 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular self-assemblies as functional nanomaterials
Eric Busseron, Yves Ruff, Emilie Moulin and Nicolas Giuseppone*
SAMS Research Group, University of Strasbourg, Institut Charles Sadron, CNRS, 23 rue du Loess, BP 84087, 67034 Strasbourg Cedex 2, France. Web: http://www-ics.u-strasbg.fr/samsE-mail: giuseppone@unistra.fr
First published on 29th May 2013
Abstract
In this review, we survey the diversity of structures and functions which are encountered in advanced self-assembled nanomaterials. We highlight their flourishing implementations in three active domains of applications: biomedical sciences, information technologies, and environmental sciences. Our main objective is to provide the reader with a concise and straightforward entry to this broad field by selecting the most recent and important research articles, supported by some more comprehensive reviews to introduce each topic. Overall, this compilation illustrates how, based on the rules of supramolecular chemistry, the bottom-up approach to design functional objects at the nanoscale is currently producing highly sophisticated materials oriented towards a growing number of applications with high societal impact.
1 Introduction
Self-assembly rests on the autonomous organization of components producing patterns or structures at any scale.1 More particularly, at the nanoscale, it reflects the information encoded in individual molecules such as shape, charge, polarizability, etc. that determine their attractive or repulsive interactions.2 Molecular self-assembly usually takes advantage of supramolecular interactions (ionic, hydrophobic, van der Waals, hydrogen and coordination bonds), but can also make use of kinetically labile covalent bonds.3 This intrinsic mobility leads to ordered nanostructures upon equilibration between aggregated and non-aggregated states, thus providing a number of interesting properties such as error correction, self-healing, and high sensitivity to external stimuli. These structuring features are nowadays well understood and can be finely controlled in order to introduce and tune functional properties of self-assembled nanomaterials used for a very wide range of applications. The goal of this review is to illustrate the current evolution of this field from fundamental to applied research, with striking implementations in biomedical sciences, information technologies, and environmental sciences. Within each of these three topics, and after a brief introduction referring to more comprehensive reviews and books, we have made a selection of the major achievements published recently (mainly during the last six years). Within these chapters, the literature has been organized depending on the functions targeted and then following a structural classification of the nano-objects involved. Some concluding remarks will try to determine what could be the next challenges of this fascinating research area. We hope that this review will constitute a useful tool for the non-specialized readers who want to get an overview of the current trends related to self-assembled nanomaterials, or for experts who want to look for a precise entry in a particular domain of application.2 Self-assembled nanomaterials for biomedical applications
Very recently, the physical and chemical properties of nanomaterials have inspired scientists to create new devices that are able to interact with biological systems at the nanoscale. While still in its early stages of development, the field of nanotechnology has already made a tremendous impact in medicine and biology in general.4,5 In particular, the field of nanobiotechnology – which refers to the use of biological systems and molecules to build nanoscale materials – is currently leading to promising applications in diagnostics and therapeutics.6,7 In this chapter we will illustrate the biomedical applications of various types of nanomaterials obtained by self-assembly (or which requires a self-assembly event with a substrate for producing the targeted function). In the first section, we will give a selection of the most recent advances in the field of diagnostics and sensing8,9 using self-assembled nanomaterials and including sensors based on optical, magnetic, and electrochemical detection. The second section will focus on recent examples of self-assembled nanomaterials for therapeutic applications. We will first highlight the use of nanomaterials for gene and drug delivery,9,10 in particular viruses, analogs of viruses, micellar systems, carbon nanotubes, inorganic/organic hybrid nanostructures, nanomaterials based on lipids or amphiphiles, and DNA. We will then describe recent applications of nanomaterials for regenerative medicine and tissue engineering.102.1 Diagnostics and sensing
Sensing of molecules for biomedical purposes requires the highest specificity and sensitivity to detect markers of diseases or active species such as drugs. Reduction of the cost and time of analysis is also an important consideration leading to increasingly miniaturized, non-destructive, and easy-to-read assays. In the following sections, we will illustrate the most recent advances in nanomaterials for biosensing applications, with detection modes based on optical, magnetic, or electrochemical assays.Gold nanoparticles. Among the various nanomaterials used for biosensing, nanoparticles are of high interest due to their inherent properties, i.e. size, high loading of signal tags, high surface area, and dynamic character for signal amplification.11,12 A popular approach rests on the self-assembly of metals, and especially gold nanoparticles,13–16 decorated by ligands interacting with biomolecules. The optical properties of gold nanoparticles are ruled by their absorption at the surface plasmon resonance wavelength. This absorption is highly dependent on the chemistry taking place at the surface of the nanoparticles (e.g. in interaction with analytes) and on particle–particle interactions.17 For example, the aggregation of gold nanoparticles results in a visible color change from red to blue due to inter-particle coupling.13,15,17
Mirkin et al. pioneered this field with the introduction of single stranded DNA (ssDNA) decorated gold nanoparticles which can be used to detect extremely low concentrations of oligonucleotides through specific DNA sequence hybridization inducing aggregation.18 When 13 nm gold nanoparticles are functionalized with DNA sequences complementary to a target DNA molecule, base pairing induces aggregation of the particle, resulting in a visible color change due to interacting particle surface plasmons and aggregate scattering properties (Fig. 1a). These complexes also have extremely sharp melting transition temperatures allowing detection of even a single base pair mismatch of the complementary ssDNA.18 The highly selective linking of nanoparticles bearing complementary ssDNA sequences can even lead in some cases to highly ordered crystalline lattices where gold nanoparticles replace the atoms in traditional molecular crystals, following a new set of rules for the self-assembly of nano-objects.19
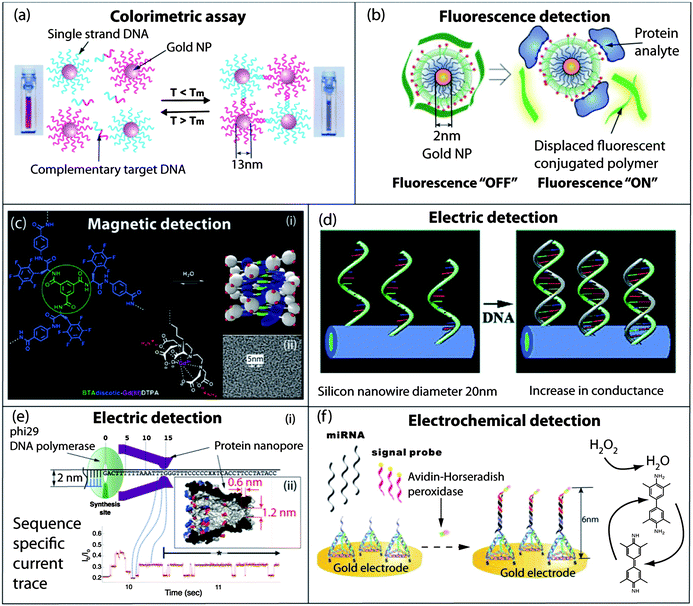 | ||
| Fig. 1 Self-assembled nanomaterials for diagnostics and sensing. (a) Colorimetric detection of single stranded DNA by hybridization with gold nanoparticles decorated with complementary single stranded DNA. The resulting aggregates, which display very sharp melting transitions, are easily observed by a visible color change or by UV-Vis spectroscopy. (b) Fluorescence detection of proteins by displacement of quenched conjugated polymers bound at the surface of a gold nanoparticle. The extent of the polymer release in solution and the resulting fluorescence are dependent on the nature of the interaction occurring between the protein and the cationic and hydrophobic groups in the nanoparticles' corona. (c) (i) Self-assembly of a fluorinated benzene-1,3,5-tricarboxamide in well-defined water soluble paramagnetic nanostructures as efficient MRI contrast agents; (ii) cryo-TEM micrograph of the self-assembled amphiphile in a serum albumin PBS buffered solution. (d) Conductance increase of silicon nanowires decorated with ssDNA upon hybridization with complementary DNA fragments. (e) (i) Self-assembled MspA protein nanopore used for the sequencing of single DNA molecules; (ii) crystal structure of the MspA protein. Reading of the sequence of the DNA template is achieved through the change in ionic current upon the translocation of the nucleobases through the pore, across a membrane separating two compartments. (f) Ultrasensitive electrochemical assay for the detection of miRNA using a self-assembled DNA tetrahedron for the spatial and accessibility control of an electrochemical probe, horseradish peroxidase. Panel (a): reproduced with permission from ref. 14; panel (b): reproduced with permission from ref. 28; panel (c): reproduced with permission from ref. 41; panel (d): reproduced with permission from ref. 47; panel (e): reproduced with permission from ref. 58; panel (f): reproduced with permission from ref. 65. | ||
The self-assembly of nanoparticles mediated by biomolecules is not limited to nucleic acid detection and has also been extended to the sensing of proteins along the formation of antibody–antigen complexes. In particular, nanoparticle probes that are reversibly encoded with a DNA tag can reach a limit of detection for a cancer marker at a concentration as low as 30 attomolars.20 In this assay, nanoparticles decorated with antibodies and heavily functionalized with double stranded DNA barcodes are used to detect immunocomplexes formed by the prostate specific antigen (PSA) and PSA monoclonal antibody functionalized magnetic microparticles, after isolation of the complexes with a magnet and dehybridization of the DNA marker. Therefore a single binding event between an antibody and the target molecules translates into the release of multiple copies of the barcode DNA. Scanometric detection of this barcode can then be achieved at a concentration higher than that of the initial analyte. Indeed, an advantage of using DNA as a barcode is that further amplification of the signal can be achieved using polymerase chain reaction, effectively lowering the detection limit to 3 attomolar.20 Recently a similar bio-barcode assay was applied to the detection of amyloid diffusible ligands, which are biomarkers for Alzheimer's disease, in the cerebrospinal fluid of 30 patients at femtomolar (fM) concentration.21
The self-assembly of gold nanoparticles has also been used for the optical detection of enzymes because their activity can either promote or suppress the interparticle supramolecular interactions.22 For example, Stevens et al. have shown that protease activity can be detected through the breaking down of peptides that maintain gold nanoparticles in an aggregated state, resulting in a visible red shift of the sample color due to the dispersion of individual nanoparticles.23 The colorimetric detection of proteins was also achieved by Liedberg et al. using specific interactions between a model protein, the human carbonic anhydrase, and a synthetic receptor present at the surface of gold nanoparticles to hinder their aggregation by self-assembly.24 A similar strategy using the interaction of a peptide derived from a viral protein and an antibody to prevent the aggregation of gold nanoparticles allowed the detection of the viral peptide with a detection limit of 10 nM.24
The reverse approach using aggregation of gold nanoparticles to detect enzymatic activity was reported by Stevens et al., who used the immuno-aggregation of nanoparticles after kinase mediated phosphorylation of tyrosine residues in peptides.25 Indirect detection of enzyme activity has also been reported.26 In this system, treatment of liposomes with phospholipase triggers the release of a peptide specifically designed to recognize a second polypeptide immobilized on gold nanoparticles. Again, this event results in a visible color change due to the aggregation of nanoparticles.26
As an alternative to colorimetric assays, the detection of cancer biomarkers using dynamic light scattering has been proposed by Huo et al.27 with the advantages that the reaction takes place directly in solution and that it requires smaller amount of samples. The quantitative determination by DLS of the ratio between antibody decorated gold nanoparticles and larger immunocomplexes obtained after addition of the prostate specific antigen (PSA) allows the detection of the antigen at very low concentrations (0.5 ng ml−1).27
Besides assays that are based on the self-assembly of nanoparticles, Rotello et al. have used the reversible character of supramolecular complexes between charged nanoparticles and oppositely charged fluorescent conjugated polymers to elaborate “chemical noses” for the detection and identification of proteins (Fig. 1b).28,29 The principle here consists in mixing a protein of interest separately with six different gold nanoparticles/conjugated polymer complexes, and in recording on a plate reader the individual fluorescence of each of the six samples. The polymers that have been displaced by the electrostatic interactions between the protein and the charged nanoparticles are responsible for the increase of fluorescence. As for the six different complexes the dissociation occurs to different extents and the resulting fluorescence pattern is specific to each protein. This assay was successfully used to identify 52 unknown protein samples with an accuracy of 94.2%.28
Supramolecular gels. As for enzyme-responsive nanoparticle aggregation–dissociation assays, enzyme triggered assembly–disassembly of small molecules can also provide a readout for the sensing of glucose oxidase30 or proteases.31 This can be achieved through the formation of a macroscopic gel using a peptide precursor containing cysteine residues and 2-cyanobenzothiazole units. This gelation occurs by the condensation of these two functions after treatment with a protease or by disulfide reduction. The gel formation can take place in vitro but also inside living cells,31 enabling for instance the imaging of the proteolytic activity of furin. A similar system was used by Liang et al. to design a sensor for caspase-3 based on the fluorescence enhancement of fluorescein–streptavidin conjugates upon mixing with self-assembled biotinylated nanospheres.32 Sensing of enzymatic activity can also be achieved indirectly upon a pH change induced by glucose oxidase, resulting in the assembly or disassembly of pH-sensitive peptide amphiphiles.30
Arrays. The adaptative change in optical properties of dynamic self-assembled small molecules upon the introduction of an analyte can also be used for detection. Arrays of self-assembled complexes have been used in colorimetric assays for the detection of carbohydrates.33 Reversible covalent bonds between carbohydrates and boronic acids allow for the dynamic rearrangement of boronic esters in carbohydrate–chromophore conjugates. For example, the addition of a polyol indicator to a library of solid-supported boronic esters of carbohydrates results in the displacement of the sugar. The rate and extent of the displacement are functions of the stability of the carbohydrate boronic ester and colorimetric arrays can be obtained to identify specific carbohydrates.34 More examples of arrays based on dynamic supramolecular complexes can be found in the following section on nanomaterials for environmental sciences.
DNA nanostructures. Self-assembled DNA nanostructures loaded with intercalator dyes have also been used as nanotags with potential use for tracking and labelling biomolecules (proteins, antibodies, nucleic acids, etc.). Indeed, when chromophores are protected and concentrated within a small volume, they display enhanced resistance to nuclease digestion and improved photostability.35 Using this approach, the co-assembly of donor and acceptor dyes has led to efficient energy transfer systems with tunable emission wavelength.35 More recently Shih et al. have described fluorescent barcodes obtained by the positioning of fluorophores on the surface of a stiff self-assembled DNA nanorod or on non-linear nanostructures.36 These nanoscale barcodes are observable by epifluorescence or total internal reflection fluorescence microscopy and can be used to tag yeast surface receptors, which suggests their potential applications as in situ imaging probes.36 Self-assembled molecular logic gates based on DNA hybridization can also be used for the optical detection of M. tuberculosis DNA.37 More examples of DNA-based logic gates are described in the following section on nanomaterials for information technology.
Iron nanoparticles. The formation of large supramolecular aggregates of super-paramagnetic iron oxide nanoparticles has a direct effect on the spin–spin relaxation time of billions of neighboring water molecules, resulting in signal amplification.38 Immuno-aggregation of super-paramagnetic iron oxide nanoparticles, decorated with specific antibodies, and multivalent bacteria or cancer cell surfaces resulted in the formation of 300 nm clusters of magnetic nanoparticles that can be used in miniaturized NMR assays to detect bacteria or cancer markers in biological fluids.39
Self-assembling paramagnetic gadolinium complexes. The incorporation of paramagnetic metal complexes into self-assembled structures is also of great interest as it allows the preparation of NMR contrast agents with well-defined morphology and dimensions.40,41 This strategy combines the benefits from low molecular weight imaging agents with the ones of macromolecular contrast agents, as self-assembled nanostructures are degradable through their ability to disassemble into their individual components, while still showing enhanced relaxivity in their assembled states. Specifically, self-assembled dendritic-like nanoparticles have been prepared by the self-assembly of a C3-symmetrical benzene-1,3,5-tricarboxamide (BTA) core that directs the self-assembly into triple hydrogen-bonded helices decorated by Gd(III)–DTPA (diethylenetriaminepentaacetic acid) chelates (Fig. 1c).40 A high degree of control on the size and shape of these supramolecular polymers was achieved in water by tuning the electrostatic repulsion between the monomers.40 The stability and potency for contrast-enhanced MRI have been demonstrated both in vivo and in vitro.41 Controlled intracellular self-assembly of paramagnetic gadolinium complexes has also been described recently by Liang et al. In this system, the condensation between 2-cyanobenzothiazole and N-terminal cysteine has been used to detect the degradation of paramagnetic peptidic precursors by furin, a protease overexpressed in tumors. The increase in relaxivity upon condensation and subsequent self-assembly were used to detect tumors in vivo (mouse model).42
Gold nanoparticles. Mirkin et al. have developed an elegant process to take advantage of the selective hybridization of a target DNA with complementary ssDNAs grafted both on a surface between electrodes and on the surface of gold nanoparticles. This leads to the localization of the gold nanoparticles between electrodes which facilitates silver deposition, thus resulting in a measurable increase in conductivity. Target DNA concentrations as low as 500 fM were detected with a point mutation selectivity factor of 100
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000:1.43
000:1.43Semiconducting nanowires. The small diameter of the nanowires allows a large change in conductance upon binding of charged biomolecules at their surface. This strong effect rests on the charge carrier depletion/accumulation within the entire diameter of the wire.12 Therefore, field-effect transistors based on such nanostructures have been extensively used for the detection of biomolecules.44–46 Lieber et al. pioneered the use of semiconducting silicon nanowires for the design of DNA sensors. They took advantage of the change of conductivity upon binding of the negatively charged ssDNA to silicon nanowires decorated with neutral peptide nucleic acid sequences (Fig. 1d).47 Concentration of ssDNA as low as 10 fM can be detected. More recently similar devices have been used for the detection of ssDNA under physiological buffer and salt concentration conditions.48
Carbon nanotubes. The conducting properties and high aspect ratio of carbon nanotubes have been used to design field-effect transistors for the sensing of biomolecules, in particular DNA, antibody–antigen complexes, and sugars.49,50 The self-assembly of the carbon nanotubes themselves represents another strategy to build electrochemical sensors.51 The selective single-walled carbon nanotubes (SWNTs) assembly onto 16-mercaptohexadecanoic acid self-assembled monolayers deposited on gold electrodes facilitates electron transfer between the electroactive species and the electrode. Sensing was achieved through the binding of the protein of interest (a tumor biomarker folate receptor) with folate molecules attached to the extremity of ssDNA. This binding prevents the degradation of the DNA strand coated carbon nanotube by the nuclease Exo1. Therefore in the presence of the protein of interest, the coated nanotube does not interact with the working surface of the gold electrode. In the absence of the supramolecular protection provided by the protein, the ssDNA coating of the nanotube is degraded by Exo1 resulting in the absorption of the SWNT on the working surface of the electrode. A detection limit of 3 pM in the folate receptor was reached using this quantitative analysis for its interaction with folate.51
Self-assembled peptide nanowires. We have seen above that electrochemical activity of electrodes can be modified by the adsorption of nanostructures like carbon nanotubes on their surfaces. The nanostructures themselves can be obtained by self-assembly. For example, Gazit et al. deposited dense arrays of well-ordered self-assembled peptide nanotubes or nanospheres, obtained from Boc-protected phenylalanine dipeptides, at the surface of gold electrodes, and characterized by chronoamperometry.52 A 17-fold improvement in the sensing performance for phenol, with respect to the uncoated electrode, was observed. More recently, a conducting peptide/polypyrrole making use of vertically aligned diphenylalanine peptide nanowires was obtained by chemical polymerization and characterized by cyclic voltammetry.53 The resulting peptide/polypyrrole coated electrodes were used as dopamine sensors with a detection limit of 3.1 μM. Interestingly, cell growth experiments proved that the modified electrodes are suitable as cell culturing platform materials.
Self-assembled nanochannels. Self-assembled protein channels with nanometric dimensions are often found in nature to control the transport of ions or molecules across membranes. As crystallisable self-assembled proteins, the structures of such nanopores can be determined with atomic precision in some cases, and can also be expressed, modified, and optimized by genetic engineering, making them ideal candidates for reproducible use in sensing devices. In particular, the application of protein or solid-state nanopores as electrosensors has a tremendous potential to lower the cost of sequencing technologies.54,55 Indeed, if a voltage is applied between two compartments separated by a lipid bilayer, and permeated only by a single pore, the residual ionic current is going to be affected by the electrophoretic transport of molecules, or macromolecules, through the nanopore. For example, the channel created by α-hemolysin incorporated in a lipid bilayer is 2.6 nm wide, 5 nm long, and with a 1.4 nm bottleneck which means that ssDNA can pass through the nanopore while the double-stranded one cannot. This allows the threading of single ssDNA molecules through the pore. At any given time, the residual ionic current will be dependent on which nucleotide is passing through the pore. The covalent attachment through the disulfide bond of a cyclodextrin into the cavity of α-hemolysin allows for even better discrimination between molecules transported across the pores and to identify nucleosides with 99.8% accuracy.56 In principle, it is therefore possible to determine the sequences of bases of the ssDNA molecule. However, under electrophoretic conditions, ssDNA moves through the nanopores at very high velocities (one nucleotide per μs), which make correct identification of a given nucleotide difficult.54,55 Recently, two groups circumvented these limitations by slowing down the translocation of the ssDNA with enzymatic motors responsible for DNA replication. Akeson et al. described the forward and reverse ratcheting of DNA templates through hemolysin at 2.5–40 nucleotides per second.57 Their approach consisted in complexing a DNA template bearing a blocking oligomer with phi29 DNA polymerase (DNAP). A similar approach was independently reported by Gundlach et al. using DNAP to pull the ssDNA template through Mycobacterium smegmatis porin A (MspA) (Fig. 1e).58 They demonstrated that the residual current levels are related to the sequence of the individual ssDNA traversing the MspA nanopore. These impressive achievements demonstrate the preparation of nanoscale self-assembled sensors incorporating molecular motors to manipulate and control the translocation of ssDNA through a self-assembled nanoscale reading unit.
DNA-based nanostructures. The self-assembly properties of DNA have nowadays reached a level of refinement allowing the predictive self-assembly of 2D or 3D hierarchical structures with a high level of complexity.59–62 In particular, the ability to use the DNA scaffold to precisely position different molecular components in three dimensions can be used to build electrochemical sensing devices.63,64
Fan et al. have described the use of the self-assembled DNA tetrahedron to engineer the working surface of gold electrodes for ultrasensitive detection of miRNA.65 In this example, the hybridization of the target miRNA acts like a linker between the rigid DNA tetrahedron spacer and an electrochemical probe, namely the avidin–horse radish peroxidase conjugate (avidin–HRP). This results in the localization of the probe at a certain distance from the electrode surface, avoiding the crowding effect and improving mass transport at the water–electrode interface (Fig. 1f). The authors have demonstrated enhanced binding recognition at the gold electrode surface and drastically improved detection sensitivity up to attomolar amounts of miRNA with single base discrimination ability. Linear constructs involving the formation of sandwich-type complexes of DNA molecules to introduce a spacer between the working electrode and avidin–HRP were also described, allowing for a detection limit of 10 copies of the target DNA strand.66
The DNA scaffold itself can also serve as self-assembled binding pockets for the detection of analytes. For example, aptamers have been used to build electrochemical sensing devices for small hydrophobic molecules of biological interest like drugs.67,68
2.2 Therapy and delivery
It is clear that recent progress in nanotechnology has deeply impacted the field of drug delivery.69 In particular, self-assembled nanostructures are highly valuable for therapeutic and delivery applications. Their size, high loading capacity, and reversible character are major assets for the controlled release of therapeutics. In addition, biomolecules and biopolymers can be efficiently used to design such nanostructured materials that are of high interest to confer them on biocompatibility and bioactivity properties.70 In recent years, all four classes of biomolecules (peptides,70–73 lipids,74–76 nucleic acids59,77–80 and sugars81–85) have been studied as building blocks for self-assembled nanostructures, yielding increasingly complex and refined therapeutic agents and nanoscale vectors.Viruses. The archetype of a highly efficient nanoscale delivery agent is a virus, as its life cycle requires specific mechanisms for cell targeting, endocytosis, and hijacking of the cellular machinery through the intracellular release of the viral genome. These organisms have been optimized by billions of years of Darwinian evolution, yielding the most efficient vectors of nucleic acid into living organisms. Their diameters are in the range of 25–30 nm for icosahedral viruses, and in the range of 6–20 nm in diameter and 300–900 nm in length for filamentous viruses.86 If native viruses aim at carrying the viral genome into healthy cells for ensuring their reproductive cycle, the genetic manipulation of these natural viruses such as lentiviruses87 has led to human therapies88 for cancer or HIV (Fig. 2a).89
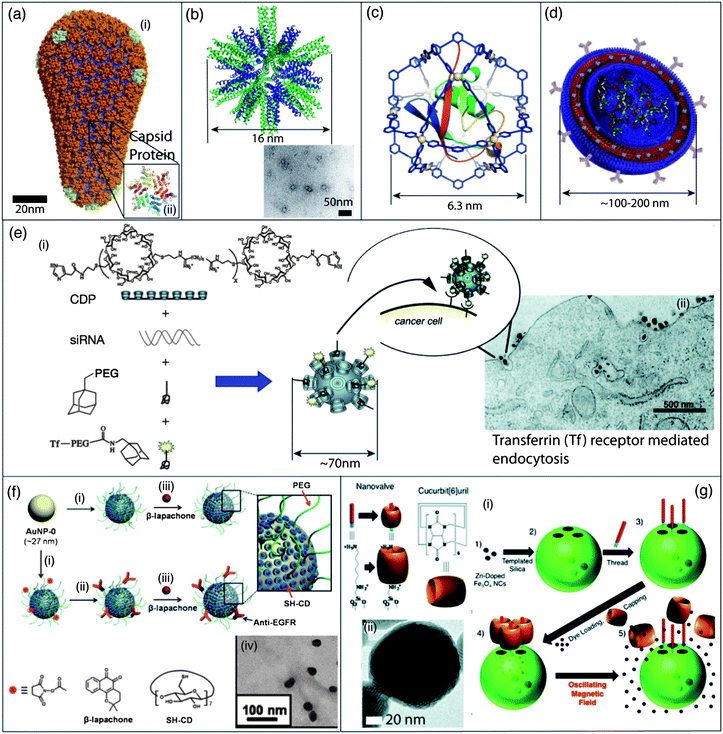 | ||
| Fig. 2 Self-assembled nanomaterials for gene and drug delivery. (a) (i) Atomic level molecular modeling of a type-1 HIV viral particle, a member of the lentivirus family;87 (ii) crystal structure in the hexameric form of disulfide type-1 HIV CA protein, one of the building blocks in the viral capsid. Self-inactivating mutants of the type-1 HIV virus are currently used for clinical gene therapies. (b) Molecular model of a peptide nanoparticle with dodecahedral symmetry formed by the self-assembly of a coiled-coil peptide with a pentameric domain (green) and a trimeric domain (blue) covalently linked to the same molecule and TEM micrograph of these peptide nanoparticles. (c) Molecular model of a discrete synthetic M12L24 nanocage containing a ubiquitin covalent conjugate. Self-assembly of Pd(II) ions (M) and additional ligands (L) results in the encapsulation of the protein into a well-defined coordination cage. (d) Schematic illustration of a polymersome decorated with antibodies for targeted delivery, and containing multiple cargos through incorporation into the hydrophobic portion of its membrane or through physical entrapment into its core. (e) (i) Schematic representation of the one-step self-assembly of four components into a targeted nanoparticle by the electrostatic interaction between siRNA and a cationic cyclodextrin containing polymer (CDP) and their inclusion complexes with adamantane (AD) derivatives on the surface of the nanoparticles; (ii) TEM micrograph of nanoparticles entering a cancer cell by receptor-mediated endocytosis. (f) (i) Functionalization of the surface of gold nanoparticles (AuNPs) with a cyclodextrin thiol derivative (SH-CD) and PEG-thiol (mPEG-SH or NHS-PEG-SH) chains; (ii) targeting was achieved through covalent decoration with anti-EGFR antibodies; (iii) a hydrophobic anti-cancer drug, β-lapachone can then be encapsulated into the hydrophobic cavity of the cyclodextrin; (iv) TEM micrograph of nanoparticles covered with cyclodextrin and PEG chains and loaded with β-lapachone. (g) (i) Preparation of mechanized silica nanoparticles loaded with a dye and zinc-doped iron oxide nanocrystals (ZnNCs). After attachment of the base of the molecular machine, the dye is loaded into the particle and the pores are blocked with the cucurbit(6)uril cap. Local heating upon the application of an oscillating magnetic field results in the dissociation of the nanovalve and release of the dye; (ii) TEM micrograph of ZnNCs encapsulated within mesoporous silica. Panel (a): reproduced with permission from ref. 87; panel (b): reproduced with permission from ref. 98; panel (c): reproduced with permission from ref. 101; panel (d): reproduced with permission from ref. 138; panel (e): reproduced with permission from ref. 136; panel (f): reproduced with permission from ref. 144; panel (g): reproduced with permission from ref. 148. | ||
Viruses can also be inactivated by the removal of their genetic information. Viral capsid proteins can be expressed in bacteria, isolated and self-assembled into homogeneous nanocarriers that have been used to develop novel drug and gene delivery agents.86 The viral capsids, self-assembled with or without a template, can be used to encapsulate enzymes, nucleic acids, or negatively charged polymers.90 Viral proteins can also be modified chemically by protein engineering and possibly by incorporation of unnatural aminoacids, to confer novel properties to these nanocontainers.86 Covalent modification of viruses with polyethylene glycol (PEG) residues and with targeting ligands for site-specific delivery is of particular interest for in vivo therapeutic applications.91 Viral particles are therefore a modular platform yielding precisely defined self-assembled icosahedral or filamentous nanoparticles that can be chemically tailored for specific biomedical applications.92
Self-assembled virus-like structures. Viral particles have several drawbacks such as their pathogenic and immunogenic properties. Expression of viruses or viral proteins also requires specialized techniques and synthetic analogs accessible by the traditional tools of organic synthesis are of high interest. What makes a nanostructure so-called “virus-like” is subject to interpretation, but very elegant approaches have been described to create self-assembled nanostructures with dimensions and architectures reminiscent of viruses.93,94 The highly complex icosahedral viral capsids are in reality composed of a limited number of C3-symmetric95 hexagonal or pentagonal protein building blocks.96 The first rational design of a C3-symmetric self-assembling β-sheet peptide to mimic the self-assembly of spherical viruses has led to the preparation of trigonal-(FKFE)2 peptides self-assembling into 20 nm nanospheres through antiparallel β-sheet formation.95 More recently, the same group reported the self-assembly of “tailor-made” hollow 30–50 nm virus-like nanocapsules, from 24-mer peptides able to self-assemble into a C3-symmetric β-annulus structure.97 In another study, regular polyhedral peptidic nanoparticles with a diameter of 16 nm were prepared by covalently linking coiled-coil peptides with C3- and C5-symmetry and applied to immunization experiments in a mouse model (Fig. 2b).98,99 Homogeneous virus-like micelles with a diameter of 20 nm were also prepared from the self-assembly of coiled-coil lipopeptides.100 The design of this peptide was optimized for the display of multiple copies of an antigen and used for the adjuvant-free stimulation of antigen-specific humoral immune responses for potential applications in vaccine development.100
Along their continuous efforts to build functional nanocages from synthetic organic ligands and metal ions (see Section 4.4), Fujita et al. have described, for the first time, the encapsulation of a native protein into a giant self-assembled nanocage with a diameter of 6.3 nm (Fig. 2c).101 When compared with viruses, these nanocages are smaller and lack several of their key functions. However, one can envision that, by an optimized design of the organic ligands, modifications will take place within or outside the nanocage in order to allow encapsulation of nucleic acid or targeted delivery.
To go one step further and design particles capable of binding nucleic acids, synthetic functional analogs of filamentous viruses have been developed by Lee et al. who designed β-sheet peptides which self-assemble into nanoribbons and form filamentous complexes with siRNA (Fig. 3a).102 These structures can be loaded with a hydrophobic dye and decorated with sugars for enhanced endocytosis in cancer cells. Another original strategy for the self-assembly of synthetic glycoviruses was described by Aoyama.103 Here, macrocyclic calixarene scaffolds were selectively decorated with sugars on one side and a hydrophobic tail on the other. These amphiphiles self-assemble irreversibly in 5 nm micelles, which can then be agglutinated by phosphate ions. The authors used this affinity for phosphate ions to complex plasmid DNA and obtained virus-like particles with a diameter of 50 nm that were able to achieve high transfection efficiency in vitro.103
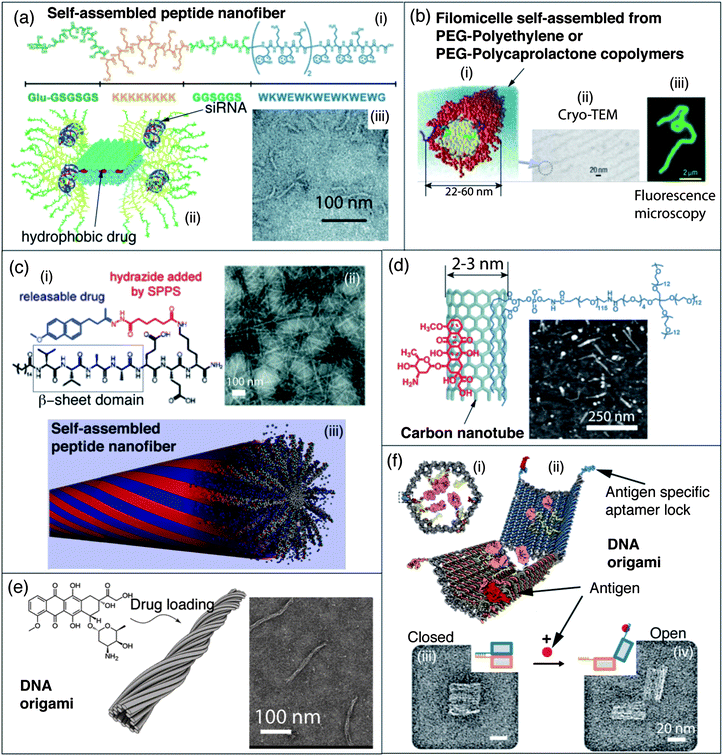 | ||
| Fig. 3 Self-assembled nanomaterials for gene and drug delivery. (a) (i) Structure of self-assembling peptide Glu-KW with a β-sheet segment (blue), flexible linker (light green), siRNA binding domain (orange) and hydrophilic targeting domain (dark green); (ii) schematic representation of artificial filamentous nanostructures used for siRNA delivery; (iii) TEM micrograph of filamentous nanostructures obtained with peptide Glu-KW. (b) (i) Schematic representation of filomicelles obtained by self-assembly of block copolymers used to probe the effect of nanostructures' shape for drug delivery in vivo; (ii) cryo-TEM micrograph of such a filomicelle, (iii) these filomicelles can be labelled with a hydrophobic fluorescent dye and observed by fluorescence microscopy. (c) (i) Molecular structure of a peptide amphiphile conjugated to a drug through a reversible covalent bond; (ii) TEM micrograph of self-assembled peptide nanofibers; (iii) this peptide amphiphile forms self-assembled peptide nanofibers in water. (d) Molecular structure of an anticancer drug (doxorubicin (DOX)) bound through stacking and hydrophobic interactions to a carbon nanotube (SWNT) dispersed by a PEGylated amphiphile, and an AFM micrograph of SWNT–DOX complexes. (e) Molecular structure of the anticancer drug doxorubicin, and schematic representation of a filamentous DNA origami (T-Nano) able to store doxorubicin, presumably via intercalation between nucleobases, and deliver it into cancer cells. A TEM micrograph of DOX loaded T-Nano DNA origami is given. (f) (i) Front view of a closed DNA origami nanorobot for the target transport of molecular payloads (antibody Fab' fragments in magenta, and gold nanoparticles in yellow); (ii) schematic representation of the open form of the nanorobot in which the aptamer lock is stabilized in a dissociated state by its antigen key (red), TEM micrographs of the corresponding (iii) closed and (iv) open states of the nanorobot. Panel (a): reproduced with permission from ref. 102; panel (b): reproduced with permission from ref. 157; panel (c): reproduced with permission from ref. 159 and 182; panel (d): reproduced with permission from ref. 168; panel (e): reproduced with permission from ref. 175; panel (f): reproduced with permission from ref. 177. | ||
Liposomes. Liposomes are closed spherical lipid bilayers obtained by the aqueous self-assembly of lipids. Their loading by therapeutic agents can be achieved by physical entrapment into the intra-liposomal aqueous phase or by diffusion within the hydrophobic section of the lipid bilayers. They were first described in 1965 (ref. 104) and are still under active clinical development today. Due to their ease of preparation, their fine tunable properties, and their functionalization through the coassembly of different lipids, liposomes are amongst the most common self-assembled nanoscale vectors for a wide range of therapeutic agents such as drugs or nucleic acids.105–107 Liposomal delivery systems have been the first self-assembled nanomaterial approved for human clinical treatment against cancer in particular.108,109 For example, Doxil® was approved in 2007 by the FDA and consists of a formulation of the anticancer drug doxorubicin in PEGylated “stealth” liposomes with a diameter around 100 nm.110
Lipoplexes. While most lipids used for liposomal drug delivery are anionic or neutral, e.g. phosphatidylcholine, the use of cationic lipids is of particular interest for the delivery of nucleic acids by liposomes, namely lipofection.111–114 The principle behind the design of these vectors is simple as it aims to take advantage of electrostatic interactions between cationic lipids and negatively charged therapeutic agents that are nucleic acids. Strong electrostatic interactions between nucleic acids and the cationic head of lipids yield complexes, i.e. lipolexes, having a wide range of architectures and liquid crystalline phases.115–117 The bioactivity of these various lipoplex structures has been studied in detail.115,116 Among the different nucleic acids which can be used therapeutically, siRNA delivery is nowadays one of the most promising therapeutic approaches for a wide range of diseases and therefore the delivery of siRNA by cationic liposomes is under intense investigation,118 in particular to develop anti-cancer treatments.119,120 Because safe and effective siRNA delivery agents are needed, the group of Langer described combinatorial approaches to identify highly active cationic lipids for lipofection by screening a library of more than 1200 lipids obtained by Michael addition of polyamines on lipid-acrylates or acrylamides.121 The lipoplexes with a diameter around 50 nm were screened both in vitro and in vivo in rat, mouse, and non-human primates to identify promising compounds for local and systemic delivery of siRNA and miRNA. Clinically relevant siRNA-mediated gene silencing was achieved in non-human primates by the delivery of specific siRNAs against apolipoprotein B (a protein associated with increased risk of coronary artery disease) using cationic PEGylated liposomes with a diameter around 77–83 nm.122 Gene silencing through the receptor specific delivery of siRNA against gp46 to hepatic stellate cells by targeted PEGylated cationic liposomes with a diameter around 120–150 nm has also been used as an antifibrotic therapy for liver cirrhosis in a rat model.123
Cationic peptides are another particular type of cationic amphiphile that is used for transfection. Short cationic amphipathic peptides,124,125 obtained by linking hydrophobic cell penetrating sequences126 derived from the fusion sequence of the HIV gp41 together with a nuclear localization sequence (KKKRKV) for nucleic acid binding, were shown to be efficient DNA, siRNA, and protein delivery agents.124 While the morphology of the complexes is not well defined, their size can be controlled by the nucleic acid/peptide ratio to obtain particles with an average diameter of 100 nm.124 These cationic peptides were also used for the intracellular delivery of proteins.124
Polyplexes. Alongside cationic lipids, cationic polymers have also been widely used to prepare nanoscale self-assembled complexes, namely polyplexes, with nucleic acids.127–129 Cationic polymers, such as polyethylene imine, are polyelectrolytes and have the advantage of interacting with polyanionic nucleic acids in a multivalent manner yielding stable complexes. The effective block copolymers commonly used thus contain a nucleic acid binding block such as polylysine, together with a neutral hydrophilic block such as PEG to prevent the aggregation of the polyplexes.129–131
Polyplexes in the form of stable and defined 45 nm polymeric micelles were prepared by Kataoka et al. by spontaneous self-assembly between poly(ethylene glycol)-block-poly(L-lysine) and siRNAs.132 Targeting was achieved by the introduction of a cyclo-Arg-Gly-Asp (cRGD) peptide on the PEG terminus. Increased gene silencing ability, improved cell uptake, and broader subcellular distribution for the complexed siRNA were demonstrated in vitro in a tumor model as well as in vivo in mice.
Recently several research groups paid particular attention to the morphology and the related mechanism of formation of polyplexes. Shinkai et al. have shown that helicoidal polysaccharides, such as β-1,3-glucans, form one dimensional hetero-triple helix complexes consisting of two chains and one polynucleotide chain with various types of nucleic acids.133 These well-defined helicoidal complexes were used for the in vitro delivery of functional oligonucleotides to various cell types and were chemically modified with a cationic or hydrophobic group to further enhance cellular uptake.133 Kataoka et al. demonstrated the quantized folding of plasmid DNA upon complexation with poly lysine–PEG block copolymers yielding mixtures of rod-like complexes with quantized length and toroids.131 Rod-like polyplexes of individual plasmid DNA molecules were described by de Vries et al.134 They used genetically engineered protein diblock copolymers composed of a DNA binding polylysine (K12) segment and a 400 amino acid hydrophilic colloidal block for the stabilization of the complexes. Although these complexes did not show transfection properties, this type of study elaborates new rules for the design of homogeneous nanoscale carriers for nucleic acids.
A good control over the morphology of polyplexes was obtained by Luijten et al.135 They were able to go from spherical complexes to elongated rod-like complexes from the same composition of the complexes, simply by changing the polarity of the solvent medium used for the complexation. Interestingly, they have shown that elongated complexes are more efficient than their spherical counterparts for in vivo gene delivery in a mouse model.135
But the most advanced supramolecular system for gene delivery was developed by the group of Davis et al. who were the first to achieve targeted siRNA silencing in human clinical trials.136,137 Before giving the injection to the patient, the therapeutic nanoparticles can be readily obtained by the spontaneous self-assembly in one pot upon mixing a solution of the siRNA and a solution of the three other components of the system: a cyclodextrin-containing polymer (CDP), a polythethylene glycol–adamantane steric stabilization agent, and a human transferring-polyethylene glycol–adamantane conjugate as a targeting ligand (Fig. 2e). The authors optimized the complex formation between cationic polycyclodextrin polymers and siRNA for the formation of homogeneous nanoparticles with a diameter around 50–70 nm for optimal uptake by the tumors.136 The modularity and the ease of formulation of these complexes demonstrate the potential of self-assembled nanomaterials for the development of functional therapeutic agents.
Polymersomes and dendrimersomes. Polymersomes are vesicles formed by the self-assembly of amphiphilic block copolymers (Fig. 2d).138 They share similar architectures and applications with their lipid counterparts,138 but they are kinetically rather stable, as their dissociation would expose large hydrophobic macromolecular segments to the aqueous phase. As for liposomes, polymersomes can be loaded with hydrophilic or hydrophobic drugs, functionalized with PEG segments for prolonged circulation times, and decorated with targeting moieties on their surface.138 While a wide range of morphologies are accessible by the self-assembly of block copolymers, Percec et al. have also demonstrated the utility of dendritic structures to obtain impressive structural diversity by the self-assembly of easily accessible amphiphilic Janus dendrimers.139 They screened combinatorial libraries of Janus dendrimers by cryo-TEM and obtained dendrimersomes, cubosomes, disks, tubular vesicles and helical ribbons, making this system of high interest for drug delivery and other biomedical applications where shape plays a crucial role in the nanoparticle function.
Gold nanoparticles. Gold nanoparticles can serve as versatile scaffolds for the delivery of drugs and nucleic acids as they can bind molecular cargos via a wide range of non-covalent interactions.16,140–142 For example, gold nanoparticles with a diameter of 2.5 nm have been decorated with an amphiphilic monolayer composed of a hydrophobic alkanethiol linked to a hydrophilic tetra(ethylene glycol).143 These constructs can transport hydrophobic drugs, such as tamoxifen, located in the dense hydrophobic corona at the gold surface, while the external PEG corona ensures the solubility of the drug loaded nanoparticles. The surface of gold nanoparticles (27 nm in diameter) can also be decorated by small receptors for hydrophobic drugs such as per-6-thio-β-cyclodextrin, by an antifouling PEG shell, and by targeting residues (Fig. 2f).144 The cyclodextrin drug pocket can then be filled with an anticancer drug (β-lapachone) and the release of the drug from the carrier surface is induced by gold–thiol ligand exchange with the glutathione present in the intracellular matrix of cancer cells.144 Gold nanoparticles are also extremely promising scaffolds for the delivery of functional nucleic acids immobilized at their surfaces by electrostatic interactions, hybridizations, or covalent thiol–gold bonds.145 Mirkin et al. recently achieved a breakthrough in topical gene silencing using such spherical nucleic acid nanoparticle conjugates (SNA-NCs) prepared by the covalent immobilization of siRNA onto gold nanoparticles.146 SNA-NCs penetrate keratinocytes with high efficiency with no associated cytotoxic effect. The application of these SNA-NCs, mixed with a commercial moisturizer, on skin leads to the knockdown of the epidermal growth factor receptor at very low concentrations in a mouse model,146 demonstrating the enormous potential of SNA-NCs in treating skin diseases by siRNA mediated gene silencing.
Silica nanoparticles. Within the toolbox of supramolecular chemistry, the mechanical bond has been the latest one to be used for designing drug delivery platforms. Stoddart et al. described the use of mechanically interlocked molecules as stimuli responsive stoppers or nanovalves at the extremity of the pores in mesoporous silica nanoparticles. The resulting mechanized silica nanoparticles can respond to redox, light, base, or acid triggers in order to release drugs such as doxorubicin in vitro or in vivo.147 A particularly elegant approach for the stimuli responsive release of cargos was achieved by loading mechanized silica nanoparticles with magnetic zinc-doped iron oxide nanocrystals. This system enables the targeted release of doxorubicin in response to local heating upon exposure to an oscillating magnetic field (Fig. 2g).148
Spherical micelles. Water soluble spherical micelles, obtained by the self-assembly of amphiphilic block copolymers, can serve as nanocontainers where hydrophobic drugs are loaded in their hydrophobic core.149 These self-assembled objects can be cross-linked by reversible or non-reversible covalent bonds to improve their stability.150 The ability of micelles to release drugs by responding to stimuli such as temperature151 has been extensively reviewed.152,153 Recently, a new class of DNA diacyllipid–molecular-beacon conjugate amphiphile has been shown to form spherical micelles displaying DNA hairpins.154 Upon hybridization with a target mRNA, DNA hairpins undergo a conformation change which results in fluorescence emission. By scavenging intracellular mRNA, these sensing devices also modulate gene expression and are therefore applicable for gene therapy.154 In another example of polymeric DNA micelles, Gianneschi et al. showed that, as for the SNA-NCs described above, the dense array of nucleic acids displayed at the micellar surface confer interesting properties such as resistance to nucleases,155 suggesting their potential for gene delivery applications.
Cylindrical micelles. One dimensional nanostructures,156 and in particular self-assembled cylindrical polymeric micelles,157 are effective carriers for drug delivery in vitro or in vivo. Discher et al. were the first to clearly show the advantage of using cylindrical micelles (or filomicelles), versus their spherical counterparts, to load and carry hydrophobic drugs to tumors in vivo (Fig. 3b).157 Other one dimensional platforms have been used for drug delivery and in particular, the Stupp group developed particularly interesting peptide nanofibers formed by the self-assembly of peptide amphiphiles (PAs).71 Upon charge screening, these PAs self-assemble into cylindrical micelles stabilized by extended β-sheet hydrogen bond networks. The PA nanofibers are degradable by cells and not immunogenic; they have been used for the controlled release of therapeutic carbon monoxide158 and the anti-inflammatory drug nabumetone159in vitro by taking advantage of reversible covalent or coordination bonds between drugs and PAs (Fig. 3c). The anticancer drug camptothecin has also been loaded into the hydrophobic core of the PA cylindrical micelles by non-covalent interactions.160 Enzyme responsive PA nanofibers were designed by incorporating a consensus substrate sequence for protein kinase A, a potential cancer biomarker. Upon phosphorylation, electrostatic repulsion between peptide monomers triggers the disassembly of the nanofibers demonstrating their dynamic and stimuli responsive character. These fibers were shown to encapsulate a hydrophobic anticancer drug, doxorubicin, and release it upon protein kinase A treatment. This property was responsible for enhanced toxicity against cancer cells known to release large amounts of protein kinase A into their media.161 The co-assembly of a cationic membrane lytic PA with a serum protective PEGylated PA helped in stabilizing the therapeutic cytotoxic peptide against protease degradation, therefore enhancing stability of the nanostructures in vitro.162 These cytotoxic nanofibers were then shown to significantly reduce tumor cell proliferation in vivo in a mouse model. Dilution by co-assembly also allows reducing the proportion of cytotoxic PAs while retaining similar tumoricidal activity in vitro.
Recently, an original approach to increase the loading of drugs into self-assembled nanocarriers was discussed by Cui et al.163 They proposed to make the drug itself an integral part of the self-assembling core of the molecules. As a proof of principle, a hydrophobic drug, camptothecin, was used to replace the hydrophobic tail in the design of a PA to obtain a drug-amphiphile (DA), allowing for higher drug content in the final self-assembled nanostructure. Cui et al. convincingly demonstrated this concept for the preparation of well-defined nanostructures from dendritic DA including nanofibers and nanotubes with different camptothecin contents. They also demonstrated the efficiency of these nanostructures against a number of cancer cell lines.163
Carbon nanotubes. Carbon nanotubes have been used as one-dimensional nanocarriers for gene164,165 and drug delivery.166–168 The use of carbon nanotubes requires dispersion in water by using covalent modification or supramolecular interaction with large amphiphiles, such as phospholipid PEG conjugates, to ensure sufficient solubility and stability. The extended aromatic surface of single wall carbon nanotubes (SWNTs) is also ideal for supramolecular π–π stacking interactions with aromatic drugs such as doxorubicin (DOX). Higher loading capacities were obtained compared to other nanocarriers like liposomes or micelles, and DOX-loaded SWNTs display an enhanced therapeutic antitumoral effect and less toxicity than the free DOX (Fig. 3d).167,168 Dai et al. have also proposed a new concept of “functionalization partitioning”167 of SWNTs which consists in the co-assembly on the same nanotube of different lipid conjugates to confer multiple properties in the targeting delivery of drugs and genes. For example, SWNTs can be loaded via non-covalent interactions with radiolabels, antibodies, or siRNA conjugated with phospholipids.165 Covalent modification of carbon nanotubes with cationic chains allows the electrostatic binding of negatively charged nucleic acids. For example, Bianco et al. have demonstrated that complexes of plasmids and ammonium-functionalized carbon nanotubes enter cells by an endosome-independent mechanism, facilitate DNA uptake, and induce gene expression.164
Gels for drug delivery. Macroscopically stable gels are suitable for some delivery applications as their porous structures allow the controlled release of a therapeutic agent over extended periods of time.169,170 In particular, self-assembled gelators are of high interest because they can potentially be formed by in situ gelation of the building blocks. This could translate into clinical applications where an injection of soluble building blocks results in the gelation of the drug delivery materials by the formation of self-assembled nanostructures. In this context, gels formed by the self-assembly of peptides or polypeptides are very popular as they can be degraded and are biocompatible.171
Recent developments can be illustrated by the encapsulation of curcumin in self-assembling β-hairpin peptide hydrogels described by Pochan et al.172 The authors have demonstrated multiple modes of interactions between the hydrophobic curcumin and the valine rich β-hairpin MAX8. They were also able to control the release rate and toxicity against cancer cells of the bioactive guest by changing the concentration of the gelating peptide. Drug release from gels prepared by the network formation of self-assembled peptide nanofibers conjugated to drugs by reversible covalent bonds has also been described.159 In particular, physical gels have been prepared using charge screening by addition of CaCl2 and the release of nabumetone was observed over more than 27 days in vitro.159
For gene delivery, the localized and long-term delivery of siRNA against cyclin B1 by hydrogels was obtained by the gelation of thermoresponsive polyplexes.173 Complexes between siRNA and L-PEI-poly(organophosphazene) have shown a temperature-dependent sol–gel phase transition. The change in temperature upon injection into a tumor xenograft model in vivo induces the aggregation of 100 nm polyplexes via hydrophobic interactions. Polyplexes are then released from the hydrogel allowing the silencing of cyclin B1 for 4 weeks around the site of injection.
DNA-based nanomaterials. In recent years, nucleic acids self-assembly has emerged as the most powerful and predictable tool for the aqueous construction of complex nanostructures. This progress, together with the fact that nucleic acids are biocompatible, degradable, and in some cases bioactive as well, make them ideal building blocks for the development of nanoscale drug delivery vehicles.174 Because DNA self-assembly allows the preparation of self-assembled nanostructures with a perfect control over the size and shape of the constructs, it represents a crucial advantage when these parameters can affect the bioactivity of the nanostructures. For example, monodisperse nanofilaments were prepared by Högberg et al. using the principles of DNA origami and loaded with doxorubicin by intercalation between the nucleobases within the DNA nanostructure (Fig. 3e).175 These one dimensional nanostructures can penetrate breast cancer cells to promote apoptosis.
To design an ideal nanoscale drug carrier, one would imagine a tridimensional structure that could be filled with a therapeutic agent or solution, closed, and then selectively reopened at the targeted delivery site. The first prototype of such a DNA nanobox, that could potentially be used as a delivery vector, was described by Gothelf and Kjems who designed a DNA origami nanocube with a controllable lid.176 This lid could be held in a closed position by a set of two locks composed of DNA duplexes with sticky-end extensions, allowing the opening of the lid by the dissociation of the duplex upon externally added “key” oligonucleotides. More recently, DNA aptamers have been used as logic gates to control the opening and closing of self-assembled targeted nanorobots (Fig. 3f).177 Here the keys that trigger the opening of a self-assembled DNA barrel are two orthogonal aptamers. Therefore, both aptamers have to bind to their target (a protein for example) in order to induce the opening of the DNA barrel and the exposure of the biologically active payload, such as antibodies. This construct, loaded with a combination of an antibody to human CD33 and an antibody to human CDw328 Fab′ fragments, is able to induce growth arrest in NKL large granular lymphocytic leukemia cells in a dose-dependent fashion.177
Nanofibers. Among all examples of self-assembled nanomaterials for tissue engineering and regenerative medicine, self-assembled peptide nanofibers are the most common.71,72,171,183–186 They are built by the self-assembly of relatively simple peptidic building blocks, readily obtained by fast and reliable synthesis, which in turn facilitates the introduction of peptidic or non-peptidic epitope for specific applications. In addition, their peptidic constitution confers them unique biocompatibility and degradability as they can in principle be used as nutrients by cells. The design of self-assembling peptides usually involves the use of short peptide sequences known to facilitate the formation of secondary structures like β-sheets or α-helices, and their conjugation to hydrophobic chains in order to promote their aggregation in water. The peptide amphiphiles (PAs) developed by the Stupp group are based on a self-assembling platform composed of an aliphatic hydrophobic chain conjugated to a peptide sequence with β-sheet propensity and rich in hydrophobic acids, such as valine, leucine and alanine.71 PAs self-assemble into high aspect ratio nanofibers that can mechanically support cells and interact with cell receptors through the display of peptidic or non-peptidic biological signals that are easily introduced at the other extremity of the molecule. PAs also form gels upon charge screening which has led to the preparation of self-assembled biomaterials for spinal cord,187 bone,188 cartilage,189 blood vessels,190 heart,191 islets192 and enamel regeneration.193
More recently, a VEGF-mimetic PA has been used to prepare nanofibers that promote proangiogenic behavior in endothelial cells in vitro, and an angiogenic response in vivo both in a chicken embryo assay and in an ischemic hind limb mouse model (Fig. 4a).194 PA nanofibers can also form highly anisotropic materials, a feature potentially useful when designing biomaterials to regenerate tissues with architectures based on cell alignment such as spinal cord, blood vessels, and muscles. For example, shear forces applied to PA nanofiber liquid crystals obtained by heating and cooling a PA solution induced the formation of highly aligned arrays of nanofibers that can be mixed with cells and stabilized by gelation with CaCl2, resulting in macroscopic monodomain gel strings, in which the nanofibers and cells are aligned (Fig. 4b).195 PA nanofibers based liquid crystals can also be organized radially in tubular constructs and used to align smooth muscle cells (SMCs), mimicking the circumferential alignment of contractile SMCs necessary for the design of artificial blood vessels.196
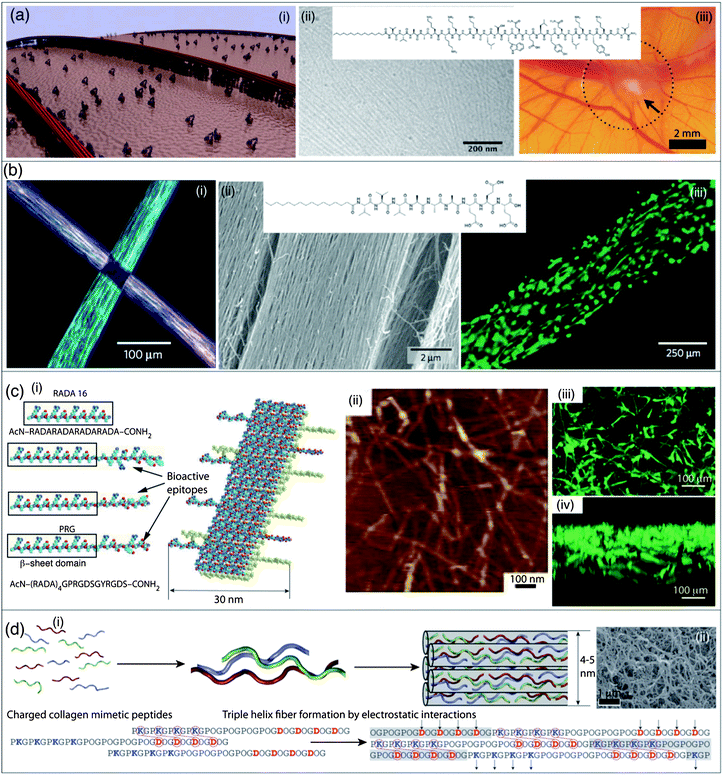 | ||
| Fig. 4 Self-assembled nanomaterials for regenerative medicine and tissue ingineering. (a) (i) Schematic representation of a self-assembled peptide nanofiber displaying high densities of biological signals that binds to receptors at the surface of a cell; (ii) molecular structure of a VEGF-mimetic peptide amphiphile and TEM micrograph of nanofibers formed by this molecule; (iii) representative image of angiogenesis from the point of contact with the VEGF-mimetic peptide amphiphile (arrow) in a chicken chorioallantoic membrane assay. (b) (i) Manual dragging of a peptide nanofiber liquid crystal onto salty media results in macroscopic strings and in the alignment of the nanofibers over macroscopic scales (cm). Light extinction between cross-polarizers at the cross-point of two strings of gel of uniformly aligned self-assembled peptide nanofibers demonstrating uniform alignment in each of them; (ii) molecular structure of a peptide amphiphile capable of forming a nanofiber liquid crystal upon thermal treatment and SEM micrograph of aligned nanofiber bundles in macroscopic strings formed by dragging thermally treated peptide amphiphile solution onto CaCl2, (iii) calcein-labelled aligned cells cultured in a string of aligned nanofiber bundles. (c) (i) Molecular models of RADA16 peptide and some of its derivatives that are able to self-assemble by the formation of β-sheets into pure or mixed nanostructures displaying biological epitopes; (ii) AFM micrograph of nanofibers formed by the co-assembly of RADA16 and PRG peptide bearing an integrin binding sequence; (iii) vertical view of a reconstructed image of 3-D confocal micrographs of MC3T3-E1 cells cultured on a mixed RADA16/PRG peptide 30/70 peptide scaffold; cells were stained with calcein, (iv) horizontal view of the same cultured scaffold showing a drastic cell migration into the 3D scaffold. (d) (i) Short collagen mimetic peptides able to form collagen-like triple helices with “sticky ends” which elongate by electrostatic interactions. Upon bundling, these triple helices self-assemble into fibrous collagen-like nanostructures, resulting in a hydrogel degradable by collagenase; (ii) SEM micrograph of a critical point dried collagen mimetic hydrogel. Panel (a): reproduced with permission from ref. 182 and 194; panel (b): reproduced with permission from ref. 195; panel (c): reproduced with permission from ref. 200; panel (d): reproduced with permission from ref. 201. | ||
Another important problem in tissue engineering and regenerative medicine is to protect grafts and cells upon transplantation. Therefore, it is of interest to design biomaterials that could encapsulate cells, protect them from the host immune system, while allowing nutrients to diffuse in and out of the capsule. Such architectures have been prepared by the Stupp group in the form of macroscopic sacs that can then be used to encapsulate cells.197 Sacs were obtained by the self-assembly of a negatively charged megadalton biopolymer and positively charged peptide amphiphiles. Upon mixing individual solutions of these two components, the formation of a diffusion layer prevents random mixing of the two solutions, and results in the formation of highly ordered membranes decorated with bundles of peptide nanofibers oriented at a 90° angle to the membrane. Such highly ordered hierarchical membranes obtained by the diffusion controlled self-assembly between a mixture of hyaluronic acid, heparin, and a heparin-binding PA, have been used recently to promote angiogenesis in the chicken chorioallantic membrane assay.198 Another class of self-assembling peptide has been introduced by Zhang et al.199 The self-assembling core of this class of peptide amphiphile is composed of repetitive sequences such as -AEAEAKAKAEAEAKAK- or -RARADADARARADADADA- that are self-complementary due to the presence of both positive and negative side chains on one side of the β-sheet, and hydrophobic side chains on the other. This self-assembly platform can be decorated with bioactive epitopes and used as 3D cell culture media for a wide range of cell types for tissue engineering.186 The self-assembling peptide scaffolds RADA16 and PRG peptide significantly enhance mouse pre-osteoblast cell proliferation and differentiation as well as stimulate cell migration into the 3D scaffold (Fig. 4c).200 More recently, Hartgerink et al. introduced a new method to recreate collagen fibers and gels from the self-assembly of simple peptides with sequences derived from the collagen's proline–hydroxyproline–glycine repeating unit. These peptides self-assemble into triple helices having electrostatic sticky ends that promote the elongation of triple helices involving salt-bridged hydrogen bonds between lysine and aspartate (Fig. 4d).201 Owing to the major role of collagen matrices in tissue engineering, this strategy is promising for the preparation of novel collagen analogs in regenerative medicine.
3 Self-assembled nanomaterials for information technologies
Information technologies based on optics and electronics play a key role in our modern society. In particular, organic (opto)electronics is a promising field of research with a high potential for the fabrication of new devices with a low cost of production. To implement this field, the bottom-up construction of electroactive nanomaterials is becoming a very popular domain of investigation thanks to its potential for miniaturizing and improving the material characteristics. In this chapter we will describe a selection of the recent literature on self-assembled nanomaterials for information technologies along two main sections. In the first one, we will describe self-assembled nanomaterials for organic electronic devices202–204 including conducting materials, organic field effect transistors, organic light emitting diodes, and organic solar cells. In the second section, we will focus on nanomaterials created for the storage and processing of information,205,206 including memory devices and Boolean logic circuits.3.1 Organic electronics
Conducting and semiconducting organic materials have recently attracted a growing number of research investigations because of their potential for the production of low cost and flexible electronic devices. Among these materials, self-assembled architectures can improve electronic properties by affording long-range ordering of molecules and by providing additional tuning parameters in order to control the devices' properties. In the following sections, we discuss some recent examples of supramolecular electroactive nanostructures displaying various electronic properties as conductive materials, field-effect transistors, light-emitting diodes, and photovoltaic devices.Nanofibers. Giuseppone and co-workers have reported the synthesis of an entirely new class of nanostructures based on triarylamine stacks, as well as their addressable self-construction between two electrodes, which represents a major progress to control supramolecular electroactive materials at the 100 nm length scale (Fig. 5a).212–214 These supramolecular triarylamine nanowires (STANWs) are generated by a simple light irradiation allowing the production of a catalytic amount of triarylammonium radicals which then piles with their neutral counterparts along nucleation–growth and self-replication processes. Using these properties, the localization and directionality of the fibers' elongation can be spatially addressed by applying an electric field directly between electrodes. An exceptional channel conductivity exceeding 5 × 103 S m−1, and an interface resistance per unit of length below 2 × 10−4 Ω m were measured for these self-constructing devices.215
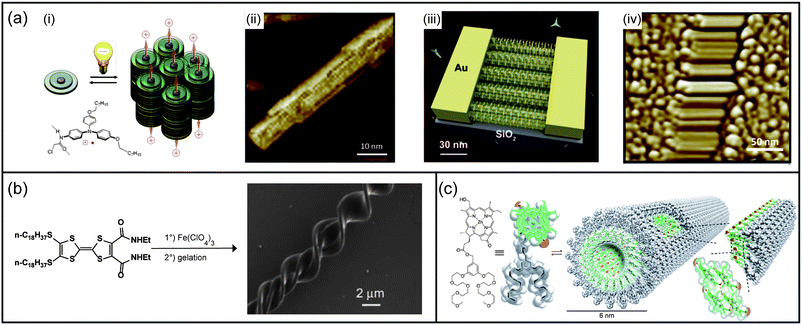 | ||
| Fig. 5 Electrically conducting self-assembled nanomaterials. (a) Light-triggered self-construction of supramolecular organic nanowires as metallic interconnects. (i) Nucleation of the fibers by supramolecular associations of a modified triarylamine upon light irradiation; (ii) AFM image of the self-assembled fiber; (iii) directed growth and insertion of the 100 nm fibers addressed by the electric field within electrodes; (iv) corresponding AFM image of the nanowires displaying high conductivity characteristics. (b) Preparation of 4,5-bis(octadecylthio)-4′,5′-bis(ethylcarba-moyl)tetrathiafulvalene ClO4−-doped coils and SEM image of resulting coiled fibers on a silicon wafer. (c) Chemical structure of a zinc chlorin derivative, the corresponding space-filling (CPK) model and a schematic model of self-assembled nanotubes formed upon injection of a concentrated solution of chlorin in methanol or THF into water. Panel (a): (i) and (ii) reproduced with permission from ref. 212, (iii) graphic by Mathieu Lejeune. (iv) Reproduced with permission from ref. 213; panel (b): reproduced with permission from ref. 218; panel (c): reproduced with permission from ref. 219. | ||
Another important class of stacked molecules extensively studied as conductive materials are tetrathiafulvalene (TTF) derivatives. Iyoda et al. have synthesized a TTF hexamer which, after oxidation to its monocationic form in chloroform, readily gives a deep green fibrous material when self-assembled upon addition of hexane.216 These nanofibers of several micrometers in length and having a diameter of 50 nm revealed a hexagonally ordered lamellar structure with a stacking distance of 3.51 Å and a conductivity of 0.11 S m−1. The conduction of the self-assembled structure is two orders of magnitude higher compared to thin-films made of the same molecule. Amabilino and co-workers studied a self-assembled organogelator composed of TTF-based nanofibers.217 Subsequent doping of these fibers with iodine leads to room-temperature conductivity of 0.5 S m−1 which could be improved by one order of magnitude after annealing. Finally, Park et al. reported a new TTF-diamine producing, after doping with perchlorate ions, mixed valence double and triple helices due to the spiral configuration of the doped nanofibers (Fig. 5b).218 A room temperature conductivity of 4.59 × 10−3 S m−1 was obtained with these nanostrutures.
Nanotubes. Würthner and co-workers studied bacteriochlorophyll c (BChl c) derivatives equipped with hydrophilic wedges and which self-assemble in water to produce well-defined nanotubes having a diameter of 6 nm (Fig. 5c).219 These nanostructures have shown conductivities up to 0.48 S m−1 and charge-carrier mobilities of 0.03 cm2 V−1 s−1. Aida et al. synthesised self-assembled nanotubes from covalent hexabenzocoronene (HBC)–C60 dyads by using a heating–cooling process.220 This structure exhibits a photovoltaic response upon light irradiation and an ambipolar character has been determined with a hole mobility of 9.7 × 10−7 cm2 V−1 s−1 and an electron mobility of 1.1 × 10−5 cm2 V−1 s−1. Co-assembly of the previous HBC–C60 dyad with another HBC derivative without fullerene also yields nanotubes. In this case, a flash-photolysis time-resolved microwave conductivity (FP-TRMC) technique221,222 was used to determine a hole mobility of 2 cm2 V−1 s−1 that is as large as the inter-sheet mobility in graphite.
Nanoribbons/nanorods. Yagai and co-workers have reported the interconvertion of oligothiophene nanorods and nanoribbons by using a supramolecular dynamic exchange.223 Highly intrinsic hole mobilities were measured by using the pulse radiolysis time-resolved microwave conductivity (PR-TRMC) technique224,225 with values of 1.3 cm2 V−1 s−1 for the nanorods and 0.57 cm2 V−1 s−1 for the nanoribbons. Zang et al. have studied other nanoribbons made from the self-assembly of perylene tetracarboxydiimide derivatives226 and which display a conductivity value of 1 × 10−3 S m−1, whereas a supplementary treatment with saturated hydrazine vapours leads to an enhanced conductivity up to 0.5–1 S m−1.
Nanofibers. Fabrication of film-type and single-nanofiber OFETs from organogels based on anthracene derivatives has been described by Lee et al. (Fig. 6a).234 The supramolecular organization in fibers leads to higher performance (field effect mobility 0.48 cm2 V−1 s−1; on–off ratio 105) compared to the film-casting versions. Samori and co-workers have shown that both processing techniques and side-chains have strong influences on the morphology of perylene tetracarboxydiimide (PDI) nanofibers and consequently, on the characteristic parameters of field-effect transistors.235 With another perylene tetracarboxylicdiimide derivative, Bao and co-workers have described the fabrication of single-crystalline wire OFETs.236 In this study, the average diameters and lengths of nanowires are respectively 519 nm and 340 μm, producing active layers with a mobility as high as 1.4 cm2 V−1 s−1. The same authors have also made use of a filtration-and-transfer method to align wires, leading to a maximum mobility of 0.24 cm−2 V−1 s−1 for multiple wire OFETs. Gong and co-workers have studied the self-assembly of a poly(p-phenylene ethynylene) derivative in crystalline nanowires with diameters comprised in the range of 5–15 nm and lengths up to hundreds of micrometers (Fig. 6b).237 Mobilities of these objects in OFET devices have shown values up to 0.1 cm2 V−1 s−1 which are several orders of magnitude higher than thin-film devices of the same polymer. Another example comes from Schenning et al. who have described the self-assembly of hydrogen-bonded oligo(p-phenylenevinylene) and perylene tetracarboxydiimide leading to thin-films with an internal rod-like morphology.238 These OFET devices have shown ambipolar transport, however with both low electron and hole mobilities (10−7 cm2 V−1 s−1).
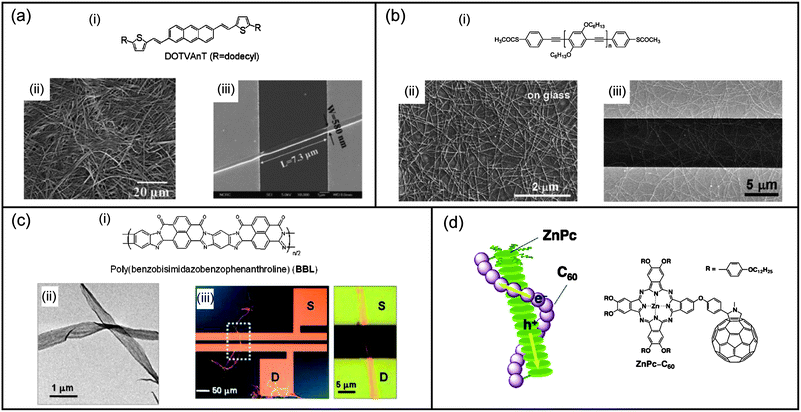 | ||
| Fig. 6 Self-assembled nanomaterials for field-effect transistors. (a) (i) Structure of a 2,6-bis(2-thienylvinyl)anthracene derivative and SEM images of (ii) its xerogel and (iii) the bottom-contact single-nanofiber transistor of the considered molecule. (b) (i) Structure of the poly(para-phenylene ethynylene) polymer with thioacetate end groups; (ii) SEM images of the self-assembled nanowires on glass substrates from THF solution and (iii) the transistor device with nanowires between the source and drain electrodes. (c) (i) Structure of poly(benzobisimidazobenzophenanthroline); (ii) TEM image of the self-assembled nanoribbons; (iii) typical single nanoribbon transistor and a close-up showing the nanoribbon bridging the source–drain electrodes. (d) Structure and proposed schematic model of the liquid crystal arrangement of the considered phthalocyanine–fullerene dyad. Panel (a) reproduced with permission from ref. 234; (b) reproduced with permission from ref. 237; (c) reproduced with permission from ref. 240; (d) reproduced with permission from ref. 247. | ||
Nanoribbons. Xia et al. have described the use of self-assembled nanoribbons from perylene tetracarboxydiimide in n-channel OFETs.239 In conjunction with p-channel hexathiapentacene nanowire OFETs, the authors have succeeded in producing a functional complementary inverter. Xia and co-workers have also reported long-term stable OFET devices based on nanobelt architectures prepared from poly(benzobisimidazo-benzophenanthroline) polymers and with widths up to 1 μm and lengths up to 150 μm (Fig. 6c).240 Electron mobilities were measured for both single and networks of nanobelts with respective values of 7 × 10−3 cm2 V−1 s−1 and 4 × 10−3 cm2 V−1 s−1. Self-assembled oligoarene nanoribbons in single-wire OFETs were studied by Pei et al.241 Mobilities up to 0.01 cm2 V−1 s−1 were obtained with an on–off ratio of 103. An improved version of these OFETs was recently reported with a mobility as high as 2.1 cm2 V−1 S−1, an on–off ratio of 2 × 105, and a threshold of −7 V.242 Self-assembled nanoribbons made of dithioperylene in OFET devices were reported by Pei et al.243 Mobility as high as 2.13 cm2 V−1 s−1 and an on–off ratio of 106 were obtained from a single-crystalline nanoribbon device.
Liquid crystals. Lee and co-workers reported a novel donor–acceptor type semiconducting copolymer poly(didodecylquaterthiophene) forming highly crystalline thin films after thermal annealing.244 High field-effect mobilities of 0.33 cm2 V−1 s−1 were obtained as well as a good environmental stability and a bias-stress stability comparable to that of amorphous silicon. Discotic quaterrylene tetracarboxyldiimide was studied by Müllen et al. as an active layer leading to ambipolar OFETs.245 Electron and hole mobilities of respectively 1.5 × 10−3 cm2 V−1 s−1 and 1 × 10−3 cm2 V−1 s−1 were obtained. After annealing, the authors showed that only the electron transport behaviour remains with a mobility of 2 × 10−4 cm2 V−1 s−1. Geng et al. have synthesised a discotic liquid crystalline phthalocyanato derivative which was used to make OFETs with mobility up to 0.017 cm2 V−1 s−1 after annealing.246 Imahori and co-workers have prepared donor–acceptor dyads from zinc phthalocyanine which self-assemble in columnar liquid crystals (Fig. 6d).247 The time-of-flight method (TOF)248 was used to determine efficient ambipolar charge-transport properties with an electron mobility of 0.26 cm2 V−1 s−1 and a hole mobility of 0.11 cm2 V−1 s−1.
Coordination complexes. Ma et al. have reported the use of two supramolecular triple decker complexes composed of tris(phthalocyanato) europium with crown ether heads and hydrophobic tails; both of them show good carrier mobilities (0.28–0.33 cm2 V−1 s−1).249 However, a noticeable difference between them concerns the direction of the current: in one case, it flows parallel to the aromatic phthalocyanine due to a J-aggregate arrangement whereas, in the second case, it flows parallel to the long axis of the complexes due to a H-aggregate arrangement.
Nanofibers. Del Guerzo et al. have described the self-assembly of blends of tetracene derivatives leading to color-tunable organogels constituted of fluorescent nanofibers (Fig. 7a).260 Under near UV light excitation, both organogels and dry films of nanofibers emit white light, which is polarized in the blue spectral region. Additional confocal fluorescence microspectroscopy experiments showed that white light is emitted by each individual nanofiber. De Cola and co-workers have reported the use of a platinium(II) complex which self-assembles in bright gelating nanofibers as a dopant for solution-processed OLEDs.261 The highest efficiency with this device reached 15.6 cd A−1 at a brightness of 203 cd m−2. The self-assembly of a rigid hexaacid compound into supramolecular polymeric nanowires with diameters between 100 and 200 nm and lengths up to hundreds of micrometers was studied by Pei et al. (Fig. 7b).262 They showed a solid-state absolute photoluminescence (PL) efficiency of 22% representing only a loss of 10% against their solvent efficiency.
 | ||
| Fig. 7 Self-assembled nanomaterials for light-emitting diodes. (a) Molecular structures and images of a 2 mM B-gel in DMSO, a 10 μM G solution in THF, and a 10 μM R solution in THF under UV light (λex 365 nm). Fluorescence intensity obtained by confocal microscopy of (i) a gel made of B and (ii) a gel made of the three components R, G, B in DMSO at 22 °C. (b) (i) and (ii) Structure of a hexaacid compound forming self-assembled polymeric nanowires and (iii) associated fluorescence micrograph. (c) Chemical structures of di-ureidopyrimidinones and schematic illustration of their use for the creation of white photoluminescence. Panel (a): reproduced with permission from ref. 260; (b) adapted with permission from ref. 262; (c) reproduced with permission from ref. 264. | ||
Supramolecular polymers. Schubert and co-workers have reported the study of π-conjugated bis(terpyridine) macroligands with poly(ε-caprolactone) on their side chains.263 The formation of supramolecular Zn(II) metallo-polymers with these macroligands leads to blue photoluminescence with high quantum yields both in diluted solution and solid state, demonstrating the suppression of the aggregation that usually occurs with metallo-polymers. Meijer et al. have also described a supramolecular copolymer resulting from the self-assembly in films of blue, green and red π-conjugated oligomers, each of them bearing complementary quadrupole hydrogen bonding ureidopyrimidone units (Fig. 7c).264 White electroluminescence was achieved with CIE (Commission Internationale de l'Energie) coordinates (−0.31; 0.45) by using an appropriate ratio of the oligomers. Other examples of supramolecular polymers using hydrogen-bond interactions instead of metal complexation were also described for the fabrication of OLED devices. Chang et al. have reported the synthesis of π-conjugated poly(triphenylamine-carbazole) which exhibits amorphous nature, high thermal stability, good hole injection, and electron-blocking ability in the solid state owing to the supramolecular cross-linking of uracil units between polymer chains.265 Trilayer OLED devices based on tris(8-hydroxyquinolinato)aluminium Alq3 and the previous polymer as a hole injection/transport layer have shown improved performance compared to PEDOT-PSS-based devices. Moreau and co-workers studied the capping of an oligophenylenevinylene imide derivative with 2,4-diamino-1,3,5-triazine derivatives by hydrogen bond interactions.266 The resulting supramolecular complex led to J-aggregates showing increased luminance compared to the non-capped oligophenylenevinylene imide derivative.
Coordination complexes. Huang and co-workers have reported a novel spirometal platinium(II) complex which was applied for OLED devices.267 It exhibits a maximum luminance of 3335 cd m−2 and a maximum external quantum efficiency of 5.2%. Iridium complexes have also attracted a lot of interest in this field and have been recently reviewed.256,258 Two examples of the state of the art iridium-based devices are shown here. Bolink et al. reported an LEC device showing an extrapolated lifetime to half the initial luminance (t1/2) of 4000 hours (initial luminance 670 cd m−2), and sub-second turn-on times in applying a pulsed current driving method.268 Su and co-workers described one of the white LEC devices showing the best performance to date with an external quantum efficiency (EQE) of 7.4% and a power efficiency of 15 lm W−1.269
Inclusion complexes. Cacialli and co-workers have reported that energy transfer, from a blue emitting cyclodextrin threaded polymer to a green emitting one, is strongly reduced by controlling the minimum intermolecular distance between emitting moieties through non-covalent encapsulation of the blue component in the cyclodextrin macrocycles.270 The measured CIE coordinates (0.282, 0.336) correspond to warm light and a power efficiency of 0.39 cd A−1.
Nanofibers. Aida and co-workers have reported different self-assembly modes between enantiopure and racemate forms of porphyrin–fullerene dyads.282 The racemate generates nanometric spheres with a mean diameter of 300 nm, whereas the enantiopure compound gives nanofibers up to 10 μm in length. A cast film of the spherical assemblies shows only a small hole mobility of 1.5 × 10−4 cm2 V−1 s−1 and no electron mobility by the time-of-flight method.248 Meanwhile, nanofibers exhibit hole and electron mobilities of 0.10 and 0.14 cm2 V−1 s−1, respectively. Shinkai et al. have described the self-assembly of electron rich quaterthiophene and electron poor perylene bisimide in order to obtain n–p heterojunction nodes (Fig. 8a).283 Interestingly, sorted formation of pure oligothiophene nanofibers and of pure perylene bismide nanofibers was observed from a 1
:1 mixture of both compounds, and a bulk heterojunction solar cell was made using a cast film of this organogel. Nuckolls et al. have reported solar cell devices based on fullerene C60 and dibenzotetrathienocoronene (6-DBTTC), which self-assembles in a network of cables after thermal annealing.284 Devices, where the donor layer was annealed followed by vapor deposition of fullerene, show a power conversion efficiency of 1.9% (JSC 6.7 mA cm−2; VOC 0.6 V). The same group has also reported solar devices based on blended films of 6-DBTTC and PC70BM.285 Supramolecular complexes are formed here in the bulk, and a 1:2 ratio of these two components gives the highest power conversion efficiency (PCE) of 2.7%. Würthner and co-workers have described the use of triaminotriazine-sustituted oligo(phenylene vinylenes) to bind perylene bisimides in order to form hydrogen bonded trimers.286 The latter then self-assemble into right-handed supercoils. Organic photovoltaic (OPV) devices show only poor diode behavior due to the lateral organization of the supercoils at the surface. Holmes et al. have reported a polypeptide generating an alpha helix structure after gelation in an organic solvent.287 In solar cell devices, blends of the polypeptide and PCBM show an improvement of PCE compared to reference compounds which do not produce any helical arrangement of chromophores. | ||
| Fig. 8 Self-assembled nanomaterials for photovoltaic devices. (a) Chemical structures of the oligothiophene and perylene organogelators with a schematic representation of their self-sorting organogel formation yielding p–n heterojunction points. (b) (i) Molecular structures of two HBCs and schematic illustrations of the preparation of a supramolecular heterojunction made of semiconducting nanotubular segments; (ii) tapping-mode AFM and (iii) scanning TEM micrograph of the nanotubular segments. (c) Molecular structures of p-oligophenyl, p-oligophenylethynyl chromophores, and naphthalenediimides, together with their corresponding supramolecular heterojunction architectures resulting in oriented multicolored antiparallel redox gradients (OMARG). (d) Overview of a J-aggregate/CdSe nanowire color-selective photodetector; (i) schematic of J-aggregate-coated nanowires contacted between electrodes; (ii) optical micrograph, showing interdigitated electrodes used to grow and test devices. The faint brown shading between electrodes is the sparse network of CdSe nanowires; (iii) detail of J-aggregate-templated CdSe nanowires with the structure of J-aggregating dyes. Panel (a): reproduced with permission from ref. 283; panel (b): adapted with permission from ref. 290; panel (c): reproduced with permission from ref. 298; panel (d): reproduced with permission from ref. 301. | ||
Nanotubes. Kamat and co-workers have used stacked-cup carbon nanotubes (SCCNTs) in a photoelectrochemical cell.288 A maximum photoconversion efficicency of 17% was reached that is lower than that of dye-sensitized solar cells but comparable to that of nanostructured semiconductor films (TiO2, ZnO, etc.). Aida et al. have synthesized photoconductive nanotubes by a controlled self-assembly of the HBC–trinitrofluorenone (TNF) covalent donor–acceptor dyad.289 Those nanotubes have a coaxial configuration in which a molecular layer of electron-accepting TNF laminates an electron-donating graphitic bilayer of π-stacked HBC. Such a structure creates an extremely wide interface for the spatially segregated redox couple, providing the electrical conduction an on–off ratio greater than 104 upon photoirradiation. The same group has also reported a supramolecular linear p–n junction composed of two different graphitic nanotubes by a stepwise self-assembly of two kinds of HBCs having different electronic structures (Fig. 8b).290 The lifetime of the charge carriers generated in the heterojunction nanotubes is five times longer than those measured for the nanotubes composed of a single component, demonstrating that the probability of the charge recombination is reduced. Hasobe and co-workers have also reported the use of a zinc meso-tetra(4-pyridyl)porphyrin to form hexagonal hollow nanotubes which were filled with C60 and C70 derivatives.291 Regenerative I−/I3− photoelectrochemical cells made with these nanotubes show, at an input power of 20 mW cm−2, a PCE of 0.63% when the nanotubes are filled with a C60 derivative, and 0.03% when kept empty.
Liquid crystals. Wong and co-workers described the use of a difluorenyl-substituted HBC which self-assembled into columnar discotic liquid crystals.292 Solar cell devices based on the blend of the previous compounds with PC60BM (1
:2) generated a short circuit current (JSC) of 2.68 mA cm−2 and PCE of 1.46%, after thermal annealing (150 °C). The same authors also modified the latter compounds by attaching oligothiophenes.293 New devices based on blends of this compound and PC70BM (1:2) gave a JSC of 2.53 mA cm−2 and PCE of 1%. More recently, diketopyrrolopyrrole units were appended and blends with PC70BM showed a JSC of 3.96 mA cm−2, a VOC of 0.86 V and PCE of 1.59%.294 A nematic liquid-crystal gel constituted of both a polymerizable electron donor and a non-polymerizable component was used by Kelly and co-workers.295 After photopolymerization and subsequent washing, an electron acceptor molecule layer was deposited to form a vertically separated distributed interface between donors and acceptors. Photovoltaic devices built from this method showed an open circuit voltage VOC of 1.1 V and a power conversion efficiency of 0.6% for a monochromatic irradiance of 45 mW cm−2. Li et al. reported the incorporation of PCBM to liquid crystalline porphyrins which form columns perpendicular to the substrate.296,297 The resulting doped blend retained the liquid crystal phase of the porphyrin derivative, allowing the formation of highly ordered nanostructured thin films which are promising for OPV devices.Zipper. Matile et al. have introduced the programmed assembly of interdigitating intra- and interlayer recognition motifs on conducting surfaces (Fig. 8c).298,299 These self-assembled zippers allow access to supramolecular coaxial n–p-heterojunctions300 with good photocurrents and high fill factors (0.50–0.60).
In addition to their interest for energy purpose, OPV devices could also be used as photodetectors by converting a light signal to an electrical one. Bawendi and co-workers reported the conception of photodetector devices based on the self-assembly of J-aggregate dyes onto crystalline inorganic nanowires, allowing the distribution of both light harvesting and charge transport functions between two materials optimized for each role (Fig. 8d).301 The deposition of strongly negative cyanine dyes by spin coating onto thiocholine-treated CdSe nanowires resulted in formation of J-aggregates at the nanowire surface. This process was shown to be reversible using exclusively solution phase processing and thus, a switchable color sensitization of the device from blue to red to green was explored. These color-sensitized devices demonstrated three specific and narrow absorption bands with a resulting photocurrent enhancement of up to 2.8-fold greater than the bare CdSe nanowire device. Samori and co-workers have also studied organic phototransistors based on perylene diimide (PDI) nanofibers.302 The experiments show that the photoresponse can be modulated by tuning the device geometry and the morphology of the semiconducting layer. An optimized PDI multifiber-based device was then built from these considerations and a strong responsivity value of 4.08 ± 1.65 × 105 A W−1 was obtained.
3.2 Information storage and processing
Extensive on-going research in molecular and supramolecular computing has been motivated by potential applications for information storage and/or processing which are of tremendous importance in our digital society. The following section has been divided into two parts: one focusing on self-assembled materials for non-linear optic applications, and the other on self-assembled Boolean logic gate materials.Nanofibers. Ishi-i et al. have reported nanofibers based on hexaazatriphenylene–triphenylamine conjugates which show moderate improvement of the two-photon absorption cross-section compared to a non-aggregative analog.310
Inclusion complexes. The inclusion of (dimethylamino)stilbazolium-type chromophores in amylose helices was studied by Kuzyk and co-workers.311 Enhancement of the second-order nonlinear optical response was clearly observed through the inclusion compared to the free chromophores.
Coordination complexes. Burke et al. have described a dicopper(I) metallocyclophane with a partial C2-symmetric structure in solution, allowing the observation of a strong second harmonic frequency response (β = (3000 ± 600) × 10−30 esu for a fundamental frequency at 800 nm).312 Kamada and co-workers reported the self-organization of two porphyrin-dimers through complexation allowing a strong two-photon absorption in the near-IR wavelength region.313 Finally, Kim et al. have shown that the formation of a double strand through coordination of bidendate ligands enhances the two-photon absorption cross-section of a conjugated porphyrin trimer, whereas the formation of a triple strand, by using a tridentate ligand, has the opposite effect.314
Based on rotaxanes. Many rotaxanes in the literature could play the role of logic gates317–320 and we only review here those which were included in functional memory storage devices. Stoddart and co-workers have constructed a molecular memory circuit incorporating bistable [2]rotaxane molecules as the data storage element (Fig. 9a).321 This 160
000-bit molecular electronic memory circuit was fabricated at a density of 1011 bits per cm2 (pitch 33 nm; memory cell size 0.0011 mm2). The working bits were then configured to form a fully functional random access memory circuit for storing and retrieving information. Gao et al. have designed a solid-state memory device based on a Langmuir–Blodgett film made of amphiphilic bistable rotaxanes.322 The system exhibits electric bistability and nanometric dots (of ca. 3 nm size) can be written over the surface using a STM tip and erased by applying a reverse voltage pulse.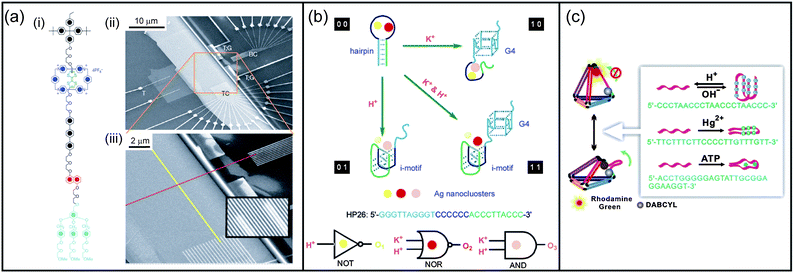 | ||
| Fig. 9 Self-assembled nanomaterials for information storage and processing. (a) (i) Structure of a rotaxane used in a memory device; (ii) SEM image of the entire nanowire crossbar memory circuit; (iii) SEM image showing the cross-point of top (red) and bottom (yellow) nanowire electrodes. Each cross-point corresponds to an ebit in memory testing. (b) Schematic diagram of logic operations based on a hairpin DNA template-tuned fluorescent Ag nanoclusters. K+ and H+ serve as two inputs to trigger the allosterism of the hairpin and modulate the fluorescence output. (c) Scheme of DNA tetrahedra that are reconfigurable by adapting dynamic sequences in one arm. Panel (a): adapted with permission from ref. 321; panel (b): reproduced with permission from ref. 343; panel (c): reproduced with permission from ref. 344. | ||
Based on self-replicating systems. Non-enzymatic reaction networks were used by Ashkenazy and coworkers to design light-triggered logic gates.323 First, a coiled-coil peptide able to catalyse its replication was caged with a photocleavable moiety preventing the autocatalytic pathway from occurring.324 Light-induced uncaging of the latter allows restoration of the self-replicative system and thus shows an AND gate behaviour with both light and the caged peptide as entries. A couple of FRET donor–acceptor moieties was then added to this system.325 Hence, fluorescence quenching of the mixture can be used to monitor the replication process. Considering fluorescence as the output signal of the logic gate, the latter becomes a light-induced NAND gate.
Based on DNA. A lot of attention has also been devoted to biocomputation with DNA.326–329 Winfree et al. have designed DNA-based logic circuits330 producing AND, OR, NOT gates, as well as signal restoration, amplification, feedback, cascading, and even a circuit able to compute the integer part of the square root of a four-bit binary number.331 These systems rely on sequence recognition and nucleic base displacement by using only single stranded DNA as inputs and outputs. Also resting on simple strand displacement, Ghadiri and co-workers have described the serial connection of DNA-based logic gates in the same solution by using fluorescence as the output.332 A half-adder was obtained from an AND gate and an XOR gate, while an OR gate was assembled through two XOR gates. DNA structures like hairpins have also been used to build logic gates. Park et al. have designed a set of two-input logic gates based on a hairpin-structure molecular probe.333 These gates can be reset by applying an additional input or a removal strand. Turberfield and co-workers have reported another DNA hairpin-based AND gate which is able to continuously re-compute its output, thus changing its response to various inputs.334 DNA sensors based on hairpin structures equipped with redox-tags and immobilized on electrode surfaces have also been reported.335,336 Another approach to design DNA-based logic gates is to use DNA molecules with biocatalytic properties mimicking enzyme functions, so-called DNAzymes.337
Willner et al. have designed an entire DNA computing circuit based on the modular coupling of DNAzyme subunits.338 This system could be tailored to generate multilayered gate cascades, fan-out gates, or parallel logic-gate applications. Other groups have reported logic gates based on ion-dependent DNAzymes.339–341 Ion-tuned DNA/Ag nanoclusters342 can also be used for the design of versatile logic gates as shown by Wang, Dong and co-workers (Fig. 9b).343 Inputs (K+ and H+ ions) induce changes of the DNA stabilizer which result in modifications of the spectral behaviour of Ag nanoclusters. Thus, multiple logic operations can be performed together via multichannel fluorescence output. Finally, Fan and co-workers designed tetrahedral DNA nanostructures as logic gates (AND, OR, XOR, INH, and a half-adder).344 These are responsive to protons, Hg2+ ions, ATP, and complementary DNA scaffolds (Fig. 9c). An example of a tetrahedra logic gate was also tested for intracellular detection of ATP in living cells.
These self-assembled materials are promising logic gates, especially DNA which could have exceptional storage capacities.345,346 However, for most of the examples in the literature, issues concerning the ways and means to integrate them in practical devices should be addressed to advance further in the field.
4 Self-assembled nanomaterials for environmental sciences
One of the main societal issues of the century concerns environment and in particular how to solve problems related to pollution and wastes. Various approaches ranging from inorganic to polymeric or carbon materials have been envisioned to overcome these issues.347 While still in their infancy, functional devices made of supramolecular self-assemblies have emerged as promising tools to tackle some of these environmental concerns.348 In this chapter, we will illustrate the variety of nanostructured objects produced by self-assembly, or involving a molecular recognition event to operate, that have been used to address these problems.349–351 In the first part, recent selected examples will highlight the various self-assembled structures used to sense chemical analytes ranging from gases to metal ions up to substances present in food or cosmetics.352–355 In the second section, supramolecular materials that are used to store gases or small molecules will be described, in particular nanoporous molecular crystals and metal organic frameworks.350,351,356 In the third part, we will first point out the importance of supramolecular ion channels as synthetic transport systems and further demonstrate the importance of nanostructured membranes and metal–organic frameworks for separation and purification of mixtures of gases or chemicals.350,351,357–360 Finally, we will show the wide diversity of nanometric objects that have been involved in the catalysis of chemical reactions.350,351,361–3634.1 Nanomaterials for detection
In recent years, many efforts have been devoted to the development of (bio)chemical sensors using the supramolecular approach.364–369 Although a large number of examples rely on the host–guest recognition of small molecules for sensing analytes,370–372 we will focus here on systems that are considered as nanomaterials, i.e. made of supramolecular self-assemblies: ribbons, pores, gels, arrays, nanoparticles, quantum dots, and metal–organic frameworks.![Self-assembled nanomaterials for detection. (a) Construction and mechanism of action of a fluorescent dye (G-coum) adsorbed on a MMT (montmorillonite)/supramolecular hydrogel 1 hybrid sensor system for polyamines; inset left: chemical structure of amino acid hydrogelator 1 and atomic force microscopy image of hydrogel 1 ([1] = 1.0 wt%/ion-exchanged water); inset right: photograph of the G-coum⊂MMT/hydrogel 1 hybrid sensor chip (λex = 365 nm) for fluorocolorimetric sensing of spermidine and spermine in artificial urine. (b) Colorimetric sensor array consisting of 36 different chemically responsive pigments which have been printed on a nonporous polyethylene terephthalate (PET) film, and images of the 36-dye colorimetric sensor array before exposure and after 2 min of exposure to ammonia at its IDLH (immediately dangerous to life or health) concentration at 298 K and 50% relatively humidity, and the generated color difference map; right: disposable low dead-volume self-sealing cartridge with the colorimetric sensor array printed directly on a PET flat. (c) Fluorogenic steroid sensing systems with cell-penetrating peptide (CPP) transporters as signal transducers and signal amplifiers (e.g., A1) that react with hydrophobic analytes (e.g., S1) to generate amphiphilic CPP activators. For cholesterol, signal generation with cholesterol oxidase (a) is followed by signal amplification with A1 (b). The obtained A1S1 conjugate and CPP (e.g., pR (polyarginine)) form pR– A1S1 transporters (c) that mediate the export of intravesicular 5(6)-carboxyfluorescein CF (d–f). Amphiphilic anions (e.g., A1S1) are CPP activators below (d–f) and CPP inactivators above their cmc (g); right: detection of active transporters as fluorescence recovery with CF-loaded vesicles. Panel (a): adapted with permission from ref. 376 and 377; (b) adapted with permission from ref. 367; (c) reproduced with permission from ref. 365 and 403.](/image/article/2013/NR/c3nr02176a/c3nr02176a-f10.gif) | ||
| Fig. 10 Self-assembled nanomaterials for detection. (a) Construction and mechanism of action of a fluorescent dye (G-coum) adsorbed on a MMT (montmorillonite)/supramolecular hydrogel 1 hybrid sensor system for polyamines; inset left: chemical structure of amino acid hydrogelator 1 and atomic force microscopy image of hydrogel 1 ([1] = 1.0 wt%/ion-exchanged water); inset right: photograph of the G-coum⊂MMT/hydrogel 1 hybrid sensor chip (λex = 365 nm) for fluorocolorimetric sensing of spermidine and spermine in artificial urine. (b) Colorimetric sensor array consisting of 36 different chemically responsive pigments which have been printed on a nonporous polyethylene terephthalate (PET) film, and images of the 36-dye colorimetric sensor array before exposure and after 2 min of exposure to ammonia at its IDLH (immediately dangerous to life or health) concentration at 298 K and 50% relatively humidity, and the generated color difference map; right: disposable low dead-volume self-sealing cartridge with the colorimetric sensor array printed directly on a PET flat. (c) Fluorogenic steroid sensing systems with cell-penetrating peptide (CPP) transporters as signal transducers and signal amplifiers (e.g., A1) that react with hydrophobic analytes (e.g., S1) to generate amphiphilic CPP activators. For cholesterol, signal generation with cholesterol oxidase (a) is followed by signal amplification with A1 (b). The obtained A1S1 conjugate and CPP (e.g., pR (polyarginine)) form pR– A1S1 transporters (c) that mediate the export of intravesicular 5(6)-carboxyfluorescein CF (d–f). Amphiphilic anions (e.g., A1S1) are CPP activators below (d–f) and CPP inactivators above their cmc (g); right: detection of active transporters as fluorescence recovery with CF-loaded vesicles. Panel (a): adapted with permission from ref. 376 and 377; (b) adapted with permission from ref. 367; (c) reproduced with permission from ref. 365 and 403. | ||
4.2 Nanomaterials for chemical storage
One classical feature of nanomaterials used in chemical storage is their structuration around nanopores, but we will see below that they are not restricted to this characteristic. The particular topic of the absorption of guest molecules on surfaces made of 2-D self-assembled nanopores will not be discussed here, although they can be used for gas storage, and readers should refer to reviews recently published in the literature.421,422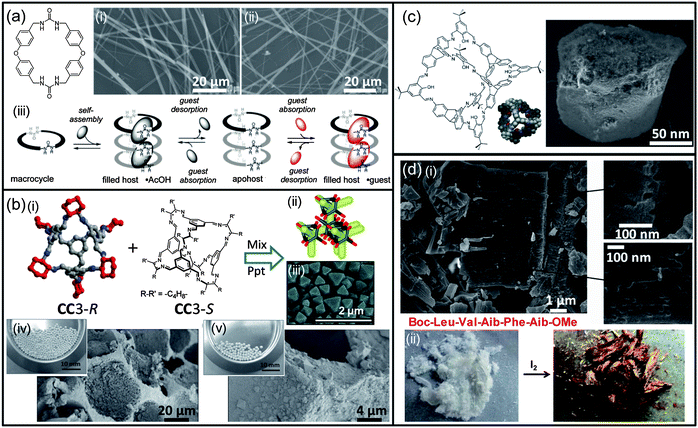 | ||
| Fig. 11 Self-assembled nanomaterials for chemical storage. (a) Bis-urea macrocycle; (i) SEM image of the filled host·AcOH; (ii) SEM image of the apohost; (iii) schematic view of self-assembly and reversible absorption of guests. (b) (i) Co-self-assembly of prefabricated intrinsically porous organic cage molecules CC3-R and CC3-S that precipitate spontaneously upon mixing in solution; (ii) scheme showing the window-to-window cage–cage packing that leads to a 3-D diamondoid interconnected pore network (in yellow) in the crystal structure of CC3-R; (iii) SEM image for racemic CC3-(R,S) crystals formed by mixing dichloromethane solutions of the homochiral CC3-R and CC3-S modules at −5 °C; (iv) photograph of porous silica beads taken at the highest cage loading and SEM image of the internal cross-section of cleaved beads. Octahedral-shaped CC3 crystals can be seen to have formed inside the macropores of the beads; (v) photograph of porous alumina–silica beads taken at the highest cage loading and SEM image of the internal cross-section of cleaved beads. (c) Structure of a salicylbisimine cage compound and its corresponding crystal structure, SEM image of the cage compound (as synthesized). (d) (i) Field emission scanning electron microscopic image of a peptide (Boc-Leu-Val-Aib-Phe-Aib-OMe) xerogel showing nanoporous morphology in the solid state; (ii) pentapeptide (Boc-Leu-Val-Aib-Phe-Aib-OMe) xerogel obtained from xylene–water showing colour change after exposure to I2 vapours. Panel (a): reproduced with permission from ref. 425; (b) adapted with permission from ref. 429, 440 and 441; (c) reproduced with permission from ref. 442; (d) reproduced with permission from ref. 447. | ||
In 2009, the group of Cooper reported on the synthesis of porous organic cages via [4 + 6]cycloimination between 1,3,5-triformylbenzene and various 1,2-diamines that further assemble in microporous organic materials (one example is displayed in Fig. 11b).437 All cages were shown to display gas sorption properties with BET surface area comprised between 530 and 620 m2 g−1. The same group then demonstrated that one of their organic cages can present different polymorphs depending on a chemical trigger, i.e. the solvent used for crystallization.438 Interestingly, these different polymorphs display different gas sorption properties that permit the selective sorption of hydrogen over nitrogen following a reversible on–off switching process. Cooper and co-workers further showed that the structure of their porous materials can indeed be predictable by using in silico simulations, leading to the discovery of the largest organic chiral cage molecule with the highest surface area observed for a material tackle by using DFT calculations.439 Based on this work, in which the authors have shown that the mixing of both CC3-R and CC3-S results in the formation of a racemic precipitate as a result of chiral recognition,440 they were able to enhance the surface area and the porosity of macroporous inorganic supports such as silica and alumina–silica beads (Fig. 11b), thereby presenting potential interest for further applications in chromatography or catalysis (see Sections 4.3 and 4.4).441 In parallel to these achievements, the group of Mastalerz has developed a series of salicylbisimine cage compounds which present very attractive properties for gas storage (Fig. 11c).429 First of all, they have shown that the cage compound depicted in Fig. 11c displays a very high BET surface area (1377 m2 g−1) which can be improved up to 2071 m2 g−1 after crystallization with a suitable solvent, yielding the highest surface area observed for cage compounds.442,443 Interestingly, when the imine bonds were reduced to amine ones, the cage compound completely lost its gas sorption property, indicating the importance of the rigidity of the structure for gas absorption. From an applicative point of view, films made of these cage compounds were deposited on the electrode surface of a quartz crystal microbalance (QCM) and could be used as sensors for common aromatic solvents.444 To complete this study, the authors have shown that the size and the shape of the cavity has a great influence on the selectivity of binding, and that [2 + 3] exo-functionalized cage compounds outperform “state-of-the-art” compounds (modified cyclodextrins) used as high affinity materials for QCMs.
600 m2 g−1.452 Interestingly, along their very high surface areas, MOFs have been shown to display excellent storage capacity for gases of importance in environmental sciences such as methane, hydrogen or carbon dioxide.453–457 For example, CO2 could be captured and recycled several times from air and flue gas with a CO2 purity exceeding 96%, and without losing the MOF capacity upon recycling.458 In another example, a partially interpenetrated MOF was used to capture gases such as CO2, N2, CH4, O2, Ar, and H2 at high pressure and to further store CO2 trapped in the nanopores when decreasing the pressure.459 MOFs have also been applied to selective sorption of organic vapors such as methanol, acetonitrile or benzene over water, thus presenting a potential interest for water purification.460 Recently, the group of Kitagawa reported one of the rare examples in which O2 and NO were selectively absorbed over other atmospheric gases by a MOF due to charge transfer interactions between the guests and the organic linkers of the MOF.461 Finally, a recent advancement in CO2 storage is the use of flexible MOFs that are not only used to absorb CO2 over other atmospheric gases but also to confirm its presence by a significant change in fluorescence due to the presence of a reporter molecule within the MOF.462 Interestingly, the fact that MOFs are able to selectively capture one gas in a mixture makes them suitable for separation and purification purposes.4554.3 Nanomaterials for separation/purification and transport
Nanopores and nanochannels. The advanced field of synthetic transport systems rests mainly on the use of nanochannels and nanopores for transporting ions, gases, or small molecules across bilayer membranes.463 Of particular interest is the work by Chui and Fyles, who have recently correlated the topology of synthetic ion channels with their class of activity, their conductance, and their opening duration, demonstrating the broad diversity in terms of structures.464 For example, the use of cholate to produce efficient ion transporters is well documented; particularly using cholapods and oligocholates.463,465 Zhao and coworkers have lately demonstrated the formation of transmembrane nanopores using four amphiphilic tri- or tetraoligocholate macrocycles through hydrophobic interactions, following a highly cooperative mechanism.466 Counter-intuitively, this system showed a more efficient transport of hydrophilic molecules in an increased hydrophobic environment and a faster transport of longer sugars versus shorter ones in a larger pore size. Recently, the group of Hou revealed new structures based on pillar[5]arenes for the efficient transport of water across bilayer membranes and demonstrated that fine tuning of the structure can block the transport of protons, like in natural water channel proteins.467,468 Another important characteristic of the recently developed nanochannels is their responsiveness to the environment. In 2006, the group of Futaki developed an artificial peptidic channel that displays ion fluxes only in the presence of Fe(III) ions and can be reversibly closed by adding EDTA as a competing ligand, thereby preventing the destabilization of the extra-membranar helical structure of the peptide, which causes the opening of the pore.469 In 2008, the groups of Satake and Kobuke took advantage of electrostatic interactions between carboxylates present at the outer side of a large artificial pore made of self-assembled oligoporphyrins and polycationic dendrimers to block a channel under low pH and allow its reopening when increasing the pH or the concentration of buffer.470 The same year, Wilson and Webb reported the first chemically responsive channel in which palladium ions were used to generate cation channels by complexing pyridyl-cholate lipids into membrane-spanning compounds.471,472 The addition of a competing ligand such as hexathia-18-crown-6 induced the closure of the channel, whereas outer decoration of the cholates with biotin moieties permits its complexation with avidin to control the ion flow across the membrane. The group of Matile has also demonstrated the possible multifunctionality of a single self-assembled system.473 Indeed, p-octiphenyl rods decorated with naphthalene units were shown to self-assemble into a quadruple π-M-helix that displays photosynthetic activity when inserted in vesicles with a well-defined environment. Addition of electron-rich ligands causes the opening of the helix into a barrel-stave ion channel selective for anions.474 This group has also taken advantage of various supramolecular interactions to develop innovative nanopores.475 In collaboration with Guichard and coworkers, they showed that oligourea/amide macrocycles can be inserted in bilayer membranes and serve as selective anion transporters.476 In that case, anion–macrodipole and macrodipole–potential interactions were shown to be responsible for anion selectivity and voltage sensitivity, respectively. In another example, by using a collection of naphthalenediimide (NDI), this group also demonstrated the relevance of anion–π interactions for the transport of anions in bilayer membranes.477,478 In particular, they have shown that π-acidification and decrowding of the active site, namely NDI, play an important role in the selective transport of chlorine over bromine and iodine ions.477 Conversely, high supramolecular organization inducing π, π-enhanced anion–π interactions was shown to account for the preferential transport of nitrate over acetate and a poorer selectivity towards halogen ions. Recently, Matile and coworkers have exploited the hydrophobicity, directionality, and strength of halogen bonds in small perfluorinated molecules, even the highly volatile trifluoroiodomethane, to transport anions across lipid bilayers.479 The absence of leakage and the observed cooperativity and selectivity make this small and simple system particularly attractive for applications in life and material sciences. Finally, it should be noted that artificial β-barrels developed by this group and mentioned in Section 4.1 have not only been used as sensors but also as transport systems across lipid membranes.403,463,464
Nanostructured membranes. One important tool in environmental science is the use of membranes to remove any contaminants that might be present in water.480,481 In 2007, Moore and coworkers used six-arm rigid star amphiphiles to produce nanofiltration membranes over a block copolymeric film that rival the best commercially available membranes for the rejection of Rhodamine Wt and As(III) together with a two- to three-fold higher water flux.482 Two years later, the group of Ichinose reported the formation of ferritin-based membranes of 30–100 nm thickness that were not only stable in organic solvents and for a wide range of pH (1.5–13) but also mechanically robust.483 This property was confirmed by the ability of the system to withstand water fluxes that are more than 1000 times the ones supported by commercially available membranes while displaying similar filtration properties. Recently, Rybtchinski and coworkers demonstrated the recyclability of a supramolecular membrane made of perylene-based amphiphilic molecules and its capacity to separate nanoparticles by size (Fig. 12a).480,484 The supramolecular membrane with an easily controlled thickness is formed after filtration over a commercial microfiltration syringe filter of a water solution of bis-perylene displayed in Fig. 12a(v). Filtration of a water solution of nanoparticles allows the direct collection of sub-5 nm nanoparticles in the filtrate and retention of the larger ones on the filter (Fig. 12a(iv)). Washing of the filter with a water–ethanol mixture disrupts the supramolecular membrane and allows the subsequent recovery of the large nanoparticles. Interestingly, this membrane could be reused up to four times without losing its properties and was also applied to the size-exclusion chromatography of sub-5 nm quantum dots with a reasonable selectivity. In 2012, the groups of Barboiu and Quemener have shown that 50 nm diameter micelles made of triblock copolymers can form nanoporous supramolecular films with tunable porosity depending on the water pressure applied.485 This adaptive behavior under an external stimulus (pressure) was confirmed by the self-healing ability of the membrane under increasing water flow pressures. The group of Barbiou has recently developed a number of adaptive membranes for the transport of gas or ions using dynamic constitutional systems and supramolecular chemistry.481,486–490 For example, they have reported one of the rare examples of a water and proton transporter491 using ureidoimidazole quartets that form barreled channels in a membrane environment, the system being inert for transporting Li+ and Na+.487
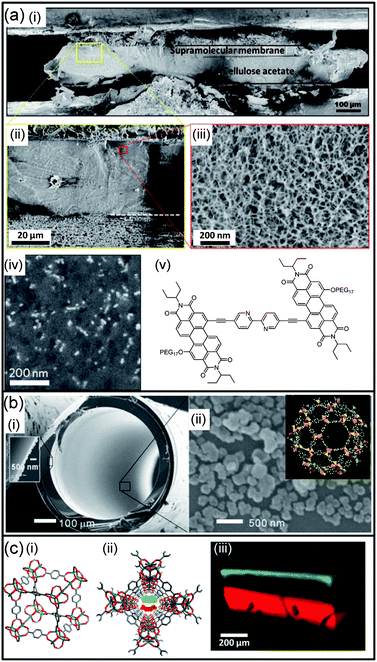 | ||
| Fig. 12 Self-assembled nanomaterials for separation and transport. (a) (i) Cryo-SEM image of the membrane's cross-section; (ii) magnified area showing the sharp border between the supramolecular membrane and its cellulose acetate support; (iii) high magnification reveals the porous three-dimensional nanostructure of the supramolecular membrane material; (iv) backscattered electron cryo-SEM image of the supramolecular membrane used for filtering a solution of gold nanoparticles. Gold nanoparticles (appearing as bright spots) have sizes ranging from 10 to 20 nm; (v) amphiphilic perylene used as a self-assembly unit for the supramolecular membrane. (b) SEM images of (i) the cross-section of MIL-101 coated capillary column (inset shows the thickness of the MIL-101 coating); and (ii) MIL-101 deposited on the inner wall of the capillary column (inset shows the structure of MIL-101). (c) (i) One unit cell of a MOF-5 crystal made of terephthalic acid struts and octahedral carboxylato Zn4O(CO2)6 secondary building units; (ii) view along the 1D channel of MOF-5. The cross-section of the channel is ∼8 × 8 Å2. The red and green molecules in the channel illustrate the mixture to be separated; (iii) 3D fluorescence confocal images of MOF-5 crystals in which the migrating dyes are resolved into separate bands: separation of pyronin B (red) and thionin (green) at 90 min. Panel (a): reproduced with permission from ref. 484; (b) reproduced with permission from ref. 496; (c) reproduced with permission from ref. 500. | ||
MOFs. MOFs have been used to selectively store gases or small molecules (see Section 4.2), so that their use in separating devices has become evident.492,493 More than five years ago, the group of De Vos reported the successful separation in the liquid phase of mixed C8 alkylaromatic compounds using MIL-47 as the absorbent, due to an efficient packing of the various isomers in the pores.494 The difference of retention observed for the xylene isomers comes from the relative alignment of their aromatic planes within a pair located in one pore. The same MOF could also be used to separate ethylbenzene from styrene with baseline-separated peaks.495 A similar separation was achieved using MIL-53 at room temperature, but the flexibility of the framework induced a loss of resolution at higher temperatures due to enthalpic considerations. Nevertheless, MIL-53 might be relevant for industrial application as it is capable of removing typical impurities (o-xylene, toluene) originating from the synthetic process. Considering the importance of aromatic molecules in industrial processes but also regarding environmental concerns, Gu and Yan reported the fabrication of a capillary column coated with MIL-101 and its application in gas-phase chromatography for the separation of the xylene isomers (Fig. 12b).496
Not only host–guest interactions, but also the presence of unsaturated coordination sites and the polarity of the MOF were shown to account for the fast and excellent separation of benzene derivatives or n-alkane mixtures. Extension of this work using other MOFs and/or surface-coating techniques for the separation of environmentally relevant samples, such as natural gas or petrol, clearly establishes MOFs as promising coating phases for high resolution gas chromatography.497,498 The groups of Bryliakov and Fedin demonstrated also the potential of a chiral MOF to efficiently separate sulfoxide enantiomers with an enantiomeric excess up to 60%.499 They have even shown the possibility of performing a one-pot oxidation/purification process on thioethers, pointing out the dual role played by the MOF as both the catalyst and stationary phase. Millimeter-sized MOF single crystals presenting nanometric pores have also been used as “chromatographic columns” to separate aromatic dyes (Fig. 12c).500 In this case, the sizes of the molecules along with their supramolecular interactions with the pores account for the selectivities observed during chromatography. Selective gas membranes have also been elaborated using MOFs either as thin films or as additives to mixed-matrix membranes.492 For example, Caro and coworkers took advantage of the difference in kinetic parameters between gases (H2, CO2, N2, CH4) to produce a membrane made of a ZIF-7 thin film over an alumina support that enabled the selective filtration of hydrogen over the other gases with a very good thermal stability.501 A similar molecular sieving effect was also observed for the separation of small gas molecules (H2, CO2) from larger ones such as methane or propane using a polymeric membrane coated with ZIF-8 due to their ability to fit into the pores of the MOF.502 For environmental applications, MOF-based materials are not far from being used in practical applications, although issues regarding their stability, scale-up and the design of the separation process remain to be addressed.492
Host–guest devices. As mentioned previously, this review is not dealing with simple host–guest recognition events. However, regarding separation and purification purposes, many devices taking advantages of this phenomenon have been reported and could be practically used for challenging separations.503,504 One particular example is the purification of single-walled carbon nanotubes (SWNTs), whose chiralities highly impact their optical, electrical, and mechanical properties.505 For more than five years, chemists have taken advantage of supramolecular interactions between SWNTs and chiral diporphyrin or DNA, for instance, to produce optically active nanotubes.506,507 Macrocylic hosts inducing supramolecular interactions with the guest have also been used for the separation of enantiomers such as fullerene C76 or various organic compounds, and could potentially be used as chiral stationary phases in HPLC columns.508,509 Finally, the reversible affinity between boronic acids and diols has been used to produce modified gel electrophoresis that can separate unambiguously saccharides and glycated from unglycated proteins, separations that cannot be achieved using classical electrophoretic techniques.510,511
4.4 Nanomaterials for catalysis
Reducing the costs and wastes of chemical reactions is one of the major environmental issues for industry and catalysts are very important tools in that direction. In living organisms, enzymes perform efficient catalytic reactions in confined and well-organized environments. Chemists have always been inspired by this structure/property relationship and they still actively try to develop small complexes or large supramolecular assemblies that can compete in these ranges of efficiency and selectivity, but also in the ability to regulate their activity using allosteric effects.512–514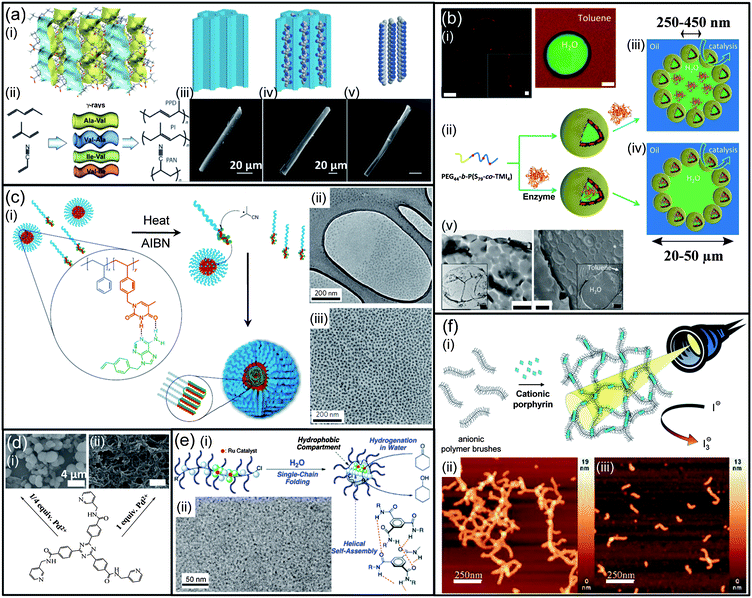 | ||
| Fig. 13 Self-assembled nanomaterials for catalysis. (a) (i) Crystal structure of the porous Ala–Val compound showing the empty channels along the c axis in blue and yellow; (ii) schematic representation of the monomers and dipeptides used for the polymerization process; SEM images of (iii) an individual Ala–Val crystal, (iv) Ala–Val/PAN crystalline adduct, (v) ladder-polymer fibril after removal of the crystal; a schematic representation of the process at the molecular level is shown above, PAN: poly(acrylonitrile). (b) (i) CLSM images of polymersome Pickering emulsions (water in toluene); left: polymersomes are stained by rhodamine B (scale bar: 20 μm); the inset is a magnified image (scale bar: 2 μm); right: FITC-Dex (4.4 kDa) and Nile red were dissolved in water and toluene, respectively (scale bar: 10 μm); (ii) representation of the crosslinking process to prepare stabilized polymersomes and schematic representation of a Pickering emulsion with the enzyme in the water phase (iii), or with the enzyme inside the polymersome lumen (iv); (v) TEM image (scale bar: 500 nm) and cryo-SEM image (scale bar: 500 nm) of polymersome Pickering emulsions; the insets are overviews at lower magnification (left inset: scale bar 2 μm, right inset: scale bar 5 μm). (c) (i) On addition of AIBN and heating, it is proposed that vinylbenzyl adenine (VBA) initiation occurs on an exchanging unimer before returning to a micelle made of the block copolymer PSt-b-PVBT (poly(styrene-b-vinylbenzyl thymine)) for continued propagation. Further dynamic VBA-loaded unimers add to the initiated micelle, from a reservoir of non-initiated micelles, to yield a stable, non-dynamic larger micelle that contains the high molecular weight, low polydispersity of the PVBA daughter polymer; (ii) TEM image of PSt115-b-PVBT18 micelles prior to VBA addition, the lighter region is the grapheme oxide coating over a hole in the lacey carbon support (darker/thicker region); (iii) TEM image of micelles obtained after the addition and polymerization of VBA in the presence of PSt115-b-PVBT18. (d) SEM images of xerogels made of a large semirigid tripodal ligand, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)-tris(N-(pyridin-3-ylmethyl)benzamide) in different Pd/L ratios of (i) 1:4 and (ii) 1:1. (e) (i) Supramolecular single-chain folding of polymers in water affording a compartmentalized catalyst for the transfer hydrogenation of ketones; (ii) cryo-TEM images of the Ru-PEGMA/BTAMA/SDP segmented terpolymer (Mw, SLS = 105000, Rh, DLS = 6.8 nm): Magnification = 62 k, PEGMA: poly(ethylene glycol) methyl ether methacrylate, BTAMA: benzene-1,3,5-tricarboxamide-bearing methacrylate, SDP: diphenylphosphinostyrene. (f) (i) Schematic representation of supramolecular structure formation between cylindrical polystyrene sulfonate (PSS) brushes and cationic tetravalent porphyrins (TMPyP) and their use as a photocatalyst; atomic force microscopy (AFM) of a TMPyP/PSS-brush sample with a charge ratio of l:0.4 deposited onto a mica surface: (ii) sample prepared in aqueous solution, (iii) sample prepared in aqueous solution with c(iodide) = 0.01 mol L−1. Panel (a): adapted with permission from ref. 531; (b) reproduced with permission from ref. 549; (c) adapted with permission from ref. 554; (d) reproduced with permission from ref. 534; (e) adapted with permission from ref. 556; (f) reproduced with permission from ref. 560. | ||
5 Conclusion
The bottom-up construction of nanostructures based on supramolecular self-assembly has been a subject of utmost fundamental interest for the past forty years and chemists have acquired a high control of objects at that scale by encoding their molecular constituents. We are currently witnessing that this field is sufficiently mature to apply its construction rules towards functional nanomaterials with novel properties related to their sizes, defined structures, and dynamics. This review illustrates how such materials afford a series of interesting responses to crucial societal problems for health, energy, and environment. Beyond this snapshot of recent advances, one can easily imagine that such a blossoming technology has still plenty of room to expand and will certainly produce even more sophisticated nanomaterials in the future. In particular, many challenges remain in the fundamental understanding of self-organization at the nanoscale when coming to the study of far from equilibrium self-assembled systems and networks. We believe, among others, that the expected progress in this challenging field related to complexity and emergence will dramatically affect the design and potential of the next generation of nanomaterials with the ultimate goal to bridge the gap, one day, between materials and life science.Acknowledgements
We thank the European Research Council under the European Community's Seventh Framework Program (FP7/2007–2013)/ERC Starting Grant agreement no. 257099. We also thank the Agence Nationale pour la Recherche (ANR-09-BLAN-034-02) and a Marie Curie Career Integration Grant for postdoctoral fellowships (E. B. and Y. R.). We acknowledge the University of Strasbourg (UdS), the Centre National de la Recherche Scientifique (CNRS), the international center for Frontier Research in Chemistry (icFRC), the Laboratory of Excellence for Complex Systems Chemistry (LabEx CSC), and the Institut Universitaire de France (IUF).Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- J.-M. Lehn, Supramolecular Chemistry, WILEY-VCH Verlag GmbH, Weinheim, 1995 Search PubMed.
- Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012 Search PubMed.
- Nanomedicine, in Nanotechnology, ed. H. Fuchs, M. Grätzel, H. Krug, G. Schmid, V. Vogel and R. Waser, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, vol. 5 Search PubMed.
- Nanomaterials for Medical Applications, ed. Z. P. Aguilar, Elsevier, Amsterdam, 2013 Search PubMed.
- NanoBiotechnology, ed. C. M. Niemeyer and C. A. Mirkin, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- Nanobiotechnology II, ed. C. M. Niemeyer and C. A. Mirkin, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007 Search PubMed.
- Z. P. Aguilar, Chapter 4-Nanobiosensors, in Nanomaterials for Medical Applications, ed. A. Zoraida, Elsevier, 2013, pp. 127–179 Search PubMed.
- Nanomaterials for Medical Diagnosis and Therapy, ed. C. S. S. R. Kumar, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007 Search PubMed.
- Nanomaterials in Drug Delivery, Imaging, and Tissue Engineering, ed. A. Tiwari and A. Tiwari, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013 Search PubMed.
- J. Lei and H. Ju, Chem. Soc. Rev., 2012, 41, 2122–2134 RSC.
- K. B. Cederquist and S. O. Kelley, Curr. Opin. Chem. Biol., 2012, 16, 415–421 CrossRef CAS.
- H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849–2866 RSC.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS.
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS.
- D. G. Georganopoulou, L. Chang, J.-M. Nam, C. S. Thaxton, E. J. Mufson, W. L. Klein and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2273–2276 CrossRef CAS.
- R. de la Rica, D. Aili and M. M. Stevens, Adv. Drug Delivery Rev., 2012, 64, 967–978 CrossRef CAS.
- A. Laromaine, L. Koh, M. Murugesan, R. V. Ulijn and M. M. Stevens, J. Am. Chem. Soc., 2007, 129, 4156–4157 CrossRef CAS.
- D. Aili, R. Selegård, L. Baltzer, K. Enander and B. Liedberg, Small, 2009, 5, 2445–2452 CrossRef CAS.
- S. Gupta, H. Andresen, J. E. Ghadiali and M. M. Stevens, Small, 2010, 6, 1509–1513 CrossRef CAS.
- D. Aili, M. Mager, D. Roche and M. M. Stevens, Nano Lett., 2010, 11, 1401–1405 CrossRef.
- X. Liu, Q. Dai, L. Austin, J. Coutts, G. Knowles, J. Zou, H. Chen and Q. Huo, J. Am. Chem. Soc., 2008, 130, 2780–2782 CrossRef CAS.
- C.-C. You, O. R. Miranda, B. Gider, P. S. Ghosh, I.-B. Kim, B. Erdogan, S. A. Krovi, U. H. F. Bunz and V. M. Rotello, Nat. Nanotechnol., 2007, 2, 318–323 CrossRef CAS.
- O. R. Miranda, B. Creran and V. M. Rotello, Curr. Opin. Chem. Biol., 2010, 14, 728–736 CrossRef CAS.
- X. Xu, B. Lin, J. Feng, Y. Wang, S. Cheng, X. Zhang and R. Zhuo, Macromol. Rapid Commun., 2012, 33, 426–431 CrossRef CAS.
- G. Liang, H. Ren and J. Rao, Nat. Chem., 2010, 2, 54–60 CrossRef CAS.
- C.-Y. Cao, Y. Chen, F.-Z. Wu, Y. Deng and G.-L. Liang, Chem. Commun., 2011, 47, 10320–10322 RSC.
- C. J. Musto and K. S. Suslick, Curr. Opin. Chem. Biol., 2010, 14, 758–766 CrossRef CAS.
- N. Y. Edwards, T. W. Sager, J. T. McDevitt and E. V. Anslyn, J. Am. Chem. Soc., 2007, 129, 13575–13583 CrossRef CAS.
- H. Özhalici-Ünal and B. A. Armitage, ACS Nano, 2009, 3, 425–433 CrossRef.
- C. Lin, R. Jungmann, A. M. Leifer, C. Li, D. Levner, G. M. Church, W. M. Shih and P. Yin, Nat. Chem., 2012, 4, 832–839 CrossRef CAS.
- E. M. Cornett, E. A. Campbell, G. Gulenay, E. Peterson, N. Bhaskar and D. M. Kolpashchikov, Angew. Chem., Int. Ed., 2012, 51, 9075–9077 CrossRef CAS.
- I. Koh, R. Hong, R. Weissleder and L. Josephson, Angew. Chem., Int. Ed., 2008, 47, 4119–4121 CrossRef CAS.
- H. Lee, E. Sun, D. Ham and R. Weissleder, Nat. Med., 2008, 14, 869–874 CrossRef CAS.
- P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS.
- P. Besenius, J. L. M. Heynens, R. Straathof, M. M. L. Nieuwenhuizen, P. H. H. Bomans, E. Terreno, S. Aime, G. J. Strijkers, K. Nicolay and E. W. Meijer, Contrast Media Mol. Imaging, 2012, 7, 356–361 CrossRef CAS.
- C.-Y. Cao, Y.-Y. Shen, J.-D. Wang, L. Li and G.-L. Liang, Sci. Rep., 2013, 3, 1024 Search PubMed.
- S.-J. Park, T. A. Taton and C. A. Mirkin, Science, 2002, 295, 1503–1506 CrossRef CAS.
- B. He, T. J. Morrow and C. D. Keating, Curr. Opin. Chem. Biol., 2008, 12, 522–528 CrossRef CAS.
- C. M. Lieber, MRS Bull., 2011, 36, 1052–1063 CrossRef CAS.
- H. Lord and S. O. Kelley, J. Mater. Chem., 2009, 19, 3127–3134 RSC.
- J.-I. Hahm and C. M. Lieber, Nano Lett., 2003, 4, 51–54 CrossRef.
- Y. L. Bunimovich, Y. S. Shin, W.-S. Yeo, M. Amori, G. Kwong and J. R. Heath, J. Am. Chem. Soc., 2006, 128, 16323–16331 CrossRef CAS.
- A. Star, E. Tu, J. Niemann, J.-C. P. Gabriel, C. S. Joiner and C. Valcke, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 921–926 CrossRef CAS.
- S. N. Kim, J. F. Rusling and F. Papadimitrakopoulos, Adv. Mater., 2007, 19, 3214–3228 CrossRef CAS.
- Z. Wu, Z. Zhen, J.-H. Jiang, G.-L. Shen and R.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 12325–12332 CrossRef CAS.
- L. Adler-Abramovich, M. Badihi-Mossberg, E. Gazit and J. Rishpon, Small, 2010, 6, 825–831 CrossRef CAS.
- L. Sasso, I. Vedarethinam, J. Emnéus, W. E. Svendsen and J. Castillo-León, J. Nanosci. Nanotechnol., 2012, 12, 3077–3083 CrossRef CAS.
- B. M. Venkatesan and R. Bashir, Nat. Nanotechnol., 2011, 6, 615–624 CrossRef CAS.
- B. N. Miles, A. P. Ivanov, K. A. Wilson, F. Dogan, D. Japrung and J. B. Edel, Chem. Soc. Rev., 2013, 42, 15–28 RSC.
- J. Clarke, H.-C. Wu, L. Jayasinghe, A. Patel, S. Reid and H. Bayley, Nat. Nanotechnol., 2009, 4, 265–270 CrossRef CAS.
- G. M. Cherf, K. R. Lieberman, H. Rashid, C. E. Lam, K. Karplus and M. Akeson, Nat. Biotechnol., 2012, 30, 344–348 CrossRef CAS.
- E. A. Manrao, I. M. Derrington, A. H. Laszlo, K. W. Langford, M. K. Hopper, N. Gillgren, M. Pavlenok, M. Niederweis and J. H. Gundlach, Nat. Biotechnol., 2012, 30, 349–353 CrossRef CAS.
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS.
- I. Willner, B. Willner and R. Tel-Vered, Electroanalysis, 2011, 23, 13–28 CrossRef CAS.
- Y. Wen, H. Pei, Y. Shen, J. Xi, M. Lin, N. Lu, X. Shen, J. Li and C. Fan, Sci. Rep., 2012, 2, 867 CrossRef.
- J. Zhang, R. Lao, S. Song, Z. Yan and C. Fan, Anal. Chem., 2008, 80, 9029–9033 CrossRef CAS.
- R. Freeman, Y. Li, R. Tel-Vered, E. Sharon, J. Elbaz and I. Willner, Analyst, 2009, 134, 653–656 RSC.
- Y. Lu, X. Li, L. Zhang, P. Yu, L. Su and L. Mao, Anal. Chem., 2008, 80, 1883–1890 CrossRef CAS.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS.
- Y.-B. Lim, K.-S. Moon and M. Lee, Chem. Soc. Rev., 2009, 38, 925–934 RSC.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- Y. Namiki, T. Fuchigami, N. Tada, R. Kawamura, S. Matsunuma, Y. Kitamoto and M. Nakagawa, Acc. Chem. Res., 2011, 44, 1080–1093 CrossRef CAS.
- Y. Zhou and T. Shimizu, Chem. Mater., 2007, 20, 625–633 CrossRef.
- M. Kwak and A. Herrmann, Chem. Soc. Rev., 2011, 40, 5745–5755 RSC.
- H. Wang, R. Yang, L. Yang and W. Tan, ACS Nano, 2009, 3, 2451–2460 CrossRef CAS.
- N. C. Seeman, Mol. Biotechnol., 2007, 37, 246–257 CrossRef CAS.
- A. Kuzuya and M. Komiyama, Nanoscale, 2010, 2, 310–322 RSC.
- Y.-B. Lim and M. Lee, Org. Biomol. Chem., 2007, 5, 401–405 CAS.
- M. Numata and S. Shinkai, Self-Assembled Polysaccharide Nanotubes Generated from β-1,3-Glucan Polysaccharides, in Self-Assembled Nanomaterials II, ed. T. Shimizu, Springer, Berlin, Heidelberg 2008, vol. 220, ch. 145, pp. 65–121 Search PubMed.
- T. Shimizu, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5137–5152 CrossRef CAS.
- N. C. Reichardt, M. Martin-Lomas and S. Penades, Chem. Soc. Rev., 2013, 42, 4358–4376 RSC.
- M. Numata and S. Shinkai, Chem. Commun., 2011, 47, 1961–1975 RSC.
- Y. Ma, R. J. Nolte and J. J. Cornelissen, Adv. Drug Delivery Rev., 2012, 64, 811–825 CrossRef CAS.
- O. Pornillos, B. K. Ganser-Pornillos and M. Yeager, Nature, 2011, 469, 424–427 CrossRef CAS.
- L. W. Seymour and A. J. Thrasher, Nat. Biotechnol., 2012, 30, 588–593 CrossRef CAS.
- J. A. Hoxie and C. H. June, Cold Spring Harbor Perspect. Med., 2012, 2 DOI:10.1101/cshperspect.a007179.
- I. J. Minten, Y. Ma, M. A. Hempenius, G. J. Vancso, R. J. M. Nolte and J. J. L. M. Cornelissen, Org. Biomol. Chem., 2009, 7, 4685–4688 CAS.
- F. M. Brunel, J. D. Lewis, G. Destito, N. F. Steinmetz, M. Manchester, H. Stuhlmann and P. E. Dawson, Nano Lett., 2010, 10, 1093–1097 CrossRef CAS.
- I. Yildiz, S. Shukla and N. F. Steinmetz, Curr. Opin. Biotechnol., 2011, 22, 901–908 CrossRef CAS.
- B. Lamarre and M. G. Ryadnov, Macromol. Biosci., 2011, 11, 503–513 CrossRef CAS.
- L. Lee, Z. Niu and Q. Wang, Nano Res., 2009, 2, 349–364 CrossRef CAS.
- K. Matsuura, K. Murasato and N. Kimizuka, J. Am. Chem. Soc., 2005, 127, 10148–10149 CrossRef CAS.
- R. Zandi, D. Reguera, R. F. Bruinsma, W. M. Gelbart and J. Rudnick, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15556–15560 CrossRef CAS.
- K. Matsuura, K. Watanabe, T. Matsuzaki, K. Sakurai and N. Kimizuka, Angew. Chem., Int. Ed., 2010, 49, 9662–9665 CrossRef CAS.
- S. Raman, G. Machaidze, A. Lustig, U. Aebi and P. Burkhard, Nanomedicine: Nanotechnology, Biology and Medicine, 2006, 2, 95–102 CrossRef CAS.
- T. A. P. F. Pimentel, Z. Yan, S. A. Jeffers, K. V. Holmes, R. S. Hodges and P. Burkhard, Chem. Biol. Drug Des., 2009, 73, 53–61 CAS.
- F. Boato, R. M. Thomas, A. Ghasparian, A. Freund-Renard, K. Moehle and J. A. Robinson, Angew. Chem., Int. Ed., 2007, 46, 9015–9018 CrossRef CAS.
- D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093 CrossRef.
- Y.-b. Lim, E. Lee, Y.-R. Yoon, M. Ä. Lee and M. Lee, Angew. Chem., Int. Ed., 2008, 47, 4525–4528 CrossRef CAS.
- Y. Aoyama, Chem.–Eur. J., 2004, 10, 588–593 CrossRef CAS.
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13, 238–252 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS.
- M. L. Tan, P. F. M. Choong and C. R. Dass, Peptides, 2010, 31, 184–193 CrossRef CAS.
- T. Lian and R. J. Y. Ho, J. Pharm. Sci., 2001, 90, 667–680 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS.
- Y. Malam, M. Loizidou and A. M. Seifalian, Trends Pharmacol. Sci., 2009, 30, 592–599 CrossRef CAS.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS.
- S. Bhattacharya and A. Bajaj, Chem. Commun., 2009, 4632–4656 RSC.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2008, 109, 259–302 CrossRef.
- J.-P. Behr, Bioconjugate Chem., 1994, 5, 382–389 CrossRef CAS.
- X.-X. Zhang, T. J. McIntosh and M. W. Grinstaff, Biochimie, 2012, 94, 42–58 CrossRef CAS.
- K. Ewert, H. M. Evans, A. Ahmad, N. L. Slack, A. J. Lin, A. Martin-Herranz and C. R. Safinya, Lipoplex Structures and Their Distinct Cellular Pathways, in Advances in Genetics, ed. M.-C. H. Leaf Huang and W. Ernst, Academic Press, 2005, vol. 53, pp. 119–155 Search PubMed.
- G. Caracciolo and H. Amenitsch, Eur. Biophys. J., 2012, 41, 815–829 CrossRef CAS.
- A. Bilalov, U. Olsson and B. Lindman, Soft Matter, 2012, 8, 11022–11033 RSC.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS.
- L. C. Gomes-da-Silva, N. A. Fonseca, V. Moura, M. C. Pedroso de Lima, S. Simões and J. N. Moreira, Acc. Chem. Res., 2012, 45, 1163–1171 CrossRef CAS.
- M. Morille, C. Passirani, A. Vonarbourg, A. Clavreul and J.-P. Benoit, Biomaterials, 2008, 29, 3477–3496 CrossRef CAS.
- A. Akinc, A. Zumbuehl, M. Goldberg, E. S. Leshchiner, V. Busini, N. Hossain, S. A. Bacallado, D. N. Nguyen, J. Fuller, R. Alvarez, A. Borodovsky, T. Borland, R. Constien, A. de Fougerolles, J. R. Dorkin, K. Narayanannair Jayaprakash, M. Jayaraman, M. John, V. Koteliansky, M. Manoharan, L. Nechev, J. Qin, T. Racie, D. Raitcheva, K. G. Rajeev, D. W. Y. Sah, J. Soutschek, I. Toudjarska, H.-P. Vornlocher, T. S. Zimmermann, R. Langer and D. G. Anderson, Nat. Biotechnol., 2008, 26, 561–569 CrossRef CAS.
- T. S. Zimmermann, A. C. H. Lee, A. Akinc, B. Bramlage, D. Bumcrot, M. N. Fedoruk, J. Harborth, J. A. Heyes, L. B. Jeffs, M. John, A. D. Judge, K. Lam, K. McClintock, L. V. Nechev, L. R. Palmer, T. Racie, I. Röhl, S. Seiffert, S. Shanmugam, V. Sood, J. Soutschek, I. Toudjarska, A. J. Wheat, E. Yaworski, W. Zedalis, V. Koteliansky, M. Manoharan, H.-P. Vornlocher and I. MacLachlan, Nature, 2006, 441, 111–114 CrossRef CAS.
- Y. Sato, K. Murase, J. Kato, M. Kobune, T. Sato, Y. Kawano, R. Takimoto, K. Takada, K. Miyanishi, T. Matsunaga, T. Takayama and Y. Niitsu, Nat. Biotechnol., 2008, 26, 431–442 CrossRef CAS.
- S. Deshayes, M. Morris, F. Heitz and G. Divita, Adv. Drug Delivery Rev., 2008, 60, 537–547 CrossRef CAS.
- A. Thomas, L. Lins, G. Divita and R. Brasseur, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 2217–2222 CrossRef CAS.
- F. Heitz, M. C. Morris and G. Divita, Br. J. Pharmacol., 2009, 157, 195–206 CrossRef CAS.
- Y. Yue and C. Wu, Biomater. Sci., 2013, 1, 152–170 RSC.
- K. Miyata, N. Nishiyama and K. Kataoka, Chem. Soc. Rev., 2012, 41, 2562–2574 RSC.
- Y. Vachutinsky and K. Kataoka, Isr. J. Chem., 2010, 50, 175–184 CrossRef CAS.
- K. Itaka, K. Yamauchi, A. Harada, K. Nakamura, H. Kawaguchi and K. Kataoka, Biomaterials, 2003, 24, 4495–4506 CrossRef CAS.
- K. Osada, H. Oshima, D. Kobayashi, M. Doi, M. Enoki, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2010, 132, 12343–12348 CrossRef CAS.
- R. J. Christie, Y. Matsumoto, K. Miyata, T. Nomoto, S. Fukushima, K. Osada, J. Halnaut, F. Pittella, H. J. Kim, N. Nishiyama and K. Kataoka, ACS Nano, 2012, 6, 5174–5189 CrossRef CAS.
- K. Sakurai, K. Uezu, M. Numata, T. Hasegawa, C. Li, K. Kaneko and S. Shinkai, Chem. Commun., 2005, 4383–4398 RSC.
- A. Hernandez-Garcia, M. W. T. Werten, M. C. Stuart, F. A. de Wolf and R. de Vries, Small, 2012, 8, 3491–3501 CrossRef CAS.
- X. Jiang, W. Qu, D. Pan, Y. Ren, J.-M. Williford, H. Cui, E. Luijten and H.-Q. Mao, Adv. Mater., 2013, 25, 227–232 CrossRef CAS.
- M. E. Davis, Mol. Pharmaceutics, 2009, 6, 659–668 CrossRef CAS.
- M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–1070 CrossRef CAS.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS.
- S. Rana, A. Bajaj, R. Mout and V. M. Rotello, Adv. Drug Delivery Rev., 2012, 64, 200–216 CrossRef CAS.
- E. C. Dreaden, M. A. Mackey, X. Huang, B. Kang and M. A. El-Sayed, Chem. Soc. Rev., 2011, 40, 3391–3404 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- C. K. Kim, P. Ghosh, C. Pagliuca, Z.-J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360–1361 CrossRef CAS.
- C. Park, H. Youn, H. Kim, T. Noh, Y. H. Kook, E. T. Oh, H. J. Park and C. Kim, J. Mater. Chem., 2009, 19, 2310–2315 RSC.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS.
- D. Zheng, D. A. Giljohann, D. L. Chen, M. D. Massich, X.-Q. Wang, H. Iordanov, C. A. Mirkin and A. S. Paller, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11975–11980 CrossRef CAS.
- M. W. Ambrogio, C. R. Thomas, Y. Zhao, J. I. Zink and J. F. Stoddart, Acc. Chem. Res., 2012, 44, 903–913 CrossRef.
- C. R. Thomas, D. P. Ferris, J.-H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J.-S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- S. V. Aathimanikandan, E. N. Savariar and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 14922–14929 CrossRef CAS.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS.
- J. Xu and S. Liu, Soft Matter, 2008, 4, 1745–1749 RSC.
- T. Chen, C. S. Wu, E. Jimenez, Z. Zhu, J. G. Dajac, M. You, D. Han, X. Zhang and W. Tan, Angew. Chem., Int. Ed., 2013, 52, 2012–2016 CrossRef CAS.
- A. M. Rush, M. P. Thompson, E. T. Tatro and N. C. Gianneschi, ACS Nano, 2013, 7, 1379–1387 CrossRef CAS.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- J. B. Matson, M. J. Webber, V. K. Tamboli, B. Weber and S. I. Stupp, Soft Matter, 2012, 8, 6689–6692 RSC.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 47, 7962–7964 RSC.
- S. Soukasene, D. J. Toft, T. J. Moyer, H. Lu, H.-K. Lee, S. M. Standley, V. L. Cryns and S. I. Stupp, ACS Nano, 2011, 5, 9113–9121 CrossRef CAS.
- M. J. Webber, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2011, 7, 9665–9672 RSC.
- D. J. Toft, T. J. Moyer, S. M. Standley, Y. Ruff, A. Ugolkov, S. I. Stupp and V. L. Cryns, ACS Nano, 2012, 6, 7956–7965 CrossRef CAS.
- A. G. Cheetham, P. Zhang, Y.-A. Lin, L. L. Lock and H. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., Int. Ed., 2004, 43, 5242–5246 CrossRef CAS.
- Z. Liu, S. M. Tabakman, Z. Chen and H. Dai, Nat. Protocols, 2009, 4, 1372–1381 CAS.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS.
- Z. Liu, A. C. Fan, K. Rakhra, S. Sherlock, A. Goodwin, X. Chen, Q. Yang, D. W. Felsher and H. Dai, Angew. Chem., Int. Ed., 2009, 48, 7668–7672 CrossRef CAS.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- A. Altunbas and D. Pochan, Peptide-Based and Polypeptide-Based Hydrogels for Drug Delivery and Tissue Engineering, in Peptide-Based Materials, ed. T. Deming, Springer, Berlin, Heidelberg, 2012, vol. 310, ch. 206, pp. 135–167 Search PubMed.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS.
- Y. Kim, M. Park and S. Song, ACS Nano, 2012, 6, 5757–5766 CrossRef CAS.
- D. Bhatia, S. Sharma and Y. Krishnan, Curr. Opin. Biotechnol., 2011, 22, 475–484 CrossRef CAS.
- Y.-X. Zhao, A. Shaw, X. Zeng, E. Benson, A. M. Nyström and B. Högberg, ACS Nano, 2012, 6, 8684–8691 CrossRef CAS.
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS.
- T. Dvir, B. P. Timko, D. S. Kohane and R. Langer, Nat. Nanotechnol., 2011, 6, 13–22 CrossRef CAS.
- E. Engel, A. Michiardi, M. Navarro, D. Lacroix and J. A. Planell, Trends Biotechnol., 2008, 26, 39–47 CrossRef CAS.
- C. Cha, W. B. Liechty, A. Khademhosseini and N. A. Peppas, ACS Nano, 2012, 6, 9353–9358 CrossRef CAS.
- S. I. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS.
- N. Stephanopoulos, J. H. Ortony and S. I. Stupp, Acta Mater., 2013, 61, 912–930 CrossRef CAS.
- E. C. Wu, S. Zhang and C. A. E. Hauser, Adv. Funct. Mater., 2012, 22, 456–468 CrossRef CAS.
- Z. Luo and S. Zhang, Chem. Soc. Rev., 2012, 41, 4736–4754 RSC.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS.
- A. Mata, Y. Geng, K. J. Henrikson, C. Aparicio, S. R. Stock, R. L. Satcher and S. I. Stupp, Biomaterials, 2010, 31, 6004–6012 CrossRef CAS.
- R. N. Shah, N. A. Shah, M. M. Del Rosario Lim, C. Hsieh, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298 CrossRef CAS.
- K. Rajangam, H. A. Behanna, M. J. Hui, X. Han, J. F. Hulvat, J. W. Lomasney and S. I. Stupp, Nano Lett., 2006, 6, 2086–2090 CrossRef CAS.
- M. J. Webber, X. Han, S. N. Prasanna Murthy, K. Rajangam, S. I. Stupp and J. W. Lomasney, J. Tissue Eng. Regener. Med., 2010, 4, 600–610 CrossRef CAS.
- J. C. Stendahl, L.-J. Wang, L. W. Chow, D. B. Kaufman and S. I. Stupp, Transplantation, 2008, 86, 478–481 CrossRef CAS.
- Z. Huang, T. D. Sargeant, J. F. Hulvat, A. Mata, P. Bringas, C.-Y. Koh, S. I. Stupp and M. L. Snead, J. Bone Miner. Res., 2008, 23, 1995–2006 CrossRef CAS.
- M. J. Webber, J. Tongers, C. J. Newcomb, K.-T. Marquardt, J. Bauersachs, D. W. Losordo and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13438–13443 CrossRef CAS.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS.
- M. T. McClendon and S. I. Stupp, Biomaterials, 2012, 33, 5713–5722 CrossRef CAS.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS.
- L. W. Chow, R. Bitton, M. J. Webber, D. Carvajal, K. R. Shull, A. K. Sharma and S. I. Stupp, Biomaterials, 2011, 32, 1574–1582 CrossRef CAS.
- Y. Yang, U. Khoe, X. Wang, A. Horii, H. Yokoi and S. Zhang, Nano Today, 2009, 4, 193–210 CrossRef CAS.
- A. Horii, X. Wang, F. Gelain and S. Zhang, PLoS One, 2007, 2, e190 Search PubMed.
- L. E. R. O'Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef CAS.
- Organic Electronics: Materials, Manufacturing and Applications, ed. H. Klauk, Wiley-VCH Verlag & Co. KGaA, Weinheim, 2006 Search PubMed.
- Organic Electronics, ed. G. Meller and T. Grasser, Advances in Polymer Science, vol. 223, 2010 Search PubMed.
- Semiconducting Polymers: Chemistry, Physics and Engineering, ed. G. Hadziioannou and G. G. Malliaras, Wiley-VCH Verlag & Co. KGaA, Weinheim, 2nd edn, 2006 Search PubMed.
- Molecular and Supramolecular Information Processing, ed. E. Katz, Wiley-VCH Verlag & Co. KGaA, Weinheim, 2012 Search PubMed.
- Nanoelectronics and Information Technology, ed. R. Waser, Wiley-VCH Verlag & Co. KGaA, Weinheim, 3rd edn, 2012 Search PubMed.
- U. Shinya, M. Takehiko and T. Toshihiro, Sci. Technol. Adv. Mater., 2009, 10, 020301 CrossRef.
- Y. Yamamoto, Sci. Technol. Adv. Mater., 2012, 13, 33001 CrossRef.
- E. Moulin, J.-J. Cid and N. Giuseppone, Adv. Mater., 2013, 25, 477–487 CrossRef CAS.
- S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766–1776 CrossRef CAS.
- M. Hasegawa and M. Iyoda, Chem. Soc. Rev., 2010, 39, 2420–2427 RSC.
- E. Moulin, F. Niess, M. Maaloum, E. Buhler, I. Nyrkova and N. Giuseppone, Angew. Chem., Int. Ed., 2010, 49, 6974–6978 CrossRef CAS.
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485–490 CrossRef CAS.
- E. Moulin, F. Niess, G. Fuks, N. Jouault, E. Buhler and N. Giuseppone, Nanoscale, 2012, 4, 6748–6751 RSC.
- N. Giuseppone, Acc. Chem. Res., 2012, 45, 2178–2188 CrossRef CAS.
- M. Hasegawa, H. Enozawa, Y. Kawabata and M. Iyoda, J. Am. Chem. Soc., 2007, 129, 3072–3073 CrossRef CAS.
- J. Puigmartí-Luis, V. Laukhin, A. Pérez del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238–241 CrossRef.
- S. Ahn, Y. Kim, S. Beak, S. Ishimoto, H. Enozawa, E. Isomura, M. Hasegawa, M. Iyoda and Y. Park, J. Mater. Chem., 2010, 20, 10817–10823 RSC.
- S. Sengupta, D. Ebeling, S. Patwardhan, X. Zhang, H. von Berlepsch, C. Böttcher, V. Stepanenko, S. Uemura, C. Hentschel, H. Fuchs, F. C. Grozema, L. D. A. Siebbeles, A. R. Holzwarth, L. Chi and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6378–6382 CrossRef CAS.
- Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051–21056 CrossRef CAS.
- J. E. Kroeze, T. J. Savenije and J. M. Warman, J. Am. Chem. Soc., 2004, 126, 7608–7618 CrossRef CAS.
- J. E. Kroeze, T. J. Savenije, M. J. W. Vermeulen and J. M. Warman, J. Phys. Chem. B, 2003, 107, 7696–7705 CrossRef CAS.
- S. Yagai, T. Kinoshita, Y. Kikkawa, T. Karatsu, A. Kitamura, Y. Honsho and S. Seki, Chem.–Eur. J., 2009, 15, 9320–9324 CrossRef CAS.
- J. M. Warman and A. M. Van De Craats, Mol. Cryst. Liq. Cryst., 2003, 396, 41–72 CrossRef CAS.
- J. M. Warman, M. P. de Haas, G. Dicker, F. C. Grozema, J. Piris and M. G. Debije, Chem. Mater., 2004, 16, 4600–4609 CrossRef CAS.
- Y. Che, A. Datar, X. Yang, T. Naddo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2007, 129, 6354–6355 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- D. M. Bassani, L. Jonusauskaite, A. Lavie-Cambot, N. D. McClenaghan, J.-L. Pozzo, D. Ray and G. Vives, Coord. Chem. Rev., 2010, 254, 2429–2445 CrossRef CAS.
- H. Imahori, T. Umeyama, K. Kurotobi and Y. Takano, Chem. Commun., 2012, 48, 4032–4045 RSC.
- B. R. Kaafarani, Chem. Mater., 2011, 23, 378–396 CrossRef CAS.
- F. S. Kim, G. Ren and S. A. Jenekhe, Chem. Mater., 2011, 23, 682–732 CrossRef CAS.
- J.-P. Hong, M.-C. Um, S.-R. Nam, J.-I. Hong and S. Lee, Chem. Commun., 2009, 310–312 RSC.
- R. C. Savage, E. Orgiu, J. M. Mativetsky, W. Pisula, T. Schnitzler, C. L. Eversloh, C. Li, K. Müllen and P. Samorì, Nanoscale, 2012, 4, 2387–2393 RSC.
- J. H. Oh, H. W. Lee, S. Mannsfeld, R. M. Stoltenberg, E. Jung, Y. W. Jin, J. M. Kim, J.-B. Yoo and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6065–6070 CAS.
- H. Dong, S. Jiang, L. Jiang, Y. Liu, H. Li, W. Hu, E. Wang, S. Yan, Z. Wei, W. Xu and X. Gong, J. Am. Chem. Soc., 2009, 131, 17315–17320 CrossRef CAS.
- P. Jonkheijm, N. Stutzmann, Z. Chen, D. M. de Leeuw, E. W. Meijer, A. P. H. J. Schenning and F. Würthner, J. Am. Chem. Soc., 2006, 128, 9535–9540 CrossRef CAS.
- A. L. Briseno, S. C. B. Mannsfeld, C. Reese, J. M. Hancock, Y. Xiong, S. A. Jenekhe, Z. Bao and Y. Xia, Nano Lett., 2007, 7, 2847–2853 CrossRef CAS.
- A. L. Briseno, S. C. B. Mannsfeld, P. J. Shamberger, F. S. Ohuchi, Z. Bao, S. A. Jenekhe and Y. Xia, Chem. Mater., 2008, 20, 4712–4719 CrossRef CAS.
- Y. Zhou, W.-J. Liu, Y. Ma, H. Wang, L. Qi, Y. Cao, J. Wang and J. Pei, J. Am. Chem. Soc., 2007, 129, 12386–12387 CrossRef CAS.
- Y. Zhou, T. Lei, L. Wang, J. Pei, Y. Cao and J. Wang, Adv. Mater., 2010, 22, 1484–1487 CrossRef CAS.
- W. Jiang, Y. Zhou, H. Geng, S. Jiang, S. Yan, W. Hu, Z. Wang, Z. Shuai and J. Pei, J. Am. Chem. Soc., 2011, 133, 1–3 CrossRef CAS.
- D. H. Kim, B.-L. Lee, H. Moon, H. M. Kang, E. J. Jeong, J.-i. Park, K.-m. Han, S. Lee, B. W. Yoo, B. W. Koo, J. Y. Kim, W. H. Lee, K. Cho, H. A. Becerril and Z. Bao, J. Am. Chem. Soc., 2009, 131, 6124–6132 CrossRef CAS.
- H. N. Tsao, W. Pisula, Z. Liu, W. Osikowicz, W. R. Salaneck and K. Müllen, Adv. Mater., 2008, 20, 2715–2719 CrossRef CAS.
- S. Dong, H. Tian, D. Song, Z. Yang, D. Yan, Y. Geng and F. Wang, Chem. Commun., 2009, 3086–3088 RSC.
- H. Hayashi, W. Nihashi, T. Umeyama, Y. Matano, S. Seki, Y. Shimizu and H. Imahori, J. Am. Chem. Soc., 2011, 133, 10736–10739 CrossRef CAS.
- S. Tiwari and N. C. Greenham, Opt. Quantum Electron., 2009, 41, 69–89 CrossRef CAS.
- Y. Gao, P. Ma, Y. Chen, Y. Zhang, Y. Bian, X. Li, J. Jiang and C. Ma, Inorg. Chem., 2009, 48, 45–54 CrossRef CAS.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS.
- M. C. Gather, A. Köhnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS.
- S. Chen, L. Deng, J. Xie, L. Peng, L. Xie, Q. Fan and W. Huang, Adv. Mater., 2010, 22, 5227–5239 CrossRef CAS.
- D. Ray, C.-K. Liang, N. D. McClenaghan and D. M. Bassani, Curr. Phys. Chem., 2011, 1, 169–180 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS.
- G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- T. Hu, L. He, L. Duan and Y. Qiu, J. Mater. Chem., 2012, 22, 4206 RSC.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- C. Giansante, G. Raffy, C. Schäfer, H. Rahma, M.-T. Kao, A. G. L. Olive and A. Del Guerzo, J. Am. Chem. Soc., 2011, 133, 316–325 CrossRef CAS.
- C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS.
- J. Luo, T. Lei, L. Wang, Y. Ma, Y. Cao, J. Wang and J. Pei, J. Am. Chem. Soc., 2009, 131, 2076–2077 CrossRef CAS.
- A. Winter, C. Friebe, M. D. Hager and U. S. Schubert, Macromol. Rapid Commun., 2008, 29, 1679–1686 CrossRef CAS.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclère, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833–843 CrossRef CAS.
- Y.-L. Chu, C.-C. Cheng, Y.-C. Yen and F.-C. Chang, Adv. Mater., 2012, 24, 1894–1898 CrossRef CAS.
- N. Delbosc, M. Reynes, O. J. Dautel, G. Wantz, J.-P. Lère-Porte and J. E. Moreau, Chem. Mater., 2010, 22, 5258–5270 CrossRef CAS.
- X.-H. Zhao, G.-H. Xie, Z.-D. Liu, W.-J. Li, M.-D. Yi, L.-H. Xie, C.-P. Hu, R. Zhu, Q. Zhao, Y. Zhao, J.-F. Zhao, Y. Qian and W. Huang, Chem. Commun., 2012, 48, 3854–3856 RSC.
- D. Tordera, S. Meier, M. Lenes, R. D. Costa, E. Ortí, W. Sarfert and H. J. Bolink, Adv. Mater., 2012, 24, 897–900 CrossRef CAS.
- H.-C. Su, H.-F. Chen, Y.-C. Shen, C.-T. Liao and K.-T. Wong, J. Mater. Chem., 2011, 21, 9653 RSC.
- S. Brovelli, F. Meinardi, G. Winroth, O. Fenwick, G. Sforazzini, M. J. Frampton, L. Zalewski, J. a. Levitt, F. Marinello, P. Schiavuta, K. Suhling, H. L. Anderson and F. Cacialli, Adv. Funct. Mater., 2010, 20, 272–280 CrossRef CAS.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS.
- W. Chen, M. P. Nikiforov and S. B. Darling, Energy Environ. Sci., 2012, 5, 8045–8074 CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS.
- D. González-Rodríguez and A. P. H. J. Schenning, Chem. Mater., 2011, 23, 310–325 CrossRef.
- M. Wang and F. Wudl, J. Mater. Chem., 2012, 22, 24297–24314 RSC.
- Y. Hizume, K. Tashiro, R. Charvet, Y. Yamamoto, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2010, 132, 6628–6629 CrossRef CAS.
- K. Sugiyasu, S.-i. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- A. a. Gorodetsky, C.-Y. Chiu, T. Schiros, M. Palma, M. Cox, Z. Jia, W. Sattler, I. Kymissis, M. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7909–7912 CrossRef CAS.
- S. J. Kang, J. B. Kim, C.-Y. Chiu, S. Ahn, T. Schiros, S. S. Lee, K. G. Yager, M. F. Toney, Y.-L. Loo and C. Nuckolls, Angew. Chem., Int. Ed., 2012, 51, 8594–8597 CrossRef CAS.
- F. Würthner, Z. Chen, F. J. M. Hoeben, P. Osswald, C.-C. You, P. Jonkheijm, J. V. Herrikhuyzen, A. P. H. J. Schenning, P. P. A. M. van der Schoot, E. W. Meijer, E. H. A. Beckers, S. C. J. Meskers and R. A. J. Janssen, J. Am. Chem. Soc., 2004, 126, 10611–10618 CrossRef.
- R. J. Kumar, J. M. MacDonald, T. B. Singh, L. J. Waddington and A. B. Holmes, J. Am. Chem. Soc., 2011, 133, 8564–8573 CrossRef CAS.
- T. Hasobe, S. Fukuzumi and P. V. Kamat, Angew. Chem., Int. Ed., 2006, 45, 755–759 CrossRef CAS.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761–1764 CrossRef CAS.
- W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340–343 CrossRef CAS.
- A. S. D. Sandanayaka, T. Murakami and T. Hasobe, J. Phys. Chem. C, 2009, 113, 18369–18378 CAS.
- W. W. H. Wong, T. B. Singh, D. Vak, W. Pisula, C. Yan, X. Feng, E. L. Williams, K. L. Chan, Q. Mao, D. J. Jones, C.-Q. Ma, K. Müllen, P. Bäuerle and A. B. Holmes, Adv. Funct. Mater., 2010, 20, 927–938 CrossRef CAS.
- W. W. H. Wong, C.-Q. Ma, W. Pisula, A. Mavrinskiy, X. Feng, H. Seyler, D. J. Jones, K. Müllen, P. Bäuerle and A. B. Holmes, Chem.–Eur. J., 2011, 17, 5549–5560 CrossRef CAS.
- W. W. H. Wong, J. Subbiah, S. R. Puniredd, B. Purushothaman, W. Pisula, N. Kirby, K. Müllen, D. J. Jones and A. B. Holmes, J. Mater. Chem., 2012, 22, 21131–21137 RSC.
- M. Carrasco-Orozco, W. C. Tsoi, M. O'Neill, M. P. Aldred, P. Vlachos and S. M. Kelly, Adv. Mater., 2006, 18, 1754–1758 CrossRef CAS.
- X. Zhou, S.-W. Kang, S. Kumar, R. R. Kulkarni, S. Z. D. Cheng and Q. Li, Chem. Mater., 2008, 20, 3551–3553 CrossRef CAS.
- L. Li, S. W. Kang, J. Harden, Q. Sun, X. Zhou, L. Dai, A. Jakli, S. Kumar and Q. Li, Liq. Cryst., 2008, 35, 233–239 CrossRef CAS.
- N. Sakai, R. Bhosale, D. Emery, J. Mareda and S. Matile, J. Am. Chem. Soc., 2010, 132, 6923–6925 CrossRef CAS.
- R. Bhosale, A. Perez-Velasco, V. Ravikumar, R. S. K. Kishore, O. Kel, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, T. Sawada, E. Vauthey, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2009, 48, 6461–6464 CrossRef CAS.
- R. Bhosale, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC.
- B. J. Walker, A. Dorn, V. Bulović and M. G. Bawendi, Nano Lett., 2011, 11, 2655–2659 CrossRef CAS.
- M. El Gemayel, M. Treier, C. Musumeci, C. Li, K. Müllen and P. Samorì, J. Am. Chem. Soc., 2012, 134, 2429–2433 CrossRef.
- S. Suresh and D. Arivuoli, Rev. Adv. Mater. Sci., 2012, 30, 243–253 CAS.
- C. J. Strachan, M. Windbergs and H. L. Offerhaus, Int. J. Pharm., 2011, 417, 163–172 CrossRef CAS.
- H. A. Collins, M. Khurana, E. H. Moriyama, A. Mariampillai, E. Dahlstedt, M. Balaz, M. K. Kuimova, M. Drobizhev, X. D. YangVictor, D. Phillips, A. Rebane, B. C. Wilson and H. L. Anderson, Nat. Photonics, 2008, 2, 420–424 CrossRef CAS.
- K.-S. Lee, R. H. Kim, D.-Y. Yang and S. H. Park, Prog. Polym. Sci., 2008, 33, 631–681 CrossRef CAS.
- S. Maruo and J. T. Fourkas, Laser Photonics Rev., 2008, 2, 100–111 CrossRef CAS.
- T. Verbiest, S. V. Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS.
- T. Ishi-i, S. Amemori, C. Okamura, K. Yanaga, R. Kuwahara, S. Mataka and K. Kamada, Tetrahedron, 2013, 69, 29–37 CrossRef CAS.
- J. Pérez-Moreno, I. Asselberghs, K. Song, K. Clays, Y. Zhao, H. Nakanishi, S. Okada, K. Nogi, O.-K. Kim, J. Je, J. Mátrai, M. De Maeyer and M. G. Kuzyk, J. Chem. Phys., 2007, 126, 1–14 CrossRef.
- L. A. Burke, G. Gonella, F. Heirtzler, H.-L. Dai, S. Jones, J. Zubieta and A. J. Roche, Chem. Commun., 2012, 48, 1000–1002 RSC.
- K. Kamada, C. Hara, K. Ogawa, K. Ohta and Y. Kobuke, Chem. Commun., 2012, 48, 7988–7990 RSC.
- S. Easwaramoorthi, S. Y. Jang, Z. S. Yoon, J. M. Lim, C.-W. Lee, C.-L. Mai, Y.-C. Liu, C.-Y. Yeh, J. Vura-Weis, M. R. Wasielewski and D. Kim, J. Phys. Chem. A, 2008, 112, 6563–6570 CrossRef CAS.
- S. Sreejith and A. Ajayaghosh, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2012, 51, 47–56 Search PubMed.
- A. P. de Silva, Molecular logic gates, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, 2012, p. 2497 Search PubMed.
- X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70–80 RSC.
- D.-H. Qu, Q.-C. Wang and H. Tian, Angew. Chem., Int. Ed., 2005, 44, 5296–5299 CrossRef CAS.
- D. A. Leigh, M. Á. F. Morales, E. M. Pérez, J. K. Y. Wong, C. G. Saiz, A. M. Z. Slawin, A. J. Carmichael, D. M. Haddleton, A. M. Brouwer, W. J. Buma, G. W. H. Wurpel, S. León and F. Zerbetto, Angew. Chem., Int. Ed., 2005, 44, 3062–3067 CrossRef CAS.
- D. H. Qu, F. Y. Ji, Q. C. Wang and H. Tian, Adv. Mater., 2006, 18, 2035–2038 CrossRef CAS.
- J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS.
- M. Feng, L. Gao, Z. Deng, W. Ji, X. Guo, S. Du, D. Shi, D. Zhang, D. Zhu and H. Gao, J. Am. Chem. Soc., 2007, 129, 2204–2205 CrossRef CAS.
- Z. Dadon, M. Samiappan, N. Wagner, N. Ashkenasy and G. Ashkenasy, Peptide-Based Computation: Switches, Gates and Simple Arithmetic, in Biomolecular Information Processing, ed. E. Katz, Wiley-VCH Verlag & Co. KGaA, Weinheim, 2012, pp. 9–32 Search PubMed.
- Z. Dadon, M. Samiappan, E. Y. Safranchik and G. Ashkenasy, Chem.–Eur. J., 2010, 16, 12096–12099 CrossRef CAS.
- M. Samiappan, Z. Dadon and G. Ashkenasy, Chem. Commun., 2011, 47, 710–712 RSC.
- E. Katz and V. Privman, Chem. Soc. Rev., 2010, 39, 1835–1857 RSC.
- M. Zhou and S. Dong, Biocomputing: Explore Its Realization and Intelligent Logic Detection, in Biomolecular Information Processing, ed. E. Katz, Wiley-VCH Verlag & Co. KGaA, Weinheim, 2012, pp. 117–132 Search PubMed.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- R. Pei, E. Matamoros, M. Liu, D. Stefanovic and M. N. Stojanovic, Nat. Nanotechnol., 2010, 5, 773–777 CrossRef CAS.
- G. Seelig, D. Soloveichik, D. Y. Zhang and E. Winfree, Science, 2006, 314, 1585–1588 CrossRef CAS.
- L. Qian and E. Winfree, Science, 2011, 332, 1196–1201 CrossRef CAS.
- N. H. Voelcker, K. M. Guckian, A. Saghatelian and M. R. Ghadiri, Small, 2008, 4, 427–431 CrossRef CAS.
- K. S. Park, M. W. Seo, C. Jung, J. Y. Lee and H. G. Park, Small, 2012, 8, 2203–2212 CrossRef CAS.
- A. J. Genot, J. Bath and A. J. Turberfield, J. Am. Chem. Soc., 2011, 133, 20080–20083 CrossRef CAS.
- Y. Xiang, X. Qian, Y. Chen, Y. Zhang, Y. Chai and R. Yuan, Chem. Commun., 2011, 47, 2080–2082 RSC.
- S. J. P. Canete and R. Y. Lai, Chem. Commun., 2010, 46, 3941–3943 RSC.
- K. Schlosser and Y. Li, Chem. Biol., 2009, 16, 311–322 CrossRef CAS.
- J. Elbaz, O. Lioubashevski, F. Wang, F. Remacle, R. D. Levine and I. Willner, Nat. Nanotechnol., 2010, 5, 417–422 CrossRef CAS.
- S. Bi, Y. Yan, S. Hao and S. Zhang, Angew. Chem., Int. Ed., 2010, 49, 4438–4442 CrossRef CAS.
- X. Chen, Y. Wang, Q. Liu, Z. Zhang, C. Fan and L. He, Angew. Chem., Int. Ed., 2006, 45, 1759–1762 CrossRef CAS.
- T. Li, E. Wang and S. Dong, J. Am. Chem. Soc., 2009, 131, 15082–15083 CrossRef CAS.
- A. Latorre and Á. Somoza, ChemBioChem, 2012, 13, 951–958 CrossRef CAS.
- T. Li, L. Zhang, J. Ai, S. Dong and E. Wang, ACS Nano, 2011, 5, 6334–6338 CrossRef CAS.
- H. Pei, L. Liang, G. Yao, J. Li, Q. Huang and C. Fan, Angew. Chem., Int. Ed., 2012, 51, 9020–9024 CrossRef CAS.
- G. M. Church, Y. Gao and S. Kosuri, Science, 2012, 337, 1628 CrossRef CAS.
- N. Goldman, P. Bertone, S. Chen, C. Dessimoz, E. M. LeProust, B. Sipos and E. Birney, Nature, 2013, 494, 77–80 CrossRef CAS.
- Hierarchically Structured Porous Materials: From Nanoscience to Catalysis, Separation, Optics, Energy, and Life Science, ed. B.-L. Su, C. Sanchez and X.-Y. Yang, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011 Search PubMed.
- Applications of Supramolecular Chemistry, ed. H.-J. Schneider, CRC Press, Boca Raton, Florida, 2012 Search PubMed.
- E. García-España, Molecular Recognition, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-V. C. H. Verlag. GmbH & Co. KGaA, Weinheim, 2012, vol. 3 Search PubMed.
- Metal-Organic Frameworks: Applications from Catalysis to Gas Storage, ed. D. Farrusseng, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011 Search PubMed.
- Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis, ed. M. Schröder, Springer, Berlin Heidelberg, 2010, vol. 293 Search PubMed.
- Chemical Sensors and Biosensors: Fundamentals and Applications, ed. F.-G. Banica, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012 Search PubMed.
- H. Maeda and P. Anzenbacher, Colorimetric Sensors, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. J. S. P. Gale, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 5, p. 2581 Search PubMed.
- M. M. Adams, L. A. Joyce and E. V. Anslyn, Uses of Differential Sensing and Arrays in Chemical Analysis, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 2, p. 709 Search PubMed.
- M. Biyikal, M. Hecht, R. Martınez-Manez, K. Rurack and F. Sancenon, Supramolecular Hybrid Nanomaterials as Prospective Sensing Platforms, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 8, p. 3669 Search PubMed.
- J. Tian, P. K. Thallapally and B. P. McGrail, Gas Storage and Separation, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, p. 3133 Search PubMed.
- S. Matile, N. Sakai and A. Hennig, Transport Experiments in Membranes, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 2, p. 473 Search PubMed.
- J. D. Lamb and N. Li, Ion Chromatography and Membrane Separations Using Macrocyclic Ligands, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 2, p. 563 Search PubMed.
- G. T. Noble, R. J. Mart, K. Ping Liem and S. J. Webb, Supramolecular Chemistry of Membranes, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 4, p. 1731 Search PubMed.
- R. Quesada, Membrane Transport, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 4, p. 1751 Search PubMed.
- D. Philp, Supramolecular Reactivity, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 4 Search PubMed.
- Supramolecular Catalysis, ed. P. W. N. M. van Leeuwen, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- Molecular Encapsulation Organic Reactions in Constrained Systems, ed. U. H. Brinker and J.-L. Mieusset, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- A. Herrmann, Chem.–Eur. J., 2012, 18, 8568–8577 CrossRef CAS.
- T. Takeuchi and S. Matile, Chem. Commun., 2013, 49, 19–29 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS.
- K. S. Suslick, Curr. Opin. Chem. Biol., 2012, 16, 557–563 CrossRef CAS.
- G. Aragay, J. Pons and A. Merkoçi, Chem. Rev., 2011, 111, 3433–3458 CrossRef CAS.
- Y. Song, W. Wei and X. Qu, Adv. Mater., 2011, 23, 4215–4236 CrossRef CAS.
- J. S. Fossey, F. D'Hooge, J. M. H. Van Den Elsen, M. P. Pereira Morais, S. I. Pascu, S. D. Bull, F. Marken, A. T. A. Jenkins, Y. B. Jiang and T. D. James, Chem. Rec., 2012, 12, 464–478 CrossRef CAS.
- C. Han and H. Li, Anal. Bioanal. Chem., 2010, 397, 1437–1444 CrossRef CAS.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- I. Yoshimura, Y. Miyahara, N. Kasagi, H. Yamane, A. OjidaItaru and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 12204–12205 CrossRef CAS.
- A. Wada, S. I. Tamaru, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5321–5330 CrossRef CAS.
- M. Ikeda, R. Ochi and I. Hamachi, Lab Chip, 2010, 10, 3325–3334 RSC.
- M. Ikeda, T. Yoshii, T. Matsui, T. Tanida, H. Komatsu and I. Hamachi, J. Am. Chem. Soc., 2011, 133, 1670–1673 CrossRef CAS.
- H. Komatsu, M. Ikeda and I. Hamachi, Chem. Lett., 2011, 40, 198–200 CrossRef CAS.
- M. Ikeda, K. Fukuda, T. Tanida, T. Yoshiia and I. Hamachi, Chem. Commun., 2012, 48, 2716–2718 RSC.
- Z. Zhu, C. Wu, H. Liu, Y. Zou, X. Zhang, H. Kang, C. J. Yang and W. Tan, Angew. Chem., Int. Ed., 2010, 49, 1052–1056 CrossRef CAS.
- N. Dave, M. Y. Chan, P. J. J. Huang, B. D. Smith and J. Liu, J. Am. Chem. Soc., 2010, 132, 12668–12673 CrossRef CAS.
- S. Yajima, K. Takami, R. Ooue and K. Kimura, Analyst, 2011, 136, 5131–5133 RSC.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490–13497 CrossRef CAS.
- Y. Choi, Y. Park, T. Kang and L. P. Lee, Nat. Nanotechnol., 2009, 4, 742–746 CrossRef CAS.
- C. W. Liu, C. C. Huang and H. T. Chang, Anal. Chem., 2009, 81, 2383–2387 CrossRef CAS.
- J. S. Lee, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 4093–4096 CrossRef CAS.
- Z. Wang, J. H. Lee and Y. Lu, Adv. Mater., 2008, 20, 3263–3267 CrossRef CAS.
- J. Lee, H. Jun and J. Kim, Adv. Mater., 2009, 21, 3674–3677 CrossRef CAS.
- P. J. Anzenbacher, P. Lubal, P. Bucek, M. A. Palaciosa and M. E. Kozelkova, Chem. Soc. Rev., 2010, 39, 3954–3979 RSC.
- S. H. Lim, L. Feng, J. W. Kemling, C. J. Musto and K. S. Suslick, Nat. Chem., 2009, 1, 562–567 CrossRef CAS.
- C. J. Musto, S. H. Lim and K. S. Suslick, Anal. Chem., 2009, 81, 6526–6533 CrossRef CAS.
- J. R. Carey, K. S. Suslick, K. I. Hulkower, J. A. Imlay, K. R. C. Imlay, C. K. Ingison, J. B. Ponder, A. Sen and A. E. Wittrig, J. Am. Chem. Soc., 2011, 133, 7571–7576 CrossRef CAS.
- P. C. Jurs, G. A. Bakken and H. E. McClelland, Chem. Rev., 2000, 100, 2649–2678 CrossRef CAS.
- K. Severin, Curr. Opin. Chem. Biol., 2010, 14, 737–742 CrossRef CAS.
- A. P. Umali and E. V. Anslyn, Curr. Opin. Chem. Biol., 2010, 14, 685–692 CrossRef CAS.
- A. P. Umali, S. E. LeBoeuf, R. W. Newberry, S. Kim, L. Tran, W. A. Rome, T. Tian, D. Taing, J. Hong, M. Kwan, H. Heymann and E. V. Anslyn, Chem. Sci., 2011, 2, 439–445 RSC.
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS.
- S. Rochat, J. Gao, X. Qian, F. Zaubitzer and K. Severin, Chem.–Eur. J., 2010, 16, 104–113 CrossRef CAS.
- N. Giuseppone and J. M. Lehn, J. Am. Chem. Soc., 2004, 126, 11448–11449 CrossRef CAS.
- E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031–1049 RSC.
- S. Otto and K. Severin, Top. Curr. Chem., 2007, 277, 267–288 CrossRef CAS.
- S. Rochat and K. Severin, J. Comb. Chem., 2010, 12, 595–599 CrossRef CAS.
- T. Takeuchi, J. Montenegro, A. Hennig and S. Matile, Chem. Sci., 2011, 2, 303–307 RSC.
- S. M. Butterfield, T. Miyatake and S. Matile, Angew. Chem., Int. Ed., 2009, 48, 325–328 CrossRef CAS.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2008, 41, 1354–1365 CrossRef CAS.
- S. Litvinchuk, H. Tanaka, T. Miyatake, D. Pasini, T. Tanaka, G. Bollot, J. Mareda and S. Matile, Nat. Mater., 2007, 6, 576–580 CrossRef CAS.
- S. Hagihara, H. Tanaka and S. Matile, J. Am. Chem. Soc., 2008, 130, 5656–5657 CrossRef CAS.
- S. M. Butterfield, D. H. Tran, H. Zhang, G. D. Prestwich and S. Matile, J. Am. Chem. Soc., 2008, 130, 3270–3271 CrossRef CAS.
- Y. Che, X. Yang, G. Liu, C. Yu, H. Ji, J. Zuo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2010, 132, 5743–5750 CrossRef CAS.
- Y. N. Wu, F. Li, W. Zhu, J. Cui, C. A. Tao, C. Lin, P. M. Hannam and G. Li, Angew. Chem., Int. Ed., 2011, 50, 12518–12522 CrossRef CAS.
- Z. Z. Lu, R. Zhang, Y. Z. Li, Z. J. Guo and H. G. Zheng, J. Am. Chem. Soc., 2011, 133, 4172–4174 CrossRef CAS.
- G. Lu, O. K. Farha, L. E. Kreno, P. M. Schoenecker, K. S. Walton, R. P. Van Duyne and J. T. Hupp, Adv. Mater., 2011, 23, 4449–4452 CrossRef CAS.
- X. Zou, J. M. Goupil, S. Thomas, F. Zhang, G. Zhu, V. Valtchev and S. Mintova, J. Phys. Chem. C, 2012, 116, 16593–16600 CAS.
- A. L. Robinson, V. Stavila, T. R. Zeitler, M. I. White, S. M. Thornberg, J. A. Greathouse and M. D. Allendorf, Anal. Chem., 2012, 84, 7043–7051 CrossRef CAS.
- B. Liu, O. Shekhah, H. K. Arslan, J. Liu, C. Wöll and R. A. Fischer, Angew. Chem., Int. Ed., 2012, 51, 807–810 CrossRef CAS.
- M. M. Wanderley, C. Wang, C.-D. Wu and W. Lin, J. Am. Chem. Soc., 2012, 134, 9050–9053 CrossRef CAS.
- Y. Takashima, V. Martínez Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef.
- X. J. Zhao, R. X. He and Y. F. Li, Analyst, 2012, 137, 5190–5192 RSC.
- M. Dionisio, G. Oliviero, D. Menozzi, S. Federici, R. M. Yebeutchou, F. P. Schmidtchen, E. Dalcanale and P. Bergese, J. Am. Chem. Soc., 2012, 134, 2392–2398 CrossRef CAS.
- M. Dionisio, J. M. Schnorr, V. K. Michaelis, R. G. Griffin, T. M. Swager and E. Dalcanale, J. Am. Chem. Soc., 2012, 134, 6540–6543 CrossRef CAS.
- T. H. Kim, H. S. Song, H. J. Jin, S. H. Lee, S. Namgung, U. K. Kim, T. H. Park and S. Hong, Lab Chip, 2011, 11, 2262–2267 RSC.
- D. Bonifazi, S. Mohnani and A. Llanes-Pallas, Chem.–Eur. J., 2009, 15, 7004–7025 CrossRef CAS.
- A. Ciesielski, C. A. Palma, M. Bonini and P. Samorì, Adv. Mater., 2010, 22, 3506–3520 CrossRef CAS.
- J. Leclaire, G. Husson, N. Devaux, V. Delorme, L. Charles, F. Ziarelli, P. Desbois, A. Chaumonnot, M. Jacquin, F. Fotiadu and G. Buono, J. Am. Chem. Soc., 2010, 132, 3582–3593 CrossRef CAS.
- K. Roy, A. C. Wibowo, P. J. Pellechia, S. Ma, M. F. Geer and L. S. Shimizu, Chem. Mater., 2012, 24, 4773–4781 CrossRef CAS.
- M. B. Dewal, M. W. Lufaso, A. D. Hughes, S. A. Samuel, P. Pellechia and L. S. Shimizu, Chem. Mater., 2006, 18, 4855–4864 CrossRef CAS.
- N. B. McKeown, J. Mater. Chem., 2010, 20, 10588–10597 RSC.
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915–920 CrossRef CAS.
- J. Tian, P. K. Thallapally and B. P. McGrail, CrystEngComm, 2012, 14, 1909–1919 RSC.
- M. Mastalerz, Chem.–Eur. J., 2012, 18, 10082–10091 CrossRef CAS.
- P. K. Thallapally, B. P. McGrail, J. L. Atwood, C. Gaeta, C. Tedesco and P. Neri, Chem. Mater., 2007, 19, 3355–3357 CrossRef CAS.
- C. Tedesco, L. Erra, M. Brunelli, V. Cipolletti, C. Gaeta, A. N. Fitch, J. L. Atwood and P. Neri, Chem.–Eur. J., 2010, 16, 2371–2374 CrossRef CAS.
- L. Erra, C. Tedesco, V. R. Cipolletti, L. Annunziata, C. Gaeta, M. Brunelli, A. N. Fitch, C. Knöfel, P. L. Llewellyn, J. L. Atwood and P. Neri, Phys. Chem. Chem. Phys., 2012, 14, 311–317 RSC.
- W. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS.
- C. G. Bezzu, J. E. Warren, M. Helliwell, D. R. Allan and N. B. McKeown, Science, 2010, 327, 1627–1630 CrossRef CAS.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS.
- J. T. A. Jones, D. Holden, T. Mitra, T. Hasell, D. J. Adams, K. E. Jelfs, A. Trewin, D. J. Willock, G. M. Day, J. Bacsa, A. Steiner and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 749–753 CrossRef CAS.
- J. T. A. Jones, T. Hasell, X. F. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS.
- T. Hasell, H. Zhang and A. I. Cooper, Adv. Mater., 2012, 24, 5732–5737 CrossRef CAS.
- M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS.
- M. W. Schneider, I. M. Oppel, H. Ott, L. G. Lechner, H. J. S. Hauswald, R. Stoll and M. Mastalerz, Chem.–Eur. J., 2012, 18, 836–847 CrossRef CAS.
- M. Brutschy, M. W. Schneider, M. Mastalerz and S. R. Waldvogel, Adv. Mater., 2012, 24, 6049–6052 CrossRef CAS.
- R. Afonso, A. Mendes and L. Gales, J. Mater. Chem., 2012, 22, 1709–1723 RSC.
- R. V. Afonso, J. Durão, A. Mendes, A. M. Damas and L. Gales, Angew. Chem., Int. Ed., 2010, 49, 3034–3036 CrossRef CAS.
- S. Maity, P. Jana and D. Haldar, CrystEngComm, 2011, 13, 3064–3071 RSC.
- J. Rabone, Y. F. Yue, S. Chong, K. Stylianou, J. Bacsa, D. Bradshaw, G. Darling, N. Berry, Y. Khimyak, A. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053–1057 CrossRef CAS.
- C. Martí-Gastaldo, J. E. Warren, K. C. Stylianou, N. L. O. Flack and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 11044–11048 CrossRef.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS.
- O. K. Farha, A. O. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. B. T. Nguyen, A. O. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS.
- T. A. Makal, J. R. Li, W. Lu and H. C. Zhou, Chem. Soc. Rev., 2012, 41, 7761–7779 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS.
- S. Yang, X. Lin, W. Lewis, M. Suyetin, E. Bichoutskaia, J. E. Parker, C. C. Tang, D. R. Allan, P. J. Rizkallah, P. Hubberstey, N. R. Champness, K. M. Thomas, A. J. Blake and M. Schröder, Nat. Mater., 2012, 11, 710–716 CrossRef CAS.
- J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2008, 130, 6010–6017 CrossRef CAS.
- S. Shimomura, M. Higuchi, R. Matsuda, K. Yoneda, Y. Hijikata, Y. Kubota, Y. Mita, J. Kim, M. Takata and S. Kitagawa, Nat. Chem., 2010, 2, 633–637 CrossRef CAS.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS.
- S. Matile, A. Vargas Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- J. K. W. Chui and T. M. Fyles, Chem. Soc. Rev., 2012, 41, 148–175 RSC.
- P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633–3647 RSC.
- H. Cho, L. Widanapathirana and Y. Zhao, J. Am. Chem. Soc., 2011, 133, 141–147 CrossRef CAS.
- W. Si, L. Chen, X. B. Hu, G. Tang, J. L. Hou and Z. T. Li, Angew. Chem., Int. Ed., 2011, 50, 12564–12568 CrossRef CAS.
- X. B. Hu, L. Chen, G. Tang, J. L. Hou and Z. T. Li, J. Am. Chem. Soc., 2012, 134, 8384–8387 CrossRef CAS.
- T. Kiwada, K. Sonomura, Y. Sugiura, K. Asami and S. Futaki, J. Am. Chem. Soc., 2006, 128, 6010–6011 CrossRef CAS.
- A. Satake, M. Yamamura, M. Oda and Y. Kobuke, J. Am. Chem. Soc., 2008, 130, 6314–6315 CrossRef CAS.
- C. P. Wilson and S. J. Webb, Chem. Commun., 2008, 4007–4009 RSC.
- C. P. Wilson, C. Boglio, L. Ma, S. L. Cockroft and S. J. Webb, Chem.–Eur. J., 2011, 17, 3465–3473 CrossRef CAS.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528–6529 CrossRef CAS.
- A. V. Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46 DOI:10.1021/ar400014r.
- A. Hennig, L. Fischer, G. Guichard and S. Matile, J. Am. Chem. Soc., 2009, 131, 16889–16895 CrossRef CAS.
- R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, S. G. T. Takeuchi, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533–538 CrossRef CAS.
- J. Mareda and S. Matile, Chem.–Eur. J., 2009, 15, 28–37 CrossRef CAS.
- A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef.
- E. Krieg and B. Rybtchinski, Chem.–Eur. J., 2011, 17, 9016–9026 CrossRef CAS.
- M. Barboiu, Chem. Commun., 2010, 46, 7466–7476 RSC.
- Y. Lu, T. Suzuki, W. Zhang, J. S. Moore and B. J. Mariñas, Chem. Mater., 2007, 19, 3194–3204 CrossRef CAS.
- X. Peng, J. Jin, Y. Nakamura, T. Ohno and I. Ichinose, Nat. Nanotechnol., 2009, 4, 353–357 CrossRef CAS.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, Nat. Nanotechnol., 2011, 6, 141–146 CrossRef CAS.
- P. Tyagi, A. Deratani, D. Bouyer, D. Cot, V. Gence, M. Barboiu, T. N. T. Phan, D. Bertin, D. Gigmes and D. Quemener, Angew. Chem., Int. Ed., 2012, 51, 7166–7170 CrossRef CAS.
- A. Cazacu, Y. M. Legrand, A. Pasc, G. Nasr, A. Van der Lee, E. Mahon and M. Barboiu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8117–8122 CrossRef CAS.
- Y. Le Duc, M. Michau, A. Gilles, V. Gence, Y. M. Legrand, A. van der Lee, S. Tingry and M. Barboiu, Angew. Chem., Int. Ed., 2011, 50, 11366–11372 CrossRef CAS.
- G. Nasr, T. Macron, A. Gilles, Z. Mouline and M. Barboiu, Chem. Commun., 2012, 48, 6827–6829 RSC.
- G. Nasr, T. Macron, A. Gilles, E. Petit and M. Barboiu, Chem. Commun., 2012, 48, 7398–7400 RSC.
- M. Barboiu and A. Gilles, Acc. Chem. Res., 2013, 46 DOI:10.1021/ar400025e.
- M. S. Kaucher, M. Peterca, A. E. Dulcey, A. J. Kim, S. A. Vinogradov, D. A. Hammer, P. A. Heiney and V. Percec, J. Am. Chem. Soc., 2007, 129, 11698–11699 CrossRef CAS.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS.
- B. Liu, J. Mater. Chem., 2012, 22, 10094–10101 RSC.
- L. Alaerts, C. E. A. Kirschhock, M. Maes, M. A. van der Veen, V. Finsy, A. Depla, J. A. Martens, G. V. Baron, P. A. Jacobs, J. F. M. Denayer and D. E. De Vos, Angew. Chem., Int. Ed., 2007, 46, 4293–4297 CrossRef CAS.
- M. Maes, F. Vermoortele, L. Alaerts, S. Couck, C. E. A. Kirschhock, J. F. M. Denayer and D. E. De Vos, J. Am. Chem. Soc., 2010, 132, 15277–15285 CrossRef CAS.
- Z. Y. Gu and X. P. Yan, Angew. Chem., Int. Ed., 2010, 49, 1477–1480 CrossRef CAS.
- N. Chang, Z. Y. Gu and X. P. Yan, J. Am. Chem. Soc., 2010, 132, 13645–13647 CrossRef CAS.
- A. S. Münch, J. Seidel, A. Obst, E. Weber and F. O. R. L. Mertens, Chem.–Eur. J., 2011, 17, 10958–10964 CrossRef.
- A. L. Nuzhdin, D. N. Dybtsev, K. P. Bryliakov, E. P. Talsi and V. P. Fedin, J. Am. Chem. Soc., 2007, 129, 12958–12959 CrossRef CAS.
- S. Han, Y. Wei, C. Valente, I. Lagzi, J. J. Gassensmith, A. Coskun, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 16358–16361 CrossRef CAS.
- Y. S. Li, F. Y. Liang, H. Bux, A. Feldhoff, W. S. Yang and J. Caro, Angew. Chem., Int. Ed., 2010, 49, 548–551 CrossRef CAS.
- M. J. C. Ordoñez, K. J. Balkus, Jr, J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2010, 361, 28–37 CrossRef.
- T. Ema, J. Inclusion Phenom. Macrocyclic Chem., 2012, 74, 41–55 CrossRef CAS.
- E. M. Pérez and N. Martín, Org. Biomol. Chem., 2012, 10, 3577–3583 Search PubMed.
- N. Komatsu and F. Wang, Materials, 2010, 3, 3818–3844 CrossRef CAS.
- X. Peng, N. Komatsu, S. Bhattacharya, T. Shimawaki, S. Aonuma, T. Kimura and A. Osuka, Nat. Nanotechnol., 2007, 2, 361–365 CrossRef CAS.
- X. Tu, S. Manohar, A. Jagota and M. Zheng, Nature, 2009, 460, 250–253 CrossRef CAS.
- Y. Shoji, K. Tashiro and T. Aida, J. Am. Chem. Soc., 2010, 132, 5928–5929 CrossRef CAS.
- T. Ema, K. Hamada, K. Sugita, Y. Nagata, T. Sakai and A. Ohnishi, J. Org. Chem., 2010, 75, 4492–4500 CrossRef CAS.
- T. R. Jackson, J. S. Springall, D. Rogalle, N. Masumoto, H. C. Li, F. D'Hooge, S. P. Perera, A. T. A. Jenkins, T. D. James, J. S. Fossey and J. M. H. van den Elsen, Electrophoresis, 2008, 29, 4185–4191 CrossRef CAS.
- M. P. Pereira Morais, J. D. Mackay, S. K. Bhamra, J. G. Buchanan, T. D. James, J. S. Fossey and J. M. van den Elsen, Proteomics, 2010, 10, 48–58 CrossRef CAS.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS.
- Y. Yin, Z. Dong, Q. Luo and J. Liu, Prog. Polym. Sci., 2012, 37, 1476–1509 CrossRef CAS.
- Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC.
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS.
- K. T. Kim, S. A. Meeuwissen, R. J. M. Nolte and J. C. M. van Hest, Nanoscale, 2010, 2, 844–858 RSC.
- M. Yoshizawa and M. Fujita, Bull. Chem. Soc. Jpn., 2010, 83, 609–618 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS.
- T. Murase, Y. Nishijima and M. Fujita, J. Am. Chem. Soc., 2012, 134, 162–164 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS.
- C. J. Hastings, M. P. Backlund, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2011, 50, 10570–10573 CrossRef CAS.
- W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–103 CrossRef CAS.
- N. Sakai, N. Sordé and S. Matile, J. Am. Chem. Soc., 2003, 125, 7776–7777 CrossRef CAS.
- S. Litvinchuk, G. Bollot, J. Mareda, A. Som, D. Ronan, M. Raza Shah, P. Perrottet, N. Sakai and S. Matile, J. Am. Chem. Soc., 2004, 126, 10067–10075 CrossRef CAS.
- M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082–12083 CrossRef CAS.
- D. Zaramella, P. Scrimin and L. J. Prins, J. Am. Chem. Soc., 2012, 134, 8396–8399 CrossRef CAS.
- J. Yang, M. B. Dewal, S. J. Profeta, M. D. Smith, Y. Li and L. S. Shimizu, J. Am. Chem. Soc., 2008, 130, 612–621 CrossRef CAS.
- S. Dawn, M. B. Dewal, D. Sobransingh, M. C. Paderes, A. C. Wibowo, M. D. Smith, J. A. Krause, P. J. Pellechia and L. S. Shimizu, J. Am. Chem. Soc., 2011, 133, 7025–7032 CrossRef CAS.
- C. H. Görbitz, Chem.–Eur. J., 2007, 13, 1022–1031 CrossRef.
- G. Distefano, A. Comotti, S. Bracco, M. Beretta and P. Sozzani, Angew. Chem., Int. Ed., 2012, 51, 9258–9262 CrossRef CAS.
- B. Escuder, F. Rodríguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044–1054 RSC.
- F. Rodríguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478–11484 CrossRef.
- Y. R. Liu, L. He, J. Zhang, X. Wang and C. Y. Su, Chem. Mater., 2009, 21, 557–563 CrossRef CAS.
- L. Ma and W. Lin, Top. Curr. Chem., 2010, 293, 175–205 CrossRef CAS.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS.
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS.
- J. M. Falkowski, C. Wang, S. Liu and W. Lin, Angew. Chem., Int. Ed., 2011, 50, 8674–8678 CrossRef CAS.
- C. Zhu, G. Yuan, X. Chen, Z. Yang and Y. Cui, J. Am. Chem. Soc., 2012, 134, 8058–8061 CrossRef CAS.
- P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS.
- M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2009, 132, 7524–7525 CrossRef.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- G. Q. Kong, S. Ou, C. Zou and C. D. Wu, J. Am. Chem. Soc., 2012, 134, 19851–19857 CrossRef CAS.
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS.
- S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688–714 CrossRef.
- D. M. Vriezema, P. M. L. Garcia, N. S. Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS.
- Z. Wang, M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, Angew. Chem., Int. Ed., 2012, 51, 10746–10750 CrossRef CAS.
- E. Moulin and N. Giuseppone, Reactions in Dynamic Self-Assemblies, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 4, pp. 1543–1574 Search PubMed.
- S. R. K. Minkler, B. H. Lipshutz and N. Krause, Angew. Chem., Int. Ed., 2011, 50, 7820–7823 CrossRef CAS.
- B. H. Lipshutz, S. Huang, W. W. Y. Leong, G. Zhong and N. A. Isley, J. Am. Chem. Soc., 2012, 134, 19985–19988 CrossRef CAS.
- J. Li, Y. Tang, Q. Wang, X. Li, L. Cun, X. Zhang, J. Zhu, L. Li and J. Deng, J. Am. Chem. Soc., 2012, 134, 18522–18525 CrossRef CAS.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS.
- M. M. Müller, M. A. Windsor, W. C. Pomerantz, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2009, 48, 922–925 CrossRef.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906–2910 CrossRef CAS.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- O. I. Wilner, Y. Weizmann, R. Gill, O. Lioubashevski, R. Freeman and I. Willner, Nat. Nanotechnol., 2009, 4, 249–254 CrossRef CAS.
- S. Frühbeißer and F. Grohn, J. Am. Chem. Soc., 2012, 134, 14267–14270 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |