DOI:
10.1039/C3NR01432K
(Paper)
Nanoscale, 2013,
5, 6491-6497
Carbon nanotube–gold nanohybrids for selective catalytic oxidation of alcohols†
Received
22nd March 2013
, Accepted 10th May 2013
First published on 15th May 2013
Abstract
Gold nanoparticles were deposited on carbon nanotubes to provide access to a nanohybrid structure which was involved in the aerobic oxidation of alcohols. The nanohybrid-catalyzed reaction was shown to be highly efficient under mild conditions (i.e. room temperature, air) and selective oxidation of alcohols to the corresponding acids or aldehydes could be achieved, depending on the reaction conditions.
Introduction
The chemoselective oxidation of alcohols is one of the key transformations in organic synthesis. While stoichiometric amounts of oxidizing reagents and harsh conditions are classically required to perform alcohol oxidation,1 recent developments have led to the discovery of highly active heterogeneous catalytic systems. Among these, supported metal nanoparticles have gained increasing interest as they provide clean, selective, and efficient reactions.2 In addition, these nanoparticulate catalysts can be easily recovered, thus permitting their reuse. Numerous metals,3 including gold,4 have been assembled onto solid supports although metallic gold has traditionally been regarded as a poor catalytic species.5 The catalytic activity of bulk gold is nevertheless dramatically enhanced when downsized to nanometric particles (AuNPs).6–8 This peculiar behaviour of nano-gold has recently boosted its use in fine chemical synthesis applied, for example, to selective hydrogenations, carbon–carbon bond formation, or oxidations.9–12 We recently contributed to the field of carbon nanotube (CNT) supported AuNPs13 and nano-gold catalyzed reactions by reporting one of the most efficient systems for silane oxidation. The system used molecular oxygen and water as oxidant sources in combination with carbon nanotube–gold nanohybrids (AuCNT, Fig. 1).14 When compared to other supports, CNTs offer some advantages that include chemical, thermal and mechanical stability, inertness (i.e. little to no interactions with the catalyzed organic transformation), high surface area, and chemically tuneable topography. Moreover, CNTs are electronically active and can contribute to the stabilization of transient higher oxidation states of the metals. In this article, we report our investigations on the selective oxidation of alcohols catalyzed by our original AuCNT nanohybrid.
 |
| | Fig. 1 Synthesis of the AuCNT assembly. | |
Experimental
General
Chemicals were purchased from Sigma-Aldrich except H218O that was obtained from Euriso-top (Saclay, France). N6-Pentacosa-10,12-diynoyl-N2,N2-bis(carboxymethyl)lysine (DANTA) was synthesized as previously described.14 Multi-walled carbon nanotubes (MWCNTs) were prepared by catalytic decomposition of methane on a Ni–Mg–O catalyst.14 Flash chromatography was carried out on a Kieselgel 60 (230–240 mesh, Merck). Electron microscopy observations were carried out on a Philips CM12 microscope operated at 100 kV. NMR spectra were recorded on a Bruker Avance DPX 400 spectrometer. Chemical shifts (δ) are given in ppm relative to the NMR solvent residual peak and coupling constants (J) in hertz.
Nanohybrid synthesis and characterization
Self-assembly and polymerization of the amphiphile on the CNTs.
Amphiphilic diacetylenic nitrilotriacetic lipid (DANTA, 20 mg) was dissolved in an aqueous Tris buffer (2 mL, pH 8) before CNTs (50 mg) were added. After 10 min of sonication with an ultra-sonic probe (5 min, 300 ms pulses per second, 25 W output power), a stable suspension was recovered and centrifuged at 5000 × g for 3 min to remove amorphous carbon. The supernatant was collected and centrifuged at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 45 min to separate the DANTA-decorated nanotubes from the excess of the amphiphile. The pellet was resuspended in a fresh buffer and centrifuged again at 11000 × g for 45 min. The final pellet was resuspended in the buffer and submitted to UV irradiation (254 nm) for 8 h to polymerize the diacetylene groups and stabilize nanoring assemblies.
000 × g for 45 min to separate the DANTA-decorated nanotubes from the excess of the amphiphile. The pellet was resuspended in a fresh buffer and centrifuged again at 11000 × g for 45 min. The final pellet was resuspended in the buffer and submitted to UV irradiation (254 nm) for 8 h to polymerize the diacetylene groups and stabilize nanoring assemblies.
Assembly of the second layer on the nanoring-coated nanotubes.
After polymerization, the CNT suspension was stirred in the presence of cationic poly(diallyldimethylammonium chloride) (PDADMAC, 700 μL of a 20% solution in H2O) for 1 h to induce the formation of the two-layer assembly. The excess of the polymer was removed by centrifugation at 11000 × g for 30 min and the pellet was resuspended in a Tris buffer. This operation was repeated twice with the Tris buffer and two more times with pure water. The final pellet was resuspended in 1 mL of water.
Assembly of gold nanoparticles.
Freshly prepared gold nanoparticles15 (1 mM colloid suspension) were added to the doubly coated CNTs and the mixture was left at RT with 1 min vortex-stirring every 30 min (4 h). The suspension was then centrifuged at 3000 × g for 5 min and the nearly colorless supernatant was discarded and replaced with a fresh gold colloid suspension. The same process was repeated 5 times to ensure optimal loading of the tubes with AuNPs. The obtained pellet (ca. 30 mg of the dry material) was washed 3 times by centrifugation/redispersion in water and finally redispersed in 4 mL of water to yield the AuCNT suspension that was used for all catalysis experiments.
Characterization and loading measurement of AuCNT nanohybrids.
The hybrids were characterized by transmission electron microscopy (TEM) to observe the morphology of the assemblies and measure the mean particle size; X-ray photoelectron spectroscopy (XPS) was used to confirm that only metallic gold was present in the hybrids; inductively coupled plasma mass spectrometry (ICP-MS) was used to measure the gold content of the suspension.
General procedure for the oxidation of alcohols and aldehydes
To a stirred solution of an alcohol/aldehyde (0.1 mmol) in a 1:1 mixture of toluene–water (0.5 mL) was added NaOH (0.3 mmol) and aqueous AuCNT (50 μL, 0.2 mol%). The reaction mixture was stirred until the complete consumption of the starting material (as monitored by TLC). After the completion of the reaction the aqueous phase was washed with Et2O (2 × 5 mL), acidified with 1 N HCl (1 mL) and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under a vacuum to afford the expected acid. For secondary alcohols, no acidic work up was needed and the aqueous layer was directly extracted with EtOAc.
Results and discussion
Synthesis of AuCNT hybrids
The carbon nanotube–gold nanohybrid catalyst was prepared by self-assembly of amphiphilic nitrilotriacetic-diyne lipid (DANTA) on carbon nanotubes (Fig. 1). This led to the formation of hemi-micelles with nanoring-like structures (Fig. 1).14,16–18 While the hydrophobic portion of DANTA is adsorbed on CNT by van der Waals interactions, its hydrophilic head is oriented toward the aqueous phase. Further stability of the rings was promoted by photo-polymerization at 254 nm of the diyne motif incorporated in the lipophilic chain. This polymerization process takes place within individual half-cylinders and reinforces the cohesion of the assembly.19 The second layer was thereafter deposited by stirring the suspended nanotubes with cationic PDADMAC which adsorbed on the tube surface by electrostatic interactions with the primary anionic layer. A solution of freshly prepared colloidal gold nanoparticles15 was then added to the doubly coated CNTs. Interaction of AuNPs with polyammonium-covered nanotubes afforded the final AuCNT nanohybrid assembly. Transmission electron microscopy showed nanotubes fully decorated with dense and uniform coating of monodisperse AuNPs (Fig. 2). Size evaluation by statistical diameter measurement indicated a mean particle diameter of ca. 3 nm. XPS analysis (see ESI, Fig. S1†) showed only the presence of metallic gold in the nanohybrid sample. The AuCNT catalyst was suspended in water and used as such in the oxidation of alcohols.
 |
| | Fig. 2 TEM picture of the AuCNT hybrid. The inset is a 250% magnification of the selected area. | |
The gold content in the aqueous suspension of the nanohybrid was measured by ICP-MS which indicated a gold concentration of 4 mM.
The catalytic activity of the nanohybrid in the aerobic oxidation of alcohols was then assessed using benzyl alcohol as a model substrate. Our primary objective was to develop a catalytic system that would be effective under the mildest and less constraining conditions20–27 since analogous supported gold catalyzed transformations usually require drastic conditions such as heating and/or oxygen pressure.28–35 Numerous reaction conditions were tested by varying the nature of the solvent (THF, toluene, water), base (K2CO3, DBU, LiOH, NaOH), and catalyst loading. We found that the optimal conditions in terms of conversion yield and reaction time were obtained using a biphasic toluene–water mixture, 0.2 mol% of AuCNT, and 3 equivalents of NaOH. More importantly, the reaction was run at room temperature and under atmospheric pressure of air (open flask). Under the above conditions, benzyl alcohol (1a) was cleanly converted to benzoic acid (2a) in 99% yield after 3 h (Scheme 1). The turnover number (TON) and turnover frequency (TOF) calculated from this experiment were 495 and 165 h−1, respectively. Although less efficient than gold-based bimetallic species (e.g. Au–Pd36 or Au–Pt37) the values obtained for our AuCNT catalyst compare favorably with most of the other nano-gold systems reported in the literature38 (for a detailed comparison, see Table S1 in the ESI†). If the kinetic values were to be calculated by taking into account only the gold-surface atoms (those in contact with the medium) and not the actual Au loading (total gold content), more flattering TOF and TON values for benzyl alcohol oxidation would be reached (TON = 1648, TOF = 549 h−1).
To highlight the superiority of CNT-supported AuNPs over other gold sources, the oxidation of benzyl alcohol to benzoic acid was also run with the same amount (0.2 mol%) of colloidal AuNPs or gold salts. However, colloidal gold NPs provided only partial conversion (15%) of benzyl alcohol after 3 h, and gold salts did not promote any formation of benzoic acid even after 5 days of reaction (Table 1). An additional control experiment was performed using the nanoassembly devoid of AuNPs (i.e. CNT/DANTA/PDADMAC only) which showed no activity in the oxidation of benzyl alcohol, even after several days. These results have to be compared to the nearly quantitative conversion of benzyl alcohol in 3 h by the AuCNT nanohybrid.
Table 1 Comparison of different gold sources to catalyze the conversion of 1a to 2aa
| Entry |
Catalyst |
Reaction outcome |
|
Conditions: 1a (0.1 mmol), gold catalyst (0.2 mol%), toluene–water 1:1 (1 mL), NaOH (3 equiv.), room temp., open flask (air).
|
| 1 |
AuCNT |
>99% conversion (3 h) |
| 2 |
Colloidal AuNPs |
15% conversion (3 h) |
| 3 |
HAuCl4 |
No reaction (5 days) |
Recyclability and heterogeneous nature of the catalyst
Recyclability of the catalyst was evaluated by performing five consecutive oxidation reactions using the same AuCNT hybrid which could be recovered by simple filtration. No drastic decrease in activity was observed throughout the experiments (Table 2) although higher reaction times (i.e. 5 h) were required for the third and fourth recyclings. The latter point suggests that some leaching of gold nanoparticles from the nanotube surface had taken place. This phenomenon could lead to an overall reduction in gold content and therefore to a “slower” catalyst. TEM analysis of the hybrid after the fourth run confirmed the partial loss of some gold nanoparticles as a few nanotubes with bare areas were detected by electron microscopy.
Table 2 Recycling of AuCNT for the conversion of 1a to 2aa
| Entry |
Cycle |
Time (h) |
Yieldb (%) |
|
Conditions: 1a (0.1 mmol), AuCNT (0.2 mol%), toluene–water 1:1 (1 mL), NaOH (3 equiv.), room temp., open flask (air).
Isolated yields.
|
| 1 |
Fresh |
3 |
99 |
| 2 |
1st recycle |
3 |
99 |
| 3 |
2nd recycle |
3 |
97 |
| 4 |
3rd recycle |
5 |
98 |
| 5 |
4th recycle |
5 |
98 |
To confirm the key role of the AuCNT nanohybrid as the active catalytic species, a standard oxidation reaction of benzyl alcohol was set up using 0.2 mol% of the catalyst. After 1 h, approximately 50% conversion into benzoic acid was reached. At this stage, the supported catalyst was removed by filtration, and the catalyst-free mixture was further stirred overnight at room temperature. However, no additional oxidation of benzyl alcohol was observed, thus confirming the heterogeneous nature of the catalytic process.
Scope and conditions of the AuCNT-catalyzed reaction
Primary alcohols.
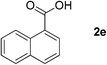
We first investigated the scope of the reaction by screening variously substituted primary alcohols. The reaction was successfully extended to aromatic alcohols bearing electron donating (1b, Table 3, entry 2) or withdrawing substituents (1c and 1d, entries 3 and 4). In both cases the expected oxidized products were produced in good to excellent yields with no significant changes in the kinetics of the oxidation, as the reaction was completed within 3 h. Naphthyl-1-methanol (1e, entry 5), indole-3-carbinol (1f, entry 6), and conjugated cinnamyl alcohol (1g, entry 7) were also efficiently oxidized into their corresponding acids but 4 h were needed for completion of the reaction in the case of the substrate 1f. Although highly efficient on aromatic substrates, the nanohybrid-mediated oxidation was not successful on aliphatic alcohols as no significant conversion of 2-phenyl-ethanol (1h, entry 8) or 1-octanol (1i, entry 9) was observed after 72 h of reaction. It should be noted that carboxylated products were readily purified by a simple acid/base workup.
Secondary alcohols.
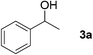
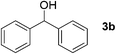
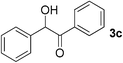
To further extend the scope of the reaction, secondary alcohols were studied under similar conditions. For instance, 1-phenylethanol (3a) was converted to acetophenone (4a) in 80% yield after 24 h of reaction (Table 4, entry 1). Secondary alcohols bearing sterically hindered groups, such as 3b, 3c, and 3d (entries 2, 3 and 4), were also readily oxidized in high yields into the corresponding ketones 4b, 4c, and 4d, respectively. Surprisingly, the reaction of 1-phenylethane-1,2-diol (3e, entry 5) under the same reaction conditions did not afford the expected ketone but led to the oxidative cleavage of the 1,2-diol unit to provide clean access to benzoic acid.‡
Reaction conditions.
Interestingly, no reaction occurred under an inert atmosphere of N2 with either primary or secondary alcohols, suggesting that molecular oxygen was needed as an activator. The same comment applies to the use of a hydroxylated base which is mandatory for the reaction to evolve. Moreover, when water was depleted from the reaction mixture, alcohol oxidation stopped at the intermediate aldehyde stage. For example, in the absence of water, benzaldehyde was produced quantitatively in 12 h starting from benzyl alcohol (Scheme 2). Upon addition of H2O to the reaction mixture, benzaldehyde (5a) got further oxidized into benzoic acid within 2 h and in 99% yield. Our catalytic system thus behaves selectively depending on the reaction conditions.
Oxidation of aldehydes.
As benzaldehyde could be readily converted into the corresponding carboxylic acid, we next turned our attention to the AuCNT-catalyzed oxidation of various aldehydes. When the oxidation was conducted with authentic benzaldehyde (5a), the reaction was completed in just 2 h in nearly quantitative yield (Table 5, entry 1). Interestingly, the nanohybrid-catalyzed oxidation of aliphatic octan-1-al (5i) afforded octanoic acid (2i) in satisfactory yield (entry 2). This result points out that the alcohol to aldehyde transformation is likely the rate determining step as the previous reaction of aliphatic alcohols was found to be lethargic (vide supra). Additionally, dialdehyde 5j (entry 3) and heteroaryl aldehyde 5k (entry 4) were also screened with regard to aerobic oxidation and afforded the corresponding acids in 85% and 78% yield, respectively, after 2 h. Under the above conditions, glucose (5l) was also smoothly oxidized to gluconic acid (2l, entry 5) in excellent yield in just 3 h.
