 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of nanoparticles as endocytic tracers in cellular microbiology†
Yuying Zhang and
Michael Hensel*
Abteilung Mikrobiologie, Fachbereich Biologie/Chemie, Universität Osnabrück, Barbarastr. 11, 49076 Osnabrück, Germany. E-mail: michael.hensel@biologie.uni-osnabrueck.de; Fax: +49 (0)541 969 3942; Tel: +49 (0)541 969 3940
First published on 24th July 2013
Abstract
The study of pathogen interactions with eukaryotic host cells requires the introduction of fluorescent probes to visualize processes such as endocytosis, intracellular transport or host cell manipulation by the pathogen. Here, three types of fluorescent nanoparticles (NPs), i.e. Rhodamine-labeled polymethacrylate (PMA) NPs, silica NPs and gold NPs, were employed to label the host cellular endolysosomal system and monitor manipulations by the pathogen Salmonella enterica. Using live cell imaging, we investigated the performance of NPs in cellular uptake, labeling of endocytic vesicles and lysosomes, as well as interaction with the pathogen. We show that fluorescent gold and silica, but not PMA NPs appropriately label host cell structures and efficiently track rearrangements of the host endosomal system by the activities of intracellular Salmonella. Silica NPs slightly aggregated and located in Salmonella-induced compartments as isolated dots, while gold NPs distributed uniformly inside such structures. Both silica and gold NPs exhibited no adverse impact on either host cells or pathogens, and are versatile tools for infection biology.
Introduction
The understanding of microbial virulence functions involves the analyses of interactions between pathogens and host cells. The cellular microbiology deploys approaches of cell biology to follow the fate of pathogens and pathogen-induced alterations to host cells with high temporal and spatial resolution. Live cell microscopy, especially epifluorescence, has evolved as a key technique, allowing the analysis of the dynamic interplay between the pathogen and the host cell. While the availability of GFP and various derivatives allows versatile labeling of pathogens and distinct host cell structures, specific probes are required for the analysis of uptake by endocytosis and the processing of such materials in infected cells.Fluorescent nanoparticles (NPs) as newly developed optical probes have received immense interest in cell and tissue imaging in recent years.1–3 Compared to traditional organic dyes, fluorescent NPs including dye-doped NPs, quantum dots and metal NPs exhibit attractive features:4,5 (i) strong signal strength, resistance to photo-bleaching, and tunable fluorescence emissions; (ii) custom fabrication in terms of different size, shape and composition; (iii) facile modification with functional groups as well as biomolecules according to specific experimental requirements. These advantages make fluorescent NPs a desirable choice for tracking endocytic processes and intracellular vesicle transport.6 Furthermore, a variety of electron-dense NPs are directly visible by electron microscopy (EM), allowing combination of fluorescence and EM imaging.7 By employing fluorescent NPs with high electron density to label specific cellular compartments, correlative light microscopy and EM obtain nanoscale spatial information after live cell analysis. Hitherto, fluorescent NPs have been rarely applied as tools in cellular microbiology, and the knowledge about interactions between intracellular microbial pathogens and fluorescent NPs is very limited.
After being internalized by eukaryotic cells, microbial pathogens, either localized in the cytosol or sequestered in membrane-bound compartments, have evolved various strategies to control their intracellular fate and secure their survival within the hosts.8 Tremendous effort has gone into investigating how bacterial pathogens interact with host cells and understanding the underlying mechanisms.9–11 Thanks to the rapid developing fluorescence labeling techniques as well as high-resolution live cell imaging approaches, dynamic host–pathogen interactions can be monitored to give a better interpretation of the infection process. For instance, the human gastrointestinal pathogen Salmonella enterica serovar Typhimurium (S. typhimurium) serves as a model system for an invasive, facultative intracellular lifestyle. S. typhimurium resides and replicates in a membrane-bound compartment of host cells following infection, which is termed Salmonella-containing vacuole (SCV) (for a model, see Fig. 1A). Intracellular Salmonella also induces the formation of extensive tubular membrane compartments, for example Salmonella-induced filaments or SIF (Fig. 1B). The biogenesis of these tubular compartments is only partially understood.12 It has long been commonly accepted that the SCV is separate from the normal endocytic pathway.13 However, recent progress by using AlexaFluor-conjugated dextran as fluid-phase tracers to label the host cell endolysosomal system together with high-resolution live cell imaging has shown that the SCV in the epithelial cell is freely accessible to both lysosomes and incoming endocytic vesicles,14,15 see Fig. 1C for an example of labeling by fluorescent fluid tracer. In spite of these observations, the precise location of extracellular tracers in Salmonella-induced membrane structures remains invisible due to the resolution limitation of optical microscopy. The other way around, using fluorescent NPs which can be detected by both light and electron microscopy as bifunctional tracers and performing a correlative study, might help to obtain ultrastructural information and provide novel insights into the biogenesis of Salmonella-induced membrane structures.
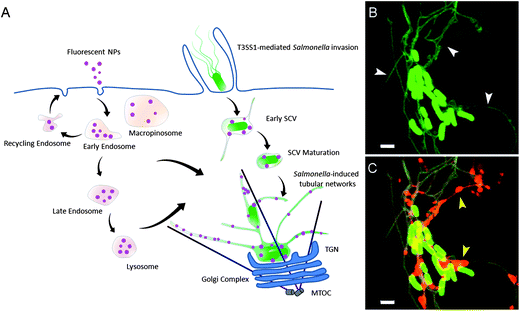 | ||
| Fig. 1 Interactions between host cell endocytic pathways and Salmonella-induced membrane structures. (A) According to previous studies, Salmonella infection results in massive remodeling of the endolysosomal system of epithelial cells. The SCV is accessible to fluid-phase tracers (purple spheres) from both lysosomes and incoming endocytic vesicles. In this study, fluorescent NPs were employed as an endocytic tracer to track the rearrangements of the host cellular endolysosomal system by Salmonella. (B) Salmonella-induced tubular structures imaged by confocal laser scanning microscopy (CLSM) (Salmonella, green rods; endosomal membranes, green tubules). (C) Localization of fluorescent fluid phase tracers (red) in Salmonella-induced tubular structures imaged by CLSM. White and yellow arrowheads indicate the tubular endosomal structures and internalized fluid phase tracers, respectively. Scale bars, 2 μm. | ||
We hypothesize that fluorescent NPs can be used as tracers to study the fate of intracellular bacteria by live cell microscopy. In this report, we describe the systematic analysis of a variety of fluorescent NPs for applications as tracers in cellular microbiology and their performance in the S. typhimurium infection model. Rhodamine-labeled gold NPs (gold NPs), polymethacrylate NPs (PMA NPs) or silica NPs (silica NPs) were applied to track the properties of host cell endosomal vesicles and the rearrangements induced by intracellular Salmonella. Gold NPs and silica NPs have been widely used for biological detection and cell imaging,16,17 and PMA NPs have been reported as polymer carriers of antibiotics for intracellular delivery into infected mice.18 In order to make the experimental results comparable, NPs in the size range 10–30 nm were investigated. S. typhimurium was used as a model bacterium, since the biogenesis of SCV and formation of SIF involve dynamic interactions with the endocytic pathway of host cells.15 The physiochemical properties of NPs and their performances in labeling of subcellular structures were compared; the fusion of incoming endocytic vesicles and late endosomes/lysosomes with SCVs and SIFs was observed; meanwhile, the effects of NPs on host cells as well as pathogens were estimated. Our results indicate that both fluorescent silica and gold NPs are promising optical probes for host cell endolysosomal vesicle labeling and long term monitoring of host–pathogen interactions.
Experimental details
Cell line and bacterial strains culture
Human epithelial cell line HeLa cells (American Type Culture Collection, ATCC no. CCL-2) were cultured in DMEM with 10% fetal calf serum (FCS) and grown at 37 °C in an atmosphere containing 5% CO2. For live cell imaging experiments, HeLa cells were transfected with the LAMP1-GFP plasmid using the calcium phosphate method.19 In brief, HeLa cells were seeded at a density of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per well in an eight well chamber glass slide (Ibidi) and allowed to adhere overnight. 1 μg of plasmid DNA was mixed with the transfection reagent and added to cells. Cells were then incubated for 4–5 h, and later, the culture medium was changed to fresh DMEM with 10% FCS. Alternatively, a Lentivirus-transfected stable HeLa cell line expressing LAMP1-GFP was used. The murine macrophage-like cell line RAW264.7 (ATCC no. TIB-71) was cultured in DMEM with 6% FCS and grown at 37 °C under 5% CO2. For the activation of RAW264.7 cells, 7.5 ng × ml−1 IFN-γ were added for 18 h and resulted in about 90% activated macrophages as judged from the cell morphology.
000 per well in an eight well chamber glass slide (Ibidi) and allowed to adhere overnight. 1 μg of plasmid DNA was mixed with the transfection reagent and added to cells. Cells were then incubated for 4–5 h, and later, the culture medium was changed to fresh DMEM with 10% FCS. Alternatively, a Lentivirus-transfected stable HeLa cell line expressing LAMP1-GFP was used. The murine macrophage-like cell line RAW264.7 (ATCC no. TIB-71) was cultured in DMEM with 6% FCS and grown at 37 °C under 5% CO2. For the activation of RAW264.7 cells, 7.5 ng × ml−1 IFN-γ were added for 18 h and resulted in about 90% activated macrophages as judged from the cell morphology.
S. enterica serovar Typhimurium NCTC 12023 was used as the wild-type (WT) strain. Mutant strains invC and ssaV defective in the Salmonella Pathogenicity island 1 (SPI1)-encoded type III secretion system (T3SS) and the SPI2-T3SS,20,21 respectively, were used as control in invasion and intracellular replication experiments. For live cell imaging, strain harboring plasmid pFPV25.1 was used for the constitutive expression of enhanced GFP. In experiments with DyLight 488-labelled NPs, strains harboring pFPV-mCherry were used for constitutive mCherry expression. Bacterial strains were routinely cultured in Luria–Bertani broth (LB) with addition of 50 μg × ml−1 carbenicillin if required to maintain plasmids. The characteristics of strains and the plasmid used in this study are listed in Table 1.
Nanoparticles
The 25 nm PMA NPs and 30 nm silica NPs were purchased from Micromod (Germany), with an emission maximum at approximately 575 nm. Each kind of NPs was obtained with three different surfaces: plain, coated with NH2 groups and with COOH groups. The 10 nm gold NPs were generated according to a previous protocol with minor modification.23 Briefly, gold NPs were prepared by the citrate and/or tannic acid reduction of chloroauric acid, then incubated with 0.2 mg × ml−1 of BSA for 30 min and centrifuged at 20000 × g for 40 min. The pellet was suspended in PBS and mixed on a Vortex mixer with 0.25 mg × ml−1 of N-hydroxysuccinimide (NHS) Rhodamine (Thermo Scientific) overnight, then dialyzed against PBS using a 12 kDa membrane with 5 changes of the buffer. Subsequently, the NPs were centrifuged twice at 20000 × g for 1 h and resuspended in a small volume of PBS containing 2 mg × ml−1 BSA. The 30 nm gold NPs were prepared using the same method but with different centrifugal forces (3000 × g, 15 min). DyLight 488-labeled gold NPs were obtained by substitution of NHS Rhodamine with DyLight 488 NHS ester (Thermo Scientific).
The size and morphology of NPs were analyzed by transmission electron microscopy (TEM, Zeiss 902). A drop of the NP suspension was added onto a copper grid with a carbon film and dried in air. The hydrodynamic diameter and surface charge of the NPs dispersed in Milli-Q water and DMEM culture medium were determined using a Malvern Zeta sizer.
Fluorescence intensity and photostability of nanoparticles
The fluorescence properties of different NPs were characterized both by a fluorescence spectrophotometer (Jasco FP-6500) and a microplate reader (Chameleon V). NPs were diluted to appropriate concentrations in PBS immediately before the determination. For the fluorescence spectrophotometer, the excitation wavelength was set at 550 nm and emission wavelength from 565 to 650 nm. For the fluorescence microplate reader, the fluorescent signal from NPs was measured with the excitation wavelength centered at 544 nm and the emission filter centered at 590 nm using appropriate band pass filters. Fluorescence intensity data of NPs during 250 rounds of excitation were collected and analyzed.An increase in fluorescence intensities was observed for gold NPs after uptake by host cells. To investigate if Rhodamine fluorescence is quenched by gold NPs, different amounts of plain gold NPs were added into Rhodamine-labeled BSA solution and the fluorescence intensity change was detected by a microplate reader. To analyze if Rhodamine is released from gold NPs after cellular uptake, Rhodamine-labeled gold NPs were suspended in different solutions (cell culture media, phosphate–citrate buffer at pH 5.8 or 4.0, PBS with different amounts of cysteine or trypsin), incubated over a period of 150 min, and fluorescence emissions were recorded by a fluorescence microplate reader with intervals of 5 min.
Cellular uptake
A Leica SP5 confocal laser-scanning microscope (CLSM) with live cell periphery was used to observe the internalization of different NPs by HeLa cells and RAW264.7 macrophages. HeLa cells were seeded in an eight well chamber glass slide at a density of 50000 per well in 300 μl DMEM containing 10% FCS and incubated overnight. The next day cells were incubated with NPs of different concentration for 3 h and then washed thrice with PBS. Subsequently, cells were incubated with the imaging medium (Eagles MEM without L-glutamine, phenol red and sodium bicarbonate, containing 30 mM HEPES, pH 7.4) for another 3 h and then subjected to live cell imaging using a Leica SP5 CLSM. RAW264.7 macrophages were seeded in an eight well chamber glass slide at a density of 100000 per well in 300 μl DMEM containing 6% FCS, and 6 h later activated by IFN-γ (7.5 ng × ml−1) overnight. The next day cells were incubated with NPs for 1 h, washed thrice with PBS, then incubated with the imaging medium and imaged using CLSM.
Cellular uptake of NPs was also quantitatively determined by flow cytometry (FACS Calibur, BD). HeLa cells were seeded in 24-well plates at a density of 150000 per well and incubated overnight to allow adhesion. The next day different NPs were added and incubated for 3 h. Then the cells were washed with PBS thrice and incubated with DMEM containing 10% FCS without NPs for another 3 h. After that, cells were detached with accutase, centrifuged, resuspended in PBS and then the average fluorescence intensity per cell was detected by flow cytometry.
Effect of NPs on cell viability
HeLa cells were seeded in a 96 well plate at a density of 10000 per well one day before the experiment. After incubation in medium with NPs for 3 h and chasing in NP-free medium overnight, 20 μl of 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide (MTT, 5 mg × ml−1) was added to each well and the cells were further cultured at 37 °C for 4 h. The dark blue formazan crystals generated by mitochondrial dehydrogenases of living cells were dissolved in dimethyl sulphoxide and then the absorbance at 590 nm was measured by a microplate reader. The cell viability was calculated as a percentage compared to control samples which were treated without adding NPs.
Effect of NPs on bacterial invasion and intracellular replication
The invasion and intracellular replication of S. typhimurium in HeLa cells were determined by gentamicin protection assay according to a previous protocol.24 HeLa cells were seeded in 24 well plates and incubated overnight to allow adhesion. 3.5 h sub-cultures of WT S. typhimurium and mutant strains invC, ssaV with a multiplicity of infection (MOI) of 2 were used for infection. For bacterial invasion assays, HeLa cells were first pulsed with NPs for 3 h, washed thrice with PBS and then chased with DMEM containing 10% FCS without NPs for another 3 h prior to invasion. After addition of bacteria into the cells, the 24 well plates were centrifuged at 500 × g for 5 min, then put back into the incubator and incubated for 25 min. Non-internalized bacteria were removed by washing with PBS twice and remaining extracellular bacteria were killed by incubation in DMEM containing 10% FCS and 100 μg × ml−1 gentamicin for 1 h. The culture medium was then changed to DMEM containing 10% FCS and 10 μg × ml−1 gentamicin and incubated for another 1 h. Cells were washed and lysed by 0.1% Triton X-100 in PBS. For intracellular replication assay, HeLa cells were first infected with different strains, incubated in culture medium containing 100 μg × ml−1 gentamicin for 1 h, and then incubated in culture medium containing 10 μg × ml−1 gentamicin and NPs for 3 h. After that, the cells were washed with PBS thrice and incubated in medium containing 10 μg × ml−1 gentamicin until lysis of the infected cells at 14 h post-infection (p.i.). 50 μl of the lysates were plated on Mueller Hinton agar using an Eddy Jet spiral plater with logarithmic dilution and incubated overnight for quantification of colony-forming units (CFU). Rates of internalization were calculated as a percentage of the CFU of the inoculum recovered as gentamicin-protected bacteria from host cells. Rates of intracellular proliferation were calculated as the ratio of gentamicin-protected bacteria recovered from host cells at 14 h p.i. to internalized bacteria.Host cell infection and NP pulse-chase
Host cells were infected according to our previously published protocol.15 For infection of HeLa cells, bacterial strains were grown overnight in LB broth, then diluted 1:31 in fresh LB broth and subcultured for 3.5 h. At this time-point, the cultures reached the late log phase and were invasive. An MOI of 100 was applied in this assay. For infection of RAW264.7 macrophages, overnight cultures were diluted and used directly at an MOI of 50. After an incubation of 30 min to allow bacterial internalization the cells were washed thrice with PBS to remove non-internalized bacteria (this time point was set as 0 h post-infection, or 0 h p.i.). Subsequently, DMEM containing 10% FCS and 100 μg × ml−1 gentamicin was added to kill non-internalized bacteria and maintained for 1 h. Then the medium was replaced by a medium containing 10 μg × ml−1 gentamicin for the rest of the incubation time.
To label the incoming endosomal vesicles and observe their fusion with the SCV and SIF, HeLa cells were first infected with S. typhimurium as described before, at 1 h p.i. NPs were added to incubate with the cells. Three hours later, cells were washed thrice with PBS and then chased in imaging medium without NPs for another 3 h. Subsequently, the cells were used for live cell imaging. For 10 nm gold NPs, cells were imaged every one hour after addition of NPs to monitor the dynamic uptake and fusion process. In order to test whether the fixation process fades the fluorescence of intracellular NPs, cells were fixed with 2.5% glutaraldehyde for 1 h and washed thrice with PBS and then subjected to imaging. Macrophages were infected, and 5 h p.i. cells were incubated with NPs for 1 h, washed thrice with PBS and chased for 3 h, and then used for live cell imaging. To measure the accessibility of the SCV and SIF to lysosome contents, the host cellular lysosomes were preloaded with NPs by 4 h co-incubation and overnight chase prior to infection. HeLa cells were then infected with S. typhimurium and incubated until 6 h p.i. for live imaging.
Results and discussion
Synthesis and characterization of nanoparticles
We compared the performance of various fluorescent NPs of use in cellular microbiology. Silica and PMA NPs were obtained from commercial suppliers, while gold NPs were generated by reduction of chloroauric acid by citrate and/or tannic acid. All NPs were conjugated with Rhodamine for direct comparison of the fluorescent properties. As shown in Fig. 2A, the synthesized gold NPs were quasi-spherical in shape, with a size of approximately 10 nm and 30 nm, respectively. BSA coating and Rhodamine labeling did not induce apparent change on their morphology and size (Fig. 2A). TEM images of PMA and silica NPs are presented in Fig. S1A and B,† respectively.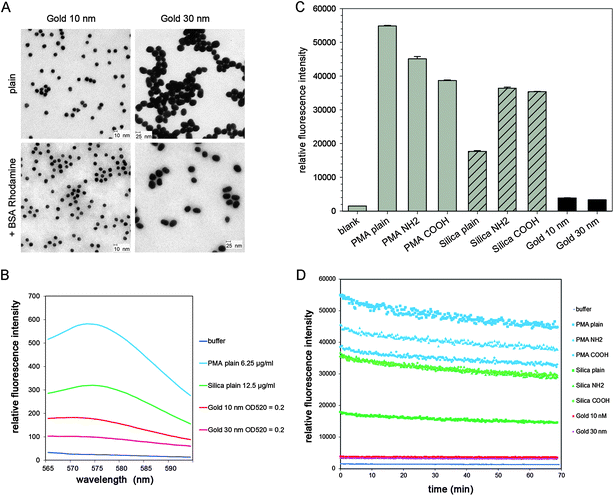 | ||
| Fig. 2 Properties of NPs used in this study. (A) TEM images of colloidal gold NPs with sizes of 10 and 30 nm. (B) Fluorescence spectra of PMA NPs, silica NPs and gold NPs detected by fluorescence spectrophotometry. (C) Fluorescence intensity of various PMA, silica and gold NPs detected by a fluorescence microplate reader. (D) Photostability of various PMA, silica and gold NPs. In (C) and (D), the concentrations of PMA and silica NPs are 100 μg × ml−1, for gold NPs OD520 = 0.2. The NPs were exposed sequentially to 250 cycles, and the fluorescence intensity was recorded by a fluorescence microplate reader. | ||
The size of various NPs used in this study was confined to a small range (10–30 nm) in order to make experimental results comparable, however, when NPs are dispersed in culture medium and applied in cell-related studies, what the cells actually “see” matters more than the characteristics of the bare material.25 Typically, the surface of NPs will be decorated by a selected group of biomolecules from culture medium to form the so-called protein ‘coronas’.26 This decoration process might alter the physiochemical properties of the NPs, such as size and surface energy, and furthermore affect the adhesion of NPs to cell membrane and cellular uptake efficiency.27 Therefore, the hydrodynamic diameters as well as the zeta potential of the NPs suspended in DMEM culture medium were detected and compared. The results are shown in Table 2. PMA NPs suspended in MilliQ water or DMEM culture medium showed similar hydrodynamic diameters, which were nearly twice the bulk NP size. Silica NPs suspended in DMEM culture medium displayed a 3–4 times increase of size compared to NPs suspended in MilliQ water, while the 10 nm and 30 nm gold NPs showed the same size when dispersed in water or culture medium. The gold NPs were previously conjugated with BSA, which is a major component of bovine serum and can stabilize gold NPs from aggregation in biological environments.23 For silica NPs, the obviously increased size implied some aggregation that occurred when the NPs were exposed to biomolecules in culture medium.
| NPs | Diameter (nm) | DDLSc in MilliQ water (nm) | DDLSc in DMEM + 10% FCS (nm) | Zeta potential in MilliQ water (mV) | Zeta potential in DMEM + 10% FCS (mV) |
|---|---|---|---|---|---|
| a Particle diameter from producer data sheet.b Particle diameter as determined by TEM.c Particle diameter according to dynamic light scattering. | |||||
| PMA plain | 25a | 45.1 ± 6.9 | 51.0 ± 4.6 | −39.0 ± 3.0 | −10.6 ± 1.6 |
| PMA NH2 | 25a | 58.5 ± 0.6 | 44.1 ± 1.3 | −28.0 ± 0.4 | −9.9 ± 1.6 |
| PMA COOH | 25a | 63.1 ± 1.9 | 42.4 ± 5.9 | −38.9 ± 3.3 | −10.1 ± 1.4 |
| Silica plain | 30a | 35.7 ± 1.2 | 127.8 ± 4.3 | −39.8 ± 1.8 | −12.3 ± 0.7 |
| Silica NH2 | 30a | 38.5 ± 2.5 | 129.6 ± 9.4 | −28.7 ± 3.5 | −12.3 ± 0.2 |
| Silica COOH | 30a | 32.6 ± 1.2 | 114.2 ± 11.8 | −33.9 ± 2.1 | −12.7 ± 1.2 |
| Gold | 9.8 ± 1.2b | 15.9 ± 0.7 | 18.8 ± 0.5 | −15.5 ± 2.5 | −7.8 ± 0.8 |
| Gold | 31.7 ± 5.4b | 34.6 ± 0.9 | 34.3 ± 0.9 | −40.0 ± 2.3 | −10.3 ± 2.9 |
The surface charge of NPs is essential for their interaction with cells. Amino or carboxyl functionalization is usually used to modify NPs in order to present positive or negative charge on the surface, respectively.28 In this study PMA NPs and silica NPs with plain surfaces, or functionalized by NH2/COOH groups were employed for labeling of the endosomal system of pathogen-infected cells. As shown in Table 2, the zeta potentials of all kinds of NPs were negative in MilliQ water, but the NH2 modified PMA and silica NPs were less negative compared to NPs with plain or COOH-modified surfaces. However, the absolute value of the zeta potentials decreased to the same level when NPs were dispersed in cell culture medium containing serum (Table 2). This effect that different initial surface charges of NPs became almost identical is likely due to adsorption of proteins which are less negatively charged under physiological conditions.26
We investigated the fluorescence properties of the NPs. The emission spectra and the fluorescence intensity detected by the microplate reader are shown in Fig. 2B and C, respectively. PMA NPs with plain surface showed much higher fluorescence emission than silica NPs with plain surface. The PMA and silica NPs surface-modified with NH2 or COOH groups exhibited lower emission. Gold NPs of 10 and 30 nm size both exhibited very low fluorescence intensity.
The photostability of NPs during repeated rounds of excitation was assessed using a fluorescence microplate reader (Fig. 2D). After 250 successive cycles of excitation, the fluorescence intensity of PMA and silica NPs decreased by about 20%, while that of the gold NPs decreased less than 10%. The decrease in emission intensity was linear for all types of NPs. These results indicated that gold NPs with Rhodamine conjugated to the surface were less sensitive to photobleaching.
Cellular uptake
A key requirement for the use of NPs in cellular microbiology is the efficient cellular uptake and accumulation in defined organelles. The internalization of the various NPs by HeLa cells was visualized by CLSM and quantified by flow cytometry (Fig. 3). We found that PMA NPs were only rarely internalized by HeLa cells regardless of their surface properties (Fig. 3A). Although the PMA NPs showed very high fluorescence intensity in solution (see Fig. 2), the cells showed almost no red fluorescence after incubation with 25 μg × ml−1 PMA NPs for 3 h. In contrast, although the gold NPs exhibited very low fluorescence in solution, they can be efficiently internalized by HeLa cells and showed bright intracellular fluorescence (Fig. 3C). After incubation with 25 μg × ml−1 silica NPs, red fluorescence was also observed inside the cells. The cells incubated with silica particles modified with NH2 or COOH showed brighter fluorescence than those incubated with silica NPs with plain surface (Fig. 3B). Of note, the concentrations of gold NPs and silica NPs used for cellular uptake were 50% and 25%, respectively, of the concentrations used for determination of fluorescence intensities.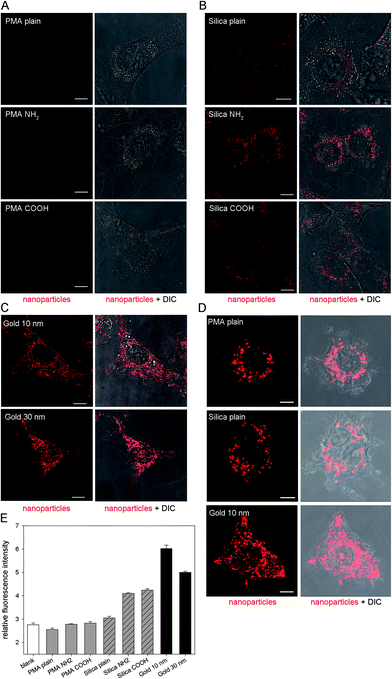 | ||
| Fig. 3 Internalization of various NPs by host cells. HeLa cells were pulsed with various PMA NPs at 25 μg × ml−1 (A), various silica NPs at 25 μg × ml−1 (B), or 10 or 30 nm gold NPs at OD520 = 0.1 (C) for 3 h and chased in medium without NPs for 3 h before live cell imaging and measurements by flow cytometry. RAW264.7 macrophages were activated overnight by IFN-γ, pulsed for 1 h with plain PMA or silica NPs at 25 μg × ml−1, or 10 nm gold NPs at OD520 = 0.1, and subsequently chased in medium without NPs for 3 h before live cell imaging. CLSM images of representative HeLa cells (A–C) and RAW264.7 macrophages (D). Nanoparticles are shown in red, and the cell morphology is indicated by differential interference contrast (DIC) microscopy. Scale bars, 10 μm. (E) Relative fluorescence intensity of HeLa cells obtained by flow cytometry. | ||
It is known that the size, shape, composition and surface properties of NPs are the main factors that dominate their cellular uptake process and intracellular distribution.29 In this study we investigated NPs with similar size (10 nm, 25 nm or 30 nm) and shape, but with different composition (PMA, silica or gold) and surface modification (plain, NH2, COOH or BSA). It has been reported that PMA NPs could be taken up by phagocytic cells and therefore used for intracellular drug transport.18 We also tested cellular uptake of various NPs by RAW264.7 macrophages. As shown in Fig. 3D, macrophages internalized the three kinds of NPs efficiently. Although the PMA NPs cannot be internalized by HeLa cells, their internalization by RAW264.7 macrophages was efficient. We conclude that PMA NPs are not suitable for intracellular imaging studies in non-phagocytic cells.
Silica and gold NPs have been reported to be readily taken up by various mammalian cells.16,30 In accordance with the previous work, we also observed a bright red fluorescence signal in HeLa cells after incubation with silica NPs or gold NPs, indicating internalization of the NPs by cells. The surface property is believed to be another essential factor affecting cellular uptake. Previous studies showed that positively charged NPs are more efficient for cellular internalization compared to neutral or negatively charged NPs, because they can effectively bind to negatively charged groups on the cell membrane.31 NPs with different surface modification were investigated here to determine whether the modification by functional groups affects the uptake behavior of NPs by HeLa cells. However, according to our results, the surface modification did not show obvious impacts. The PMA NPs with plain, NH2 or COOH modified surfaces could not be taken up by HeLa cells, while the silica NPs with three kinds of surface and the gold NPs with BSA surface could be internalized efficiently. The cells incubated with the NH2- or COOH-modified silica NPs displayed similar intracellular fluorescence intensities, much higher than those incubated with plain silica NPs. This should be attributed to their better signal strength compared to the silica NPs with a plain surface (Fig. 3B). The uptake efficiency of NPs by HeLa cells were not affected by NH2 or COOH functionalization, a reasonable explanation is that the surface charge of all NPs was adjusted to the same value (about −10 mV) due to protein adsorption.25 The average fluorescence intensities of cells determined by flow cytometry (Fig. 3E) were in agreement with the observations made by CLSM. After incubation with PMA NPs (25 μg × ml−1) for 3 h, the average fluorescence intensity per cell showed no increase compared to the blank control, and the same result was obtained at an even higher NP concentration (100 μg × ml−1) (data not shown).
For cells incubated with silica NPs (25 μg × ml−1) and gold NPs (OD520 = 0.1), the average fluorescence intensity per cell increased significantly compared to the control. Especially the gold NPs, although their fluorescence intensity in PBS were much lower than PMA NPs and silica NPs, were highly internalized by HeLa cells and showed the highest average fluorescence intensity per cell. We first speculated that the fluorescence intensity increase was induced by the acidic environment in late endosome/lysosomes, however, dispersal of gold NPs in buffers at pH 4.0 or pH 5.8 did not have any effect on fluorescence intensities (Fig. 4B). Recent research found that gold NPs could cause distance-dependent fluorescence quenching,32 and gold nanoparticle conjugates can be destabilized by thiol-containing small molecules.33 Therefore, we supposed that the fluorescence of Rhodamine-labeled BSA was first quenched by gold NPs when BSA molecules were tightly bound to gold NPs in PBS. After incubation with cells, some BSA molecules may be substituted by other proteins or amino acids, or degraded in lysosomes which are rich in hydrolytic enzymes, thereby detaching Rhodamine from gold surfaces and causing elevated fluorescence intensity. Two control experiments were performed to test this speculation. First, different amounts of bare gold NPs were added to Rhodamine labeled BSA solution to test whether gold NPs quench the fluorescence of BSA molecules. As shown in Fig. 4A, addition of gold NPs induced a dose-dependent drop of fluorescence intensity. Second, the Rhodamine-labeled gold NPs were suspended in various cell culture media or PBS with varying amounts of cysteine or trypsin, and the fluorescence intensity was recorded over time. The results are shown in Fig. 4B. Dithiothreitol (DTT), which is able to efficiently displace thiolated compounds from gold surfaces, was used here as a control.33 Exposure to DTT induced a substantial increase of fluorescence intensity. Gold NPs dispersed in PBS were stable over 150 min, as well as NPs dispersed in imaging medium or buffer containing 0.1 μg × ml−1 trypsin. However, dispersion in PBS containing cysteine or a higher concentration of trypsin caused a substantial increase of fluorescence intensity. Gold NPs suspended in DMEM also showed some increase of fluorescence intensity, likely due to the presence of 200 μM cysteine included in this culture medium. According to a previous study,34 glutathione, which is a thiol-containing molecule derived from cysteine, is present in concentrations ranging from 1 to 10 mM in the cytosol.
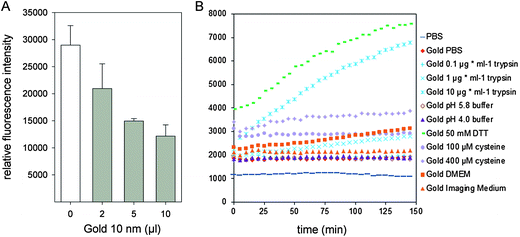 | ||
| Fig. 4 Fluorescence properties of gold NPs. (A) Quenching effect of gold NPs on the fluorescence of Rhodamine-labeled BSA. Aliquots of 2 μl, 5 μl or 10 μl of a suspension of gold NPs (OD520 = 3.5) were added to 100 μl Rhodamine-labeled BSA in PBS (7.2 μg × ml−1) and the fluorescence emission was detected by a microplate reader. (B) Fluorescence intensities of gold NPs in various solutions as a function of time. Gold NPs were dispersed in PBS, DMEM, imaging medium, phosphate–citrate buffer at pH 5.8 or 4.0, or PBS containing 50 mM DTT, or various amounts of cysteine or trypsin as indicated. The fluorescence emission of Rhodamine was recorded over 150 min by a microplate reader with intervals of 5 min. | ||
In summary, we demonstrate that the apparent elevation of the fluorescence intensity of gold NPs after cellular internalization should be attributed to the quenching effect and detachment of BSA molecules from gold NPs as well as degradation by intracellular enzymes.
Cytotoxicity of NPs
Recent research on the toxicity of NPs revealed that the effect of NPs on eukaryotic cells is size-dependent and also cell type-specific.35,36 Gold NPs larger than 4 nm and silica NPs larger than 60 nm were reported to be of low cytotoxicity to human leukemia cells and endothelial cells, respectively.35,37 In our study the cytotoxicity of NPs was evaluated by the MTT assay, and the results are shown in Fig. 5. For each kind of NPs, three concentrations were tested, and the lowest concentration was used for the uptake and host cell structure labeling experiment. The viability of cells showed no significant decrease after treatment with different NPs: for cells incubated with PMA plain, PMA NH2 and PMA COOH NPs (100 μg × ml−1), the cell viabilities were 107.8 ± 6.3%, 112.2 ± 6.8% and 104.8 ± 22%, respectively; for cells incubated with silica plain, silica NH2 and silica COOH NPs (100 μg × ml−1), the cell viabilities were 120.7 ± 8.6%, 103.0 ± 5.0% and 100.2 ± 9.3%, respectively; for cells incubated with 10 nm and 30 nm gold NPs (OD520 = 2), the cell viabilities were 102.2 ± 9.4% and 104.1 ± 8.3%, respectively. In a former study, PMA NPs were applied as a polymer carrier for drug delivery and no cytotoxicity was observed on human monocyte-like U937 cells,18 and in this study, we also found that PMA NPs exhibited no obvious effect on the viability of HeLa cells. Gold and silica NPs are widely used in biomedical imaging and diagnostic tests. Recent research indicated that fibroblasts, epithelial cells, macrophages, and melanoma cells are sensitive to gold NPs of 1.4 nm in size, while gold NPs of 15 nm in size are nontoxic at up to 60-fold higher concentrations.36 In accordance with previous studies,35,36 gold NPs of 10 nm and 30 nm in size and silica NPs of 30 nm in size used in this study showed good biocompatibility in HeLa cells. According to our results, NPs used in this work have negligible effects on HeLa cells and are suitable for following labeling experiments.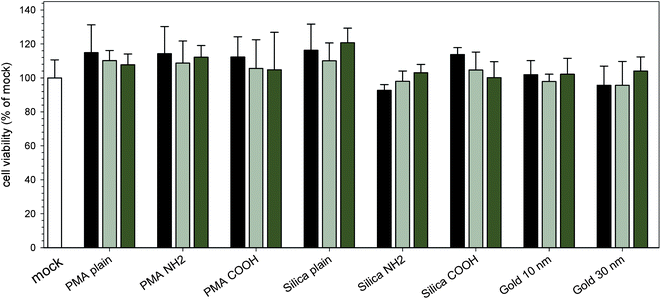 | ||
| Fig. 5 Effect of internalization of various NPs on cell viability. HeLa cells were pulsed for 3 h with various PMA, silica or gold NPs. PMA and silica NPs were applied at concentrations of 25 μg × ml−1 (black bars), 50 μg × ml−1 (light grey bars) or 100 μg × ml−1 (dark grey bar), and gold NPs were applied at concentrations of OD520 of 0.1 (black bars), 1 (light grey bars) or 2 (dark grey bars). Subsequently, the viability of NP-exposed cells was determined by the MTT assays. The values were normalized to those of cells incubated without NPs. The graphs show means and standard deviations of assays performed in triplicate and datasets representative of three independent experiments are shown. | ||
Photostability of internalized nanoparticles
Since a main application of NPs in cellular microbiology is the use in live cell imaging and vesicle transport analyses, the stability of fluorescence signals of NPs during prolonged periods of excitation is a very important parameter. The photostability of internalized silica and gold NPs was compared under continuous excitation by a 543 nm laser using a CLSM. The fluorescence images were acquired with the corresponding intensity curves recorded. As displayed in Fig. 6A and B, after 20 min intensive excitation, the intracellular red fluorescence signal of both NPs remained stable. Quantitative analysis of the images (Fig. 6C) indicated that the 20 min excitation caused less than 5% decrease of fluorescence intensity for both NPs. The results suggested a successful application of these NPs in further intracellular labeling and long term live cell imaging experiments.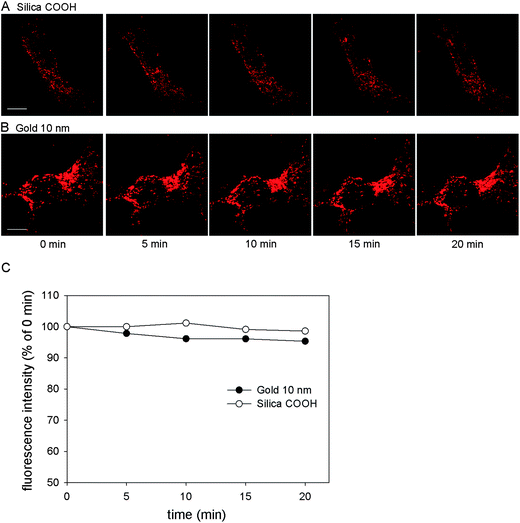 | ||
| Fig. 6 Photostability of internalized silica and gold NPs. HeLa cells were incubated with (A) silica COOH NPs (25 μg × ml−1) or (B) 10 nm gold NPs (OD520 = 0.1) for 3 h and chased with NP-free medium for 3 h, then imaged using CLSM with 20 min continuous laser excitation. Scale bar, 10 μm. (C) Quantification of signal intensities in corresponding images. The fluorescence emission is expressed as the percentage of the initial emission at 0 min. | ||
Effects of NPs on bacterial invasion and intracellular replication
Some carbon-based NPs or oxide NPs have been reported to have adverse impacts on microorganisms.38,39 Since NPs will be applied in cellular microbiology studies, their effects on pathogen behaviors such as invasion and intracellular phenotypes are of relevance. We deployed the infection model of S. typhimurium interaction with mammalian cells. This Gram-negative pathogen has the ability to invade non-phagocytic eukaryotic cells by injection of effector proteins through the SPI1-encoded T3SS system and adopts an intracellular lifestyle depending on the function of a second T3SS encoded by SPI2.40 The effects of various NPs on invasion and intracellular replication of Salmonella in HeLa cells are shown in Fig. 7. After preincubation with various NPs, infection by Salmonella was performed. For WT Salmonella, about 1–1.5% of the inoculum was internalized by HeLa cells. The ssaV strain showed invasion ability similar to WT, while the invC strain defective in T3SS-1 could not invade HeLa cells. Pre-incubation without or with any of the NPs did not affect internalization, indicating that NPs have almost no effect on Salmonella invasion (Fig. 7A). We next quantified the intracellular replication of Salmonella in HeLa cells. In this assay, HeLa cells were first infected by S. typhimurium, and then incubated without or with various NPs for 3 h to determine if bacterial proliferation was affected by the NPs. In HeLa cells without NPs, WT Salmonella showed about 30-fold replication over 14 h of intracellular presence, while the ssaV strain defective in T3SS-2 showed only 8-fold replication (Fig. 7B). In HeLa cells incubated with NPs similar results were obtained for WT Salmonella, and about 30-fold intracellular replication was observed after 14 h infection.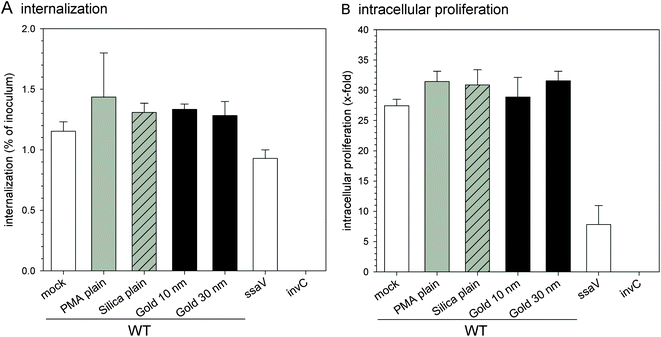 | ||
| Fig. 7 Effect of NPs on virulence functions of Salmonella. Internalization of S. typhimurium by HeLa cells (A) and intracellular proliferation of S. typhimurium in HeLa cells (B) were analyzed after pre-incubation without or with various NPs. Isogenic WT, SPI2-deficient (ssaV) and SPI1-deficient strains (invC) were used to infect HeLa cells at an MOI of 2. Cells were lysed by addition of 0.1% Triton X-100 in PBS at 2 h or 14 h p.i. and plated onto agar plates to determine the amounts of intracellular bacteria. The internalization is expressed as the percentage of the bacterial inoculum applied for invasion; and the x-fold replication is the ratio of the intracellular bacteria at 14 h divided by the 2 h values. The graphs show means and standard deviations of assays performed in triplicate and datasets representative of three independent experiments are shown. | ||
These results demonstrate that the NPs used in this study have no adverse effects on bacterial phenotypes such as invasion and intracellular replication, and therefore are suitable for live cell imaging studies of host–pathogen interactions.
Interactions between the endosomal system and pathogens
After internalization, Salmonella resides in a specialized membrane compartment termed Salmonella-containing vacuole or SCV and the induction of long tubular endosomal compartments, so-called Salmonella-induced filaments or SIF, was observed (for a model, see Fig. 1) (for review, see ref. 12 and 40). A characteristic feature of the SCV as well as SIF is the presence of various late endosomal/lysosomal markers on their membrane, for example lysosomal glycoproteins such as LAMP1. Therefore, HeLa cells transfected with a vector for LAMP1-GFP expression were used for infection, allowing the visualization of SCVs and SIFs by live cell fluorescence microscopy. Previous work indicated that biogenesis of SCV and formation of SIF involve sequential interactions with the endosomal system of host cells.14,15 Hereby, fluorescent NPs were applied to label the host cell endolysosomal vesicles, and the interactions between the vesicles and SCV or SIF were investigated.Labeling of incoming endocytic vesicles
HeLa cells were first infected with S. typhimurium, and at 1 h p.i. different NPs were added to the medium as a marker of the incoming endocytic content. As shown in after 3 h co-incubation and 3 h chase, silica NPs were internalized by HeLa cells and some NPs could be observed inside the SCV or SIF. Note that LAMP1-GFP labels the cytoplasmic face of SCV and SIF membranes, while NPs are located in the lumen of these compartments. In higher magnification fluorescence images (Fig. 8A–C), silica NPs can be readily found located in the vicinity of Salmonella or inside the SIF lumen, which verified the interaction of incoming endosomal traffic with SCVs and SIFs. The NPs appeared as separate dots around Salmonella or inside SIFs, and there was no apparent difference between NPs with plain surface and NPs modified with NH2 or COOH groups. A larger amount of NPs were observed to be fused with SCVs or SIFs in the images of the cells labeled with gold NPs (Fig. 8D and E). The 10 nm gold NPs appeared uniformly distributed inside SIF tubules or surrounding Salmonella (Fig. 8D). In the 568 nm channel, some rod-shaped structures could be observed to be formed by the gold NPs around Salmonella, but located inside the SCV. The precise colocalization of LAMP1-GFP and gold NPs indicate the excellent labeling of the host cell structure by gold NPs, and demonstrate the massive remodeling of the host cell endosomal system by the activities of S. typhimurium. Gold NPs with a diameter of 30 nm were also used for the intracellular labeling experiment and exhibited similar behavior to that of 10 nm gold NPs (Fig. 8E). Vesicles containing this marker fused efficiently with SCVs and SIFs and dispersed homogeneously inside these host cell-derived structures. We hypothesize that the aggregation of silica NPs, but not gold NPs, in culture medium should be responsible for the distinct intracellular performance of silica NPs and gold NPs with respect to dispersal. Furthermore, the release of Rhodamine from gold NPs under intracellular conditions (compare Fig. 4) may contribute to the uniform distribution of fluorescence signals within endosomal compartments.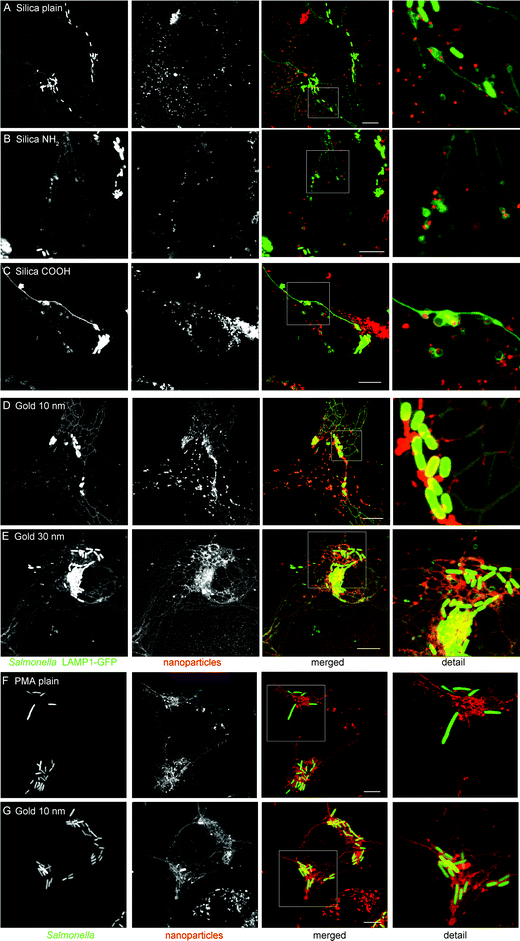 | ||
| Fig. 8 Labeling of pathogen-induced host cell structures by silica and gold NPs. (A–E) HeLa cells constitutively expressing the late endosomal/lysosomal marker LAMP1-GFP (green) were infected with S. typhimurium WT expressing GFP (green) and incubated for 3 h with various NPs (red) as follows: 25 μg × ml−1 of silica plain (A), silica NH2 (B), silica COOH (C), 10 nm gold at OD520 = 0.1 (D), or 30 nm gold at OD520 = 0.1 (E). Subsequently, cells were chased in NP-free medium for 3 h, and analyzed by live cell imaging using a Leica SP5 CLMS. (F and G) RAW264.7 macrophages were activated by incubation with IFN-γ overnight, then pulsed with plain PMA NPs at 25 μg × ml−1, plain silica NPs at 25 μg × ml−1, or 10 nm gold NPs at OD520 = 0.1 for 1 h, and chased in medium without NPs for 3 h before imaging. Expression of LAMP1-GFP by transfection of RAW264.7 cells was not possible. Scale bars, 10 μm. | ||
PMA NPs were not endocytosed by non-infected HeLa cells (Fig. 3A). To investigate whether pathogen infection causes abnormal uptake behavior by host cells, Salmonella-infected HeLa cells were pulsed with PMA NPs for 3 h, chased for 3 h and observed by CLSM. The results presented in Fig. S2† indicate that PMA NPs were not internalized by Salmonella-infected HeLa cells, demonstrating that Salmonella infection did not cause aberrant internalization of NPs. On the other hand, Salmonella-induced phenotypes in host cells were not affected by the presence of non-internalized NPs. Although all three kinds of NPs could be taken up by RAW264.7 macrophages, only PMA NPs and gold NPs were found to be distributed inside intracellular tubular structures (Fig. 8F and G). Silica NPs in RAW264.7 cells were mostly located in isolated phagosomes (Fig. S2†).
To investigate the uptake kinetics of 10 nm gold NPs and their interaction with host cell structures, live cell imaging of Salmonella-infected cells was performed (Fig. 9). At 1 h p.i., gold NPs pulsed for 3 h. The late endosomal/lysosomal compartments are marked by green fluorescence due to GFP labeling. After 1 h of pulse (2 h p.i.), Rhodamine-positive vesicles were observed inside HeLa cells, indicating the uptake of NPs by HeLa cells. Some vesicles emitted red fluorescence, others showing a yellow color indicated maturation of endosomes and location of gold NPs in late endosomal/lysosomal compartments. At 3 h p.i., SIF started to form inside HeLa cells, while NPs were internalized continually and enriched in the vicinity of Salmonella. Red fluorescence was also visible inside the newly formed SIF, indicating some gold NPs were located inside SIF tubular structures. At 5 h p.i., gold NPs could be found to be uniformly distributed surrounding Salmonella inside the SCV as well as inside complex SIF networks. At 6 h p.i., the SCV and SIF showed intensive labeling by gold NPs and became even brighter at later time points. Time lapse movies were recorded at 4 h (ESI Movie 1†) and 8 h (ESI Movie 2†) and demonstrate the incorporation of gold NPs in highly dynamic endosomal structures.
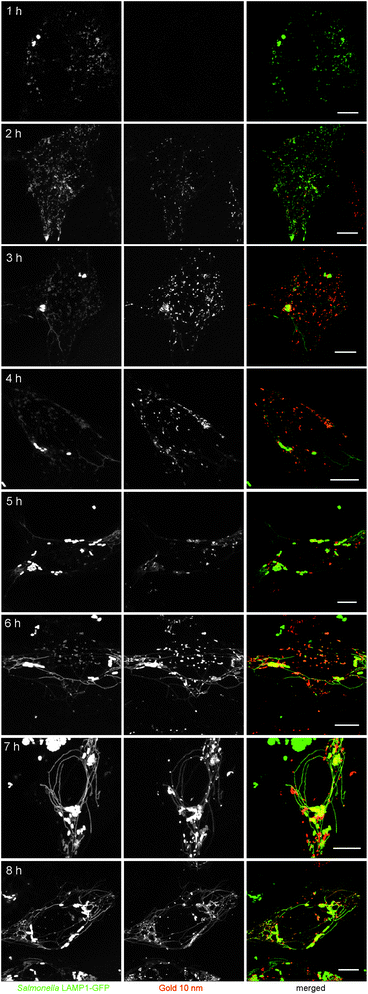 | ||
| Fig. 9 Dynamics of pathogen-induced host cell structures analyzed by labeling with gold NPs. HeLa cells were infected with S. typhimurium WT for 1 h, incubated with 10 nm gold NPs (OD520 = 0.1) for up to 3 h and then cultured in NP-free medium for the rest of the experiment. Live cell imaging was performed at intervals of 1 h after addition of NPs and the time point after infection is indicated. Scale bars, 10 μm. | ||
Labeling of lysosomes
We finally investigated the stability of NPs for labeling of lysosomes. In order to preload lysosomes with NPs, HeLa cells were incubated with silica NPs or gold NPs for 4 h, followed by overnight chase. Subsequently, cells were infected with S. typhimurium and processed for live cell imaging. As shown in Fig. S3† pre-incubation and overnight chase resulted in the localization of all silica NPs in lysosomes. Salmonella infection resulted in the efficient recruitment of NP-containing lysosomes to the SCV and SIF. As a consequence, most of the silica NPs were located in SCV compartments adjacent to Salmonella or inside SIF at 6 h p.i. (Fig. 10A–C). No apparent difference was observed among silica NPs with plain surface and NH2- or COOH-functionalized surface. As presented in Fig. 10D, lysosomes preloaded with 10 nm gold NPs also fused efficiently with SCVs and SIFs, therefore the entire lumen of the SCV and SIF was labeled with the tracer.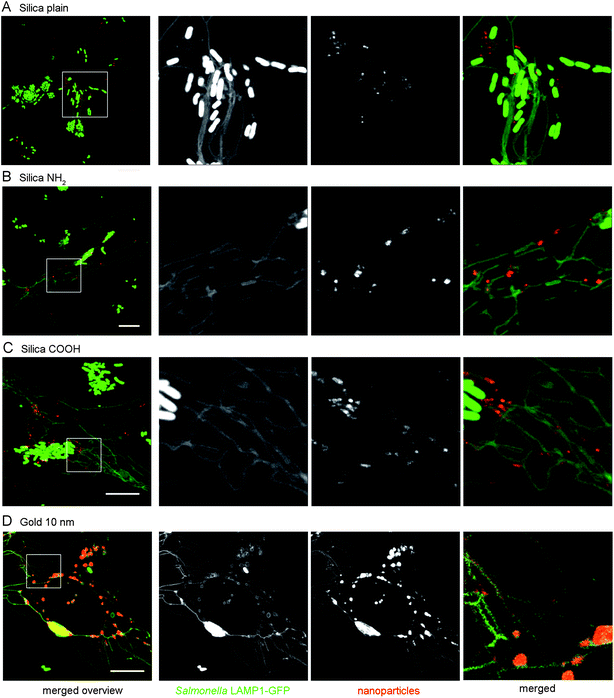 | ||
| Fig. 10 Fusion of the SCV and SIF with lysosomes preloaded with silica NPs or gold NPs. HeLa cells were pre-incubated with 25 μg × ml−1 of silica plain NPs (A), silica NH2 NPs (B), silica COOH NPs (C), or 10 nm gold NPs (OD520 = 0.1) (D) for 4 h and chased with NP-free medium overnight. Subsequently, cells were infected with S. typhimurium and live cell imaging was performed at 6 h p.i. Scale bars, 10 μm. Merged higher magnifications indicate the presence of NPs within the lumen of tubular membrane compartments. | ||
AlexaFluor-conjugated dextran has been reported recently as a fluid-phase tracer to track the interactions between Salmonella and host cells and showed good labeling of SCV and SIF.14,15 However, the fluorescence signal of the tracer lost more than 90% during fixation and processing steps.14 The retention ability of gold NPs in SCV and SIF after cell fixation by 2.5% glutaraldehyde was investigated and the result is presented in Fig. S4.† After fixation and washing with PBS, the fluorescence signal of gold NPs remained very strong and it was easy to distinguish the SCV and SIF structures. Longer lifespan in living cells and better retention in fixed or permeabilized cells make fluorescent NPs more flexible and adaptable in bioimaging studies.6 Furthermore, fluorescence emission of NPs can be adjusted through labeling with different fluorophores. As shown in Fig. S5,† gold NPs coated with BSA and conjugated with DyLight 488 fused with SCV and SIF and labeled these host cell structures distinctly.
Conclusions
In summary, fluorescent PMA NPs, silica NPs and gold NPs were applied for labeling of the endosomal system and monitoring the arrangement of endolysosomal vesicles by the intracellular pathogen S. typhimurium. The performance of the various NPs is concluded in Table 3. The NPs had no detectable toxicity on HeLa cells and no apparent impact on virulence traits of the bacteria. PMA NPs were not able to enter HeLa cells before or after infection, and therefore are not suitable for intracellular labeling. Silica NPs with different surfaces and gold NPs were readily internalized by HeLa cells. The NPs located in incoming endocytic vesicles as well as preloaded in lysosomes fused efficiently with the SCV and SIF and as a consequence accumulated inside these host cell structures. Gold NPs dispersed uniformly surrounding intracellular Salmonella in the SCV or along with SIFs, while silica NPs distributed in the SCV or SIF as isolated vesicles, likely because of aggregation. Our results indicate that fluorescent silica NPs and gold NPs are appropriate and efficient optical probes for host cell structure labeling and host–pathogen interaction monitoring and therefore have great potential to be broadly applied for live cell imaging studies in infection biology.| NPs | Fluorescent intensityb | Photo-stabilityc | Cyto-toxicityd | Effects on pathogense | Cellular uptakef | Dispersity in SCV and SIFg |
|---|---|---|---|---|---|---|
| a ++, very good; +, good; Ø, none; n.d., not determined.b Fig. 2 and 3.c Fig. 6.d Fig. 5.e Fig. 7.f Fig. 3 and 8.g Fig. 8–10. | ||||||
| PMA plain | ++ | + | Ø | Ø | Ø | n.d. |
| PMA NH2 | ++ | + | Ø | Ø | Ø | n.d. |
| PMA COOH | ++ | + | Ø | Ø | Ø | n.d. |
| Silica plain | ++ | + | Ø | Ø | + | + |
| Silica NH2 | ++ | + | Ø | Ø | + | + |
| Silica COOH | ++ | + | Ø | Ø | + | + |
| 10 nm gold | + | ++ | Ø | Ø | ++ | ++ |
| 30 nm gold | + | ++ | Ø | Ø | ++ | ++ |
Acknowledgements
This work was supported by the DFG through Sonderforschungsbereich 944 ‘Physiology and Dynamics of Cellular Microcompartments’, project Z and DFG priority programme 1580, grant HE 1964/18-1.Notes and references
- A. M. Coto-Garcia, E. Sotelo-Gonzalez, M. Fernandez-Arguelles, R. Pereiro, J. M. Costa-Fernandez and A. Sanz-Medel, Anal. Bioanal. Chem., 2011, 399, 29–42 CrossRef CAS.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesner, Nano Lett., 2005, 5, 113–117 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- H. Shi, X. He, Y. Yuan, K. Wang and D. Liu, Anal. Chem., 2010, 82, 2213–2220 CrossRef CAS.
- K. Cortese, A. Diaspro and C. Tacchetti, J. Histochem. Cytochem., 2009, 57, 1103–1112 CrossRef CAS.
- A. P. Bhavsar, J. A. Guttman and B. B. Finlay, Nature, 2007, 449, 827–834 CrossRef CAS.
- L. Y. Gao and Y. A. Kwaik, Trends Microbiol., 2000, 8, 306–313 CrossRef CAS.
- E. Campoy and M. I. Colombo, Biochim. Biophys. Acta, Mol. Cell Res., 2009, 1793, 1465–1477 CrossRef CAS.
- B. B. Finlay and P. Cossart, Science, 1997, 276, 718–725 CrossRef CAS.
- N. Schroeder, L. J. Mota and S. Meresse, Trends Microbiol., 2011, 19, 268–277 CrossRef CAS.
- F. Garcia-del Portillo and B. B. Finlay, Trends Microbiol., 1995, 3, 373–380 CrossRef CAS.
- D. Drecktrah, L. A. Knodler, D. Howe and O. Steele-Mortimer, Traffic, 2007, 8, 212–225 CrossRef CAS.
- R. Rajashekar, D. Liebl, A. Seitz and M. Hensel, Traffic, 2008, 9, 2100–2116 CrossRef CAS.
- S. Santra, Methods Mol. Biol., 2010, 624, 151–162 CAS.
- R. A. Sperling, P. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- M. Paul, R. Durand, Y. Boulard, T. Fusai, C. Fernandez, D. Rivollet, M. Deniau and A. Astier, J. Drug Targeting, 1998, 5, 481–490 CrossRef CAS.
- R. Brent, F. M. Ausubel, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith and K. Struhl, Current Protocols in Molecular Biology, Wiley, New York , 1987 Search PubMed.
- R. G. Gerlach, N. Claudio, M. Rohde, D. Jackel, C. Wagner and M. Hensel, Cell. Microbiol., 2008, 10, 2364–2376 CrossRef CAS.
- J. E. Shea, M. Hensel, C. Gleeson and D. W. Holden, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2593–2597 CrossRef CAS.
- R. H. Valdivia and S. Falkow, Science, 1997, 277, 2007–2011 CrossRef CAS.
- J. W. Slot and H. J. Geuze, Eur. J. Cell Biol., 1985, 38, 87–93 CAS.
- S. U. Hölzer and M. Hensel, PLoS One, 2012, 7, e33220 Search PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Aberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS.
- S. Kralj, M. Rojnik, R. Romih, M. Jagodic, J. Kos and D. Makovec, J. Nanopart. Res., 2012, 14, 1151–1164 CrossRef.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang and C. Chen, Small, 2011, 7, 1322–1337 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS.
- Z. G. Yue, W. Wei, P. P. Lv, H. Yue, L. Y. Wang, Z. G. Su and G. H. Ma, Biomacromolecules, 2011, 12, 2440–2446 CrossRef CAS.
- G. Schneider, G. Decher, N. Nerambourg, R. Praho, M. H. Werts and M. Blanchard-Desce, Nano Lett., 2006, 6, 530–536 CrossRef CAS.
- T. A. Larson, P. P. Joshi and K. Sokolov, ACS Nano, 2012, 6, 9182–9190 CrossRef CAS.
- M. Oishi, J. Nakaogami, T. Ishii and Y. Nagasa, Chem. Lett., 2006, 1046–1047 CrossRef CAS.
- D. Napierska, L. C. Thomassen, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, J. A. Martens and P. H. Hoet, Small, 2009, 5, 846–853 CrossRef CAS.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS.
- S. Kang, M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2009, 43, 2648–2653 CrossRef CAS.
- R. Brayner, R. Ferrari-Iliou, N. Brivois, S. Djediat, M. F. Benedetti and F. Fievet, Nano Lett., 2006, 6, 866–870 CrossRef CAS.
- A. Haraga, M. B. Ohlson and S. I. Miller, Nat. Rev. Microbiol., 2008, 6, 53–66 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr01550e |
| This journal is © The Royal Society of Chemistry 2013 |