 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile route to model catalysts: the synthesis of Au@Pd core–shell nanoparticles on γ-Fe2O3 (0001)†
Robert J. Davies*,
Michael Bowker,
Philip R. Davies and
David J. Morgan
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: daviesrj11@cf.ac.uk; Tel: +44 (0)29 20877051
First published on 31st July 2013
Abstract
A straightforward method of synthesising Au@Pd core–shell particles on a well characterised γ-Fe2O3 (0001) substrate has been developed which will enable fundamental studies into the surface chemistry of these catalytically interesting systems. Au and Pd were sequentially deposited onto a γ-Fe2O3 (0001) substrate in ultra high vacuum by metal vapour deposition and probed by LEIS and STM. Deposition of Au followed by heating at 573 K formed nanoparticles of 5 to 10 nm in diameter whereas subsequent deposition of Pd produced smaller nanoparticles of 2 to 4 nm diameter. At this stage, LEIS shows both metals to be present but heating the combined system to 573 K resulted in the loss of the Au signal in the LEIS and disappearance of the smaller particles from the STM images indicating the formation of Au@Pd core–shell structures.
Introduction
Alloy or bimetallic catalysts find applications in a wide range of industrial processes due to the increased activity, selectivity and stability which can be achieved by mixing two metals.1 Combining Au and Pd to create a bimetallic catalyst can create a synergistic system with significantly altered characteristics from its individual component parts. As such, the activity of a bimetallic catalyst will depend on factors which include the distribution of electron density over the particle/cluster, the positions of the different component species on/in the catalyst, and changes in geometry caused by the interaction of the two metals. For example, Venezia et al. showed that dilution by Au of a supported Pd catalyst results in an increase in activity for the hydrodesulphurisation of thiophene. This behaviour was attributed to the Au reducing the size of the active Pd ensemble, and therefore inhibiting the formation of Pd sulphide.2 The promotional effect of Au was also highlighted in a study of the acetoxylation of ethene to vinyl acetate. The active species in the reaction had been identified as suitably spaced Pd monomers, and it was observed that the key role of Au was to isolate these Pd units which facilitated the adsorption and coupling of ethane and acetic acid to form vinyl acetate.3Supported Au–Pd alloys have been shown by Hutchings et al. to be excellent catalysts for the direct synthesis of hydrogen peroxide.4–6 In a study comparing catalysts for both CO oxidation and H2O2 synthesis, the iron oxide supported Au catalyst which is active for CO oxidation was found to be inactive for H2 oxidation. Addition of Pd to this catalyst significantly improves its performance for the synthesis of H2O2, and conversely the Au–Pd bimetallic catalyst showed little activity for CO oxidation.6 Similarly it has been demonstrated that addition of Au to an oxide supported Pd catalyst caused significant increase in the yield of H2O2 compared to Au or Pd only catalysts. Electron microscopy studies showed nanoparticles of 2 to 10 nm in size which were shown to be Au–Pd alloys. Analysis of the catalysts by XP spectroscopy suggested that these particles were rich in Pd at the surface.7 A more recent study of the effects of heat treatment and acid pre-treatment of the support on catalyst performance and structure highlighted the importance of the oxidation state of Pd on the catalyst surface. A high concentration of Pd2+ was found to facilitate the formation of H2O2 and minimize its decomposition by hydrogenation.8
The aim of this work is to synthesise a model catalyst of supported bimetallic Au–Pd under ultra high vacuum. These can then be studied using surface science techniques with a view to better understanding the interaction of Au and Pd in the real bimetallic catalysts. The primary techniques employed are low energy ion scattering (LEIS) which due to its top layer sensitivity allows us to carefully follow core–shell formation; and scanning tunnelling microscopy (STM) which is used to analyse surface topography.