Role played by water and oxidation mechanism
To elucidate the role of water in the oxidative process and assign the origin of the oxygen atom incorporated in the carboxylic group, an AuCNT oxidation reaction of benzyl alcohol was set up by replacing H2O by 18O-labeled water (Scheme 3). Benzoic acid was recovered after 3 h and mass spectra analysis indicated that the carboxylic acid was labeled with one 18O atom (80% isotopic enrichment). To confirm that 18O incorporation did take place during the oxidation process and not by exchange of oxygen atoms between the newly formed acid and H218O, a control experiment was performed. Non-labeled benzoic acid was stirred with H218O under the above conditions. The solution was stirred at room temperature for 3 h after which the product was recovered and analyzed by mass spectrometry which indicated that no oxygen exchange had taken place. This experiment thus designates water as the prominent oxygen source.
 |
| | Scheme 3
18O-labeling experiment to explore the role of water. | |
Based on previous reports,39 alcohol oxidation over CNT-supported AuNPs proceeds by means of an aldehyde intermediate which gets further oxidized into the corresponding acid. Although not fully understood yet, one can suggest a plausible sequential mechanism for this transformation (Scheme 4): (i) the initial step is the deprotonation of the alcohol and the formation of a gold-alcoholate intermediate with concomitant adsorption of oxygen onto the gold surface which produces a transient hydroperoxide. This step is followed by (ii) the sodium hydroxide-mediated dehydrogenation of the activated alcohol into the corresponding aldehyde with the departure of hydrogen peroxide. In the presence of both water and sodium hydroxide, (iii) hydration of the newly formed aldehyde takes place, and finally, (iv) reiteration of steps (i) and (ii) on the hydrate intermediate ultimately delivers the expected carboxylic acid. As demonstrated above, oxygen incorporation originated from water but not from O2. The exact nature of the catalytic sites remains unclear although it has been investigated on related systems by others.40
Conclusion
In conclusion, we have shown that the CNT-supported gold nanohybrid is a promising catalyst for the aerobic oxidation of diversified alcohols under very mild conditions. The reported system is effective without any added O2 and requires no heating. While alcohols and aldehydes were chemoselectively oxidized to the corresponding acids, under anhydrous conditions, primary alcohols could also be converted to the corresponding aldehydes. Finally, the role of water in the oxidation process was elucidated using 18O-labeled water. Our CNT-based system compares favorably to other supported gold nanoparticles in terms of recyclability, catalytic activity, selectivity, and mildness of the operating conditions with regard to the oxidation of alcohols.
Acknowledgements
Support from the Indo-French Centre for the Promotion of Advanced Research (IFCPAR)/Centre Franco-Indien pour la Promotion de la Recherche Avancée (CEFIPRA) is gratefully acknowledged. R. K. thanks the French Embassy in India for the award of a Charpak fellowship. The International Relations Division (DRI) of the CEA is also acknowledged for support. We thank the TEM-team platform of the CEA for transmission electron microscopy pictures. The “Service de Chimie Bioorganique et de Marquage” belongs to the Laboratory of Excellence in Research on Medication and Innovative Therapeutics (LabEx LERMIT).
Notes and references
-
M. B. Smith and J. March, in March's Advanced Organic Chemistry, Wiley-Interscience, New York, 5th edn, 2001, pp. 1514–1518 and p. 1537 Search PubMed.
- For example, see: K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS.
- J. John, E. Gravel, I. N. N. Namboothiri and E. Doris, Nanotechnol. Rev., 2012, 1, 515 Search PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469 CrossRef CAS.
- B. Hvolbæk, T. V. W. Janssens, B. S. Clausen, H. Falsig, C. H. Christensen and J. K. Nørskov, Nano Today, 2007, 2, 14 CrossRef.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC.
- Y. Mikami, A. Dhakshinamoorthy, M. Alvaro and H. García, Catal. Sci. Technol., 2013, 3, 58 CAS.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467 CrossRef CAS.
