 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomistic modelling of CVD synthesis of carbon nanotubes and graphene
James A.
Elliott
*a,
Yasushi
Shibuta
b,
Hakim
Amara
c,
Christophe
Bichara
d and
Erik C.
Neyts
e
aDepartment of Materials Science and Metallurgy, University of Cambridge, Pembroke Street, Cambridge, CB2 3QZ, UK. E-mail: jae1001@cam.ac.uk
bDepartment of Materials Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: shibuta@material.t.u-tokyo.ac.jp
cLaboratoire d'Etudes des Microstructures, ONERA-CNRS, BP 72, 92322 Châtillon Cedex, France. E-mail: hakim.amara@onera.fr
dAix-Marseille Université, CNRS, CINaM UMR 7325, 13288 Marseille, France. E-mail: xtof@cinam.univ-mrs.fr
eDepartment of Chemistry, PLASMANT Research Group, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk-Antwerp, Belgium. E-mail: erik.neyts@ua.ac.be
First published on 6th June 2013
Abstract
We discuss the synthesis of carbon nanotubes (CNTs) and graphene by catalytic chemical vapour deposition (CCVD) and plasma-enhanced CVD (PECVD), summarising the state-of-the-art understanding of mechanisms controlling their growth rate, chiral angle, number of layers (walls), diameter, length and quality (defects), before presenting a new model for 2D nucleation of a graphene sheet from amorphous carbon on a nickel surface. Although many groups have modelled this process using a variety of techniques, we ask whether there are any complementary ideas emerging from the different proposed growth mechanisms, and whether different modelling techniques can give the same answers for a given mechanism. Subsequently, by comparing the results of tight-binding, semi-empirical molecular orbital theory and reactive bond order force field calculations, we demonstrate that graphene on crystalline Ni(111) is thermodynamically stable with respect to the corresponding amorphous metal and carbon structures. Finally, we show in principle how a complementary heterogeneous nucleation step may play a key role in the transformation from amorphous carbon to graphene on the metal surface. We conclude that achieving the conditions under which this complementary crystallisation process can occur may be a promising method to gain better control over the growth processes of both graphene from flat metal surfaces and CNTs from catalyst nanoparticles.
 James A. Elliott | James Elliott is a Reader in Macromolecular Materials in the University of Cambridge, where he carries out research on multiscale computational modelling of soft matter systems, including coarse-grained and molecular modelling of polymers, carbon nanotubes and their composites. He obtained his MA in Natural Sciences (Physics) from Cambridge, and his PhD in Polymer Physics at the University of Bristol. He was a JSPS Invitation Fellow and Visiting Professor at the University of Tokyo in 2008, and collaborates with several groups working on CNT synthesis and thermal properties of CNT-polymer composites. http://www.elliotts.org.uk |
 Yasushi Shibuta | Yasushi Shibuta has been a Lecturer at the University of Tokyo (UT) since 2011. He received his PhD from UT in 2004. After being a JSPS Postdoctoral Research Fellow, he joined the Department of Materials Engineering, UT as an Assistant Professor at 2005. He was a Visiting Fellow at Fitzwilliam College, Cambridge, UK in 2005 and 2006. His recent research focuses on understanding the nature of phase transitions during the synthesis of materials by numerical modelling. His chosen systems of interest range from base materials such as iron and steel to advanced materials such as carbon nanotubes and graphene. http://www.mse.t.u-tokyo.ac.jp/shibuta/ |
 Hakim Amara | Hakim Amara is a senior scientist working at ONERA (French Aerospace Lab) who has received his PhD in Material Science in 2005. His main interest lies in theoretical condensed matter physics. He has an expertise in the development of numerical computations at the atomic scale using tight-binding and ab initio techniques. Current research focuses on modelling of nucleation and growth mechanisms of carbon nanotubes and graphene from metallic catalysts and also electronic properties (STM images) of defects in carbon nanostructures. |
 Christophe Bichara | Christophe Bichara is Research Director at CNRS with an expertise in Condensed Matter Physics, Chemical Physics and Computational Materials Science. He graduated in Chemical Engineering (Toulouse – 1979) and obtained a “Doctorat ès Science” (Marseille – 1987). His current interest lies in the study of disorder, liquid matter, non-crystalline materials as well as the growth mechanisms of carbon nanostructures. |
 Erik C. Neyts | Erik Neyts is appointed as tenure track professor at the University of Antwerp (UA). After his PhD in chemistry on molecular dynamics (MD) simulations for the deposition of diamond-like carbon layers, he investigated the deposition of metal oxide layers by MD and Monte Carlo (MC) simulations as a postdoctoral researcher. His current work focuses on MD/MC simulations for the growth of nanostructured materials, including carbon nanotubes, graphene and silicon oxide nanowires. Recently, new research directions were launched focusing on the study of plasma catalysis and plasma medicine by MD/MC simulations as well. |
Introduction
The first single-wall carbon nanotubes (SWCNTs) were synthesised by high temperature techniques (e.g. laser ablation and arc discharge1), all of which involve a metal catalyst. In these methods, carbon and transition metal (TM) elements (such as Co, Ni, Fe, etc.) are vapourised at temperatures above 3000 K and then condensed at lower temperatures in an inert gas (He or Ar) flow. SWCNTs and multi-wall carbon nanotubes (MWCNTs) are now commonly produced by catalytic chemical vapour deposition (CCVD) at much lower temperatures, ranging from 600–1300 K, or by plasma-enhanced CVD (PECVD), in which the gas is not activated by high temperatures as in thermal CCVD, but rather by applying a sufficiently strong voltage, causing gas breakdown. There is currently great interest in adapting these techniques for production of high-quality single- and few-layer graphene. Here, we restrict our attention to CNTs and graphene produced by CVD methods, which are widely used due to their low cost and ease of scaling to industrial production. We begin by briefly reviewing existing experimental CVD synthesis methods and proposed mechanisms for nucleation and growth, before summarizing previous computational work. We then conclude with a comparative analysis of our own recent simulation results of carbon on nickel, based on which we propose a complementary heterogeneous nucleation step for the transformation of amorphous carbon to graphene on the metal surface.Experimental CVD synthesis methods
For 50 years, the formation of filamentous carbon by catalytic decomposition of gases has been intensively studied, mainly due to its role in industrial chemical processes.2 Much effort has since been devoted to adapting CCVD methods to the synthesis of SWCNTs, MWCNTs, and lately graphene. In the case of CNTs, it is known that the nanotube grows from a TM nanoparticle that is either attached to a substrate, often silica or alumina, or floating in a gas phase reactor. Depending on the growth temperature, different products are obtained. Although there is no absolute rule, the general tendency is to obtain MWCNTs at medium temperatures (between 600 and 1000 K) and SWCNTs at higher temperatures (between 900 and 1300 K) although there are some exceptions to this discussed below. The process is complex, but it is accepted that the metal nanoparticle acts as a catalyst to favour the decomposition of the carbon-bearing precursor (typically C2H2, C2H4, CH4 or alcohols). The nanoparticle also serves as a heterogeneous surface on which the initial nanotube cap can grow, leading to the formation of long tubes. In the case of graphene, for metals with low carbon solubility such as Cu, nucleation is initiated at impurities or defects on the metal surface, and growth proceeds from the edges of the nascent sheet, typically leading to single- or few-layer graphene. For metals with higher carbon solubility, such as Ni, the dominant mechanism is surface segregation and precipitation of dissolved carbon, typically leading to multi-layer graphene.Although the products obtained from the medium temperature methods have a lower quality (as measured by their crystalline order) than those produced by the high temperature routes, the CCVD method has numerous advantages especially in terms of control of the growth conditions. In the case of CNTs, growth can be precisely localised using a lithographically patterned catalyst.3 In this respect, vertically-oriented SWCNT carpets or forests grown by CCVD have received enormous attention because of their suitability in a number of important technological applications.4,5 To promote growth and avoid any poisoning effect that prevents it, water has been used as a protective agent against coating by amorphous carbon. The development of water-assisted CCVD growth (also referred to as “super growth”) revealed that the activity and lifetime of the metal catalysts are enhanced by introducing a limited amount of H2O, resulting in the fast growth of highly dense and vertically aligned SWCNT carpets of high purity.6
The number of variables involved in CCVD synthesis is quite significant (e.g., temperature, pressure, catalyst composition, and nature of feedstock or substrate). Efficient catalysts such as Fe, Co and Ni and alloys have often been selected empirically. When liquid, these metals can dissolve large amounts of carbon. Upon solidification, they undergo an almost complete segregation, although what dissolved carbon remains may be significant for the synthesis mechanism. These properties have been applied to the synthesis of carbon fibres. How this applies to the CCVD synthesis of CNTs and graphene, however, is still not fully explained. According to Deck and Vecchio,7 efficient catalysts for CCVD should have non-zero but limited (0.5–1.5 wt%) carbon solubility in the solid bulk state. Nevertheless, it seems possible to grow SWCNTs on noble metals,8 or even non-metallic nanoparticles.9 Similarly, single-layer graphene can be grown on copper substrates despite its low carbon solubility.10 The temperature, pressure and composition of the gaseous reactants flow must also be optimised according to the catalyst decomposition reaction considered. For instance, CCVD synthesis of SWNCTs at exceptionally low temperatures (down to 623 K) has been reported using low partial pressures of acetylene.11 The gas pressure and feeding rate are reported to have an impact on the diameter distribution of the SWCNTs.12,13 Similarly, both experimental and modelling studies have shown a strong dependence of Cu-catalysed graphene nucleation on the pressure of hydrogen14,15 and carbon feedstock16 gases. Finally, it should be noted that even the substrate can have an important role. Although the growth of CNT forests on metallic substrates has been demonstrated, the catalyst and CNTs are usually on insulating support layers such as SiO2 or Al2O3.17,18 This latter is known to make the production of dense carpets of vertically aligned SWCNTs possible.19
Accurately controlling the location, diameter, chiral angle and length of SWCNTs produced by CCVD is one of the critical issues for an effective use of the unique properties of these nanoscale objects in many applications such as electronic devices. Using CCVD synthesis, it would be highly desirable to obtain structural control directly during the growth. Although some experimental conditions have been proven to lead to partial chirality selectivity, the reasons for this selectivity are not well understood.20–24
In plasma-enhanced CVD (PECVD), the gas is not activated by a high temperature as in thermal CCVD, but rather by applying a sufficiently strong voltage, causing gas breakdown. Indeed, this leads to the creation of a plethora of reactive species, including electrons and ions, neutral molecules, molecular radicals and atoms, and photons, in addition to the presence of electromagnetic fields. All of these factors contribute to a number of advantages of PECVD growth over thermal growth.25 For example, since the gas activation in PECVD does not require a high temperature, there is no high dehydrogenation barrier to be overcome, and therefore growth may be accomplished at a lower temperature. This was convincingly demonstrated by Hofmann and co-workers,26–28 who reported MWCNT growth at temperatures as low as 400 K in a dc plasma using an C2H2–NH3 gas mixture at a pressure of 1.5 mbar. The activation energy for the growth rate on Ni was found to be 0.23 eV, which is much lower than the value of 1.2–1.5 eV typically found for thermal CCVD.26
Furthermore, PECVD has also been shown to be capable of allowing the growth of freestanding, vertically aligned single-wall CNTs by Kato and co-workers.29,30 This is a direct consequence of the presence of an electric field, aligning the tubes, as also found in atomistic simulations.31 The formation of freestanding vertically aligned SWCNTs is not possible using thermal CCVD. Furthermore, the plasma in PECVD growth may also have an influence on the catalyst. Indeed, the plasma may induce a different surface structure, possibly resulting in epitaxial growth. This has been envisaged as a possible route for chirality-selective synthesis.21,32,33 Finally, the presence of etchant species in the plasma may also affect the growth. For instance, in a study by Ghorannevis et al.34 a narrow diameter and chirality distribution was found, which was explained by etching of larger diameter structures by the plasma. Also, Zhang and co-workers found that metallic tubes could be preferentially etched over semiconducting tubes in the case of medium diameter tubes.35 Recently, Neyts et al. showed that ion bombardment in a suitable energy range allows for enhanced nucleation of CNT cap due to defect healing.36
In conclusion, both CCVD and PECVD methods are versatile methods for potentially large-scale production of high quality CNTs and graphene films. The key challenge in both cases is to obtain better control over the structural order and (in the case of CNTs) alignment of graphene sheet to improve properties, e.g. large area, few-layer graphene sheets10,37,38 and CNTs with narrow distributions of diameter and chiral angle.34,39 Although post-synthesis processing techniques exist (for example, solution-purification of CNTs40,41), these can add additional expense to the production process, are non-scalable, and can damage or shorten the tubes. A better understanding of how synthesis process affects final structure presents the best possible opportunity for achieving a simple one-step procedure for high quality product with large yields. With this in mind, we now briefly review the existing proposed synthesis mechanisms for CNTs and graphene via CVD on metal surfaces.
Proposed synthesis mechanisms
The role of the catalytic metals during the synthesis of CNTs and graphene has been widely studied. Although it is not feasible to discuss all of the published studies in full, we briefly summarise the essential details here. Several recent review articles25,33,42–49 also provide further information. In earlier times, when laser ablation50 and arc-discharge1 were the main techniques used for CNT growth, most of the growth models focused on the role of isolated metal atoms, as typified by the scooter model.50 On the other hand, Yudasaka et al.51–53 focused on the aggregate of molten carbon–metal alloy obtained from laser ablation in the early stages, and proposed a model in which a graphenic network segregates from the carbon-saturated alloy as a pathway to produce CNTs. Once the CCVD technique54–56 was shown to be a promising synthesis method for CNTs, the above concept of graphene segregation from carbon-saturated alloys has been used to explain the growth process of CNTs via a CCVD technique as follows. In the first step, the deposition of pyrolysed C atoms or carbon source molecules (which are then catalytically decomposed) on metal nanoparticles occurs, followed by diffusion of the C atoms within the metals, and their segregation and graphenisation once the metal nanoparticle is supersaturated with carbon. This process is regarded as similar to the vapour–liquid–solid (VLS) mechanism, which was originally proposed to explain the formation of silicon whiskers57 and continuous carbon filaments.2,58Although the VLS mechanism has been widely accepted as the basis for explaining the growth of SWCNTs from catalytic metal nanoparticles, there have been several CCVD techniques reported using solid or non-metallic nanoparticles in which the VLS mechanism is not thought to apply since the nanoparticles are solid during the synthesis process. Such models may also be applicable to catalytic deposition of graphene on metallic surfaces with low carbon solubility. For example, the group of Homma successfully synthesised SWCNTs via the CCVD technique using semiconductors, such as SiC, Ge and Si nanoparticles.9 Page et al.59 then proposed a vapour–solid–solid (VSS) model for the growth of SWCNTs from SiO2 nanoparticles on the basis of computer simulation results, in which a solid SiC shell is formed at the surface of the SiO2 nanoparticle and continued addition of carbon to the surface leads to the formation of the CNTs. On the other hand, a surface-mediated growth mechanism was proposed by Hofmann et al.,26,60 in which the carbon atoms diffuse over the surface of the solid Ni particle instead of bulk diffusion in the liquid nanoparticle. Recently, the initial cap formation process has been directly observed via in situ environmental transmission electron microscopy (TEM).61,62
Since the VLS/VSS models provide no explicit information on how the synthesis mechanisms operate at the atomic scale, there is still much debate about this issue. In particular, the term ‘epitaxy’ is often used to describe the interaction between the graphene network and the catalyst surface. While epitaxy is only strictly possible in the case of a solid catalyst (VSS model), there is some evidence to suggest that, even for a liquid catalyst (VLS model), certain specific surface ordering may affect the structure of CNTs and graphene grown on metal surfaces. For example, Zhu and co-workers63 analysed transmission electron microscopy (TEM) images of SWCNTs seeded from catalytic cobalt nanoparticles and concluded that the chirality of the SWCNT relates to the angle of the step-edge on the Co(111)fcc plane of the catalytic nanoparticle with respect to the growth direction of SWCNT. Koziol et al.39 focused on the orientational relationship between the nanotube layer and an iron carbide catalyst particle in MWCNTs grown by a CCVD technique, and discussed the importance of reconstruction of the surface layers of the carbide particle in contact with the growing end of the tubes. Moreover, Ding et al.64 focused on the axial screw dislocation in the CNT and proposed that the growth rate is proportional to the Burgers vector of dislocation, and therefore to the chiral angle of CNTs. Recent measurements of growth rates by in situ Raman spectroscopy appear to confirm this prediction, and suggest the importance of growth stage in determining distribution of chiral angle.65 Although the mechanism of control is not yet fully resolved, the atomic structure of the contacting edge or surface of the CNT to the metal nanoparticle should affect the chiral angle of CNTs. Previous studies have demonstrated that graphene with a commensurate orientational relationship with close-packed metal surfaces can have significant barriers to translational motion.66 Therefore, the possibility of 1D edge or 2D planar ‘epitaxy’ between the graphene network and catalytic metals may be important even in liquid nanoparticles where such effects are transient and not related to the bulk structure of the particle.
The epitaxy between the graphene network and crystalline metal surfaces is also thought to play an important role in the growth of the graphene via a CCVD process. Although such epitaxy has been suggested in the field of surface science prior to the discovery of CNTs and graphene,67,68 recently, the epitaxial growth of the single- or few-layers of graphene on close-packed metal surfaces has become a reality using various metals such as Ir,69 Ni,70–72 Cu,70,73 Co (ref. 71) and Ru.73 Regarding the growth mechanism, low-energy electron microscopy (LEEM) revealed nucleation and subsequent growth processes of the graphene on the metal surface,74 in which the nucleation starts at impurities or defects at the metal surface such as steps and bumps.49 Therefore, the enhancement of crystallinity of the metal surface decreases the nucleation density, which results in the formation of the large single-crystalline graphene.75 Moreover, Li and co-workers70 used carbon isotope labelling to track carbon atoms during the graphene growth on Ni and Cu surface and proposed two formation paths for the graphene growth: carbon diffusion, segregation and precipitation for the Ni (high carbon solubility metal) and surface adsorption and edge growth on the Cu (low carbon solubility metal). However, a recent modelling study by Neyts et al.76 has suggested that single layer graphene deposition on Ni is theoretically possible under high flux conditions. The effect of the carbon diffusion into Ni layers on the graphene growth is also discussed on the basis of a kinetic model72,77 and, in relation to the discussion of carbon solubility in metals, it is useful to apply knowledge from the field metallurgy.78
Some open questions and challenges are as follows. Are the existing models sufficient to explain how control over the growth process may be achieved? None of the models proposed to date gives a full description of the growth process, starting from nucleation through continuous growth through to termination, including the action of catalyst. Is full control over the growth process possible, even in principle, given the high temperatures involved and disordered structure of catalyst surface or bulk of particles? The answers to these questions have important implications for high quality commercial production necessary for device applications, in particular the manufacture of square-centimetre area continuous sheets of single layer graphene, and SWCNTs exceeding one micron in length with uniform chiral angle and negligible defect concentrations.
Review of modelling methods and previous applications to graphene and CNT growth
We classify the modelling methods used for studying graphene and CNT growth into three main categories: ab initio (or first principles), semi-empirical and empirical, based on the stage at which data from experiment and/or lower level theory are introduced to determine any free parameters in the Hamiltonian. Of course, such a division is somewhat ambiguous, and often more than one type of method is employed to tackle situations in which phenomena occur over multiple time and length scales.79,80 Nevertheless, it serves as a convenient framework in which to discuss previous modelling studies.So-called ab initio or first principles methods, based on Molecular Orbital (MO) or Density Functional Theory (DFT) formulations of quantum chemistry (QC), have been widely used to study theoretically the carbon–metal interaction due to their ability to describe different bonding environments at an electronic level. On the other hand, essentially because of the large computational cost of these approaches, direct ab initio studies of the nucleation or growth of CNTs are rather scarce. For this reason, tight-binding81 or semi-empirical molecular orbital methods82 are often used, in which certain approximations (requiring parameterization) are made to simplify the calculation of electronic interactions. Since they still possess an intrinsically quantum mechanical character, they can describe correctly the bonding in a carbon–metal system and can handle a few hundreds of atoms evolving over a period of a few hundred picoseconds. However, in order to model thousands or millions of atoms over tens or hundreds of nanoseconds, it is necessary to eliminate a direct treatment of electrons altogether by moving to empirical bond-order type potentials, such as Finnis–Sinclair81,83 (for metals), Brenner84,85 (for hydrocarbons) or ReaxFF86 (for both), whose form is justified by the second moment expansion of tight-binding potential.
First principles modelling methods
An important first step is to check the reliability of MO or DFT-based methods to correctly account for carbon–metal interactions. Discrepancies were noticed for small clusters,87–89 but DFT was later shown to give reliable heats of solution and diffusion barriers for C in Ni.90,91 The interaction of individual C atoms or dimers has been widely studied on different TMs (Ni, Co, Fe, Pd) and noble metals (Cu, Ag, Au), possibly relevant for nanotube growth.92–98 Different surface or subsurface sites have been considered, and C adsorption or incorporation in metal is generally less favourable than C incorporation in an infinite graphene layer. The stability difference is of the order of 1 eV per C atom for the adsorption of a C atom on Ni, Pd and Pt, and in the 2–4 eV per C atom range for Cu, Ag and Au. For Ni and Co, semi-octahedral (100) surface and subsurface sites are more favourable for C atoms than compact (111) facets. Adsorbed carbon dimers are found to be more stable on noble metal surfaces than individual atoms,95 and tight-binding calculations showed that dimers can also be stabilised in neighbouring subsurface sites of Ni.99 Summarizing these results, one can conclude that the interaction of C with late TMs is strong but leads to limited carbon solubility and less effective catalytic properties, while it is even stronger, favouring carbide formation (which may in some cases be detrimental for catalysis) for early TMs.For high catalytic activity, having atomic carbon interacting with metal in the correct energy range is a necessary condition, but not the full story. To sustain CNT growth, one has to avoid two major causes of growth termination. The first one is the closure of the nanotube open end in contact with the catalyst. As pointed out by Ding et al.,100 clusters of Fe, Ni and Co, that are known as efficient catalysts for CNT growth, interact more strongly with CNT edges than Au, Cu and Pd, and effectively prevent the tube closure and detachment. The second one is the encapsulation of the catalyst nanoparticle by graphene-like carbon layers, forming onions. Such a situation depends not only on the growth conditions, but also on the adhesion energy of the graphene layer with the metal catalyst. This problem has essentially been studied within the context of graphene growth where it is of crucial importance since too large an electronic coupling between the metal substrate and the graphene layer will strongly affect the peculiar electronic properties of the latter. As discussed by Batzill,48 a linear dispersion at the Dirac point is observed for Cu, Ag, Au, Ir and Pt, indicating an electronic decoupling between the metal and the graphene layer, while the separation between them is around 3 Å, characteristic of a weak, van-der-Waals-like interaction. On the other hand Co, Ni, Ru, Rh and Re show a stronger interaction and shorter separations. DFT-based calculations, for example those described in review by Wintterlin and Bocquet,45 are pushed close to their limits due to their neglect of non-local electronic contributions to the bonding. The crudest Local Density Approximation (LDA) generally results in overbinding, while the various Generalised Gradient Approximations (GGA) sometimes find no binding at all.101 Recent contributions have tackled this difficult problem of including non-local effects in the calculations, either via a “van der Waals functional”,102 or using the Random Phase Approximation (RPA).103,104 As shown in Fig. 1, the different methods disagree on the presence, depth and number of energetic minima, which complicates the interpretation of the growth mechanism. However, recently, the group of Irle and Morokuma performed quantum chemical molecular dynamics simulation on the evolution of graphene network and showed that step edge or molecular template has an important role of the graphene formation.105 They also demonstrated the formation of a carbon precursor beneath a Ni(111) step-edge in the case of high sub-surface carbon concentrations.106
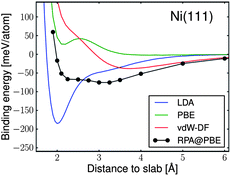 | ||
| Fig. 1 Ab initio DFT calculations showing that graphene (in top fcc configuration) on 2D hexagonal Ni(111)fcc surface using different functionals (LDA, GGA, vdW-corrected and RPA) disagree on the presence, depth and number of energetic minima (adapted with permission from Olsen et al.103). The graphene lattice parameter was scaled to fit the surface unit cell of metal. | ||
With regard to CNT growth studies, ab initio calculations have been used from an early stage to support growth models, including those with metal catalysts. Maiti et al.107,108 calculated ab initio the electric field required to stabilise growth of an open-ended CNT. A number of similar studies subsequently followed, either to calculate carbon cap stability,109 or growth mechanisms in the absence of catalysts.110,111 Charlier et al.112 showed that the open end of a SWCNT spontaneously closes in the absence of a metal catalyst while the open end of a double wall tube is stabilised by the lip–lip interaction between the tubes. Comparing the interaction of a Ni atom with armchair or zigzag tube edges, Lee et al.113 concluded that the former would be energetically favoured, a similar result to that of Ding et al.64 who proposed a growth model based on dislocation theory, discussed earlier and supported by DFT calculations. Nucleation mechanisms of a carbon cap on a Ni surface were proposed by Fan et al.114 Because of its utmost importance, the chirality issue has been addressed by DFT calculations.115,116 These calculations correspond to highly idealised situations that may not be representative of the actual growth conditions. In particular, the growth dynamics corresponding to successive incorporation of C atoms is not at all taken into account in the above cited works. We should note that the average growth rate is actually extremely slow, less than 1 nm per second as observed by Lin et al.,117 as compared to the time scale that can be spanned by Molecular Dynamics (MD), typically 100 ps for currently affordable DFT-based calculations and 100 ns for classical MD on carbon–metal systems. Nevertheless, insightful results could be gained from direct attempts to simulate elementary growth processes by DFT-based MD, a selection of which are illustrated in Fig. 2. Gavillet et al.118 found evidence for carbon segregation upon cooling down a Co–C liquid cluster (Fig. 2a), and could observe the diffusion of carbon towards the edge of a carbon cap sitting on a Co surface, thus supporting a root growth mechanism. Later on, Raty et al.119 observed the carbon diffusion on a Fe55 cluster (Fig. 2b), leading to sp2 carbon structures forming a cap that weakly adheres to its surface and can lift off. Interestingly, this calculation does not indicate any diffusion of C inside the Fe nanoparticle. Whether this finding is realistic or is an artefact of the very short time scale (10 ps) of the simulation, the neglect of spin polarization for Fe, or even the use of a “magic size” cluster with a special stability is not yet established. More recently, Kim et al.120 have used QC MD simulations to show that zigzag SWCNTs are significantly inferior in maintaining their chirality during growth compared to armchair SWCNTs.
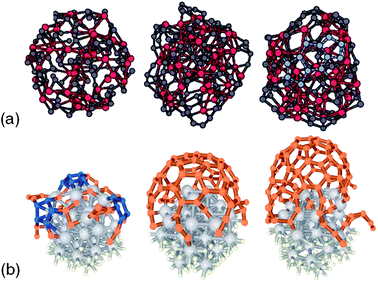 | ||
| Fig. 2 Comparison of cap and tube structures generated from ab initio modelling approaches: (a) segregation process in a cluster containing 51 Co atoms (larger red spheres) and 102 C atoms (smaller grey spheres) as the temperature is reduced from 2000 (right) to 1500 K (left) (adapted with permission from Gavillet et al.118), (b) early stages of SWCNT growth on a 1 nm Fe55 catalyst nanoparticle during Car-Parinello MD simulation using PBE functional (adapted with permission from Raty et al.119). | ||
Semi-empirical modelling methods
In the semi-phenomenological tight-binding scheme, it is conventional to split the total energy of the system into two parts: a band structure term (attractive contribution) that describes the formation of an energy band when atoms are put together, and a repulsive term that empirically accounts for the ionic and electronic repulsion.121,122 Such models depend on adjustable parameters to build the Hamiltonian matrix of the interactions and to describe the empirical repulsive term that is always present. All parameters are usually fitted to ab initio or experimental data. This fitting procedure is complex and difficult, thus limiting the number of groups developing such models, especially for a carbon–metal system.To avoid treating the full complex binary system within a tight-binding scheme (TMs such as Fe, Co, or Ni and C) at the first stage, the pioneering works in this area were focused on modelling the non-catalytic growth of CNTs. As an example, extensive MD simulations based on the tight-binding (TB) formalism have been performed to study the cap formation processes suggesting that the growth of armchair nanotubes is energetically more favourable than zigzag tubes.123 In addition, Zhang and Crespi have proposed a mechanism for SWCNT nucleation based on the rolling of a pre-existing graphene double layer into a tube.124
To go beyond this stage, a few groups have developed specific TB potentials for TMs, carbon and TM carbides in order to simulate the nucleation and growth mechanisms of SWCNTs. As mentioned above, the price to pay is generally rather high in terms of parameters to be fitted and of computational cost. In this respect, Andriotis et al., have developed a self-consistent TB-MD method to incorporate effects of charge transfer efficiently into the parameters.125 According to their calculations, the Ni atom repairs defects in the nanotube, binds to the defect, and then catalyses the incorporation of a gas phase carbon atom to anneal the Ni-stabilised defects, freeing the Ni atom to be available to migrate to a new defect site,126 as shown in Fig. 3a. One can also mention the group of Irle and Morokuma, who have employed a DFT–TB method for the carbon–metal system and revealed the evolution of hexagonal network at the wall of the open-ended (5,5) nanotube or the carbon cap in contact with an Fe cluster,127–129 as shown in Fig. 3b. An alternative approach related to self-consistent TB is the semi-empirical molecular orbital (SEMO) method,82 based on the neglect of diatomic differential overlap (NDDO) approximation, and originally developed for main group elements.130 However, recent parameterization for d-block elements, such as in the AM1* (ref. 131) and PM6 (ref. 132) Hamiltonians, has enabled the use of such methods for studying fullerene-encapsulated metal nanoclusters133 relevant to catalytic CNT growth. The results from such models for TMs such as cobalt and nickel134 compares favourably with DFT calculations on TM–organic complexes135 using hybrid functionals at a fraction of the computational cost.
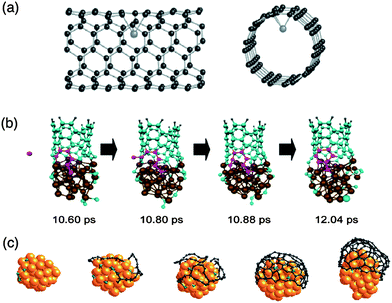 | ||
| Fig. 3 Comparison of cap and tube structures generated from tight-binding modelling approaches: (a) catalytic Ni atom moving into the interior of the nanotube leaving a carbon vacancy on the wall resulting in distortions of the SWCNT (adapted with permission from Andriotis et al.126), (b) Fe-catalysed SWCNT growth using DFTB (adapted with permission from Ohta et al.128), and (c) Grand Canonical Monte Carlo (GCMC) simulations showing the nucleation of a cap from Ni nanoparticle (adapted with permission from Amara et al.137). | ||
A simpler and much less computationally demanding TB scheme was derived has been proposed,136 where the recursion method has been used to calculate the local electronic density of states on each site. Using Monte Carlo simulations in the grand canonical ensemble (GCMC), the nucleation of SWCNTs in the particular case of Ni catalyst was explained,137 as shown in Fig. 3c. More recently, the formation of graphene from carbon atoms diffusing on a Ni surface has also been demonstrated.138 The role of the nanoparticle catalyst is to confine C atoms close to its surface (C2 dimers are found stable in subsurface region99), until a critical surface concentration is reached, where chains and then sp2 carbon structures, weakly interacting with the surface, become more stable and nucleation takes place. Recently, the healing mechanisms of defected carbon structures has been discussed in the case of graphene sheet139 and also nanotubes140 leading to perfect tube with a well-defined chirality.
Empirical modelling methods
Besides the simulations described above, based on quantum mechanics, classical atomistic simulations have also been performed to study CNT growth. The first classical MD simulations were carried out by Shibuta and co-workers, based on their own bond-order potential,141–143 and illustrated in Fig. 4b. The effect of pressure on the nucleation and growth was studied, concluding that an optimum carbon addition rate must exist for SWCNT nucleation to take place. Conversely, when too high a C addition rate was used, the formation of an amorphous network covering the catalyst surface was observed.142 The same authors also studied the effect of the catalyst particle size on the growth, concluding that a minimum size is required for SWCNT nucleation.142 This was later confirmed by simulations performed by Ding and co-workers, employing a different interatomic potential,144 as shown in Fig. 4c. Indeed, below a certain threshold particle size, the curvature energy is too high for a SWCNT cap to nucleate.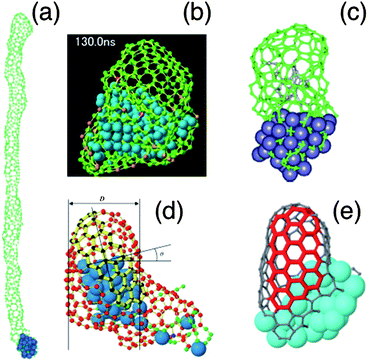 | ||
| Fig. 4 Comparison of cap and tube structures generated by empirical force field methods: (a) growth of a 13 nm long SWCNT at 600 K from hypothetical catalyst particle with a work of adhesion of 50 meV per C atom (adapted with permission from Ribas et al.148), (b) growth of SWCNT cap structure from Ni108 nanoparticle after 130 ns at 2500 K, with arrows showing supply of C from exposed metal surface (adapted with permission from Shibuta33), (c) nucleation and growth of a SWCNT on the surface of an iron carbide cluster at 1000 K (adapted with permission from Ding et al.154), (d) growth of SWCNT cap structure with well-defined (12,4) chiral indices (adapted with permissions from Neyts et al.150), (e) growth of SWCNT on surface-bound Ni40 cluster showing evolution of chiral indices towards (7,7) as highlighted by red portion of tube (adapted with permission from Neyts et al.151). | ||
Ding et al.145 also studied the effect of temperature on the growth. It was found that a temperature gradient is not required for SWCNT growth to take place on small nanoparticles. Also, it was found that carbon diffusion mostly occurs via surface diffusion at low temperatures and, at higher temperatures, through the bulk of the particle146 in agreement with experimental results from Hofmann and co-workers.28 Balbuena and co-workers modified the Shibuta potential to further study the nucleation process.147 A large number of MD simulations were carried out by the same group to investigate the interaction between the growing carbon network and the catalytic particle.148,149 These authors concluded that either a low work of adhesion, fast carbon diffusion, or a high temperature is required for SWCNT growth to occur, as illustrated in Fig. 4a.
Employing the ReaxFF bond-order potential, Neyts and co-workers studied the effect of longer time scale effects on the growth process, by coupling a Monte Carlo method to the MD model.150,151 It was found that far less defective structures than those typically obtained in pure MD simulations could be obtained. In fact, these simulations constituted the first simulations in which a tube with specific chiral angle could be obtained, as shown in Fig. 4d. Furthermore, evolution of chiral indices towards high angle configurations was found,151 as shown in Fig. 4e, confirming earlier predictions that near-armchair structures are preferred.110 Others have also studied the formation of graphene on metal surfaces using molecular dynamics simulation with the ReaxFF potential152 or the Morse-type potential.153 Neyts et al. recently also used the ReaxFF potential to investigate PECVD specific effects on SWCNT growth. In agreement with the experimental results of Kato and co-workers,30 it was found that applying a suitable electric field strength results in the growth of vertically aligned SWCNTs.31 Finally, Neyts et al. also found that ion bombardment in a suitable energy range enhances SWCNT cap formation, again in agreement with experimental results.36
Comparative modelling of graphene stability on Ni(111) surface
Having reviewed the current state of the art in modelling of graphene and CNT growth, we now present the results from our own comparative modelling study in order to answer two fundamental questions: (i) is it possible to achieve ‘epitaxial’-like stabilization of graphene on the surface of bulk metals or nanoparticles, even at temperatures where these are either highly disordered or liquid, and (ii) given the differences between modelling methodologies discussed in previous section, can these methods give consistent predictions regarding such a mechanism?We have chosen for our model system the adsorption of a 2D quasi-periodic carbon sheet, in amorphous and crystalline forms, onto flat Ni surfaces generated from an amorphous melt and the close-packed (111) facet of an fcc Ni-crystal. This choice was made in order to eliminate complications of excessive C dissolution in metal, carbide formation and curvature effects from using nanoparticle, although such effects may be present in real systems. The computational methods used to optimise the system are the reactive bond order potential of Shibuta and Maruyama,155 the ReaxFF potential80 employing the parameterisation by Mueller et al.,156 the tight-binding potential of Amara et al.,136 and the AM1* semi-empirical molecular orbital Hamiltonian.131,134 Due to the systems sizes involved (300 Ni atoms, 100 C atoms), and the averaging required in systems with amorphous Ni/C, it was not feasible to carry out the required number of calculations using DFT to enable a full comparative analysis.
It is important to understand that the different modelling methodologies employed will produce different structures upon minimisation of the total energy. Strictly, the enthalpy of formation, ΔHf, specified per carbon atom, n(C), by eqn (1.1), is defined between reactants (isolated C and Ni) and products (C + Ni in association) in their lowest enthalpy states, which are calculated from the structures optimised using each method, Hopt. However, since these structures are all slightly different, it is problematic to compare directly their enthalpies of formation. This applies especially for amorphous structures, but is also the case for the simplest example of graphene on crystalline nickel. Therefore, we have also computed the interaction energies, ΔE, specified per carbon atom, n(C), by eqn (1.2), of identical structures, all optimised using ReaxFF, calculated from single-point energies, ESP, using the different methods of the C + Ni complexes relative to crystalline Ni and graphene structures. This allows for a fairer comparison of the relative stability of the same structures predicted by the different methods.
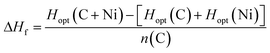 | (1.1) |
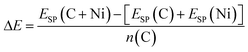 | (1.2) |
Fig. 5a shows a section of graphene sheet (100 C atoms, periodic in x–y plane) adsorbed on a crystalline Ni(111) surface (300 atoms, periodic in x–y plane, 6 layers in thickness, with lowest 3 layers hidden for clarity) in two different low energy configurations: hollow site (Rosei) and top fcc site. In Fig. 5b and c, the interaction energy (defined by eqn (1.2)) for each configuration is displayed as a function of separation between graphene and Ni surface for the four different methods. In each case, the only degree of freedom allowed for the graphene–Ni complex is the separation between centres of C and Ni atoms in 1st nearest layer, with all other relative atomic positions and orientations fixed by optimisation with ReaxFF method. Despite this, it can be seen from Fig. 5b and c that there is a wide range of relative energies, although all methods predict a stable bound state for the complex in both hollow and top fcc site configurations.
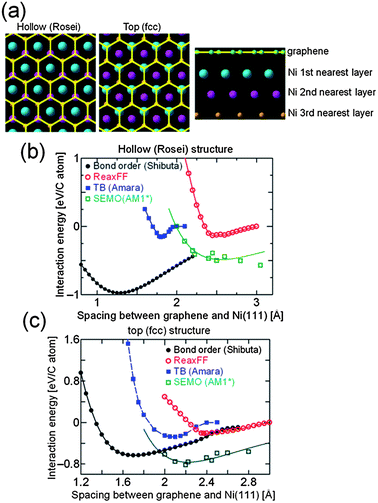 | ||
| Fig. 5 (a) Graphene (yellow) on crystalline Ni(111) (coloured according to distance from graphene) in hollow and top fcc site configurations, (b) interaction energy of hollow site structure as function of separation of graphene sheet and Ni surface calculated using different methods, and (c) interaction energy of top fcc site structure as function of separation of graphene sheet and Ni surface calculated using different methods. | ||
Apart from the bond order potential of Shibuta and Maruyama,155 all methods predict that the top fcc site configuration for graphene on Ni(111) is more energetically favourable than the hollow site configuration, with an optimum separation between the graphene sheet and metal surface ranging from 2.1 to 2.4 Å, in good agreement with DFT results shown in Fig. 1. Although the optimum separations predicted by the bond order potential are unphysically small, the top fcc site configuration is still lower in energy than hollow site configuration for separations greater than 2.2 Å. The binding energies at optimum separations for the top fcc site configuration range from 200 to 800 meV per C atom, depending on method used, which is somewhat higher than those predicted from DFT, although the lower limit (from fully optimised ReaxFF structure) is rather close to that from LDA functional. In general, there is better agreement between the energies predicted by four different methods for the top fcc site configuration than the hollow site configuration, and so the former was chosen as reference state for graphene on crystalline Ni for comparison with amorphous structures in the further calculations below.
Fig. 6 shows example configurations of C–Ni systems (optimised using ReaxFF) used for comparative analysis of stability, where Fig. 6a and e correspond to graphene on crystalline Ni (hereafter referred to as Gra + cNi), Fig. 6b and f correspond to amorphous carbon on crystalline Ni (aC + cNi), Fig. 6c and g correspond to graphene on amorphous Ni (Gra + aNi), and Fig. 6d and h correspond to amorphous carbon on amorphous Ni (aC + aNi). A number of graphene structures with high defect densities were generated by performing TB GCMC simulations at 2500 K. For the present case, the carbon chemical potential chosen to obtain amorphous carbon (aC) was around −2.0 eV atom−1. Under such conditions, defects have a very high probability to be formed, and generally correspond to non-hexagonal cycles. Since there are a great many possible structures with a similar defect density, the results for aC + cNi were averaged over 10 independent aC configurations to avoid bias. The amorphous Ni (aNi) structures were generated by liquefying a crystalline Ni(111) surface using force-biased MC at 5000 K in order to remove any long-range order, followed by an MD quench at 1 K and energy minimisation. Again, to avoid any bias in selection, the results for Gra + aNi structures were averaged over 3 independent aNi configurations, and those for aC + aNi over 30 independent configurations (i.e. 10 × 3, for aC and aNi structures, respectively). It can be seen from Fig. 6f–h that the aC and graphene structures contain significant out-of-plane distortions compared to graphene on crystalline Ni(111).
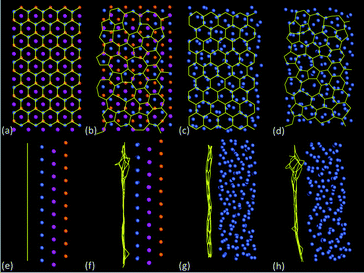 | ||
| Fig. 6 Example configurations of C–Ni systems optimised with ReaxFF, with (a)–(d) top views and (e)–(h) side views: (a) and (e) graphene on Ni(111) in top fcc configuration, (b) and (f) amorphous C (one of 10 configurations) on Ni(111), (c) and (g) graphene on amorphous Ni (one of 10 configurations) and (d) and (h) amorphous C (one of 10 configurations) on amorphous Ni (one of 3 configurations). Carbon bonds are indicated in yellow, whereas cNi atoms in (a)–(b) and (e)–(f) are coloured according to distance of layers from carbon sheet according to key in Fig. 5a, and aNi atoms in (c)–(d) and (g)–(h) are coloured blue-grey. | ||
Fig. 7 shows the enthalpies of formation (defined by eqn (1.1)) for each type of structure shown in Fig. 6, plus the hollow site Gra + cNi configuration for comparison, after full optimisation using each method, arranged in decreasing order of stability according to ReaxFF method. The dashed lines are intended only as a guide to the eye – there is no continuous reaction co-ordinate connecting the different structures. All methods predict that the Gra + cNi structure (in either top fcc or hollow site configuration) is more stable than any other combination of Gra/aC or cNi/aNi. With the exception of SEMO (AM1*), all methods predict that aNi + aC is least stable. In fact, with the exceptions of bond order potential (Shibuta) and SEMO (AM1*), the methods predict that only Gra + cNi has negative enthalpy of formation. The Δ(ΔH) between Gra + cNi and aNi + aC structures ranges from −0.6 to −1.4 eV per C atom, which would favour the formation of the crystalline structures even at the high temperatures present in CVD synthesis. For example, at 1300 K, any negative configurational or vibrational entropy changes would have to exceed 100 J mol−1 K−1 in order to counteract the favourable enthalpy change. We are therefore confident that our predictions of relative thermodynamic stability from optimisation at zero Kelvin are still relevant to the conditions for in situ CVD synthesis. Of course, there may well be kinetic barriers that strongly affect the rate of graphenisation, but we do not consider these explicitly here.
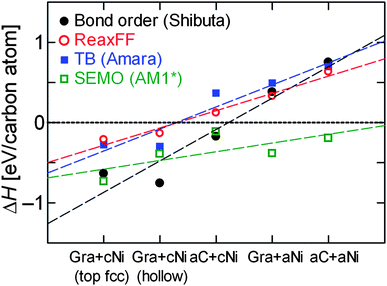 | ||
| Fig. 7 Enthalpy of formation of structures shown in Fig. 6 optimised using different methods: (black filled circles) reactive bond order potential of Shibuta and Maruyama,155 (red open circles) ReaxFF potential,86 (blue filled squares) tight-binding potential of Amara et al.,136 and (open green squares) semi-empirical molecular orbital (AM1* Hamiltonian131,134). | ||
The results in Fig. 7 from ReaxFF and TB potential agree remarkably well, which is mainly due to the close correspondence between the aC structures predicted by each method after full structural optimisation. Fig. 8 shows a comparison of aC structures from ReaxFF and TB optimised on both cNi (Fig. 8a) and aNi (Fig. 8b), where the two structures have been aligned so as to minimise the root-mean square deviation (RMSD) between the sets of C and Ni atoms. There is a slightly larger difference between the aC network predicted by ReaxFF and TB methods on aNi (Fig. 8b) than on cNi (Fig. 8a), as might be expected due to the higher overall disorder on the amorphous metal surface.
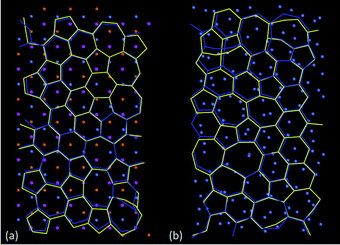 | ||
| Fig. 8 Comparison of the aligned, superimposed structures of amorphous carbon (aC) optimised by the ReaxFF (yellow) and TB (blue) methods, highlighting the similarity between them: (a) aC on crystalline Ni(111) (RMSD = 0.88 Å per atom), (b) aC on amorphous Ni (RMSD = 0.97 Å per atom). In (a), cNi atoms are coloured according to distance of layers from carbon sheet according to key in Fig. 5a, whereas aNi atoms in (b) are coloured blue-grey. | ||
Despite the close agreement between some of the structures relaxed using the different methods, it is possible that the trends observed in Fig. 7 are affected by any remaining differences. In order to check this, the interaction energies (defined by eqn (1.1)) of the structures optimised by ReaxFF were calculated using each method, without further optimisation, and plotted in Fig. 9. They show a similar trend to those in Fig. 7 (with results for ReaxFF being identical), namely that the Gra + cNi structure (in either top fcc or hollow site configuration) is more stable than any other combination of Gra/aC or cNi/aNi. The data in Fig. 9 also appear less scattered than those in Fig. 7, which is due to the greater consistency between the structures used to calculate the energies. However, since none of the structures are minimised with respect to any other method apart from ReaxFF, then the results in Fig. 9 cannot be used to draw any quantitative conclusions about their relative thermodynamic stability. Nevertheless, both Fig. 7 and 9 confirm the robustness of prediction that there is a strong driving force to convert aC to graphene, even on a disordered Ni surface.
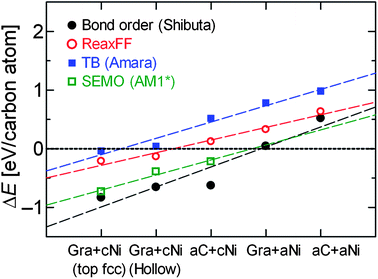 | ||
| Fig. 9 Interaction energy of structures optimised with ReaxFF calculated using different methods: (black filled circles) reactive bond order potential of Shibuta and Maruyama,155 (red open circles) ReaxFF potential,86 (blue filled squares) tight-binding potential of Amara et al.,136 and (open green squares) semi-empirical molecular orbital (AM1* Hamiltonian131,134). | ||
From thermodynamic considerations, discussed above, we therefore expect that amorphous carbon deposited on a TM particle will spontaneously begin to graphenise, via a series of intermediate structures (e.g. chains, rings or sp2 carbon assemblies) that will depend on the available kinetic pathways and chemical potential, and this in turn will stabilise the formation of a crystalline metal facet in contact with the nascent graphene layer. Once a fully graphenised layer is formed in contact with the Ni(111) surface, this structure is then thermodynamically stable against reversion to the amorphous structures of either phase. The question naturally arises as to the minimum size of graphene flake that is thermodynamically stable against reversion to amorphous carbon, i.e. a critical nucleus.
This can be addressed by constructing an expression for the work of transformation, W(a), of a 2D hexagonal graphene nucleus, side-length a, on the surface of metal, which is given by eqn (1.3):
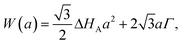 | (1.3) |
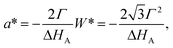 | (1.4) |
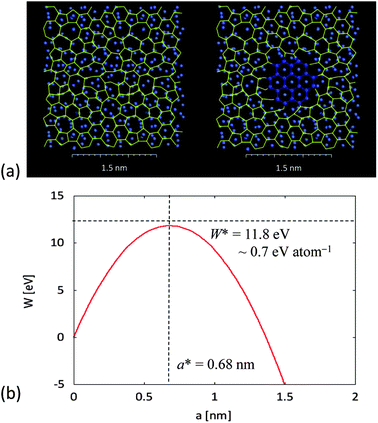 | ||
| Fig. 10 2D classical nucleation model of graphene layer on crystalline facet from amorphous carbon on amorphous metal layer, showing: (a) the pre-nucleation state of aC (yellow) on aNi (blue-grey), and (b) formation of a graphene nucleus (dark blue) on an island of cNi. In (c), the work of nucleation, W, given by eqn (1.3), is shown as a function of nucleus size, a, with enthalpy of stabilization per unit area, ΔHA, taken from Fig. 7 (with energies calculated according to TB method) and aC–graphene energy per unit length, Γ, calculated from energy difference of defective graphene sheet relative to perfect graphene divided by perimeter (again, using TB). The resulting critical size of hexagonal graphene nucleus, a*, and critical work of nucleation, W*, are calculated from eqn (1.4). | ||
Note that this calculation assumes that the co-crystallisation of graphene and metal occurs spontaneously in a region close to the surface of metal particle (less than 4 layers from surface), but it does not require bulk crystallization of metal nanoparticle. It also assumes that the liquid–solid interfacial energy of metal (about 0.281 J m−2 for Ni (ref. 157)) is much smaller than the amorphous carbon–graphene surface energy (estimated as 4.7 J m−2 for stack of graphene sheets with turbostratic alignment). Although we have substituted the enthalpy for free energy in eqn (1.3), as argued above, this will be the dominant term in the transformation. The resulting size of the critical nucleus is in good agreement with kinetic arguments from Ding et al. about the number of complete pentagons (minimum of 5) required to achieve stable cap structure for continued growth of CNTs, although our argument is based on more general thermodynamic considerations and does not specify in detail the atomic structure of the nucleus. In particular, our model of complementary 2D heterogeneous nucleation could apply equally well to the formation of graphene on flat metal surface or stable CNT cap on a curved metal nanoparticle.
Conclusions
In summary, we have reviewed the application of a wide range of computational modelling techniques to the synthesis of carbon nanotubes (CNTs) and graphene by catalytic chemical vapour deposition (CCVD) and plasma-enhanced CVD (PECVD). Although none of the models proposed to date gives a full description of the growth process, from nucleation through to termination, it is clear that they do have many consistent features and that modelling has made a significant contribution to advancing our understanding of this process. Despite the different physical assumptions underlying quantum mechanical and classical modelling techniques, there is a remarkable consistency in their predictions of the relative stability of ordered and disordered phases of carbon–metal system.In particular, our most recent simulations shown that the principle of ‘epitaxial’ structural templating seen in the vapour–solid–solid (VSS) model for non-metallic catalyst particles can be applied even in the case of liquid metallic catalyst particles (VLS model), provided that the complementary interactions with carbon sheet are taken into account. The catalytic process is thus seen not as the formation of graphene on a static template, but rather as a means for both carbon and metal phases to lower their free energies by complementary crystallisation. Since the relative orientation of graphene sheet and ordered metal surface will strongly influence any further growth, achieving the conditions under which this complementary crystallisation process can occur may be a promising method to gain better control over both graphene grown from flat metal surfaces and CNTs from catalyst nanoparticles.
Acknowledgements
The authors gratefully acknowledge helpful discussions with F. Ducastelle. YS acknowledges the Japan Society for Promotion of Science (JSPS) for the Grant-in-Aid for Young Scientists (a) (no. 24686026). JAE acknowledges Accelrys Ltd for use of Materials Studio 6.0 software package. HA and CB acknowledge the financial support of the GDRI GNT (Graphene and Nanotube) and the Agence Nationale de la Recherche (ANR) (Grant no. ANR-09-NANO-028:”SOS Nanotubes”).Notes and references
- C. Journet, W. K. Maser, P. Bernier, A. Loiseau, M. L. delaChapelle, S. Lefrant, P. Deniard, R. Lee and J. E. Fischer, Nature, 1997, 388, 756–758 CrossRef CAS.
- R. T. K. Baker, M. A. Barber, R. J. Waite, P. S. Harris and F. S. Feates, J. Catal., 1972, 26, 51–62 CrossRef CAS.
- G. Y. Zhang, D. Mann, L. Zhang, A. Javey, Y. M. Li, E. Yenilmez, Q. Wang, J. P. McVittie, Y. Nishi, J. Gibbons and H. J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16141–16145 CrossRef CAS.
- Y. Murakami, S. Chiashi, Y. Miyauchi, M. H. Hu, M. Ogura, T. Okubo and S. Maruyama, Chem. Phys. Lett., 2004, 385, 298–303 CrossRef CAS.
- A. A. Puretzky, G. Eres, C. M. Rouleau, I. N. Ivanov and D. B. Geohegan, Nanotechnology, 2008, 19, 055605 CrossRef CAS.
- P. B. Amama, C. L. Pint, L. McJilton, S. M. Kim, E. A. Stach, P. T. Murray, R. H. Hauge and B. Maruyama, Nano Lett., 2009, 9, 44–49 CrossRef CAS.
- C. P. Deck and K. Vecchio, Carbon, 2006, 44, 267–275 CrossRef CAS.
- D. Takagi, Y. Homma, H. Hibino, S. Suzuki and Y. Kobayashi, Nano Lett., 2006, 6, 2642–2645 CrossRef CAS.
- D. Takagi, H. Hibino, S. Suzuki, Y. Kobayashi and Y. Homma, Nano Lett., 2007, 7, 2272–2275 CrossRef CAS.
- X. S. Li, W. W. Cai, J. H. An, S. Kim, J. Nah, D. X. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS.
- M. Cantoro, S. Hofmann, S. Pisana, V. Scardaci, A. Parvez, C. Ducati, A. C. Ferrari, A. M. Blackburn, K. Y. Wang and J. Robertson, Nano Lett., 2006, 6, 1107–1112 CrossRef CAS.
- C. Lu and J. Liu, J. Phys. Chem. B, 2006, 110, 20254–20257 CrossRef CAS.
- T. Hiraoka, S. Bandow, H. Shinohara and S. Iijima, Carbon, 2006, 44, 1853–1859 CrossRef CAS.
- W. Zhang, P. Wu, Z. Li and J. Yang, J. Phys. Chem. C, 2011, 115, 17782–17787 CAS.
- H. Mehdipour and K. Ostrikov, ACS Nano, 2012, 6, 10276–10286 CrossRef CAS.
- H. Kim, C. Mattevi, M. R. Calvo, J. C. Oberg, L. Artiglia, S. Agnoli, C. F. Hirjibehedin, M. Chhowalla and E. Saiz, ACS Nano, 2012, 6, 3614–3623 CrossRef CAS.
- G. F. Zhong, T. Iwasaki, J. Robertson and H. Kawarada, J. Phys. Chem. B, 2007, 111, 1907–1910 CrossRef CAS.
- G. Zhong, J. H. Warner, M. Fouquet, A. W. Robertson, B. Chen and J. Robertson, ACS Nano, 2012, 6, 2893–2903 CrossRef CAS.
- K. Hata, D. N. Futaba, K. Mizuno, T. Namai, M. Yumura and S. Iijima, Science, 2004, 306, 1362–1364 CrossRef CAS.
- S. M. Bachilo, L. Balzano, J. E. Herrera, F. Pompeo, D. E. Resasco and R. B. Weisman, J. Am. Chem. Soc., 2003, 125, 11186–11187 CrossRef CAS.
- W. H. Chiang and R. M. Sankaran, Nat. Mater., 2009, 8, 882–886 CrossRef CAS.
- M. He, A. I. Chernov, P. V. Fedotov, E. D. Obraztsova, J. Sainio, E. Rikkinen, H. Jiang, Z. Zhu, Y. Tian, E. I. Kauppinen, M. Niemela and A. O. I. Krauset, J. Am. Chem. Soc., 2010, 132, 13994–13996 CrossRef CAS.
- M. Fouquet, B. C. Bayer, S. Esconjauregui, R. Blume, J. H. Warner, S. Hoffmann, R. Schlogl, C. Thomsen and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235411 CrossRef.
- M. S. He, H. Jiang, B. L. Liu, P. V. Fedotov, A. I. Chernov, E. D. Obraztsova, F. Cavalca, J. B. Wagner, T. W. Hansen, I. V. Anoshkin, E. A. Obraztsova, A. V. Belkin, E. Sairanen, A. G. Nasibulin, J. Lehtonen and E. I. Kauppinen, Sci. Rep., 2013, 3, 1460 Search PubMed.
- E. C. Neyts, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2012, 30, 030803 Search PubMed.
- S. Hofmann, C. Ducati, J. Robertson and B. Kleinsorge, Appl. Phys. Lett., 2003, 83, 135–137 CrossRef CAS.
- S. Hofmann, C. Ducati, B. Kleinsorge and J. Robertson, Appl. Phys. Lett., 2003, 83, 4661–4663 CrossRef CAS.
- S. Hofmann, B. Kleinsorge, C. Ducati, A. C. Ferrari and J. Robertson, Diamond Relat. Mater., 2004, 13, 1171–1176 CrossRef CAS.
- T. Kato, G. H. Jeong, T. Hirata, R. Hatakeyama, K. Tohji and K. Motomiya, Chem. Phys. Lett., 2003, 381, 422–426 CrossRef CAS.
- T. Kato and R. Hatakeyama, Chem. Vap. Deposition, 2006, 12, 345–352 CrossRef CAS.
- E. C. Neyts, A. C. T. van Duin and A. Bogaerts, J. Am. Chem. Soc., 2012, 134, 1256–1260 CrossRef CAS.
- S. Reich, L. Li and J. Robertson, Chem. Phys. Lett., 2006, 421, 469–472 CrossRef CAS.
- Y. Shibuta, Diamond Relat. Mater., 2011, 20, 334–338 CrossRef CAS.
- Z. Ghorannevis, T. Kato, T. Kaneko and R. Hatakeyama, J. Am. Chem. Soc., 2010, 132, 9570–9572 CrossRef CAS.
- G. Y. Zhang, P. F. Qi, X. R. Wang, Y. R. Lu, X. L. Li, R. Tu, S. Bangsaruntip, D. Mann, L. Zhang and H. J. Dai, Science, 2006, 314, 974–977 CrossRef CAS.
- E. C. Neyts, K. Ostrikov, Z. J. Han, S. Kumar, A. C. T. Van Duin and A. Bogaerts, Phys. Rev. Lett., 2013, 110, 065501 CrossRef CAS.
- A. Reina, X. T. Jia, J. Ho, D. Nezich, H. B. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 30–35 CrossRef CAS.
- S. Bae, H. Kim, Y. Lee, X. F. Xu, J. S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim, Y. I. Song, Y. J. Kim, K. S. Kim, B. Ozyilmaz, J. H. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS.
- K. K. K. Koziol, C. Ducati and A. H. Windle, Chem. Mater., 2010, 22, 4904–4911 CrossRef CAS.
- M. S. Arnold, A. A. Green, J. F. Hulvat, S. I. Stupp and M. C. Hersam, Nat. Nanotechnol., 2006, 1, 60–65 CrossRef CAS.
- M. C. Hersam, Nat. Nanotechnol., 2008, 3, 387–394 CrossRef CAS.
- A. C. Dupuis, Prog. Mater. Sci., 2005, 50, 929–961 CrossRef CAS.
- F. Banhart, Nanoscale, 2009, 1, 201–213 RSC.
- S. Irle, Y. Ohta, Y. Okamoto, A. J. Page, Y. Wang and K. Morokuma, Nano Res., 2009, 2, 755–767 CrossRef CAS.
- J. Wintterlin and M. L. Bocquet, Surf. Sci., 2009, 603, 1841–1852 CrossRef CAS.
- G. D. Nessim, Nanoscale, 2010, 2, 1306–1323 RSC.
- C. Journet, M. Picher and V. Jourdain, Nanotechnology, 2012, 23, 142001 CrossRef.
- M. Batzill, Surf. Sci. Rep., 2012, 67, 83–115 CrossRef CAS.
- N. C. Bartelt and K. F. McCarty, MRS Bull., 2012, 37, 1158–1165 CrossRef CAS.
- A. Thess, R. Lee, P. Nikolaev, H. J. Dai, P. Petit, J. Robert, C. H. Xu, Y. H. Lee, S. G. Kim, A. G. Rinzler, D. T. Colbert, G. E. Scuseria, D. Tomanek, J. E. Fischer and R. E. Smalley, Science, 1996, 273, 483–487 CAS.
- M. Yudasaka, F. Kokai, K. Takahashi, R. Yamada, N. Sensui, T. Ichihashi and S. Iijima, J. Phys. Chem. B, 1999, 103, 3576–3581 CrossRef CAS.
- M. Yudasaka, R. Yamada, N. Sensui, T. Wilkins, T. Ichihashi and S. Iijima, J. Phys. Chem. B, 1999, 103, 6224–6229 CrossRef CAS.
- M. Yudasaka, Y. Kasuya, F. Kokai, K. Takahashi, M. Takizawa, S. Bandow and S. Iijima, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 377–385 CrossRef CAS.
- H. Dai, A. G. Rinzler, P. Nikolaev, A. Thess, D. T. Colbert and R. E. Smalley, Chem. Phys. Lett., 1996, 260, 471–475 CrossRef CAS.
- P. Nikolaev, M. J. Bronikowski, R. K. Bradley, F. Rohmund, D. T. Colbert, K. A. Smith and R. E. Smalley, Chem. Phys. Lett., 1999, 313, 91–97 CrossRef CAS.
- J. E. Herrera, L. Balzano, A. Borgna, W. E. Alvarez and D. E. Resasco, J. Catal., 2001, 204, 129–145 CrossRef CAS.
- R. S. Wagner and W. C. Ellis, Appl. Phys. Lett., 1964, 89, 89–90 CrossRef.
- R. T. K. Baker, Carbon, 1989, 27, 315–323 CrossRef CAS.
- A. J. Page, K. R. S. Chandrakumar, S. Irle and K. Morokuma, J. Am. Chem. Soc., 2011, 133, 621–628 CrossRef CAS.
- S. Hofmann, G. Csanyi, A. C. Ferrari, M. C. Payne and J. Robertson, Phys. Rev. Lett., 2005, 95, 036101 CrossRef CAS.
- S. Hofmann, R. Sharma, C. Ducati, G. Du, C. Mattevi, C. Cepek, M. Cantoro, S. Pisana, A. Parvez, F. Cervantes-Sodi, A. C. Ferrari, R. Dunin-Borkowski, S. Lizzit, L. Petaccia, A. Goldoni and J. Robertson, Nano Lett., 2007, 7, 602–608 CrossRef CAS.
- H. Yoshida, S. Takeda, T. Uchiyama, H. Kohno and Y. Homma, Nano Lett., 2008, 8, 2082–2086 CrossRef CAS.
- H. W. Zhu, K. Suenaga, J. Q. Wei, K. L. Wang and D. H. Wu, J. Cryst. Growth, 2008, 310, 5473–5476 CrossRef CAS.
- F. Ding, A. R. Harutyunyan and B. I. Yakobson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2506–2509 CrossRef CAS.
- R. Rao, D. Liptak, T. Cherukuri, B. I. Yakobson and B. Maruyama, Nat. Mater., 2012, 11, 213–216 CrossRef CAS.
- Y. Shibuta and J. A. Elliott, Chem. Phys. Lett., 2012, 538, 112–117 CrossRef CAS.
- R. Rosei, M. Decrescenzi, F. Sette, C. Quaresima, A. Savoia and P. Perfetti, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 1161–1164 CrossRef CAS.
- Y. Gamo, A. Nagashima, M. Wakabayashi, M. Terai and C. Oshima, Surf. Sci., 1997, 374, 61–64 CrossRef CAS.
- J. Coraux, A. T. N'Diaye, C. Busse and T. Michely, Nano Lett., 2008, 8, 565–570 CrossRef CAS.
- X. Li, W. Cai, L. Colombo and R. S. Ruoff, Nano Lett., 2009, 9, 4268–4272 CrossRef CAS.
- S. Yoshii, K. Nozawa, K. Toyoda, N. Matsukawa, A. Odagawa and A. Tsujimura, Nano Lett., 2011, 11, 2628–2633 CrossRef CAS.
- R. S. Weatherup, B. C. Bayer, R. Blume, C. Baehtz, P. R. Kidambi, M. Fouquet, C. T. Wirth, R. Schloegl and S. Hofmann, ChemPhysChem, 2012, 13, 2544–2549 CrossRef CAS.
- P. W. Sutter, P. M. Albrecht and E. A. Sutter, Appl. Phys. Lett., 2010, 97, 213101 CrossRef.
- K. F. McCarty, P. J. Feibelman, E. Loginova and N. C. Bartelt, Carbon, 2009, 47, 1806–1813 CrossRef CAS.
- H. Ago, Y. Ogawa, M. Tsuji, S. Mizuno and H. Hibino, J. Phys. Chem. Lett., 2012, 3, 2228–2236 CrossRef CAS.
- E. C. Neyts, A. C. T. van Duin and A. Bogaerts, Nanoscale, 2013 10.1039/c3nr00153a.
- R. S. Weatherup, B. Dlubak and S. Hofmann, ACS Nano, 2012, 6, 9996–10003 CrossRef CAS.
- A. R. Harutyunyan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7992 CrossRef.
- J. A. Elliott, Int. Mater. Rev., 2011, 56, 207–225 CrossRef CAS.
- K. Raji and C. B. Sobhan, Nanotechnol. Rev., 2013, 2, 73–105 Search PubMed.
- M. Finnis, Interatomic Forces in Condensed Matter, Oxford University Press, New York, 2003 Search PubMed.
- T. Bredow and K. Jug, Theor. Chem. Acc., 2005, 113, 1–14 CrossRef CAS.
- M. W. Finnis and J. E. Sinclair, Philos. Mag. A, 1984, 50, 45–55 CrossRef CAS.
- D. W. Brenner, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9458–9471 CrossRef CAS.
- D. W. Brenner, O. A. Shenderova, J. A. Harrison, S. J. Stuart, B. Ni and S. B. Sinnott, J. Phys.: Condens. Matter., 2002, 14, 783–802 CrossRef CAS.
- A. C. T. van Duin, S. Dasgupta, F. Lorant and W. A. Goddard III, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- C. Rey, M. M. G. Alemany, O. Dieguez and L. J. Gallego, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 12640–12643 CrossRef CAS.
- G. E. Froudakis, M. Muhlhauser, A. N. Andriotis and M. Menon, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 241401 CrossRef.
- R. C. Longo, M. M. G. Alemany, B. Fernandez and L. J. Gallego, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 167401 CrossRef.
- D. J. Klinke, S. Wilke and L. J. Broadbelt, J. Catal., 1998, 178, 540–554 CrossRef CAS.
- D. J. Siegel and J. C. Hamilton, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 094105 CrossRef.
- L. Gracia, M. Calatayud, J. Andres, C. Minot and M. Salmeron, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 033407 CrossRef.
- G. Kalibaeva, R. Vuilleumier, S. Meloni, A. Alavi, G. Ciccotti and R. Rosei, J. Phys. Chem. B, 2006, 110, 3638–3646 CrossRef CAS.
- W. J. Sun and M. Saeys, J. Phys. Chem. A, 2008, 112, 6918–6928 CrossRef CAS.
- O. V. Yazyev and A. Pasquarello, Phys. Rev. Lett., 2008, 100, 156102 CrossRef.
- G. E. Ramirez-Caballero, J. C. Burgos and P. B. Balbuena, J. Phys. Chem. C, 2009, 113, 15658–15666 CAS.
- F. Cinquini, F. Delbecq and P. Sautet, Phys. Chem. Chem. Phys., 2009, 11, 11546–11556 RSC.
- S. Riikonen, A. V. Krasheninnikov and R. M. Nieminen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125459 CrossRef.
- M. Moors, H. Amara, T. V. de Bocarme, C. Bichara, F. Ducastelle, N. Kruse and J. C. Charlier, ACS Nano, 2009, 3, 511–516 CrossRef CAS.
- F. Ding, P. Larsson, J. A. Larsson, R. Ahuja, H. M. Duan, A. Rosen and K. Bolton, Nano Lett., 2008, 8, 463–468 CrossRef CAS.
- M. Fuentes-Cabrera, M. I. Baskes, A. V. Melechko and M. L. Simpson, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 035405 CrossRef.
- M. Vanin, J. J. Mortensen, A. K. Kelkkanen, J. M. Garcia-Lastra, K. S. Thygesen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 081408 CrossRef.
- T. Olsen, J. Yan, J. J. Mortensen and K. S. Thygesen, Phys. Rev. Lett., 2011, 107, 156401 CrossRef.
- F. Mittendorfer, A. Garhofer, J. Redinger, J. Klimes, J. Harl and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 201401 CrossRef.
- Y. Wang, A. J. Page, Y. Nishimoto, H.-J. Qian, K. Morokuma and S. Irle, J. Am. Chem. Soc., 2011, 133, 18837–18842 CrossRef CAS.
- H.-B. Li, A. J. Page, Y. Wang, S. Irle and K. Morokuma, Chem. Commun., 2012, 48, 7937–7939 RSC.
- A. Maiti, C. J. Brabec, C. M. Roland and J. Bernholc, Phys. Rev. Lett., 1994, 73, 2468–2471 CrossRef CAS.
- A. Maiti, C. J. Brabec, C. Roland and J. Bernholc, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 14850–14858 CrossRef CAS.
- S. Reich, L. Li and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 165423 CrossRef.
- D. A. Gomez-Gualdron and P. B. Balbuena, Nanotechnology, 2008, 19, 485604 CrossRef.
- Q. Wang, M. F. Ng, S. W. Yang, Y. H. Yang and Y. A. Chen, ACS Nano, 2010, 4, 939–946 CrossRef CAS.
- J. C. Charlier, A. DeVita, X. Blase and R. Car, Science, 1997, 275, 646–649 CrossRef CAS.
- Y. H. Lee, S. G. Kim and D. Tomanek, Phys. Rev. Lett., 1997, 78, 2393–2396 CrossRef CAS.
- X. Fan, R. Buczko, A. A. Puretzky, D. B. Geohegan, J. Y. Howe, S. T. Pantelides and S. J. Pennycook, Phys. Rev. Lett., 2003, 90, 145501 CrossRef CAS.
- Y. Y. Liu, A. Dobrinsky and B. I. Yakobson, Phys. Rev. Lett., 2010, 105, 235502 CrossRef.
- D. A. Gomez-Gualdron, J. Zhao and P. B. Balbuena, J. Chem. Phys., 2011, 134, 014705 CrossRef.
- M. Lin, J. P. Y. Tan, C. Boothroyd, K. P. Loh, E. S. Tok and Y. L. Foo, Nano Lett., 2006, 6, 449–452 CrossRef CAS.
- J. Gavillet, A. Loiseau, C. Journet, F. Willaime, F. Ducastelle and J. C. Charlier, Phys. Rev. Lett., 2001, 87, 275504 CrossRef CAS.
- J. Y. Raty, F. Gygi and G. Galli, Phys. Rev. Lett., 2005, 95, 096103 CrossRef.
- J. Kim, A. J. Page, S. Irle and K. Morokuma, J. Am. Chem. Soc., 2012, 134, 9311–9319 CrossRef CAS.
- F. Ducastelle, Order and Phase Stability in Alloys, North-Holland, Amsterdam, 1991 Search PubMed.
- D. Pettifor, Bonding and Structure in Molecules and Solids, Oxford University Press, Oxford, 1995 Search PubMed.
- D. H. Oh and Y. H. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7407–7411 CrossRef CAS.
- P. H. Zhang and V. H. Crespi, Phys. Rev. Lett., 1999, 83, 1791–1794 CrossRef CAS.
- A. N. Andriotis and M. Menon, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15942–15949 CrossRef CAS.
- A. N. Andriotis, M. Menon and G. Froudakis, Phys. Rev. Lett., 2000, 85, 3193–3196 CrossRef CAS.
- Y. Ohta, Y. Okamoto, S. Irle and K. Morokuma, ACS Nano, 2008, 2, 1437–1444 CrossRef CAS.
- Y. Ohta, Y. Okamoto, S. Irle and K. Morokuma, J. Phys. Chem. C, 2009, 113, 159–169 CAS.
- A. J. Page, S. Irle and K. Morokuma, J. Phys. Chem. C, 2010, 114, 8206–8211 CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- P. Winget, C. Selcuki, A. H. C. Horn, B. Martin and T. Clark, Theor. Chem. Acc., 2003, 110, 254–266 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS.
- J. A. Elliott and Y. Shibuta, Mol. Simul., 2008, 34, 891–903 CrossRef CAS.
- H. Kayi and T. Clark, J. Mol. Model., 2010, 16, 29–47 CrossRef CAS.
- M. Walther, B. M. Alzubi, R. Puchta, G. Linti, R. Meier and R. Van Eldik, J. Coord. Chem., 2011, 64, 18–29 CrossRef CAS.
- H. Amara, J. M. Roussel, C. Bichara, J. P. Gaspard and F. Ducastelle, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 014109 CrossRef.
- H. Amara, C. Bichara and F. Ducastelle, Phys. Rev. Lett., 2008, 100, 056105 CrossRef CAS.
- S. Haghighatpanah, A. Borjesson, H. Amara, C. Bichara and K. Bolton, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 205448 CrossRef.
- S. Karoui, H. Amara, C. Bichara and F. Ducastelle, ACS Nano, 2010, 4, 6114–6120 CrossRef CAS.
- M. Diarra, H. Amara, C. Bichara and F. Ducastelle, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 245446 CrossRef.
- Y. Shibuta and S. Maruyama, Phys. B, 2002, 323, 187–189 CrossRef CAS.
- Y. Shibuta and S. Maruyama, Chem. Phys. Lett., 2003, 382, 381–386 CrossRef CAS.
- S. Maruyama, Y. Murakami, Y. Shibuta, Y. Miyauchi and S. Chiashi, J. Nanosci. Nanotechnol., 2004, 4, 360–367 CrossRef CAS.
- F. Ding, A. Rosen and K. Bolton, J. Chem. Phys., 2004, 121, 2775–2779 CrossRef CAS.
- F. Ding, K. Bolton and A. Rosen, Appl. Surf. Sci., 2006, 252, 5254–5258 CrossRef CAS.
- F. Ding, A. Rosen and K. Bolton, Carbon, 2005, 43, 2215–2217 CrossRef CAS.
- J. Zhao, A. Martinez-Limia and P. B. Balbuena, Nanotechnology, 2005, 16, S575–S581 CrossRef CAS.
- M. A. Ribas, F. Ding, P. B. Balbuena and B. I. Yakobson, J. Chem. Phys., 2009, 131, 224501 CrossRef.
- J. C. Burgos, H. Reyna, B. I. Yakobson and P. B. Balbuena, J. Phys. Chem. C, 2010, 114, 6952–6958 CAS.
- E. C. Neyts, Y. Shibuta, A. C. T. van Duin and A. Bogaerts, ACS Nano, 2010, 4, 6665–6672 CrossRef CAS.
- E. C. Neyts, A. C. T. van Duin and A. Bogaerts, J. Am. Chem. Soc., 2011, 133, 17225–17231 CrossRef CAS.
- L. Meng, Q. Sun, J. Wang and F. Ding, J. Phys. Chem. C, 2012, 116, 6097–6102 CAS.
- G. Barcaro, B. Zhu, M. Hou and A. Fortunelli, Comput. Mater. Sci., 2012, 63, 303–311 CrossRef CAS.
- F. Ding, A. Rosen and K. Bolton, Chem. Phys. Lett., 2004, 393, 309–313 CrossRef CAS.
- Y. Shibuta and S. Maruyama, Comput. Mater. Sci., 2007, 39, 842–848 CrossRef CAS.
- J. E. Mueller, A. C. T. van Duin and W. A. Goddard III, J. Phys. Chem. C, 2010, 114, 4939–4949 CAS.
- Z. Y. Jian, K. Kuribayashi and W. Q. Jie, Mater. Trans., 2002, 43, 721–726 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |