific surface area of the coated gold nanoparticles will increase the amount of active gold atoms, which will increase the activity of the catalyst, finally leading to a higher reaction rate. In the following the relationship between the apparent order of the reduction reaction and the coated gold nanoparticle coverage amount will be discussed. In Fig. 5 we plotted n for all data above-mentioned as a function of the coated AuNP coverage amount. In general, an increase in AuNP coverage amount on composites leads to an increase in the apparent order of reaction, n. Under all experiments studied, the highest apparent reaction order of 7th for the reduction reaction was obtained at the gold nanoparticle coverage of 9.7619 × 10−12 moles. The greater the gold nanoparticle coverage on the modified PGMA sphere, the more active the gold atoms become, and the more reactive sites are available. A greater AuNP coverage also correlates with more AuNP/PGMA interfaces, which serve as reservoirs supplying a large number of enriched electrons, facilitating electron transfer to the adsorbed reactant p-NP.38
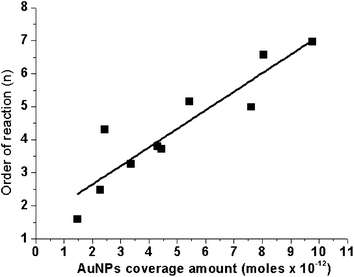 |
| | Fig. 5 Plot of the order of reaction, n as a function of the gold nanoparticle coverage amount. | |
3.7. Mechanism of catalytic activity enhancement
The as-prepared composite catalyst with enhanced catalytic activity can be attributed to the active epoxy groups of the PGMA spheres and a large adsorption of p-nitrophenol anion reactants onto the positively-charged PAH-modified PGMA spheres (seen in Scheme 1). Borohydride ions are adsorbed onto the surface of gold nanoparticles to react and transfer electrons to the gold nanoparticle surface. The carbons in the epoxy groups are very reactive electrophiles, so epoxy groups of the PGMA spheres have a tendency to attract electrons and act as electron acceptors. Thus charge distribution occurs between the gold nanoparticles and PGMA spheres. Therefore, electrons leave the gold nanoparticles and end up with an electron-enriched region at the interface of gold nanoparticles and PGMA spheres. Meanwhile, the p-nitrophenolate anion reactants can be easily adsorbed onto the positively charged PAH-modified PGMA spheres due to electrostatic interactions. The existence of the surplus electrons added to the PGMA spheres facilitates the uptake of electrons by the adsorbed p-nitrophenol molecules, which leads to the reduction of p-nitrophenol into the p-aminophenol products. Detachment of the product p-aminophenol creates a free surface and the catalytic cycle can start again.
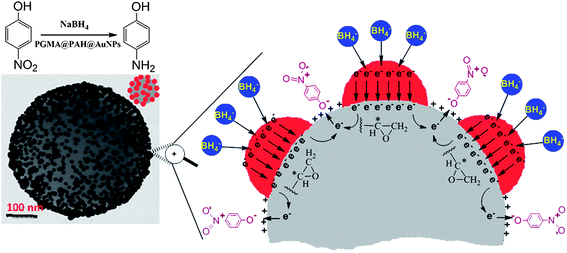 |
| | Scheme 1 Graph of p-nitrophenol reduction at the gold nanoparticle surface/PGMA interface. | |
4. Conclusions
A detailed kinetic study of the model catalytic reduction reaction p-nitrophenol/sodium borohydride on the PGMA@PAH@AuNP composite particles has been conducted. Experimental data show a non-(pseudo-)first-order mechanism for the studied reaction system, based on the fact of the non-linear relationship between Ln(CA/CA0) and reduction time t. The nth-order kinetic equation (n > 1) was obtained by a simple surface-controlled catalytic modeling, which was used to fit the time-dependent p-nitrophenol concentration change curves. The results indicate the reaction order number is closely correlated with the surface coverage amount of the coated gold nanoparticles on the modified polymer spheres. The catalytic activity of the as-prepared composite particles was enhanced by the supporting materials containing functional epoxy groups and positive electric charges. More importantly, such an enhanced reaction activity could be tuned specifically via the surface modifier PAH concentration, the volume ratio of the polymer spheres to gold nanoparticles, and the solution pH values for the composite synthesis. Last but not least, a shorter induction period (t0 < 30 s) was found on the model reaction as well, which could be ascribed to the catalytic-induced reconstruction of the surface of the heterogeneous composite catalyst.
Acknowledgements
We acknowledge St. John's University for the Seed Grant support. And we thank Kim Kisslinger and Lihua Zhang from Brookhaven National Laboratory for the help with TEM imaging. Research was carried out in part at the Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of energy, Office of Basic Energy Sciences, under Contract no. DE-AC02-CH10886.
References
- Z. Jin, M. Xiao, Z. Bao, P. Wang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 6406–6410 CrossRef CAS PubMed.
- Y. Sun and C. Lei, Angew. Chem., Int. Ed., 2009, 48, 6824–6827 CrossRef CAS PubMed.
- K. d. O. Santos, W. C. Elias, A. M. Signori, F. C. Giacomelli, H. Yang and J. B. Domingos, J. Phys. Chem. C, 2012, 116, 4594–4604 CAS.
- J. Li, C.-y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CAS.
- N. Pradhan, A. Pal and T. Pal, Langmuir, 2001, 17, 1800–1802 CrossRef CAS.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
- H. Zhang, X. Li and G. Chen, J. Mater. Chem., 2009, 19, 8223–8231 RSC.
- N. Sahiner, S. Butun, O. Ozay and B. Dibek, J. Colloid Interface Sci., 2012, 373, 122–128 CrossRef CAS PubMed.
- S. Zhang, W. Wu, X. Xiao, J. Zhou, J. Xu, F. Ren and C. Jiang, Asian J. Chem., 2012, 7, 1781–1788 CrossRef CAS PubMed.
- K. Kuroda, T. Ishida and M. Haruta, J. Mol. Catal. A: Chem., 2009, 298, 7–11 CrossRef CAS.
- S. Wu, J. Dzubiella, J. Kaiser, M. Drechsler, X. Guo, M. Ballauff and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 2229–2233 CrossRef CAS PubMed.
- J. Huang, S. Vongehr, S. Tang, H. Lu and X. Meng, J. Phys. Chem. C, 2010, 114, 15005–15010 CAS.
- M. Beija, E. Palleau, S. Sistach, X. Zhao, L. Ressier, C. Mingotaud, M. Destarac and J.-D. Marty, J. Mater. Chem., 2010, 20, 9433–9442 RSC.
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2009, 26, 2885–2893 CrossRef PubMed.
- Y. Du, H. Chen, R. Chen and N. Xu, Appl. Catal., A, 2004, 277, 259–264 CrossRef CAS.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- G. A. Somorjai, A. M. Contreras, M. Montano and R. M. Rioux, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10577–10583 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- M. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739–746 CrossRef CAS PubMed.
- Y. Liu, M. Li and G. Chen, J. Mater. Chem. A, 2013, 1, 930–937 CAS.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237–243 CrossRef CAS PubMed.
- K. Kuroda, T. Ishida and M. Haruta, J. Mol. Catal. A: Chem., 2009, 298, 7–11 CrossRef CAS.
- B. Dong, D. L. Miller and C. Y. Li, J. Phys. Chem. Lett., 2012, 3, 1346–1350 CrossRef CAS PubMed.
- F.-h. Lin and R.-a. Doong, J. Phys. Chem. C, 2011, 115, 6591–6598 CAS.
- K. B. Narayanan and N. Sakthivel, Bioresour. Technol., 2011, 102, 10737–10740 CrossRef CAS PubMed.
- Y.-C. Chang and D.-H. Chen, J. Hazard. Mater., 2009, 165, 664–669 CrossRef CAS PubMed.
- A. Azetsu, H. Koga, A. Isogai and T. Kitaoka, Catalysts, 2011, 1, 83–96 CrossRef CAS.
- S. Tang, S. Vongehr and X. Meng, J. Mater. Chem., 2010, 20, 5436–5445 RSC.
- K. Laszlo, P. Podkoscielny and A. Dabrowski, Langmuir, 2003, 19, 5287–5294 CrossRef CAS.
- G. Wei, J. S. Wainright and R. F. Savinell, J. New Mater. Electrochem. Syst., 2000, 3, 121–129 CAS.
- W. Xu, J. S. Kong, Y.-T. E. Yeh and P. Chen, Nat. Mater., 2008, 7, 992–996 CrossRef CAS PubMed.
- M. B. Cortie and E. van der Lingen, Mater. Forum, 2002, 26, 1–14 CAS.
- N. R. Jana, Z. L. Wang and T. Pal, Langmuir, 2000, 16, 2457–2463 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a