Experimental
Experiments were carried out in a custom designed Omicron Multiprobe UHV system comprising four stainless steel chambers pumped by three turbo pumps and four ion pumps giving a typical base pressure of 1 × 10−9 mbar. The preparation (P) chamber contains a fast entry lock and an Omicron ISE ion gun for sputtering. The central (C) chamber contains two separate metal vapour depositors – one an Au bead (from foil, Goodfellow, 99.99%) formed on a W coil, the other a Pd wire (Goodfellow, 99.95%) wrapped around a W coil. The analysis (A) chamber is equipped with Omicron SpectaLEED apparatus, an Omicron ISE fine focus ion gun for low energy ion scattering (LEIS), and an energy analyser used with LEIS. Adjacent to the preparation chamber is the SPM chamber which houses an Omicron variable temperature scanning probe microscope used predominantly for scanning tunnelling microscopy (STM). The SPM chamber maintains a base pressure of ∼1 × 10−10 mbar when isolated from the rest of the system by a gate valve. The substrate used as a support for the Au and Pd was a γ-Fe2O3 (0001) single crystal which was mounted on a standard Omicron sample plate which could be transported between the chambers using three horizontal manipulators and a linear horizontal probe. Sample heating can be achieved on each manipulator using a resistive heating filament and temperature was recorded through a thermocouple attached to the manipulator in close proximity to the sample. The sample was cleaned by cycles of Ar+ ion bombardment followed by annealing in vacuum at 873 K. Au and Pd were dosed at an evaporation current of 2.5 A at room temperature. LEIS spectra were recorded with an ion beam current of 1 μA, and a beam energy of 1 keV at a He pressure of 1 × 10−7 mbar. The small LEIS peak observed at ∼460 eV in Fig. 2c is assigned to surface contamination by fluorine which occasionally segregated from the bulk, particularly after heating. The small Pd trace in the LEIS spectra in Fig. 2b and c is due to Pd from previous experiments which could not be removed completely by sputter anneal cycles.Results
The γ-Fe2O3 (0001) surface was prepared by cleaning by Ar ion bombardment followed by annealing in vacuum for 20 minutes at 873 K, which gave a hexagonal (2 × 2) LEED structure. STM images of the (2 × 2) surface are shown in Fig. 1 in which large terraces of up to 50 nm are observed separated by steps of ∼0.5 nm in height. The LEIS spectrum of this surface is shown in Fig. 2a with the O peak observed at ∼420 eV and the Fe peak at ∼780 eV kinetic energy. All subsequent work was carried out on this (2 × 2) surface, of which a full characterisation is given elsewhere.9 | ||
| Fig. 1 STM images of the clean γ-Fe2O3 (2 × 2) surface prepared by sputtering and annealing in vacuum for 20 minutes at 873 K. | ||
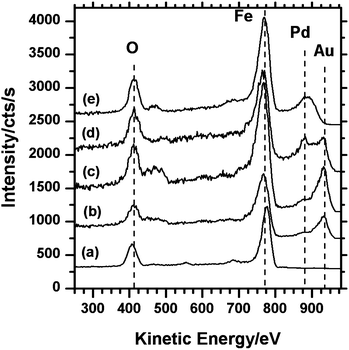 | ||
| Fig. 2 LEIS spectra showing the sequential deposition of Au and Pd on the γ-Fe2O3 surface and evidence for the formation of Au@Pd core–shell particles: (a) clean γ-Fe2O3 surface; (b) Au on γ-Fe2O3; (c) Au/γ-Fe2O3 heated at 573 K for 1 minute; (d) Pd on Au/γ-Fe2O3; (e) Pd/Au/γ-Fe2O3 heated at 573 K for 1 minute. | ||
Evaporation of Au
Differentiating between nanoparticles of different composition is not easily achieved with STM, our experimental strategy therefore was to create Au and Pd particles with different and distinct sizes. Initial experiments showed that Au tends to form bigger particles on the iron oxide surface than Pd, therefore our first step was to deposit Au followed by heating to form particles with widths in the range 5 to 10 nm before depositing Pd which would be identifiable as much smaller particles.10 The LEIS spectrum after Au was evaporated for 10 seconds is shown in Fig. 2b, where the Au peak can be observed at ∼930 eV with the Fe peak at 780 eV and the oxygen signal at 420 eV. A small increase in the width of the Fe scattering peak on the adsorption of Au may be due to multiple scattering effects.11 STM images of this surface (Fig. 3) shows Au particles of up to 4 nm in width with the majority in the range of 1 to 3 nm (Fig. 4a). The particles are distributed fairly evenly across the surface with no apparent preference for step edge sites. After heating this surface at 573 K for 1 minute, an increase in average particle width to between 5 and 10 nm was observed as shown in the STM images (Fig. 5) and the particle size distribution (Fig. 4b). The LEIS spectrum after heating showed no significant change in the intensity of the Au peak relative to that of the oxygen (Fig. 2c) suggesting that the gold coverage over the surface has not changed despite the change in island distribution. However, assuming the particle shapes are approximately spherical sections, the particle size distribution in Fig. 4 indicate a significant increase in the volume of gold present on annealing. This suggests that before annealing a significant volume of gold present at the surface is not identified in the STM images possibly because it is present as very small particles (<1 nm) or “hidden” at step edges.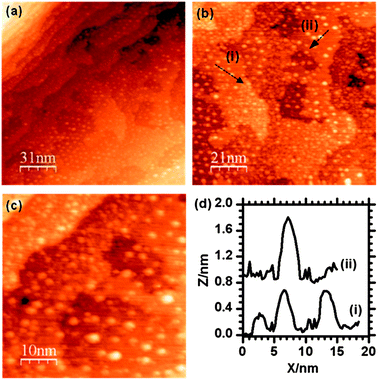 | ||
| Fig. 3 STM images of the γ-Fe2O3 (2 × 2) surface after deposition of Au. The majority of particles are in the range of 1 to 3 nm in diameter with larger particles being up to 4 nm. The particle size distribution is summarised in Fig. 4. | ||
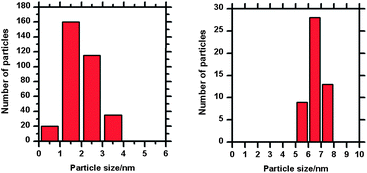 | ||
| Fig. 4 Au nanoparticle size distributions for Fig. 3b and 5b (before and after heating to 573 K for 1 minute). Heating has resulted in the sintering of small gold nanoparticles and the formation of a smaller number of larger particles. | ||
 | ||
| Fig. 5 STM images of the γ-Fe2O3 (2 × 2) surface after deposition of Au and subsequent heating to 573 K for 1 minute. | ||
Addition of Pd
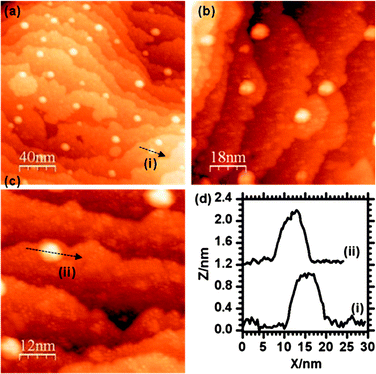
Adding palladium to the Au/γ-Fe2O3 surface by metal vapour deposition (MVD) produced small particles of up to 3 nm in size which could be identified on flat areas of the surface between the existing Au particles (Fig. 6). Another noticeable result from the addition of Pd was the formation of pits on the surface which were measured to be fairly shallow, typically up to 5 Å deep, and between 5 and 10 nm in diameter. These may be due to a local reduction in the iron oxide substrate and the oxidation of the deposited palladium. The LEIS spectrum after deposition (Fig. 2d) shows the Pd peak at ∼880 eV and the Au peak at 930 eV indicating both species are present. In the STM, there is no apparent change to the size or shape of the Au particles after the Pd was introduced and in the LEIS, the relative height of the Au peak to the oxygen peak is also unchanged. Since palladium deposition should decrease the LEIS oxygen peak intensity, the constant Au/O ratio suggests an equal probability of palladium deposition on the gold surface as on the oxide, although this is not discernible from the STM.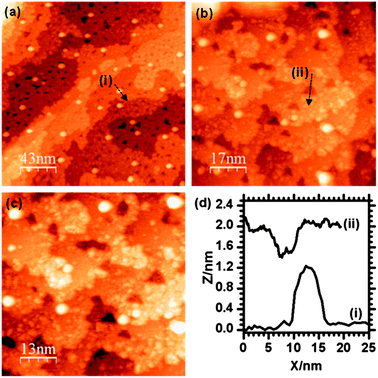 | ||
| Fig. 6 STM images taken after the deposition of Pd onto the Au/γ-Fe2O3 surface. Particles with widths of ∼10 nm can be assigned to Au, smaller particles are due to the Pd. | ||
On heating the surface containing both Au and Pd nanoparticles to 573 K for 1 minute, the Au signal was lost from the LEIS spectrum but a broad signal due to the Pd at 880 eV remained (Fig. 2e). STM images (Fig. 7) show that the small particles observed before heating and attributed to the palladium (Fig. 6) have largely been lost whereas the larger particles (5 to 10 nm and previously attributed to Au) can still be seen. The small pit like depressions seen in Fig. 6 have also apparently decreased in number.
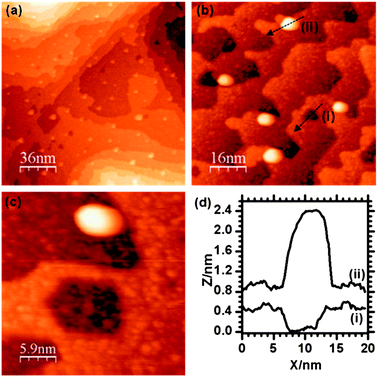 | ||
| Fig. 7 STM images of the Pd/Au/γ-Fe2O3 surface after heating at 573 K for 1 minute. | ||
Discussion
The complimentary information provided by LEIS and STM gives a useful insight into the behaviour of the Pd/Au/Fe2O3 system. For the initial deposition of gold and the subsequent deposition of palladium, the nanoparticles that are evident in the STM can be unambiguously assigned on the basis of their size: the annealed gold nanoparticles showing typical widths of 5 to 10 nm whilst the subsequently dosed palladium nanoparticles are typically 1 nm in width and gathered in clusters on bare areas of the substrate. The LEIS indicates no loss of Au signal intensity (relative to the substrate peaks) after the addition of palladium, implying that the nucleation of the palladium is at least as fast on the Fe2O3 surface as it is on the gold nanoparticles. An interesting feature of this LEIS spectrum (Fig. 2d) is the relatively high intensity between the Au and Pd peaks at 930 and 880 eV respectively. This may be due to multiple scattering effects which have been reported previously for situations where an adlayer is present on a metal surface.11After heating, STM images of the mixed Au and Pd surface show only the larger type of particle (5 to 10 nm) remaining, these were previously assigned to the sintered Au. However, the LEIS spectrum shows no evidence of gold, the only peaks present being those due to the iron oxide substrate and Pd at 880 eV. Since, in the absence of palladium, the Au remains on the surface on heating (Fig. 2c and 5), its absence from the LEIS spectra in this case suggests it is being hidden from the LEIS by the Pd implying the latter has migrated to the Au particles creating Au–Pd core–shell structures. It is estimated from the difference between Au and Au–Pd particle sizes that the thickness of the Pd shell is 1 to 2 nm. Under ideal conditions gold/palladium clusters are expected to form gold shell–palladium core structures (Pd@Au).12–14 However, palladium shell structures have frequently been observed experimentally in situations where (as in the present case) kinetic control dominates over thermodynamics15–17 or else, where oxidation of the palladium occurs.18 The present case is unusual because of the stability of the palladium shell on annealing in vacuum to 573 K. However, although oxygen is not present in the gas phase there is a viable supply of oxygen from the substrate and oxidation of the palladium would account for the vacancies observed in the iron oxide terraces after palladium dosing. Studies of Au and Pd deposition on SiO2 supports have shown cluster surface enrichment in Au even without annealing19 or when the deposition sequence is varied.20 The difference between these and our results can be explained in terms of the reducibility of the oxide support. In our case the support can readily supply oxygen thus facilitating the formation of PdO, whereas in the case of SiO2 this is unlikely to occur. Interestingly, Freund and co-workers have shown that Au also segregates to the surface of Au–Pd nanoparticles even when supported on thin film reducible oxide supports such as Fe3O4 and CeO2 (ref. 21) when treated in a reducing atmosphere. Cycles of O2 and CO treatments were performed at elevated temperatures. They do not report the state of the nanoparticles when exposed to only an oxidizing atmosphere.
Iron oxide is known to have a strong promotional effect on metal catalyst particles; it has been used for example as promoter for CO oxidation over Pt/alumina catalysts where it was suggested to act as a source of oxygen for the reaction.22 Similar effects have been observed for Pd based systems such as the enhancement of water gas shift activity of Pd/ceria catalyst by addition of iron oxide. The improvement in performance required the iron oxide, which was impregnated onto the ceria in preparation, to be in contact with the Pd and it was suggested that the key role of the iron oxide was to transfer oxygen to the Pd.23 A third example comes from the work of Liu et al.24 who showed that the iron oxide, as support, acted as an oxygen reservoir for the oxidation of CO over Pd and Pt. In previous work from our own laboratories we found that an iron oxide thin film formed on a Fe (111) support could act as an oxygen reservoir which could replenish a reduced surface via a short anneal.25 It is suggested that in our Au/Pd system, palladium undergoes oxidation at the Pd/Fe2O3 interface and subsequently forms a Au@Pd–O core–shell particle.
Conclusions
Using a combination of STM and LEIS spectroscopy we have demonstrated the fabrication of Au/Pd bimetallic nanoparticles supported on γ-Fe2O3 using metal vapour deposition under ultra high vacuum. Au and Pd particles were sequentially deposited onto the iron oxide substrate and after brief thermal treatment the two species alloyed to form Au–Pd core–shell particles. These supported particles could be used as models for real Au–Pd catalysts to explore mechanisms which are highly active for numerous reactions.Notes and references
- V. Ponec, Appl. Catal., A, 2001, 222, 31–45 CrossRef CAS.
- A. M. Venezia, V. La Parola, V. Nicolì and G. Deganello, J. Catal., 2002, 212, 56–62 CrossRef CAS.
- M. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS.
- J. K. Edwards, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2009, 48, 8512–8515 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. Herzing, M. Watanabe, C. J. Kiely and G. J. Hutchings, J. Mater. Chem., 2005, 15, 4595–4600 RSC.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1917–1923 RSC.
- J. K. Edwards, J. Pritchard, M. Piccinini, G. Shaw, Q. He, A. F. Carley, C. J. Kiely and G. J. Hutchings, J. Catal., 2012, 292, 227–238 CrossRef CAS.
- M. Bowker, G. Hutchings, P. R. Davies, D. Edwards, R. Davies, S. Shaikhutdinov and H.-J. Freund, Surf. Sci., 2012, 606, 1594–1599 CrossRef CAS.
- P. Stone, R. D. Smith and M. Bowker, Faraday Discuss., 2004, 125, 379–390 RSC.
- W. Soszka, D. Sitko and M. Soszka, Surf. Sci., 1995, 336, 199–204 CrossRef CAS.
- H. B. Liu, U. Pal, R. Perez and J. A. Ascencio, J. Phys. Chem. B, 2006, 110, 5191–5195 CrossRef CAS.
- I. V. Yudanov and K. M. Neyman, Phys. Chem. Chem. Phys., 2010, 12, 5094–5100 RSC.
- L. Reinaudi and O. A. Oviedo, Electrochim. Acta, 2012, 76, 424–429 CrossRef.
- A. F. Lee, C. J. Baddeley, C. Hardacre, R. M. Ormerod, R. M. Lambert, G. Schmid and H. West, J. Phys. Chem., 1995, 99, 6096–6102 CrossRef CAS.
- Y. Kobayashi, S. Kiao, M. Seto, H. Takatani, M. Nakanishi and R. Oshima, Hyperfine Interact., 2004, 156–157, 75–79 CrossRef.
- Y. W. Lee, M. Kim, Z. H. Kim and S. W. Han, J. Am. Chem. Soc., 2009, 131, 17036–17037 CrossRef CAS.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- A. R. Haire, J. Gustafson, A. G. Trant, T. E. Jones, T. C. Q. Noakes, P. Bailey and C. J. Baddeley, Surf. Sci., 2011, 605, 214–219 CrossRef CAS.
- K. Luo, T. Wei, C.-W. Yi, S. Axnanda and D. W. Goodman, J. Phys. Chem. B, 2005, 109, 23517–23522 CrossRef CAS.
- H. L. Abbott, A. Aumer, Y. Lei, C. Asokan, R. J. Meyer, M. Sterrer, S. Shaikhutdinov and H.-J. Freund, J. Phys. Chem. C, 2010, 114, 17099–17104 CAS.
- X. Liu, O. Korotkikh and R. Farrauto, Appl. Catal., A, 2002, 226, 293–303 CAS.
- X. Wang and R. Gorte, Appl. Catal., A, 2003, 247, 157–162 CrossRef CAS.
- L. Liu, F. Zhou, L. Wang, X. Qi, F. Shi and Y. Deng, J. Catal., 2010, 274, 1–10 CrossRef CAS.
- R. Davies, D. Edwards, J. Gräfe, L. Gilbert, P. R. Davies, G. Hutchings and M. Bowker, Surf. Sci., 2011, 605, 1754–1762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr03047d |
| This journal is © The Royal Society of Chemistry 2013 |