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350 RSC.
- S. Carrettin, M. Carmen Blanco, A. Corma and A. S. K. Hashmi, Adv. Synth. Catal., 2006, 348, 1283 CrossRef CAS.
- E. Gravel, S. Foillard, H. B. Zhang, H. Li and E. Doris, Sci. China: Chem., 2010, 53, 2015 CrossRef CAS.
- J. John, E. Gravel, A. Hagège, H. Li, T. Gacoin and E. Doris, Angew. Chem., Int. Ed., 2011, 50, 7533 CrossRef CAS.
- D. G. Duff, A. Baiker and P. P. Edwards, Langmuir, 1993, 9, 2301 CrossRef CAS.
- C. Thauvin, S. Rickling, P. Schultz, H. Celia, S. Meunier and C. Mioskowski, Nat. Nanotechnol., 2008, 3, 743 CrossRef CAS.
- C. Richard, F. Balavoine, P. Schultz, T. W. Ebbesen and C. Mioskowski, Science, 2003, 300, 775 CrossRef CAS.
- N. Mackiewicz, G. Surendran, H. Remita, B. Keita, G. Zhang, L. Nadjo, A. Hagège, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2008, 130, 8110 CrossRef CAS.
- D. J. Ahn and J.-M. Kim, Acc. Chem. Res., 2008, 41, 805 CrossRef CAS.
- H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151 CrossRef CAS.
- S. Kim, S. W. Bae, J. S. Lee and J. Park, Tetrahedron, 2009, 65, 1461 CrossRef CAS.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Adv. Synth. Catal., 2009, 351, 1890 CrossRef CAS.
- K. Kaneda, T. Mitsudome, T. Mizugaki and K. Jitsukawa, Molecules, 2010, 15, 8988 CrossRef CAS.
- Y. Yuan, N. Yan and P. J. Dyson, Inorg. Chem., 2011, 50, 11069 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374 CrossRef CAS.
- S. Kanaoka, N. Yagi, Y. Fukuyama, S. Aoshima, H. Tsunoyama, T. Tsukuda and H. Sakurai, J. Am. Chem. Soc., 2007, 129, 12060 CrossRef CAS.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812 CrossRef CAS.
- A. Abad, C. Almela, A. Corma and H. García, Tetrahedron, 2006, 62, 6666 CrossRef CAS.
- A. Abad, A. Corma and H. García, Pure Appl. Chem., 2007, 79, 1847 CrossRef CAS.
- N. Zheng and G. D. Stucky, Chem. Commun., 2007, 3862 RSC.
- J. Yang, Y. Guan, T. Verhoeven, R. Santen, C. Li and E. J. M. Hensen, Green Chem., 2009, 11, 322 RSC.
- W. Fang, Q. Zhang, J. Chen, W. Deng and Y. Wang, Chem. Commun., 2010, 46, 1547 RSC.
- P. Liu, Y. Guan, R. A. Santen, C. Li and E. J. M. Hensen, Chem. Commun., 2011, 47, 11540 RSC.
- N. Asao, N. Hatakeyama, Menggenbateer, T. Minato, E. Ito, M. H. Y. Kim, Y. Yamamoto, M. Chen, W. Zhang and A. Inoue, Chem. Commun., 2012, 48, 4540 RSC.
- M. Alhumaimess, Z. Lin, W. Weng, N. Dimitratos, N. F. Dummer, S. H. Taylor, J. K. Bartley, C. J. Kiely and G. J. Hutchings, ChemSusChem, 2012, 5, 125 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS.
- K. Kaizuka, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2010, 132, 15096 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC.
- For example see: B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74 CrossRef CAS.
- A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and supplementary figures. See DOI: 10.1039/c3nr01432k |
| ‡ This reaction likely involves phenyl glyoxalic acid as the intermediate as treatment of authentic phenyl glyoxalic acid with the AuCNT nanohybrid under classical reaction conditions (biphasic mixture, air, room temp., NaOH) also cleanly provided access to benzoic acid. |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence