 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust and specific ratiometric biosensing using a copper-free clicked quantum dot–DNA aptamer sensor†
Haiyan Zhanga, Guoqiang Fengb, Yuan Guo*a and Dejian Zhou*a
aSchool of Chemistry and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK. E-mail: y.guo@leeds.ac.uk; d.zhou@leeds.ac.uk
bKey Laboratory of Pesticide and Chemical Biology of Ministry of Education, College of Chemistry, Central China Normal University, 152 Luoyu Road, Wuhan 430079, P.R. China
First published on 15th August 2013
Abstract
We report herein the successful preparation of a compact and functional CdSe–ZnS core–shell quantum dot (QD)–DNA conjugate via highly efficient copper-free “click chemistry” (CFCC) between a dihydro-lipoic acid–polyethylene glycol–azide (DHLA–PEG–N3) capped QD and a cyclooctyne modified DNA. This represents an excellent balance between the requirements of high sensitivity, robustness and specificity for the QD-FRET (Förster resonance energy transfer) based sensor as confirmed by a detailed FRET analysis on the QD–DNA conjugate, yielding a relatively short donor–acceptor distance of ∼5.8 nm. We show that this CFCC clicked QD–DNA conjugate is not only able to retain the native fluorescence quantum yield (QY) of the parent DHLA–PEG–N3 capped QD, but also well-suited for robust and specific biosensing; it can directly quantitate, at the pM level, both labelled and unlabelled complementary DNA probes with a good SNP (single-nucleotide polymorphism) discrimination ability in complex media, e.g. 10% human serum via target-binding induced FRET changes between the QD donor and the dye acceptor. Furthermore, this sensor has also been successfully exploited for the detection, at the pM level, of a specific protein target (thrombin) via the encoded anti-thrombin aptamer sequence in the QD–DNA conjugate.
Introduction
The unique, size-dependent, highly stable and bright fluorescence of quantum dots (QDs) make them powerful tools in broad ranges of bio-related applications.1–4 In particular, their broad absorption and narrow and symmetric emission are extremely well-suited for Förster resonance energy transfer (FRET) based sensing, because these spectral characteristics enable a wide selection of excitation wavelengths to minimise direct excitation of the acceptor, reducing the background and hence improving the sensitivity.1,2 Indeed, numerous QD-FRET based biosensors have been reported.3,4 Despite these, the sensitivity and specificity of the QD-FRET based biosensors have largely been limited by challenges in preparing compact and functional QD-bioconjugates that are stable and effectively resist non-specific adsorption.2–4 For example, water-soluble QDs prepared by ligand exchange are compact, but they often show low stability in biologically relevant buffers and resistance to non-specific adsorption, limiting their sensing specificity and robustness.2 Whereas those prepared by physical encapsulation with amphiphilic polymers and/or PEGylated lipids (on which most commercial water-soluble QDs are based) are stable and can resist non-specific adsorption, but their large size (with hydrodynamic radii often greater than the R0 of most QD–dye FRET pairs even prior to bioconjugation)2 can greatly limit their FRET efficiency (sensitivity). Although the FRET efficiency can be enhanced by increasing the ratio of acceptors on each QD, such designs are inefficient at low target to QD ratios.2 Therefore for biosensing, it is important to balance the requirements of sensitivity and robustness because they are often incompatible. In this regard, QDs capped with PEGylated small-molecule ligands appeared to be highly attractive; they are relatively compact yet display good stability and, more importantly, effective resistance to non-specific adsorption of biomolecules.2Besides surface capping, a robust and efficient QD-bioconjugation chemistry that can offer high bioactivity without compromising the QD fluorescence is also important. In this regard, the Cu(I) catalysed azide–acetylene cycloaddition, best known as the “click chemistry” (CuCC),5a is highly powerful and versatile; it offers exquisite functional group selectivity and high yield. It has been used successfully in preparing a wide range of functional nanoparticle bioconjugates (e.g. gold, magnetic, silica and polymer nanoparticles) for sensing and biomedical applications.5 However, the CuCC is unsuitable for the QD, because the Cu(I) catalyst used in the CuCC can efficiently and irreversibly quench the QD fluorescence.6 The Cu-free “click chemistry” (CFCC) between strained cyclooctynes and azides happens rapidly and efficiently, and moreover, it does not require any Cu catalyst.7 Therefore, the CFCC appears to be ideal for efficient QD-bioconjugation without compromising the QD fluorescence.7a Indeed, the CFCC has been successfully used to make functional QD–protein/small-molecule conjugates recently for live virus labelling/imaging and intra-cellular trafficking studies.8 Despite such developments, the QDs used in these studies were all capped with polymer based ligands (and hence of relatively big sizes) because the QD sizes here are less critical for such applications.8 To our knowledge, the CFCC has yet to be used to develop QD-FRET based biosensors where the compact size of the QD-bioconjugate is known to be of critical importance. Herein, we report the successful preparation of the first compact and functional QD–DNA conjugate via the CFCC between a dihydrolipoic acid–polyethylene glycol–azide (DHLA–PEG–N3) capped CdSe–ZnS core–shell QD and a cyclooctyne modified DNA, giving a good balance between the requirements of high sensitivity, specificity and robustness. This is supported by a FRET analysis showing a relatively short QD-dye distance of ∼5.8 nm for the QD–DNA FRET system. Moreover, the CFCC clicked QD–DNA conjugate is found not only to retain the native fluorescence quantum yield (QY) of the parent QD, but also well-suited for robust biosensing; it can directly quantitate, at the pM level, both labelled and unlabelled complementary DNA probes with a good SNP (single-nucleotide polymorphism) discrimination ability even in complex media, e.g. 10% human serum, on a conventional fluorimeter. It can also directly detect, at the pM level, a specific protein via the encoded DNA aptamer sequence.
Results and discussion
CFCC based QD–DNA conjugation and sensing principle
Scheme 1 shows our approach to the QD–DNA conjugate via the CFCC and its use in label- and label-free-detection of DNA and protein targets via target binding induced changes in the QD sensitized dye FRET signals. First, a multi-functional ligand, containing a dihydrolipoic acid (DHLA, for strong QD binding) head group, a polyethylene glycol moiety of a molecular weight of 600 (PEG600, for providing good water-solubility and effective resistance to non-specific adsorption of biomolecules) and a terminal azide group (for efficient DNA conjugation via the CFCC), DHLA–PEG600–N3, was prepared (see the ESI† for details).9,10 A PEGylated DHLA ligand was used as the QD surface capping ligand here because it represented an excellent balance between the requirements of high stability and resistance to non-specific adsorption (for robust biosensing) and the structural compactness (for high sensitivity).2 Then a hydrophobic CdSe–ZnS core–shell QD (λEM ∼ 605 nm, QY ∼ 20%, capped with hydrophobic trioctyl-phosphine oxide/trioctylphosphine) was made water-soluble by ligand exchange with the DHLA–PEG600–N3 in a mixed solvent of CHCl3–ethanol using our previously established procedures,3l yielding the QD–DHLA–PEG600–N3 which was readily soluble in polar solvents. Its fluorescence QY was found to decrease to ∼6.0% (and hence a decrease of ca. 70%), which is in good agreement with most other reports in the literature where most hydrophobic CdSe–ZnS core–shell QDs typically showed a QY decrease of 50–80% following the ligand exchange and transfer to aqueous media.3,4 A single-stranded (ss) target DNA encoded with a 29 mer anti-thrombin (TB) aptamer sequence with strong affinity for TB (Kd ∼ 0.5 nM, modified with a C6-amine at 5′, H2N–TBA, see Table 1)11 was reacted with an N-hydroxysuccinimide (NHS) ester activated cyclooctyne to yield TBA–cyclooctyne, which was then reacted with the QD–DHLA–PEG600–N3 in a mixed solvent of ethanol–water at a molar ratio of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This led to QD–TBA covalent conjugation via the efficient CFCC approach. Approximately 20 strands of TBAs were found to be conjugated to each QD, denoted as QD–TBA20 hereafter, this gave a DNA conjugation efficiency of ∼67%. The detailed experimental procedures for the ligand synthesis and QD–DNA conjugation are given in the ESI.† The QY of the resulting QD–TBA20 was determined as ∼5.9% using rhodamine 6G in ethanol as the calibration standard (QY 95%),3b which is effectively the same as that of the QD–DHLA–PEG600–N3 (ca. 6.0%).
:1. This led to QD–TBA covalent conjugation via the efficient CFCC approach. Approximately 20 strands of TBAs were found to be conjugated to each QD, denoted as QD–TBA20 hereafter, this gave a DNA conjugation efficiency of ∼67%. The detailed experimental procedures for the ligand synthesis and QD–DNA conjugation are given in the ESI.† The QY of the resulting QD–TBA20 was determined as ∼5.9% using rhodamine 6G in ethanol as the calibration standard (QY 95%),3b which is effectively the same as that of the QD–DHLA–PEG600–N3 (ca. 6.0%).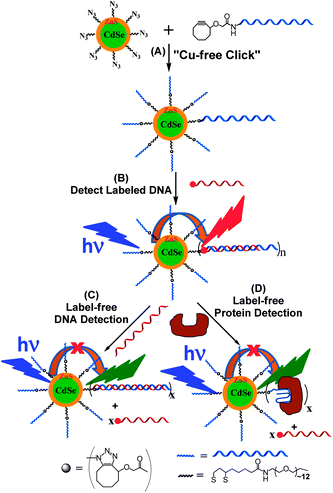 | ||
| Scheme 1 (A) Schematic approach to the Cu-free “clicked” QD–DNA conjugate. (B) Hybridization of a complementary dye-labelled DNA probe with the QD–DNA conjugate leads to QD sensitized dye FRET signals as a readout for labelled DNA detection. (C) Incubation of the QD–double-stranded (ds) DNA conjugate formed in (B) with a longer, unlabeled DNA displaces the shorter labelled DNA reporter, reducing the QD to dye FRET for label-free DNA detection. (D) Incubation of the QD–dsDNA conjugate (B) with a target protein that binds to the encoded aptamer sequence in the QD–dsDNA conjugate displaces the reporter DNA, leading to reduction of QD to dye FRET for label-free protein detection. The block arrows give the FRET directions. | ||
| DNA code | Sequence |
|---|---|
| TBA | 5′-TTAGTCCGTGGTAGGGCAGGTTGGGGTGACT-3′ |
| DNA29 | 3′-TCAGGCACCATCCCGTCCAACCCCACTGA-5′ |
| DNA18 | 3′-AATCAGGCACCATCCCGT-5′ |
| DNA15 | 3′-AATCAGGCACCATCC-5′ |
| DNA12 | 3′-TCAGGCACCATC-5′ |
| DNA12-SM | 3′-TCAGACACCATC-5′ |
| DNA-NC | 3′-TAGTCC CGATT TCTCACG-5′ |
The QD–TBA20 was found to be highly soluble and stable in aqueous media. It showed no change of physical appearance or fluorescence after being stored in a fridge at 4 °C for over two months. More importantly, the QD–TBA20 was found to have effectively retained the native QY of the parent QD–DHLA–PEG600-N3. In contrast, conjugation of the H2N–TBA to a water-soluble, glutathione capped QD (the same batch of QD) by using 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC)–NHS mediated covalent coupling resulted in significantly reduced QY. The fluorescence intensity of the former was ∼6.4 times as strong as the latter (see ESI, Fig. S1†) despite that the latter exhibited a higher QY in pure water (∼18%). Moreover, the number of TBA strands conjugated to each QD by the CFCC (20) was also 4 times that of the latter (∼5). All these demonstrate that the CFCC based QD–DNA conjugation approach developed herein is far superior over the EDC–NHS mediated coupling, a conventional widely used QD-bioconjugation method,3 in terms of both the DNA conjugation efficiency and ability of maintaining a high QY of the QD.
FRET analysis of the CFCC clicked QD–DNA conjugate
Prior to using the CFCC clicked QD–DNA conjugate for sensing, a FRET analysis on the CFCC clicked QD–TBA20 after hybridisation with a complementary strand (DNA29) of different molar ratios was carried out to ensure a relatively small donor–acceptor distance (r) for high sensitivity. This is because the FRET efficiency (E) decreases dramatically with the increasing r value following the Förster dipole–dipole interaction formula:| E = 1/[1 + (r/R0)6] | (1) |
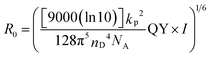 | (2) |
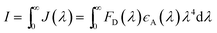 | (3) |
I is the integral of the donor–acceptor spectral overlap over all wavelengths λ, FD(λ) is the normalized donor emission and εA is the absorption extinction coefficient of the acceptor.
The normalised absorption and emission spectra of the QD and Atto647N and the corresponding spectral overlap function are shown in the ESI, Fig. S2.† A significant overlap between the QD (donor) emission and Atto647N (acceptor) absorption is clear, suggesting that they can have efficient FRET. The spectral overlap, I, can be calculated from Fig. S2B,† giving a value of 6.68 × 1015 M−1 cm−1 nm4. These combined with the QY of the QD (5.9%) and the parameters above yielded a R0 value of 4.25 nm for the QD–Atto647N FRET pair (at 1:1 molar ratio).
For a single-donor (QD here) simultaneously FRET with n identical acceptor systems, E is given by the following equation:
| E = nR06/[r6 + nR06] | (4) |
| Apparent E = IA/[IA + ID] | (5) |
The FRET study was carried out with 2 nM of the QD–TBA20 sample after hybridization with different molar equivalents of DNA29 (3′-labelled with an Atto647N acceptor, see Table 1). Hybridization of DNA29 with the QD–TBA20 should bring the Atto647N acceptor in the close proximity to the QD, leading to the QD sensitized Atto-647N FRET signal upon excitation of the QD (Scheme 1B). For eqn (4) to be valid, all DNA29 strands introduced (and Atto647N labels) should bind to the QD–TBA20. Hence the longest DNA29 probe which forms the most stable duplex with the QD–TBA20 was used.
Fig. 1A clearly shows that with the increasing molar ratio of the DNA29:QD–TBA20, the QD fluorescence is quenched while the Atto647N FRET signal is increased progressively, suggesting efficient FRET between the QD and the Atto647N dye. Moreover, the resulting E and DNA29/QD molar ratio can be fitted well by the single-QD donor FRET with multiple identical acceptor models (R2 = 0.991) with a relatively short donor–acceptor distance r of 5.82 ± 0.01 nm. This result confirms that the CFCC clicked QD–DNA conjugate FRET system is indeed compact, and moreover, all Atto647N labels on the DNA29 strands are bound to the QD at an identical spatial separation (the same r value).
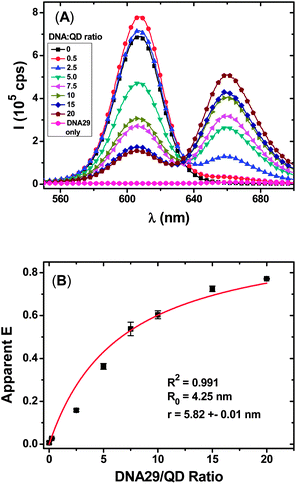 | ||
| Fig. 1 (A) Fluorescence spectra of QD–TBA20 (2 nM) hybridised with different molar equivalents of DNA29 with a 7 μM His6–Cys peptide. The DNA29 only curve has a concentration of 60 nM. (B) A plot of the apparent E as a function of DNA29/QD molar ratio; data were fitted using eqn (4), yielding a r value of 5.82 ± 0.01 nm. | ||
Detection of DNA29 using the CFCC clicked QD–TBA20
The sensitivity of the QD–TBA20 based FRET sensor in detecting complementary DNA probes was further evaluated by using DNA29. Hybridization of DNA29 with the QD–TBA20 should bring the Atto647N dye (acting as the FRET acceptor) in the close proximity to the QD, leading to the QD sensitized Atto647N FRET signal upon excitation of the QD. An advantage of the FRET based signal readout over other approaches (e.g. donor quenching or life time change) is its ratiometric signal, which can be effectively insensitive to signal fluctuation and instrument noise, allowing for more reliable and accurate detection.2 Moreover, since FRET only happens over short distances (ca. < 10 nm), any free, unbound species will be too far to participate in FRET with the QD donor and hence undetected, allowing for convenient probe detection to be carried out in a separation-free format.2,3n Interestingly, the QD–TBA20 (CQD = 2 nM) fluorescence in PBS (10 mM sodium phosphate, 150 mM NaCl, pH 7.40) was found to be enhanced significantly after treatment with a cysteine–histidine6 short peptide and/or bovine serum albumin (BSA, see ESI, Fig. S3A†), presumably because these molecules can bind or adsorb onto the QD–TBA20 to enhance the QD fluorescence QY as reported previously.3a They may also adsorb onto sample tubes to reduce the non-specific adsorption and/or salt-induced aggregation of the QD. Moreover, the added peptide/BSA was also found to improve the FRET efficiency of the QD–TBA/DNA29 system considerably (see ESI, Fig. S3B†), and the effect became saturated at ∼7 μM. With the peptide/BSA being added, the hybridized QD–TBA20/DNA29 FRET system was found to be highly stable, no significant change of the QD fluorescence or Atto647N FRET signals was observed after being stored for 18 h in PBS (see ESI, Fig. S4†). This is important for biosensing, allowing the experiments to be carried out at ease without the need to worry about the stability of the sensor (from our own experience, most small-molecule ligand capped QDs, including glutathione, 3-mercaptopropinoic acid (MPA) and DHLA, showed rather limited stability in PBS, and therefore all sensing measurements should be performed within 1 h after target addition to avoid a significant decrease of the QD fluorescence).3l–n,4d,e All subsequent sensing experiments were carried out with 7 μM added peptide/BSA on a conventional fluorimeter with a low QD concentration of 2 nM.Fig. 2A shows that in general the QD fluorescence (peaking at ∼605 nm) was quenched progressively together with a concurrent simultaneous significant increase of the Atto647N FRET signal (peaking at ∼665 nm) with the increasing DNA29 concentration, [DNA29], suggesting efficient QD-sensitised dye FRET from hybridisation of the DNA29 with QD–TBA20. A careful examination of the Atto647N emission spectra over the 640–700 nm range revealed that the Atto647N emission obtained from direct excitation of 60 nM DNA29 was actually weaker than that of the QD-sensitized FRET signal for 0.25 nM DNA29 (see ESI, Fig. S5†), suggesting that the QD sensitized FRET is at least 240 times as efficient as direct excitation. To our knowledge, this has been the highest ratio of FRET-sensitized signals over that of direct excitation for the QD-FRET systems reported so far (most reported ratios in the literature typically ranged from ∼2.5 to 40).2,3 This is presumably because λEX = 450 nm, corresponding to the λabs minimum of the Atto647N, was used here to minimise the direct excitation of the Atto647N acceptor. Moreover, the CFCC conjugated QD–TBA20 here retained a much higher QY of the QD than those prepared via EDC–NHS coupling (see ESI, Fig. S1†), as a result, the sensing experiments were able to be performed at 2 nM QD, ∼10 to 500 fold lower than those reported previously (see Table 2).2,3 Such a high FRET-sensitised signal over the direct excitation background is highly advantageous for biosensing, which can effectively eliminate the need for background correction from direct acceptor excitation, making data analysis easy and straightforward.
![(A) Fluorescence spectra of the QD–TBA20 (CQD = 2 nM) after hybridization with different amounts of DNA29 for 2 h in PBS excited at 450 nm, the λabs minimum of Atto647N. (B) A plot of the integrated donor/acceptor fluorescence ratio, IDye/IQD, as a function of [DNA29]. The data were fitted to a two-stage linear relationship with fitting parameters of y = −0.539 + 0.1121x, R2 = 0.9978 over 10–40 nM, and y = −0.000395 + 0.0354x, R2 = 0.9947 over 0–5 nM (shown in the inset), in which the detection limit is based on. (C) Plot of the IDye/IQD ratios as a function of concentration of different length complementary DNA probes. (D) The IDye/IQD ratios for different length DNA probes at 40 nM.](/image/article/2013/NR/c3nr02897f/c3nr02897f-f2.gif) | ||
| Fig. 2 (A) Fluorescence spectra of the QD–TBA20 (CQD = 2 nM) after hybridization with different amounts of DNA29 for 2 h in PBS excited at 450 nm, the λabs minimum of Atto647N. (B) A plot of the integrated donor/acceptor fluorescence ratio, IDye/IQD, as a function of [DNA29]. The data were fitted to a two-stage linear relationship with fitting parameters of y = −0.539 + 0.1121x, R2 = 0.9978 over 10–40 nM, and y = −0.000395 + 0.0354x, R2 = 0.9947 over 0–5 nM (shown in the inset), in which the detection limit is based on. (C) Plot of the IDye/IQD ratios as a function of concentration of different length complementary DNA probes. (D) The IDye/IQD ratios for different length DNA probes at 40 nM. | ||
| Detection system | CQD (nM) | Target/length | Specificity | SNP DR | LOD (nM) | Ref. |
|---|---|---|---|---|---|---|
| a BRET: bioluminescence resonance energy transfer; LOD: limit of detection; specificity: the FRET ratio between the full- and non-complementary DNA probes; SNP DR: SNP discrimination ratio. | ||||||
| QD-BRET | 20 | DNA/22 mer | ? | ? | 20 | 3o |
| QD-FRET | 1000 | DNA/19 mer | <2 | No | 40 | 3g |
| QD-FRET | 60 | DNA/24 mer | <2 | No | 12 | 3g |
| QD-FRET | 100 | DNA/25 mer | ∼3 | No | 200 | 3p |
| QD-FRET | ? | DNA/18–32 mer | 2–3 | No | ∼5 | 3q |
| QD-FRET | 100 | DNA/30 mer | Yes | No | ∼1 | 3l |
| QD-FRET | 4 | DNA/30 mer | 34 | No | 0.5 | 3m |
| QD-FRET | 2 | DNA/12–29 mer | 816 | 3.3 (12 mer) | 0.091 (29 mer) | This work |
| Microcantilever | / | DNA/12 mer | ? | ? | 75 | 12a |
| Microcantilever | / | DNA/12 mer | ∼3 | ? | ∼0.010 | 12b |
| Electrochemical | / | DNA/24 mer | ? | ? | 0.01 | 12c |
| Electrochemical | / | DNA/34 mer | Yes | 1.5–2.0 | 0.05 | 12d |
| Direct SPR | / | DNA/16 mer | ? | ? | 10 | 12e |
| Direct QCM | / | DNA/509 mer | ? | ? | 10 | 12f |
Despite that the QD fluorescence does not always follow a simple trend of progressive quenching with the increasing [DNA29], especially at low [DNA29] shown in Fig. 2A, possibly due to a slight increase of the QD QY as DNA29 is hybridized, this has no impact on the ratiometric based data analysis employed here. In fact, the ratio of the integrated fluorescence intensity between the acceptor and the donor, IDye/IQD (see the ESI† for the detailed calculation method)3l displayed a two-stage linear dependence with the increasing [DNA29]: a slow increasing phase over low [DNA29] (0–5 nM, slope: 0.0353 nM−1, R2 = 0.9947) and a more rapid phase at higher concentrations (10–40 nM, slope: 0.1121 nM−1, R2 = 0.9978, Fig. 2B). As [DNA29] was increased to above 40 nM, the IDye/IQD value showed a little further increase, suggesting that the hybridization reached saturation. Thereafter, any extra added DNA29 strands were unable to hybridize with the QD–TBA20 and would remain free. Since FRET only takes place over short distances (ca. < 10 nm), such free DNA29 strands are unable to participate in the FRET process and hence undetected. Note here that 40 nM corresponds to the total [TBA] in the 2 nM QD–TBA20 conjugate, suggesting that all TBAs conjugated to the QD are functional and available for hybridization. The detection limit (DL) for DNA29, based on 3 times the standard deviation/slope of linear calibration over the lower concentration range (3σ/slope), is estimated as ∼91 pM,3h making it one of the most sensitive QD-FRET based sensors for direct DNA quantification without probe pre-amplification using conventional fluorescence spectroscopy (see Table 2). Moreover, this level of sensitivity is also comparable to or better than many other sensitive direct DNA detection methods without probe pre-amplification, such as the optimised, sensitive microcantilever sensors (∼10 pM),12b electro-chemical detection (10 pM),12c surface plasmon resonance (SPR, 10 nM)12d and quartz crystal microbalance (QCM, 10 nM)12e detection (see Table 2 for details).
Theoretically, a linear correlation between the IDye/IQD and the probe concentration, [DNA29], is expected if all introduced DNA probes are hybridized with the QD–TBA20 at identical positions (the same QD–dye distance, r).13 The excellent linear relationship observed here clearly confirmed that all DNA29 strands were hybridized with the QD at identical spatial separation between the QD donor and the dye acceptor. This result is also in good agreement with the earlier FRET analysis where E can be fitted very well (R2 = 0.991) by the single-donor FRET with multiple identical acceptor models.2 The two-stage linear dependence observed here may indicate two different phases of DNA hybridization: the slower increase over the 0–5 nM range is likely due to incomplete hybridization of DNA29 with the QD–TBA20, arising presumably from the low, sub-Kd levels of [DNA29], whereas the faster increase over the 10–40 nM range may be attributed to more effective, complete binding of the introduced DNA29 to the QD–TBA20 under such conditions. Given that Kd values of 17, 19 and 41 nM have been reported for 25,14a 20,14b and 12 (ref. 12a) mer dsDNAs, respectively, we believe such explanations here are highly plausible.
The CFCC clicked QD–DNA FRET sensor was found to be highly specific; incubation of the QD–TBA20 with a non-complementary probe (DNA-NC, also 3′-Atto647N labelled, see Table 1) under identical conditions (with 10 μM added BSA) produced effectively non-detectable FRET. The IDye/IQD ratios for the DNA29 and DNA-NC (both at 30 nM) were determined as 2.563 and 0.00314, respectively, yielding an outstanding signal discrimination ratio of 816 between the full- and non-complementary DNA probes (see ESI, Fig. S7†). The discrimination ratio here is 24–400 fold higher than previously reported QD-FRET based DNA sensors (see Table 2), demonstrating an excellent DNA sensing specificity. Moreover, the QD-FRET based DNA sensor was highly robust, it worked pretty well even in clinically relevant media, e.g. 10% human serum (see ESI, Fig. S8†). It should be noted that despite that numerous QD-FRET based DNA sensors have been reported in the literature, few have demonstrated the working function in serum, one of the most frequently used clinical media. These results clearly demonstrated an excellent sensing specificity and robustness of the CFCC clicked QD–DNA sensor, which we attribute to the excellent stability, and more importantly, the outstanding resistance toward non-specific adsorption of biomolecules afforded by the PEGylated capping ligands on the QD surface.9,10
Detection of different length DNA probes and SNP (single-nucleotide polymorphism) discrimination
Besides offering high discrimination between complementary and non-complementary DNA probes, the CFCC clicked QD–DNA sensor can effectively discriminate complementary DNA probes of different lengths. As shown in Fig. 2C, although the IDye/IQD ratios increased with the increasing concentration for all probes, the rates of increase were significantly different, with DNA29 being the fastest while DNA12-SM being the slowest. A general trend here is that the IDye/IQD increase rate showed a positive correlation with the length of the DNA probe, e.g. DNA29 > DNA18 > DNA15 > DNA12 > DNA12-SM. Moreover, DNA18 also showed a two-stage IDye/IQD–[DNA] linear increase similar to that for DNA29, while for DNA15 and DNA12, this became much less clear, and DNA12-SM effectively displayed a single linear dependence. Such differences may reflect the different Kds of the different length probes toward the common TBA target: only those with Kds that span across the [DNA] range studied here may display two-stage dependence.The slopes of the rapidly increasing IDye/IQD phase (over 10–40 nM range) were found to be 0.112, 0.091, 0.062, 0.036 and 0.011 nM−1 for DNA29, DNA18, DNA15, DNA12 and DNA12-SM, respectively. Therefore the slope of the IDye/IQD increase rate for the DNA29 is ∼3 times that of DNA12, while that for DNA12 is a further ∼3.3 times that of DNA12-SM, the same length (12 mer) probe containing just a single base mismatch with TBA, equivalent to single nucleotide polymorphism (SNP). The CFCC clicked QD–DNA sensor can therefore offer a SNP discrimination ratio of ∼3.3 for the 12 mer DNA probes. Similar levels of probe length dependence and SNP discrimination ratio (ca. > 3 between DNA12 and DNA12-SM) were also obtained from the IDye/IQD ratios at 40 nM probe concentration (Fig. 2D). More interestingly, the discrimination between DNA12 and DNA12-SM was found to be unaffected by the presence of complex media, such as 10% human serum. In fact, the discrimination ratio actually increased to 6.1 (against ∼3 in PBS, see ESI, Fig. S9†), demonstrating good potential for SNP based clinical diagnosis. It should be noted that despite several QD-FRET based DNA sensors have been reported in the literature, most of which displayed rather low discrimination ratios between full- and non-complementary DNA probes, few have displayed the SNP discrimination ability (see Table 2). Since SNPs are known to be closely associated with a number of important human diseases, such as cancer, neurodegenerative diseases and diabetes, etc.,15 the excellent specificity, sensitivity and robust SNP discrimination ability in complex media may make the CFCC clicked QD-DNA sensor potentially suitable for clinical applications.
Detection of unlabelled DNA probes
The ability of detecting unlabeled DNA probes is more useful for potential clinical applications, avoiding the need for the probe labelling step which can be complex, expensive and sometimes even impossible. In this regard, a new DNA displacement assay is developed here: a longer unlabeled probe (e.g. DNA29-NL, with the same sequence as DNA29 but without the Atto647N label) that forms more stable duplex with TBA can effectively displace a shorter labelled DNA (e.g. DNA12-SM, acting as a FRET reporter) pre-hybridized with the QD–TBA20, leading to a decreased FRET as the unlabelled probe readout signal (Scheme 1C).Fig. 3 reveals that this is indeed feasible, where the Atto647N FRET signal at ∼665 nm was almost diminished accompanied by a concurrent significant recovery of the QD fluorescence at 605 nm as the [DNA29-NL] was increased, suggesting a successful displacement of the DNA12-SM reporter strand by the DNA29-NL, leading to a significant increase (∼21 fold) of the I605/I665 ratio (from 1.32 ± 0.06 to 28.6 ± 2.2 as the [DNA29-NL] increased from 0 at 100 nM, see Fig. 3B). Interestingly, replacing the DNA12-SM with DNA12 as the FRET reporter strand led to a much smaller increase of the I605/I665 ratio under identical conditions (from 0.66 ± 0.02 to 3.48 ± 0.09, an increase of ∼5.3 fold, see ESI, Fig. S10† for details), suggesting that a high stability difference between the reporter and the probe DNAs for the common target is key to achieve efficient reporter strand displacement and hence the greatly increased I605/I665 ratio. The I605/I665 response curve as a function of [DNA29-NL] was found to be non-linear (Fig. 3B), where 500 pM [DNA29-NL] produced a signal consistently above the background (Fig. 3B, inset), suggesting that this sensor can readily detect 500 pM DNA29-NL without probe amplification. Therefore this signal-on DNA sensing approach developed here can readily detect ∼500 pM unlabelled DNA probes together with a maximum ratiometric signal enhancement of ∼21 fold, which is already competitive against some other more established DNA sensing approaches, such as molecular beacons (ca. 10–20 fold signal enhancement with single-quenchers, nM sensitivity)11b,c and a recently optimised electrochemical DNA sensor (ca. 8-fold).12d An advantage of our approach here is its ratiometric signal, which is insensitive to instrument noise and signal fluctuation, allowing more reliable target detection. In addition, the DNA displacement assay was found to work equally efficiently in complex media, such as PBS with a large excess of BSA (10 μM) and in 10% human serum, suggesting that it may have good potential for clinical applications.
![(A) Typical fluorescence spectra of QD–TBA20 (2 nM) pre-hybridized with DNA12-SM (60 nM) after addition of different [DNA29-NF] for 2 h (B) A plot of the corresponding fluorescence intensity ratio at 605 and 665 nm (I605/I665) as a function of [DNA29-NF], the inset shows I605/I665 responses in the sub-nM range.](/image/article/2013/NR/c3nr02897f/c3nr02897f-f3.gif) | ||
| Fig. 3 (A) Typical fluorescence spectra of QD–TBA20 (2 nM) pre-hybridized with DNA12-SM (60 nM) after addition of different [DNA29-NF] for 2 h (B) A plot of the corresponding fluorescence intensity ratio at 605 and 665 nm (I605/I665) as a function of [DNA29-NF], the inset shows I605/I665 responses in the sub-nM range. | ||
Detection of unlabelled proteins
The CFCC clicked QD–TBA20 can be readily extended for label-free protein sensing via the anti-thrombin DNA aptamer sequence encoded within the TBA sequence, where the formation of thrombin (TB)–TBA complex can effectively displace the pre-hybridised reporter DNA12-SM, leading to the FRET decrease (and hence an increase of the I605/I665 ratio, see Scheme 1D). Fig. S11 (ESI†) reveals that this was indeed true, where the Atto647N FRET signal gradually decreased while the QD fluorescence increased concurrently as the target [TB] was increased, leading to the increased I605/I665 ratio (see Fig. 4). The maximum I605/I665 ratio obtained at 100 nM TB here (∼3.1) was not as high as that obtained in DNA29-NF detection (∼29), suggesting that the TB binding here is less efficient in displacing the DNA12-SM reporter strands from the QD–TBA20 conjugate as compared to DNA29-NF. Given that the binding affinity between the 29 mer anti-TB aptamer and TB (Kd ∼ 0.5 nM)11a is as strong as that of the TBA/DNA29 duplex (most likely to be in the low nM range as described above) here, the relatively low efficiency in displacing the reporter strands observed for TB here is therefore attributed to the significantly greater size of the TB–aptamer complex as compared to the TBA/DNA29 duplex, leading to steric hindrance and reduced accessibility for TB binding on the QD–DNA conjugate, especially under high [TB] conditions. Similar to the DNA29-NL based displacement assay above, a non-linear response curve between the I605/I665 signal and [TB] was also observed (Fig. 4). Moreover, the amplified response curve over the 0–2 nM [TB] range revealed that 500 pM [TB] produced a signal consistently above the background (Fig. 4, inset), suggesting that the CFCC clicked QD–DNA aptamer sensor can detect 500 pM TB directly without target pre-amplification. This sensitivity achieved here is among those of the most sensitive QD-FRET based label-free TB sensors using direct target detection without pre-amplification (see ESI, Table S1†). Moreover, this sensitivity is also comparable to those of other more established electrochemical sensing methods for TB detection (∼1 nM detection limit).12g![Label-free detection of thrombin using the CFCC clicked QD–DNA aptamer sensor using 2 nM QD–TBA20 pre-hybridized with DNA12-SM (60 nM) in PBS containing 20 μM BSA. A typical calibration curve showing the I605/I665 ratio as a function of thrombin concentration [TB] (inset: the response over 0–2 nM of [TB]).](/image/article/2013/NR/c3nr02897f/c3nr02897f-f4.gif) | ||
| Fig. 4 Label-free detection of thrombin using the CFCC clicked QD–DNA aptamer sensor using 2 nM QD–TBA20 pre-hybridized with DNA12-SM (60 nM) in PBS containing 20 μM BSA. A typical calibration curve showing the I605/I665 ratio as a function of thrombin concentration [TB] (inset: the response over 0–2 nM of [TB]). | ||
Conclusion
In summary, we have successfully developed a reliable CFCC approach for the preparation of a compact and stable QD–DNA/aptamer conjugate that can retain the native fluorescence QY of the parent QD. The resulting QD–DNA conjugate is relatively compact and can effectively resist non-specific adsorption. It has been successfully exploited for robust, sensitive and ratiometric quantitation of specific DNA probes directly with pM sensitivity even in complex media, such as 10% human serum. This QD–DNA FRET sensor has offered an excellent signal discrimination (>800 fold) between the full- and non-complementary DNA probes, which is the highest for the QD-FRET based sensors. Moreover, it can discriminate between the perfect-match and the SNP targets in 10% serum. The sensor has also been exploited for sensitive label-free detection, at the pM level, of thrombin via the anti-thrombin aptamer sequence encoded in the QD–DNA conjugate. This QD–DNA/aptamer sensor can be readily extended for detection of other DNA and protein targets by clicking other specific DNA/aptamer sequences against such targets.16 Given its high stability, specificity, robustness and sensitivity, the CFCC clicked QD–DNA/aptamer sensor appears to have good potential for a wide range of biosensing and diagnostic applications.Acknowledgements
We would like to thank the University of Leeds, the Leeds Biomedical Health Research Centre and the Leeds Cancer Research UK Centre for supporting this work. HZ thanks the Engineering and Physical Sciences Research Council (EPSRC, UK) for providing a research studentship to support her study at the University of Leeds. YG thanks the Wellcome Trust for providing a Career Re-entry Fellowship (grant number: 097354/Z/11/Z). GF thanks the National Natural Science Foundation of China (Grant No. 21172086) for supporting his research.Notes and references
- (a) P. Alivisatos, Nat. Biotechnol., 2004, 22, 47 CrossRef CAS PubMed; (b) X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS PubMed; (c) I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435 CrossRef CAS PubMed; (d) R. C. Somers, M. G. Bawendi and D. G. Nocera, Chem. Soc. Rev., 2007, 36, 579 RSC.
- (a) I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17 RSC; (b) W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825 CrossRef CAS PubMed; (c) H. Mattoussi, G. Palui and H. B. Na, Adv. Drug Delivery Rev., 2012, 64, 138 CrossRef CAS PubMed; (d) D. J. Zhou, Biochem. Soc. Trans., 2012, 40, 635 CrossRef CAS PubMed; (e) Z. Jin and N. Hildebrandt, Trends Biotechnol., 2012, 30, 394 CrossRef CAS PubMed.
- (a) T. Pons, I. L. Medintz, X. Wang, D. S. English and H. Mattoussi, J. Am. Chem. Soc., 2006, 128, 15324 CrossRef CAS PubMed; (b) D. J. Zhou, J. P. Piper, C. Abell, D. Klenerman, D.-J. Kang and L. M. Ying, Chem. Commun., 2005, 4807 RSC; (c) M. Suzuki, Y. Husimi, H. Komatsu, K. Suzuki and K. T. Douglas, J. Am. Chem. Soc., 2008, 130, 5720 CrossRef CAS PubMed; (d) Q. Wang, Y. Liu, Y. Ke and H. Yan, Angew. Chem., Int. Ed., 2008, 47, 316 CrossRef CAS PubMed; (e) H. Peng, L. Zhang, T. H. M. Kjallman, C. Soeller and J. Travas-Sejdic, J. Am. Chem. Soc., 2007, 129, 3048 CrossRef CAS PubMed; (f) D. E. Prasuhn, J. B. Blanco-Canosa, G. J. Vora, J. B. Delehanty, K. Susumu, B. C. Mei, P. E. Dawson and I. L. Medintz, ACS Nano, 2010, 4, 267 CrossRef CAS PubMed; (g) W. R. Algar and U. J. Krull, Anal. Chim. Acta, 2007, 581, 193 CrossRef CAS PubMed; (h) J. Liu, J. H. Lee and Y. Lu, Anal. Chem., 2007, 79, 4120 CrossRef CAS PubMed; (i) H. Yao, Y. Zhang, F. Xiao, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2007, 46, 4346 CrossRef CAS PubMed; (j) I. L. Madintz, L. Berti, T. Pons, A. F. Grimes, D. S. English, A. Alessandrini, P. Facci and H. Mattoussi, Nano Lett., 2007, 7, 1741 CrossRef PubMed; (k) R. Freeman, T. Finder and I. Willner, Angew. Chem., Int. Ed., 2009, 48, 7818 CrossRef CAS PubMed; (l) D. J. Zhou, L. M. Ying, X. Hong, E. A. Hall, C. Abell and D. Klenerman, Langmuir, 2008, 24, 1659 CrossRef CAS PubMed; (m) D. J. Zhou, Y. Li, E. A. H. Hall, C. Abell and D. Klenerman, Nanoscale, 2011, 3, 201 RSC; (n) H. Y. Zhang, P. G. Stockley and D. J. Zhou, Faraday Discuss., 2011, 149, 319 CAS; (o) K. A. Cissell, S. Campbell and S. K. Deo, Anal. Bioanal. Chem., 2008, 391, 2577 CrossRef CAS PubMed; (p) J. Lee, Y. Choi, J. Kim, E. Park and R. Song, ChemPhysChem, 2009, 10, 806 CrossRef CAS PubMed; (q) H. Peng, L. Zhang, T. H. M. Kjallman, C. Soeller and J. Travas-Sejdic, J. Am. Chem. Soc., 2007, 129, 3048 CrossRef CAS PubMed; (r) G.-I. Kim, K.-W. Kim, M.-K. Oh and Y.-M. Sung, Nanotechnology, 2009, 20, 175503 CrossRef PubMed.
- (a) M. Levy, S. F. Cater and A. D. Ellington, ChemBioChem, 2005, 6, 2163 CrossRef CAS PubMed; (b) C. Y. Zhang, H. C. Yeh, M. T. Kuroki and T. H. Wang, Nat. Mater., 2005, 4, 826 CrossRef CAS PubMed; (c) E. Oh, M. Y. Hong, D. Lee, S. H. Nam, H. C. Yoon and H. S. Kim, J. Am. Chem. Soc., 2005, 127, 3270 CrossRef CAS PubMed; (d) Y. Zhang, H. Y. Zhang, J. Hollins, M. E. Webb and D. J. Zhou, Phys. Chem. Chem. Phys., 2011, 13, 19427 RSC; (e) H. Y. Zhang and D. J. Zhou, Chem. Commun., 2012, 48, 5097 RSC; (f) H. Lu, O. Schöps, U. Woggon and C. M. Niemeyer, J. Am. Chem. Soc., 2008, 130, 4815 CrossRef CAS PubMed; (g) D. Geißler, L. J. Charbonnière, R. F. Ziessel, N. G. Butlin, H.-G. Löhmannsröben and N. Hildebrandt, Angew. Chem., Int. Ed., 2010, 49, 1396 CrossRef PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) E. Lallana, E. Fernandez-Megia and R. Riguera, J. Am. Chem. Soc., 2009, 131, 5748 CrossRef CAS PubMed; (c) N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717 RSC; (d) E. Boisselier, L. Salmon, J. Ruiz and D. Astruc, Chem. Commun., 2008, 5788 RSC; (e) L. Nebhani and C. Barner-Kowollik, Adv. Mater., 2009, 21, 3442 CrossRef CAS; (f) G. von Maltzahn, Y. Ren, J.-H. Park, D.-H. Min, V. R. Kotamraju, J. Jayakumar, V. Fogal, M. J. Sailor, E. Ruoslahti and S. N. Bhatia, Bioconjugate Chem., 2008, 19, 1570 CrossRef CAS PubMed.
- (a) A. Bernardin, A. Cazet, L. Guyon, P. Delannoy, F. Vinet, D. Bonnaffé and I. Texier, Bioconjugate Chem., 2010, 21, 583 CrossRef CAS PubMed; (b) G. Beaune, S. Tamang, A. Bernardin, P. Bayle-Guillemaud, D. Fenel, G. Schoehn, F. Vinet, P. Reiss and I. Texier, ChemPhysChem, 2011, 12, 2247 CrossRef CAS PubMed.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed; (b) J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793 CrossRef CAS PubMed; (c) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef CAS PubMed.
- (a) T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878 CrossRef CAS PubMed; (b) C. Schieber, A. Bestetti, J. P. Lim, A. D. Ryan, T. L. Nguyen, R. Eldridge, A. R. White, P. A. Gleeson, P. S. Donnelly, S. J. Williams and P. Mulvaney, Angew. Chem., Int. Ed., 2012, 51, 10523 CrossRef CAS PubMed; (c) P. Zhang, S. Liu, D. Gao, D. Hu, P. Gong, Z. Sheng, J. Deng, Y. Ma and L. Cai, J. Am. Chem. Soc., 2012, 134, 8388 CrossRef CAS PubMed.
- (a) K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987 CrossRef CAS PubMed; (b) B. C. Mei, K. Susumu, I. L. Medintz and H. Mattoussi, Nat. Protoc., 2009, 4, 412 CrossRef CAS PubMed.
- (a) K. L. Prime and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 10774 CrossRef; (b) D. Zhou, A. Bruckbauer, L. M. Ying, C. Abell and D. Klenerman, Nano Lett., 2003, 3, 1517 CrossRef CAS; (c) D. J. Zhou, A. Bruckbauer, C. Abell, D. Klenerman and D. J. Kang, Adv. Mater., 2005, 17, 1243 CrossRef CAS.
- (a) Y. Kim, Z. Cao and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5664 CrossRef CAS PubMed; (b) C. J. Yang and W. Tan, J. Am. Chem. Soc., 2005, 127, 12772 CrossRef CAS PubMed; (c) S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303 CrossRef CAS PubMed.
- (a) R. McKendry, J. Zhang, Y. Arntz, T. Strunz, M. Hegner, H. P. Lang, M. K. Baller, U. Certa, E. Meyer, H.-J. Güntherodt and C. Gerber, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9783 CrossRef CAS PubMed; (b) J. Zhang, H. P. Lang, F. Huber, A. Bietsch, W. Grange, U. Certa, R. Mckendry, H.-J. Güntherodt, M. Hegner and C. Gerber, Nat. Nanotechnol., 2006, 1, 214 CrossRef CAS PubMed; (c) C. Fan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9134 CrossRef CAS PubMed; (d) A. A. Rowe, K. N. Chuh, A. A. Lubin, E. A. Miller, B. Cook, D. Hollis and K. W. Plaxco, Anal. Chem., 2011, 83, 9462 CrossRef CAS PubMed; (e) L. He, M. D. Musick, S. R. Nicewarner, F. G. Salinas, S. J. Benkovic, M. J. Natan and C. D. Keating, J. Am. Chem. Soc., 2000, 122, 9071 CrossRef CAS; (f) A. K. Deisingh and M. Thompson, Analyst, 2001, 126, 2153 RSC; (g) R. Polsky, R. Gill, L. Kaganovsky and I. Willner, Anal. Chem., 2006, 78, 2268 CrossRef CAS PubMed.
- For a single QD FRET to n identical acceptors, the FRET efficiency, E, is given by: E = nR06/[nR06 + r6] = 1/[1 + r6/(nR06)], where R0 is the Förster radius and r is donor–acceptor distance. E can also be measured via the enhanced acceptor emission by the following equation: E = IDye/[IDye + IQD] = 1/[1 + IQD/IDye]. The combination of the two equations gives: 1/[1 + IQD/IDye] = 1/[1 + r6/(nR06)], and therefore IDye/IQD = nR06/r6, e.g. IDye/IQD should increase linearly with n, the number of acceptors on each QD ([DNA29] here under a fixed [QD]) if all of the acceptors are hybridised to the QD with identical spatial configuration (identical r value).
- (a) A. W. Peterson, L. K. Wolf and R. M. Georgiadis, J. Am. Chem. Soc., 2002, 124, 14601 CrossRef CAS PubMed; (b) D. J. Zhou, K. Sinnah, C. Abell and T. Rayment, Angew. Chem., Int. Ed., 2003, 42, 4934 CrossRef CAS PubMed.
- (a) M. K. Halushka, J. B. Fan, K. Bentley, L. Hsie, N. Shen, A. Weder, R. Cooper, R. Lipshutz and A. Chakravarti, Nat. Genet., 1999, 22, 239 CrossRef CAS PubMed; (b) Y. Zhang, Y. Guo, P. Quirke and D. J. Zhou, Nanoscale, 2013, 5, 5027 RSC.
- (a) D. H. J. Bunka and P. G. Stockley, Nat. Rev. Microbiol., 2006, 4, 588 CrossRef CAS PubMed; (b) J. Liu, Z. H. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis, purification and characterisation of the DHLA–PEG600–N3, cyclooctyne–DNA, and QD–TBA20 conjugates as well as all supporting figures and tables. See DOI: 10.1039/c3nr02897f |
| This journal is © The Royal Society of Chemistry 2013 |