 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ball milling: a green mechanochemical approach for synthesis of nitrogen doped carbon nanoparticles†
Tan
Xing
a,
Jaka
Sunarso
b,
Wenrong
Yang
*c,
Yongbai
Yin
d,
Alexey M.
Glushenkov
a,
Lu Hua
Li
a,
Patrick C.
Howlett
b and
Ying
Chen
*a
aARC Centre of Excellence for Functional Nanomaterials, Institute for Frontier Materials, Deakin University, Waurn Ponds, Victoria 3216, Australia. E-mail: ying.chen@deakin.edu.au
bARC Centre of Excellence for Electromaterials Science, Institute for Frontier Materials, Deakin University, Burwood, Victoria 3125, Australia
cSchool of Life and Environmental Sciences, Deakin University, Waurn Ponds, Victoria 3216, Australia. E-mail: wenrong@deakin.edu.au; Fax: +61 3 52271040; Tel: +61 3 5227 2932
dSchool of Physics, The University of Sydney, New South Wales 2006, Australia
First published on 26th June 2013
Abstract
Technological and scientific challenges coupled with environmental considerations have attracted a search for robust, green and energy-efficient synthesis and processing routes for advanced functional nanomaterials. In this article, we demonstrate a high-energy ball milling technique for large-scale synthesis of nitrogen doped carbon nanoparticles, which can be used as an electro-catalyst for oxygen reduction reactions after a structural refinement with controlled thermal annealing. The resulting carbon nanoparticles exhibited competitive catalytic activity (5.2 mA cm−2 kinetic-limiting current density compared with 7.6 mA cm−2 on Pt/C reference) and excellent methanol tolerance compared to a commercial Pt/C catalyst. The proposed synthesis route by ball milling and annealing is an effective process for carbon nanoparticle production and efficient nitrogen doping, providing a large-scale production method for the development of highly efficient and practical electrocatalysts.
Introduction
Precious Pt-based materials have been used as an effective electrocatalyst for oxygen reduction reactions (ORRs),1 yet large-scale commercial use has been precluded by high cost, limited supply, and weak durability of platinum. Recent intensive research efforts directed at reducing or replacing Pt-based electrodes in fuel cells have led to the development of new ORR electrocatalysts.2,3 In particular, the state-of-the-art nitrogen-doped carbon nanomaterials have been proved to have better resistance to poisoning,1,4,5 and are relatively cheaper to fabricate6 than Pt-based catalysts. Carbon nanomaterials exhibit some unusual physical, chemical, and mechanical properties, such as light absorption, mechanical stiffness, and high conductivity.7 Thus, nitrogen-doped carbon materials have been generally accepted as potential candidates for replacing Pt to achieve the commercialization of fuel cell technology. Nevertheless, most of the current methods of synthesizing nitrogen doped carbon materials face some issues like low production yield and use of toxic gas (NH3 for example). Therefore, the development of a new fabrication method is still required.Ball milling is a process, in which the moving balls apply their kinetic energy to the milled material, break chemical bonding and produce fresh surfaces by fracturing material particles. The dangling bonds on the newly created surfaces are usually chemically reactive. Also, it has been reported that the impact of a high-energy ball milling process sometimes can create a local high temperature above 1000 °C8 and/or high pressure up to several GPa.9 Due to this nature of the ball milling process, the process has been used as a mechano-chemical synthesis method8,10 for the fabrication of unique nanostructures with new chemical properties. It has been reported that ball milling can trigger solid–solid reactions to synthesize PbTe alloy from PbO and Te.11 Zhang et al. reported that solid–liquid reactions happened during milling of titanium in the benzene solution of pyrazine with the formation of intermediate Ti2N.12 Solid–gas reactions were also investigated systematically by milling of titanium and zirconium in a nitrogen-containing atmosphere.13–15 Other than these, ball milling has also been used recently to fabricate a nanostructured material that has unique properties.16 Graphite has a layered structure of aromatic rings. Numerous strong bonds are broken upon disruption of the layers, which produces free valences along the fresh borders. This would open the way for the formation of carbon nanostructures with new functionalities. In the previous work,17 we have demonstrated that a nanoporous carbon structure can be produced in the graphite samples after ball milling at ambient temperature. Formation of the nanoporous structure is associated with the disordered graphite structure. The disordered and nanoporous structure is probably fullerene-like in nature. Subsequently, it has also been showed that annealing of a disordered carbon nanostructure at 1400 °C produced carbon nanotubes.18 Except the disordered structure, ball milling has been demonstrated by different groups to produce graphene by exfoliating graphite.19–21
In this article, we report that ball milling can be used as a powerful green method to synthesize nitrogen-doped carbon nanoparticles (NDCPs), which can be used as an electrochemical catalyst for ORRs (Scheme 1 and Fig. S1†). The resulting carbon nanoparticles exhibited competitive catalytic activity (5.2 mA cm−2 kinetic-limiting current density at −0.5 V compared with 7.6 mA cm−2 on Pt/C reference) and excellent methanol tolerance compared to a commercial Pt/C catalyst.
 | ||
| Scheme 1 Fabrication of nitrogen-doped carbon nanoparticles (NDCPs) and their ORR catalytic properties. | ||
Materials and methods
Graphite (powder, particle size <20 micron, synthetic) was obtained from Sigma Aldrich® (Sydney, Australia) and used as the starting material. Nitrogen (99.99% purity) and a nitrogen–15% hydrogen mixture were used as atmospheric gases.Sample preparation
NDCPs were prepared by ball milling of pristine graphite powders in a high-energy rolling ball mill in the presence of nitrogen gas (300 kPa) at room temperature. The set-up of the ball mill is described elsewhere.22 The volume of the ball mill jar is about 300 mL. In a typical experiment, 2 g of the graphite powders were placed into a milling jar with four Ø 2 cm hardened steel balls. The sample to ball mass ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :132. The milling process lasted for 24 hours with a rotation speed of 150 rpm. The milled samples were then heated in a horizontal tube furnace from room temperature to 700 °C at a rate of 25 °C min−1 and then annealed for 3 hours. The sample was cooled down to room temperature. N2–15% H2 mixture gas was used as the annealing atmosphere with a flow rate of 0.05 L min−1.
:132. The milling process lasted for 24 hours with a rotation speed of 150 rpm. The milled samples were then heated in a horizontal tube furnace from room temperature to 700 °C at a rate of 25 °C min−1 and then annealed for 3 hours. The sample was cooled down to room temperature. N2–15% H2 mixture gas was used as the annealing atmosphere with a flow rate of 0.05 L min−1.
Instrumentation
The sample structure was characterized using X-ray diffraction (PANalytical X'Pert Pro diffractometer) with Cu Kα radiation (λ = 0.15418 nm). The sample morphologies were studied using a scanning electron microscope (SEM, Supra 55VPP) at 3 kV and a working distance of 4.6 mm. The particle size was measured with a dynamic light scattering method (Malvern ZETASIZER). 0.5 mg of the samples were added into 5 mL deionized water to make dispersions. The dispersions were ultrasonically treated for 30 min before the test.Ultraviolet-visible (UV-Vis) spectroscopy was used to characterize the dispersive ability of the samples in water. The dispersions were made by adding 20 mg of samples to a 5 mL volume of deionized water. Then, dispersions were ultrasonically treated for 30 min. After that, they were diluted by 50 times to make them adequately transparent for UV-Vis tests. The scans were run from 200 nm to 800 nm with a scan rate of 600 nm min−1 and a scan step of 1 nm. Water and a completely opaque cell were used as 100% and 0% transmittance standards, respectively, to correct the baseline.
X-ray photoelectron spectroscopy (XPS) (model: SPECS-XPS, from SPECS, Germany) was used to analyze the concentrations of elements and binding energies. The XPS system was equipped with a high sensitivity PHOIBOS 150-9 MCD energy analyzer. An Al K-alpha X-ray source (1486.74 eV) was used for the analysis. Transmission electron microscopy (TEM) images were taken with a JEOL 2100F instrument. The accelerating voltage used was 200 kV in the experiment. Thermogravimetric analysis (TGA) was done with a NETZSCH STA 409 PC. Samples were heated to 700 °C from room temperature with a heating rate of 10 °C min−1. A N2–15%H2 mixture was used as atmosphere. The flow rate was 10 mL min−1.
Thin film electrode preparation
The catalyst suspension was prepared by sonication (Soniclean 160T, Soniclean Pty Ltd) of the sample with tetrahydrofuran (THF, 99.9+%, Sigma-Aldrich) for 1 h, resulting in ink with final concentrations of 5 mg sample per mL. An aliquot of 4 μL of suspension was drop-cast onto a glassy carbon (GC) disk electrode (4 mm diameter, 0.126 cm2 area, rotating ring-disk electrode (RRDE) Pt ring/GC disk electrode, ALS Co., Ltd). The electrode was previously polished with 0.05 μm alumina slurry (Buehler) on a clean polishing cloth (Buehler), sequentially rinsed with distilled water and acetone, and then dried with lint-free tissue paper. After slow evaporation of solvent, 1 μL of Nafion® solution (5 wt%, Fluka) was drop-cast on the electrode surface and dried slowly overnight inside a closed beaker to attach the catalyst particles onto the glassy carbon disk electrode. Based on the density of Nafion® solution of ∼0.874 g cm−3 and the density of the Nafion® film of ∼2.0 g cm−3,23 an estimated Nafion® film thickness of ∼1.74 μm was obtained which is reasonably thin to render a negligible O2 transport resistance within the film.24Electrochemical characterization
Voltammetric experiments were performed with a Biologic VMP2/Z multi-channel potentiostat. Rotating disk electrode (RDE) voltammograms were obtained in 0.1 M potassium hydroxide (KOH) prepared from deionized water and potassium hydroxide pellets (KOH, 99.99%, Sigma-Aldrich) at a scan rate of 20 mV s−1 in either a N2 or O2 (ultrahigh purity grade, BOC) saturated atmosphere at room temperature (which corresponds to an electrolyte temperature of ∼17 ± 1 °C). Voltammograms for the reductions were obtained using a three-electrode arrangement in a 100 mL glass cell vial utilizing an RRDE-3A rotating ring/disk electrode apparatus (ALS Co., Ltd, Japan). This consists of a glassy carbon disk electrode (0.126 cm2, ALS Co., Ltd, Japan), a platinum wire counter electrode and an Ag|AgCl (3M NaCl) reference electrode (RE-1B, ALS Co., Ltd, Japan).The background capacitive current contribution (obtained from N2 saturated experiment) is subtracted from each voltammogram curve. Three catalyst films were prepared for each sample to check the reproducibility of the film quality and performance; the results of those vary within the experimental error. The current density values were obtained assuming an effective disk surface area of 0.1257 cm2 (diameter of 4 mm) for all samples.
Results and discussion
The preparation of NDCPs and their ORR catalytic properties are shown in Scheme 1. Ball milled graphite (C2) was fabricated by milling the commercial graphite (C1) in a nitrogen atmosphere. Then, C2 was annealed to produce NDCPs (C4) with catalytic properties. For comparison purposes, C1 was also annealed under the same conditions to produce annealed commercial graphite (C3).Ball milling has been widely used for mechano-chemical synthesis.8,10 We have shown that nitrogen can be effectively doped with materials by ball milling in the presence of nitrogen gas.25,26 It was demonstrated that mechanical energy of ball milling can break the chemical bonds in solid materials and produce dangling bonds, which are very active for chemical reactions.10,22 Simultaneously, gas atoms react with the solid atoms with dangling bonds. SEM images (Fig. 1) show a total breakdown of the graphitic layer structure. Furthermore, XRD patterns (Fig. 2) confirm the crystalline structure breakdown from a significant widening of the diffraction peaks of graphite.27 These results imply a wide introduction of dangling bonds and the potential to trigger the gas–solid reaction.
 | ||
| Fig. 1 Typical SEM images of (a) starting graphite, (b) milled graphite, (c) heated graphite without milling treatment and (d) heated graphite after milling treatment. | ||
 | ||
| Fig. 2 XRD diffraction patterns of graphite after different treatments. | ||
The high-resolution SEM images show the different morphologies of four samples. Starting commercial graphite (Fig. 1a, C1) had a flake-like shape. Annealing treatment did not change this morphology, as shown in Fig. 1c, C3. However, ball milling treatment has changed the morphology substantially of C1. There is no typical flake-like structure in the milled graphite sample (Fig. 1b and d). Instead, the milled samples contain irregular sphere-like structures with a wide distribution of particle sizes. After annealing of the milled graphite, abundant edges are found in C4, as high resolution TEM images of C2 and C4 are shown in Fig. S2.†
XRD patterns of the four samples are consistent with the SEM imaging results. The starting (C1) and heated (C3) graphite samples have similar XRD patterns, showing most intensive (002) peaks at ∼26.5 degrees and some other weak peaks at ∼45 degrees (100, 101), 55 degrees (004) and 77 degrees (110). However, after ball milling, the XRD pattern of C2 shows a much broadened peak of (002), which suggests a structural distortion and the reduction of crystal size. Comparing C4 with C2, the FWHM of peaks becomes narrower after annealing, indicating that annealing refines the graphitic structure. This structure refinement is expected to increase the sample's conductivity.28
XPS was used to characterize the element contents of the samples. As expected, in the XPS analysis of sample C1, the C 1s peak (for carbon) was the only strong peak found with a very small O1s peak, corresponding to approximately 2.2% oxygen. After ball milling treatment (sample C2), approximately 7.5% oxygen and 6.3% nitrogen were found. The high nitrogen content in the milled sample indicates that the milling process led to nitrogen incorporation into the graphite. The rise in the oxygen level in C2 might be caused by residual oxygen in the milling chamber or atmosphere contamination before XPS measurement. Both nitrogen and oxygen contents were reduced down to approximately 1.0% for nitrogen and 0.7% for oxygen (sensitivity of the analysis was about 0.1%) (Fig. S3 and S4†) after the annealing treatment (sample C4). As a comparison, direct annealing of commercial graphite in the N2–H2 mixture (sample C3) did not introduce detectable nitrogen using XPS. The possible mechanism of the ball milling in a nitrogen atmosphere to act as an effective process in the synthesis of a N-doped graphite catalyst is that the mechanical ball milling process introduces active sites such as unpaired bonds during the milling, resulting in nitrogen covalent bonding to the carbon surface.8
XPS N1s spectra were analysed to characterize the bonding nature of the nitrogen in the catalysts since different states of nitrogen could result in different catalytic properties.29 The N 1s spectrum of sample C2 (Fig. 3a) can be fitted by 4 components at 398.8 eV, 399.7 eV, 401.1 eV and 403.6 eV, corresponding to the nitrogen in pyridinic (N1), pyrrolic (N2), graphitic (N3) and nitric oxide (N4) structures.6,30 After annealing (sample C4, Fig. 3b), these three components remained while the pyrrolic structure (N2) disappeared and the relative amount of the graphitic structure (N3) increased from 43% to 66%. This phenomenon was consistent with previous research that the graphitic structure (N3) had better thermal stability than pyridinic and pyrrolic structures.29
 | ||
| Fig. 3 High resolution N1s XPS of (a) C2 and (b) C4. The signal of C2 can be fitted with 4 components at 398.8, 399.7, 401.1, 403.6 eV, which correspond to pyridinic N (N1), pyrrolic N (N2), graphitic N (N3), and nitric oxide (N4). After annealing, pyrrolic N (N2) disappears and nitric oxide (N4) peak shifts to 403.1 eV. | ||
The particle size distribution was measured to characterize how the particle size changed after annealing (Fig. 4). C2 had an average particle size as 210 nm; the particle size became 348 nm after annealing (C4). This phenomenon might be caused by the agglomeration and the refinement of the graphitic structure during high temperature annealing, which could be seen from XRD patterns.31
 | ||
| Fig. 4 Particle size measurement of (a) C2 and (b) C4. The average particle size is 210 nm for C2 and 348 nm for C4. | ||
Fig. S4† shows the thermogravimetric analysis (TGA) of the ball milled graphite. The heating process was programmed to be the same as the annealing process. C1 shows little weight loss during the annealing. Both C2 and C4 show a weight loss about 3% below 100 °C, which might be contributed by the absorbed moisture. We noticed C2 has steady mass loss during the annealing while C4 shows little significant weight loss after 100 °C. This difference might be due to the reduction of carbon oxide groups and the loss of thermal unstable nitrogen doping of C2.
UV-Vis absorption spectroscopy was used to characterize the dispersive and hydrophilic property of samples (Fig. 5). C1 and C3 could not form a stable dispersion in water. However, C2 and C4 could be dispersed in water, as reflected by the presence of a UV-Vis absorption band of the aromatic system in graphite.32 This enhanced dispersive ability and hydrophilicity are likely to be due to the changed surface property and the decreased average particle size created by ball milling.33 Indeed, the introduction of N and O doping26 has been proved to be able to increase the hydrophilicity. This increased hydrophilicity may be responsible for the improved catalytic property by enabling enhanced mass exchange rates and the stability of the three-phase zone.34,35
 | ||
| Fig. 5 UV-Vis absorption of four samples after ultrasonic treatment. C1 and C3 quickly precipitate after ultrasonic treatment and indicate no UV-Vis absorption (inset: C1–C4 from left to right taken after 30 min of ultrasonication). | ||
Fig. 6 shows ORR in 0.1 M KOH using thin films of C1, C2, C3 and C4 deposited on the RDE rotated at 1600 rpm. As a control experiment, Fig. 7 was obtained under the same conditions confirming the absence of reduction currents for typical carbons e.g. C2 and C4 in the absence of oxygen. The ORR activity is enhanced in the order of C3, C1, C2 and C4. The ORR enhancement is evident in both the current onset and density e.g. a sample showing more positive onset potential value exhibits a larger current density. C4, in particular exhibits a similar onset potential value and higher current density in excess of −0.6 V with respect to Pt/C (40 wt% Pt), the standard benchmark for ORR in conventional fuel cells, implying the remarkable potential of the proposed doping method to synthesize alternative ORR catalysts. This is due to the N doping and the breaking of the graphite basal plane structure. Nitrogen doped carbon materials have been reported to have unique ORR catalytic property both theoretically and experimentally.1,4,5,29,36–39 At the same time, it has been shown that the basal-plane aromatic ring structure of graphite is very inert to chemical reactions and electron transfer,40–43 which implies that defects and boundary carbon atoms are the real active sites where chemical reactions and catalytic processes take place. As demonstrated, ball milling could result in a large quantity of structural defects and even change the morphology, which would increase the quantity of active catalytic sites. However, it is interesting to compare the RDE results of C1, C2 and C4. In fact, the current density of ORR does not change much after only the ball milling process, while the potential onset shifts to be more positive than C1. The onset shift is due to the nitrogen doping, while the reason for the low current may be that the damaged structure leads to reduced conductivity. The reduced conductivity causes a limitation of catalytic current and more energy consumption when charge transfer occurred in the material, which could compensate the energy saving brought by the catalytic process. Another possible reason is that although the ball milling induced nitrogen doping, the doped nitrogen is not catalytically active. XPS uses bonding energy to differentiate different nitrogen doping. It is possible that some nitrogen doping or absorbed nitrogen have not any catalytic contribution (which had bonding energy within the fitting area) to the extent that the catalytic nitrogen doping might be less than the fitting showed. After annealing, the catalytic property enhanced even though the nitrogen content was much lower, reduced from 6.3% to 1%. Previous research suggests that the chemical structures of nitrogen doping with active catalytic properties are pyridinic and graphitic nitrogen.29 However, in this case, XPS shows that the content of all kinds of nitrogen doping decreases after annealing. Thus, in this work, the catalytic property of the sample is not quantitatively related to the nitrogen content.
 | ||
| Fig. 6 Rotating disk electrode (RDE) voltammograms of C1–C4 and Pt/C reference at 1600 rpm in O2 saturated 0.1 M KOH at a scan rate of 20 mV s−1. | ||
 | ||
| Fig. 7 RDE voltammograms of C2 and C4 in 0.1 M KOH saturated with N2 or O2 at a rotating rate of 1600 rpm. | ||
To determine the ORR characteristics, the relationship between the current density and the rotation rate was examined. The typical ORR current density profile versus rotation rate is shown in Fig. 8 for sample C4. Fig. 9 plots the typical inverse of the current density as a function of the inverse of the root square of the rotation rate e.g. the Koutecky–Levich plots.44,45 The slope seems to vary between C3/C1 and C2/Pt/C/C4. Considering the widely established facts that ORR on Pt/C occurs through a four-electron mechanism whereas conventional carbon through a two-electron mechanism,44,46 it is most likely that (within the experimental error), ORRs on C3 and C1 follow a two-electron mechanism while ORRs on C2, Pt/C and C4 follow a four-electron mechanism pathway. The shift of the mechanism from two-electron to four-electron is consistent with the ORR activity enhancement trend observed above. Also, the kinetic current density (Jk) could be calculated from the y-axis intercept of Koutecky–Levich plots. The Jk increased in the order of C3, C1, C2 and C4, the same with the order of potential onset increase. The Jk of C3 was 4.2 mA cm−2 and 5.2 mA cm−2 for C4. C4 showed a high kinetic current density approaching that of Pt/C (7.5 mA cm−2). These results suggest that ball milling has introduced catalytic nitrogen doping in the graphite. After ball milling, the mechanism of ORR changed from a less efficient 2-electron mechanism of graphite to a mixture of 2-electron and 4-electron mechanisms, which is due to the nitrogen doping.29 On the other hand, the refinement of annealing could also be seen from both the increase of Jk and the more preferential 4-electron mechanism of C4. The increase of Jk may be caused by the refined graphitic structure.
 | ||
| Fig. 8 Rotating disk electrode (RDE) linear sweep voltammograms of C4 in O2 saturated 0.1 M KOH with different rotation rates at a scan rate of 20 mV s−1. | ||
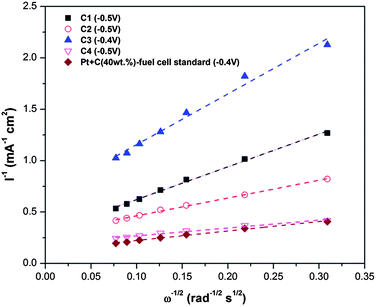 | ||
| Fig. 9 Koutechy–Levich plots derived from RDE voltammograms (at 1600 rpm) of different materials. | ||
Furthermore, we did not see the change of catalytic current of C2 compared with commercial graphite, but was significantly improved after annealing. Thus, the annealing is considered as a structure refined step of the milled samples. Our results showed ball milling of graphite in nitrogen could distort the graphitic structure and reduce the size of the (0 0 2) plane. As graphite is an anisotropic material, which has significant anisotropic electro-conductivity, the distorted graphitic structure and the tailored (0 0 2) plane may lead to an increased electronic resistance, which reduces the performance and limits the catalytic current. The annealing, in this case, is used to refine the graphitic structure and improve the conductivity. After annealing, as our results show, the FWHM of the (0 0 2) peak was smaller, which suggests the refinement of the graphitic in-planar structure. In the annealing atmosphere, N2–15%H2 is not compulsory. In fact, the annealed samples in pure N2 were also tested and proved to have catalytic properties. However, as shown in XPS, the milled samples contain oxygen. In order to reduce the oxygen level, we chose a reducing atmosphere.
In the real fuel cells, the organic compounds like methanol at the anode would penetrate the membrane and reach the Pt cathode, which could change the chemical reaction of oxygen reduction to the oxidation of those organic compounds.47 This phenomenon is called the poisoning of catalyst. The advantage of carbon-based materials becomes apparent in terms of its methanol resistance. The chronoamperometric response was obtained at −0.35 V (Fig. 10). We found that introducing methanol leads to slight deterioration of the ORR current for C4. For Pt/C, however, methanol oxidation occurs and causes a total loss of cathodic current. This indicates a much better stability of reaction sites in C4 than Pt/C in an alkaline environment, of interest for advanced fuel cells.
 | ||
| Fig. 10 Chronoamperometric responses of C4 and Pt/C at −0.35 V in O2 saturated 0.1 M KOH without and with methanol at a rotating rate of 1600 rpm. | ||
Conclusions
High-performance ORR electrocatalysts can be synthesized based on nitrogen-doped carbon nanoparticles via ball milling and annealing processes. The nitrogen doped carbon nanostructures exhibit a high activity and excellent tolerance to methanol in comparison to other non-precious metal catalysts. The synthesis strategy demonstrated here exploits novel carbon-based structures, and could represent a general approach to highly efficient electrocatalysts for a range of important electrochemical reactions, which is low-cost, and easy to scale up, providing a feasible method for development of large-scale practical electrocatalysts.Notes and references
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2012, 51, 3892–3896 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- J. Cao, Q. Wang and H. Dai, Nat. Mater., 2005, 4, 745–749 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- X. Chen, H. Yang, G. Wu, M. Wang, F. Deng, X. Zhang, J. Peng and W. Li, J. Cryst. Growth, 2000, 218, 57–61 CrossRef CAS.
- G. Kaupp, CrystEngComm, 2009, 11, 388–403 RSC.
- H. Rojas-Chávez, S. Díaz-de la Torre, D. Jaramillo-Vigueras and G. Plascencia, J. Alloys Compd., 2009, 483, 275–278 CrossRef.
- F. Zhang, W. Kaczmarek, L. Lu and M. Lai, J. Alloys Compd., 2000, 307, 249–253 CrossRef CAS.
- Y. Chen, J. Williams and G. Wang, J. Appl. Phys., 1996, 79, 3956–3962 CrossRef.
- Y. Chen and J. S. Williams, J. Mater. Res., 1996, 11, 1500 CrossRef CAS.
- Y. Chen, Z. Li and J. Williams, J. Mater. Sci. Lett., 1995, 14, 542–544 CrossRef CAS.
- Y. Chen, J. D. Fitz Gerald, J. S. Williams and S. Bulcock, Chem. Phys. Lett., 1999, 299, 260–264 CrossRef CAS.
- Y. Chen, J. D. Fitz Gerald, L. T. Chadderton and L. Chaffron, Appl. Phys. Lett., 1999, 74, 2782–2784 CrossRef CAS.
- Y. Chen, M. J. Conway, J. D. Fitz Gerald, J. S. Williams and L. T. Chadderton, Carbon, 2004, 42, 1543–1548 CrossRef CAS.
- W. Zhao, M. Fang, F. Wu, H. Wu, L. Wang and G. Chen, J. Mater. Chem., 2010, 20, 5817–5819 RSC.
- I.-Y. Jeon, H.-J. Choi, S.-M. Jung, J.-M. Seo, M.-J. Kim, L. Dai and J.-B. Baek, J. Am. Chem. Soc., 2013, 135(4), 1386–1393 CrossRef CAS.
- V. León, M. Quintana, M. A. Herrero, J. L. Fierro, A. de la Hoz, M. Prato and E. Vázquez, Chem. Commun., 2011, 47, 10936–10938 RSC.
- Y. Chen, T. Halstead and J. S. Williams, Mater. Sci. Eng., A, 1996, 206, 24–29 CrossRef.
- U. Paulus, T. Schmidt, H. Gasteiger and R. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- T. Schmidt, H. Gasteiger, G. Stäb, P. Urban, D. Kolb and R. Behm, J. Electrochem. Soc., 1998, 145, 2354 CrossRef CAS.
- Y. Chen, M. Conway, J. Williams and J. Zou, J. Mater. Res., 2002, 17, 1896–1899 CrossRef CAS.
- L. Li, L. H. Li, Y. Chen, X. J. Dai, T. Xing, M. Petravic and X. Liu, Nanoscale Res. Lett., 2012, 7, 417 CrossRef CAS.
- T. Shen, W. Ge, K. Wang, M. Quan, J. Wang, W. Wei and C. Koch, Nanostruct. Mater., 1996, 7, 393–399 CrossRef CAS.
- Y. Kuga, M. Shirahige, Y. Ohira and K. Ando, Carbon, 2002, 40, 695–701 CrossRef CAS.
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- L. Wang, X. Yue, F. Zhang and R. Zhang, J. Alloys Compd., 2010, 497, 344–348 CrossRef CAS.
- D. Long, J. Y. Hong, W. Li, J. Miyawaki, L. Ling, I. Mochida, S. H. Yoon and J. Jang, ACS Nano, 2011, 5, 6254–6261 CrossRef CAS.
- W. Wu and G. H. Nancollas, Adv. Colloid Interface Sci., 1999, 79, 229–279 CrossRef CAS.
- K. Horita, Y. Nishibori and T. Ohshima, Carbon, 1996, 34, 217–222 CrossRef CAS.
- S. Shanmugam and A. Gedanken, J. Phys. Chem. B, 2006, 110, 2037–2044 CrossRef CAS.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith and M. Jaroniec, J. Am. Chem. Soc., 2011, 133, 20116–20119 CrossRef CAS.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS.
- S. Kattel, P. B. Atanassov and B. Kiefer, J. Phys. Chem. C, 2012, 116, 17378–17383 CAS.
- C. E. Banks, T. J. Davies, G. G. Wildgoose and R. G. Compton, Chem. Commun., 2005, 829–841 RSC.
- R. J. Bowling, R. T. Packard and R. L. McCreery, J. Am. Chem. Soc., 1989, 111, 1217–1223 CrossRef CAS.
- K. K. Cline, M. T. McDermott and R. L. McCreery, J. Phys. Chem., 1994, 98, 5314–5319 CrossRef CAS.
- K. Ray III and R. L. McCreery, Anal. Chem., 1997, 69, 4680–4687 CrossRef.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1263–B1268 CrossRef CAS.
- S. Treimer, A. Tang and D. C. Johnson, Electroanalysis, 2002, 14, 165–171 CrossRef CAS.
- L. Jörissen, J. Power Sources, 2006, 155, 23–32 CrossRef.
- W. Chen, G. Sun, J. Guo, X. Zhao, S. Yan, J. Tian, S. Tang, Z. Zhou and Q. Xin, Electrochim. Acta, 2006, 51, 2391–2399 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Demonstration of mass fabrication of the catalyst, high-resolution TEM images, XPS survey of samples, and TGA of the ball milled graphite. See DOI: 10.1039/c3nr02328a |
| This journal is © The Royal Society of Chemistry 2013 |