 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoparticle-enhanced luminescence of silicon quantum dots co-encapsulated in polymer nanoparticles
Noor Aniza Harunab, Matthew J. Benningc, Benjamin R. Horrocks*a and David A. Fulton*a
aChemical Nanoscience Laboratory, School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: b.r.horrocks@ncl.ac.uk; d.a.fulton@ncl.ac.uk
bDepartment of Chemistry, Universiti Malaysia Terengganu, 21030, Kuala Terengganu, Terengganu, Malaysia
cSchool of Mechanical and Systems Engineering, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
First published on 5th March 2013
Abstract
The preparation of two-component polymer composite nanoparticles encapsulating both Si quantum dots (SiQDs) and Au nanoparticles (AuNPs) by a single step miniemulsion polymerization of divinylbenzene is described. This simple and robust method affords well-defined polymer composite nanoparticles with mean diameters in a range of 100–200 nm and with narrow polydispersity indices as determined by dynamic light scattering and transmission electron microscopy. The successful encapsulation of AuNPs within poly(divinylbenzene) was confirmed by UV-visible spectroscopy and from TEM images. Plasmon-enhanced fluorescence of the luminescence of the SiQDs by AuNPs encapsulated within the polymer composite nanoparticles was evaluated by confocal microspectroscopy, and luminescence enhancements of up to 15 times were observed. These observations indicate that the luminescence of the SiQDs is enhanced by the proximity of the AuNPs. The polymer composite nanoparticles were successfully ink-jet printed onto a glass substrate, which demonstrates that these composites are processable in printing applications.
Introduction
Quantum dots (QDs) are inorganic fluorophores that possess unique photophysical properties which are useful in a diverse range of applications.1–8 The combination of different nanoparticles possessing different functionalities within a composite nanoparticle presents an exciting approach to expand the range of applications of inorganic nanoparticles. Multicomponent composite nanoparticles can lead to improvements and enhancements in the properties of individual inorganic nanoparticles, whilst benefiting from the superior physical and chemical properties of the polymeric materials used in the fabrication of the nanocomposite. In particular the surface derivatization of the polymer is typically much more facile than of the inorganic nanoparticles.We recently showed that silicon quantum dots (SiQDs) can be encapsulated inside polymeric nanoparticles, resulting in a nanocomposite which possesses the luminescence of the SiQDs and the processability of the polymer matrix.9 SiQDs are of interest on account of their size-dependent luminescence characteristics; they exhibit intense room-temperature luminescence from the radiative recombination of quantum-confined excitons.10 The intensity of luminescence is enhanced in SiQDs by the quasi-direct nature of the transitions involved.11 The emission frequencies can be tuned through part of the visible and near-infrared spectrum by varying QDs size12 or surface termination.13 The most significant advantage of SiQDs over other QDs for applications in the life sciences is the absence of heavy metal ion leaching, which results in reduced toxicity.14 However, it is well noted that the overall brightness of SiQDs can be restricted by a low radiative emission rate as a consequence of the indirect band gap of bulk silicon,10,15,16 which limits the possible utility of this class of nanoparticle.
We were intrigued by the possibility of enhancing the fluorescent properties of SiQDs by combining them with metallic nanoparticles within a nano-sized polymer matrix, as metallic nanoparticles of gold, silver and aluminium offer a possible method to enhance the brightness of SiQDs. These noble-metallic nanoparticles possess interesting plasmonic properties which arise because of the collective oscillation of the conduction electrons when they are in resonance with electromagnetic radiation incident on the surface of the particles.17–20 This phenomenon is confined near the boundary between the metal nanostructures and their surrounding dielectric matrix, resulting in a range of optical signal enhancements, such as surface-enhanced Raman scattering (SERS), surface-enhanced fluorescence, and surface-enhanced infrared absorption.21–24 Metal-enhanced fluorescence occurs when metal nanoparticles are located near to a fluorophore and the plasmon mode is resonant with the incident light. Gold nanoparticles (AuNPs) in particular are very attractive candidate nanoparticles to facilitate these fluorescence enhancements because of their high chemical stabilities, high homogeneities, good biocompatibilities25 and low toxicities.1 Furthermore, the surface chemistry of AuNPs is well-understood and numerous chemistries are available to facilitate the surface modification of AuNPs. The incident radiation must also be resonant with the absorption transition of the fluorophore. SiQDs are convenient in this case because they possess a featureless absorption spectrum with an absorbance that rises monotonically above the gap (∼2 eV in our SiQDs) and therefore it is only necessary to match the excitation wavelength to the plasmon mode of the metal.
Previously, there have been a number of reports involving the preparation of polymer nanoparticles encapsulating multiple inorganic NPs.26–28 AuNPs have also been successfully fabricated with other fluorophores to prepare nanocomposites with optically active properties, especially in metal-enhanced fluorescence, surface-enhanced Raman scattering, surface plasmon-coupled emission and plasmon-controlled fluorescence systems.29 Furthermore, there have been several recent impressive reports describing the fabrication of AuNPs with fluorescent dyes30 or CdSe-type quantum dots31,32 in various particular research areas.
When considering synthetic approaches to nanocomposite materials, miniemulsion polymerization has been reported as a convenient and powerful technique to encapsulate a range of different type of materials, including organic and inorganic nanoparticles33–40 inside polymeric nanoparticles.41–48 Encapsulation of inorganic species inside polymer matrices affords composite materials which benefit from the outstanding properties associated with synthetic polymers e.g. their excellent mechanical, chemical, electrical, rheological, magnetic and optical properties.33 The hydrophobic nature of inorganic species can facilitate their direct dispersion inside a suitable monomer system, which is conventionally also hydrophobic, and results in successful encapsulation of the inorganic species upon polymerization of miniemulsion droplets. We anticipated that the intense field near the AuNPs surface can produce substantial fluorescence enhancements of nearby SiQDs.8,16,49–52 To the best of our knowledge, there are no reports of composite nanoparticles prepared by miniemulsion polymerization encapsulating both AuNPs and SiQDs within polymer nanoparticles. Most of the methods reported where QDs are combined with plasmonic materials often involve the immobilization of the QDs upon films or substrates16,53–57 which may limit some of advantages that come from co-encapsulation with a nanoparticle matrix. For example, Swihart et al.53 investigated the energy transfer within a micelle platform where SiQDs were co-encapsulated with an anthracene-based dye in the hydrophobic core of micelles prepared from 1,2-distearoyl-sn-glycero-3-phosphoethanolaminate-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG). This work indicated that the luminescence of SiQDs from the core of a 150 nm phospholipid micelles is enhanced by more than 80% upon addition of anthracene dye. This polymer composite colloidal solution can serve as a template for improving the QDs emission intensity, especially in biological and solar applications.
In our previous work,9 we have successfully synthesized polymer composite nanoparticles composed of styrene and 4-vinylbenzaldehyde monomers which encapsulated SiQDs via miniemulsion polymerization methods. In this work we describe a synthetic route to prepare polymer composite nanoparticles which encapsulate plasmonic metal AuNPs and highly fluorescent SiQDs using miniemulsion polymerization techniques, and demonstrate that the PL of the SiQDs is enhanced as a consequence of their co-encapsulation with AuNPs. This composite nanoparticle is expected to widen the scope of applications for both silicon and noble metal-nano devices.
Experimental section
Materials and general experimental
Water was deionized before use (nominal resistivity 18 MΩ cm, Nanopure™ purification system, Barnstead). All chemicals were purchased from Sigma Aldrich or Alfa Aesar and were used as received without further purification. Divinylbenzene was refrigerated and used without purification. Membranes for purification by dialysis of polymer composite nanoparticles were obtained from Spectrum Laboratories (Spectra/Por® Dialysis Membrane Tubing, molecular weight cut-off: 3500 Da, width: 18 mm and diameter: 11.5 mm). Toluene was dried over sodium and distilled as required.Preparation of silicon quantum dots (SiQDs)
Photoluminescent SiQDs were prepared according to a modification of the procedure described by Lie et al.58 The porous silicon layers were formed by electrochemical etching of a boron-doped p-Si〈100〉 oriented wafer (1–10 Ω cm resistivity, Compart Technology, Peterborough, UK) in an electrolyte comprised of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v solution of 48% aqueous HF and EtOH. The Si wafers were cut into squares approximately 1.2 cm2 using a diamond scribe. A porous silicon layer was obtained at high current density (10 min at 250 mA cm−2) supplied by a programmable power supply (Keithley 2601), and the resulting porous layer luminesced orange when observed under a handheld UV lamp (λ = 365 nm). The dry porous Si chips were then refluxed in 25 mL of dry PhCH3 solution containing 0.4 mL of 1-undecene under an atmosphere of N2 for 3 h. The resulting solution emitted an orange-coloured luminescence under an UV light (λ = 365 nm). This fluorescent solution was filtered to remove undissolved silicon particles and then the solvent and unreacted 1-undecene were removed under reduced pressure. The product SiQDs were obtained as an oily/waxy residue which luminesces under UV light (λ = 365 nm) and was soluble in nonpolar solvents e.g., THF, CH2Cl2 and PhCH3. It was estimated that 100 μg of alkyl Si-QDs were typically produced per Si chip.
:1 v/v solution of 48% aqueous HF and EtOH. The Si wafers were cut into squares approximately 1.2 cm2 using a diamond scribe. A porous silicon layer was obtained at high current density (10 min at 250 mA cm−2) supplied by a programmable power supply (Keithley 2601), and the resulting porous layer luminesced orange when observed under a handheld UV lamp (λ = 365 nm). The dry porous Si chips were then refluxed in 25 mL of dry PhCH3 solution containing 0.4 mL of 1-undecene under an atmosphere of N2 for 3 h. The resulting solution emitted an orange-coloured luminescence under an UV light (λ = 365 nm). This fluorescent solution was filtered to remove undissolved silicon particles and then the solvent and unreacted 1-undecene were removed under reduced pressure. The product SiQDs were obtained as an oily/waxy residue which luminesces under UV light (λ = 365 nm) and was soluble in nonpolar solvents e.g., THF, CH2Cl2 and PhCH3. It was estimated that 100 μg of alkyl Si-QDs were typically produced per Si chip.Preparation of citrate stabilized AuNPs
Aqueous-AuNPs were synthesized using a citrate reduction method described in the literature.59 To boiling distilled water (400 mL) was added HAuCl4·3H2O (0.16 g, 0.41 mM) to afford a yellow solution. A solution of 3.28 mM sodium citrate (34 mL) was added to the gold(III) chloride solution and the colour changed from light yellow to dark purple. The reaction mixture was boiled for 30 min, and the resulting aqueous dispersion of AuNPs possessed a ruby-red colour. The average diameter of the resulting aqueous-AuNPs was measured to be 13–30 nm by TEM and DLS analysis.Synthesis of thiol-terminated PS by RAFT polymerization
Thiol-terminated polystyrene was prepared by RAFT polymerization of styrene followed by aminolysis to convert the dithioester end group into a thiol.38 The RAFT agent (methyl-2-phenyl-2-(phenyl-carbonothioylthio)) (1 eq.), AIBN (0.022 g) and a magnetic stirrer bar were added to a Schlenk flask which was then sealed. The reaction mixture was degassed by three freeze–pump–thaw cycles, and then heated with stirring at 110 °C for 80 h under an atmosphere of N2. The product was precipitated into cold MeOH, filtered and dried under vacuum to afford polystyrene as a pink powder (5.18 g, 74%) (Mn = 13500, Mw = 15300, PDI = 1.13). A solution of polymer (1.06 g) in 20 mL THF was subjected to five freeze–pump–thaw cycles. Hexylamine (3.5 eq.) was then added using a N2-purged syringe, and the reaction mixture was heated at 60 °C for 30 min, and then stirred under an atmosphere of N2 at room temperature for 20 h. The resulting thiol-terminated polystyrene was purified by precipitation in cold MeOH to afford a white powder (0.88 g, 82%). 1H NMR (400 MHz, CDCl3): δ 1.45 (br, CHCH2, polymer backbone), δ 1.88 (br, CHCH2, polymer backbone), δ 3.49 (br, OCH3, of the chain terminus), δ 6.59 (br, Ar, polymer backbone), δ 7.06 (br, Ar, polymer backbone).Preparation of polymer-grafted AuNPs
Thiol-terminated polystyrene (10 mg) was dissolved in inhibitor free THF (100 mL) and mixed with an aqueous solution of AuNPs (100 mL) in a separating funnel, resulting in formation of a dark-purple coloured solution. CHCl3 (50 mL) was added and the organic and aqueous layers separated. The organic layer was collected and evaporated to dryness to afford a dark purple film. This film was then re-dissolved in a small volume of inhibitor free THF and then subjected to centrifugation (7000 rpm for 30 min) to afford the product (PS-AuNPs) as dark purple solid.38General procedure for miniemulsion polymerizations
Composite polymer nanoparticles were prepared following the oil-in-water miniemulsion polymerization method described by Hawker and van Berkel.38 In brief, 2,2′-azobis-(2-amidinopropane) dihydrochloride (V-50) (7.5 mg) and cetyltrimethylammonium bromide (CTAB) (1.3 mg) were dissolved in 2 mL of deionized water. PS-AuNPs (3 mg, 6 mg, 12 mg, 18 mg or 24 mg) were each dispersed in the monomer phase (55 mg of divinylbenzene containing dispersed SiQDs), and the resulting mixture was added into the aqueous solution and emulsified by stirring vigorously (600 rpm) for 30 min. The emulsion mixture was then subjected to sonication over an ice bath for 15 min using a high intensity ultrasonic processor at 30% amplitude (VC750220, Fisher Scientific, tapered microtips, power: 750 W). The reaction vessel was fitted with a condenser and a N2 inlet and the polymerization was performed at 50 °C under an atmosphere of N2, with continuous stirring for 4 h. The resulting polymer nanocomposites were purified by dialysis against deionized water (3–4 days) and the purified polymer composites were stored as dispersions in aqueous solution. It has to be noted that, because of the very low quantities obtained from the SiQDs production, it is not possible to control the exact amount of the SiQDs in each formulation. Therefore, to ensure a constant concentration of SiQDs in each formulation, multiple batches of SiQDs were dispersed in CH2Cl2 (60 mL) and the resulting solution divided to six equal proportions which were subsequently used for each polymer synthesis. For composite polymer nanoparticles encapsulating only SiQDs (P2[Si]), a similar procedure was applied as above, without the addition of PS-AuNPs inside the monomer phase system. The complete protocols are given in Table 1.| Polymer composite | PS-AuNPsb (mg) | Droplet and particles sizes characterizationa | |||
|---|---|---|---|---|---|
| Droplets | Purified latex | ||||
| Dh (nm) | PDI | Dh (nm) | PDI | ||
| a Determined by dynamic light scattering (DLS).b Total mass of PS-AuNPs used in the miniemulsion protocol. All experiments were performed with total monomer content of DVB = 55 mg; CTAB (cationic surfactant) = 1.5 mg; V-50 (initiator) = 7.5 mg; deionized H2O = 2 mL; T = 50 °C; stirring rate = 500 rpm; time = 4 h; sonication condition = 15 min, 30% amplitude, 750 W, tapered microtips. The amount of SiQDs is constant for all of the polymer synthesis. | |||||
| P1[Au] | 12 | 111.3 ± 2.9 | 0.24 ± 0.05 | 110.0 ± 0.8 | 0.29 ± 0.03 |
| P2[Si] | 0 | 110.4 ± 1.1 | 0.23 ± 0.07 | 118.2 ± 2.7 | 0.20 ± 0.02 |
| P3[Au–Si] | 3 | 144.0 ± 1.7 | 0.20 ± 0.08 | 147.4 ± 1.7 | 0.17 ± 0.03 |
| P4[Au–Si] | 6 | 139.1 ± 2.4 | 0.29 ± 0.02 | 146.4 ± 1.8 | 0.22 ± 0.01 |
| P5[Au–Si] | 12 | 151.3 ± 2.8 | 0.23 ± 0.03 | 160.7 ± 1.2 | 0.20 ± 0.03 |
| P6[Au–Si] | 18 | 162.6 ± 0.6 | 0.21 ± 0.04 | 158.7 ± 2.2 | 0.20 ± 0.02 |
| P7[Au–Si] | 24 | 166.2 ± 3.6 | 0.19 ± 0.01 | 161.5 ± 1.9 | 0.13 ± 0.02 |
Preparation of Au–Si polymer composite nanoparticle – containing ink for ink-jet printing
A colloidal suspension of P6[Au–Si] was subjected to vacuum centrifugation to afford 2 mg of solid powder. Printing inks were prepared by adding 1 mL of cyclohexanone to a powder of P6[Au–Si] (2 mg) and the resulting suspension then sonicated in an ultrasonic bath for 30 min.Ink-jet printing of Au–Si polymer composite nanoparticles microdots
The prepared ink (P6[Au–Si]) was loaded into a cartridge (Dimatrix Material Catridges) fitted with a 10 picoliter piezo print head (Fujifilm Dimatix Inc., DMC-11610 series, USA). Arrays were obtained by printing the ink using a materials printer (Fujifilm Dimatix Inc., DMP-2831 series, USA). 100 × 100 droplet arrays were printed onto untreated glass slides with the diameter of each droplet in a range of 25 μm and at spacing of 15 μm. The cartridges were viewed ejecting droplets under a stroboscope at 1 kHz and 5 kHz ejection frequencies using the DMP-2831 on-board drop watcher facility. Manual droplet optimisation was performed using a single orifice resulting in the following print settings: print orifice = 8, firing frequency = 1 kHz, cartridge temperature = 30 °C, meniscus set point = 4 inch of water, piezo voltage = 18.5 V, wave form = multi step trapezoidal with a pulse width of 12.101 μs, drop velocity = 6 ms−1, print head substrate clearance = 1000 μm. Prior to printing, the print heads were cleaned using the standard “spit-purge-blot” procedure and several dot arrays were then printed in order to increase the likelihood of generating a regular deposition of droplets for post-process viewing.Characterization of polymer nanoparticles
Results and discussion
Polymer “grafting to” method
An important requirement in the preparation of polymer composite nanoparticles by miniemulsion polymerization methods is that the inorganic nanoparticles disperse well in the hydrophobic monomer system. Only by meeting this requirement can the hydrophobic inorganic species be encapsulated within the polymer nanoparticle upon polymerization of the miniemulsion droplets.60 Alkylated SiQDs prepared in our laboratory can be readily dispersed in hydrophobic monomer systems on account of the solubilising 11-carbon-thick alkyl monolayers surrounding their Si cores. In order to disperse AuNPs in hydrophobic organic monomers the surface of AuNPs must be modified to improve hydrophobicity. There are numerous published procedures61–69 describing the synthesis of hydrophobic AuNPs, most of which typically use long chain alkyl amines or thiols to alter the size, shapes and colloidal stability of the resulting AuNPs. These small-molecule ligands, however, have been demonstrated to be ineffective in promoting encapsulation into polymer matrices,38,70 and also oxidize easily at elevated temperatures.36An alternative route towards increasing the hydrophobicity of AuNPs is by the covalent “grafting to” approach,71,72 where hydrophobic polymers featuring sulphur-containing functional groups such as dithioester, trithioester, thiol, thioether or disulphide groups either at their termini or in the middle of the polymer chains, are grafted onto the surface of AuNPs to enable their dispersion in hydrophobic solvents and monomers. The grafting of polymer chains onto the surface of the AuNPs can discourage the aggregation of AuNPs,73,74 and it has also been suggested that the use of polymeric stabilizers for metal nanoparticles can enhance long term stabilities as well as promoting their compatibility and processability.71 In this work, the “grafting to” approach was utilized successfully to conjugate polystyrene thiol chains onto the surface of AuNPs to afford hydrophobic AuNPs which can be easily re-dispersed in organic solvents, including the desired monomer system required for miniemulsion polymerization.
Thiol-terminated polystyrene was synthesized (Scheme 1a) via reversible addition fragmentation chain transfer (RAFT) polymerization,75–78 a convenient method for the synthesis of a wide range of polymers of controlled architecture, molecular weight and narrow polydispersity indices. The dithioester end group of the RAFT polymer was smoothly converted to the thiol through an aminolysis process and the resulting end-functionalized polymers were then grafted onto the surface of AuNPs to afford hydrophobic PS-thiol AuNPs (PS-AuNPs).38
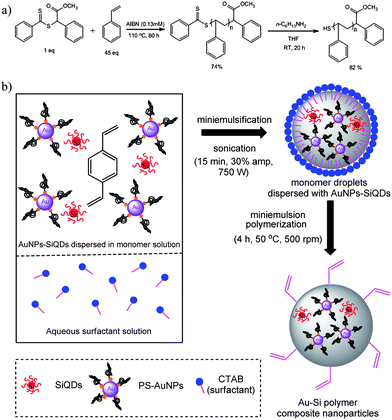 | ||
| Scheme 1 (a) Synthesis of thiol-terminated polystyrene (b) miniemulsion polymerization of Au–Si polymer composite nanoparticles. | ||
Preparation of Au–Si polymer composite nanoparticles via miniemulsion polymerization
Miniemulsion polymerization is a versatile method for polymer nanoparticle formation because of its robustness and synthetic ease allowing good control of particle size and particle size distribution. To prepare polymer composite nanoparticles co-encapsulating both SiQDs and AuNPs, the miniemulsion polymerization procedures described by Hawker et al. were employed38 (Scheme 1b). AuNPs grafted with PS-thiol ligands (PS-AuNPs) and SiQDs were dispersed in divinylbenzene monomer, and subjected to miniemulsion polymerization. Divinylbenzene was chosen as a monomer as it has been demonstrated to successfully encapsulate PS-AuNPs to afford polymer–AuNPs composite particles.26,38 It is worth noting that although conventional miniemulsion polymerization utilizes a co-stabilizer (e.g., hexadecane, cetyl alcohol, and polymeric co-stabilizer) in order to stabilize the emulsion droplets against coalescence and Ostwald ripening (diffusional degradation),79 no co-stabilizer was required here because hydrophobic PS-AuNPs appear themselves to sufficiently accomplish the function of co-stabilizing agent.38,80 A major concern arising in miniemulsion polymerizations are the issues of low levels of encapsulation and uneven distribution of inorganic particles throughout the polymer matrix. By scaling-down the total volume at which the miniemulsion is performed, ensuring a relatively higher ratio of inorganic nanoparticles to monomer, it was possible to achieve good distribution of inorganic species within the polymer nanoparticles (see section on transmission electron microscopy characterization on the following page).To explore the effects of different encapsulated ratios of AuNPs:SiQDs upon the fluorescence properties of the polymer composite nanoparticles, six batches of composite particles were prepared (Table 1) which contained different masses of PS-AuNPs but with constant masses of SiQDs. Because of the low masses of SiQDs obtained in our electrochemical etching method, it is not possible to easily determine the precise mass of SiQDs contained within each polymerization reaction, however, by dividing a SiQDs solution into six equal portions, it was possible to ensure a constant mass of SiQDs within each polymerization. Because we did not measure the molar mass of the AuNPs, we simply report the quantity of Au as the total number of mg of PS-AuNPs used in each preparation.
Particle size characterization
The particle sizes and size distributions (PDI) of the monomer droplets and their corresponding resultant polymer composite nanoparticles were measured by dynamic light scattering (DLS) (Table 1 and Fig. 1). The measurements involving monomer droplets were performed immediately after the reaction mixtures were emulsified by sonication, whilst for the polymer composite nanoparticles, the measurements were performed after the polymerizations were completed and purified by dialysis. The results obtained show that the size of monomer droplets was essentially identical to the size of the resulting polymer nanoparticles and suggests that the distribution of Au–Si in the polymer nanoparticles reflects their homogeneous distribution in the monomer solution.9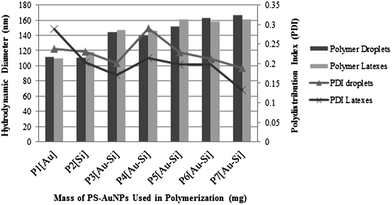 | ||
| Fig. 1 Hydrodynamic diameters (Dh) and PDI of Au–Si polymer composite nanoparticles containing different masses of PS-AuNPs. | ||
To gauge the possible effects of the inorganic nanoparticles upon the outcomes of the miniemulsion polymerizations, control samples were prepared which encapsulated only PS-AuNPs (P1[Au]) or SiQDs (P2[Si]). These controls indicated both the monomer droplets and polymer nanoparticles possessed mean diameters of 110–120 nm, and relatively narrow PDIs of 0.20–0.29, indicating no significant differences between these control samples in terms of particle size and particle size distributions. It should be noted that, the particles size of citrate stabilized AuNPs and PS-AuNP are 13.9 ± 0.7 nm and 13.4 ± 0.7 nm respectively as measured by DLS, while the size of alkyl capped SiQDs is approximately 5.0 nm.81 The average hydrodynamic diameters for the series of Au–Si polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]) (Fig. 1) displayed an upward trend as the quantity of PS-AuNP increased, suggesting that increasing the mass of AuNPs present in the miniemulsion polymerization did have a small effect upon the resultant particle sizes of the polymer composites.
The PDIs for both the monomer droplets and corresponding Au–Si polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]) showed fluctuating trends. The PDIs of the monomer droplets are generally higher (0.19–0.29) than the PDIs of the corresponding polymer composite nanoparticles (0.13–0.29). The low PDIs values of Au–Si polymer composites nanoparticles (P3[Au–Si]–P7[Au–Si]) may be attributed to the traces of 1-undecene contaminants which are leftover from the synthesis of SiQDs, and which might help to improve the PDIs and the stability of the polymer composite nanoparticles.9 In general, the PDIs values observed for the Au–Si polymer composite nanoparticles are slightly broad (P3[Au–Si]–P7[Au–Si]), in the range of 0.13–0.29. However, it has been noted previously that the incorporation of inorganic materials can result in inhomogeneous dispersions on account of their strong interparticle interactions which promotes agglomeration and hence broadening the size distribution.36 Moreover, the requirement to scale down the total monomer content and volume of water may also contribute towards the increase of the PDIs.
UV-visible spectroscopy characterization
UV-visible spectroscopy (Fig. 2) was used to compare the plasmon resonance bands of colloidal AuNPs (aq-AuNPs), PS-AuNPs and the Au–Si polymer composite nanoparticles (P1[Au] and P3[Au–Si]–P7[Au–Si]). The absorbance of the PS-AuNPs (530 nm) is red-shifted relative to the citrated-stabilized AuNPs (525 nm), whilst the Au–Si polymer composite nanoparticles (P1[Au] and P3[Au–Si]–P7[Au–Si]) are further red-shifted to approximately 540 nm. The likely reason for this red-shifting is because the surface plasmon resonance band of AuNPs is sensitive to the changes in the dielectric environment of the surrounding medium.32 This red-shifting phenomenon is therefore expected; the surface plasmon resonance energy of the Au decreases as the refractive index of the medium surrounding the metal nanoparticles increases.82 In contrast for P7[Au–Si], a very broad surface plasmon resonance spectrum was observed, which is likely a consequence of the self-aggregation of PS-AuNPs during the polymerization process, suggesting incomplete encapsulation of AuNPs within the polymer matrix as confirmed by the TEM characterization (as discussed in the following section).![Comparison of UV-vis spectra of aqueous-AuNPs, PS-AuNPs (in THF) and P1[Au] and P3[Au–Si]–P7[Au–Si].](/image/article/2013/NR/c3nr00421j/c3nr00421j-f2.gif) | ||
| Fig. 2 Comparison of UV-vis spectra of aqueous-AuNPs, PS-AuNPs (in THF) and P1[Au] and P3[Au–Si]–P7[Au–Si]. | ||
Transmission electron microscopy characterization
Transmission electron microscopy (TEM) micrographs (Fig. 3) were obtained of Au–Si polymer composite nanoparticles (P3[Au–Si], P5[Au–Si], and P7[Au–Si]), and compared to those of citrate-stabilized AuNPs (aq-Au) and PS-AuNPs. Fig. 3a shows typical TEM images of PS-AuNPs cast onto a carbon coated TEM grid, showing uniform distribution of nanoparticles upon the TEM grid. The polystyrene grafts upon the AuNPs cannot be observed under TEM on account of their low contrast between carbon-coated grid and the polymer chains, and more importantly, the large contrast compared to the AuNPs.75 In contrast however, the citrate-stabilized AuNPs (Fig. 3a (inset)) are clearly visible and tend to aggregate during sample preparation after evaporation of the aqueous solvent. The even distribution of AuNPs within the PS-AuNPs sample may be attributable to the polymer grafts upon the surface of AuNPs, which prevents aggregation between PS-AuNPs.75 The particle sizes of citrate stabilized AuNPs and PS-AuNPs are of 13–30 nm as measured by TEM, and showed reasonable agreement with the hydrodynamic diameter of the nanoparticles obtained by DLS.†![Transmission electron (TEM) images of (a) PS-thiol grafted AuNPs in THF (130 000× magnification). Inset shows TEM images of citrate-stabilized AuNPs in water (13 000× magnification) (b) P3[Au–Si] (64 000× magnification) (c) P5[Au–Si] (64 000× magnification) (d) P7[Au–Si] (64 000× magnification). Because of the low scattering factor of Si, TEM cannot confirm the presence or absence of SiQDs within the polymer matrix.](/image/article/2013/NR/c3nr00421j/c3nr00421j-f3.gif) | ||
| Fig. 3 Transmission electron (TEM) images of (a) PS-thiol grafted AuNPs in THF (130000× magnification). Inset shows TEM images of citrate-stabilized AuNPs in water (13000× magnification) (b) P3[Au–Si] (64000× magnification) (c) P5[Au–Si] (64000× magnification) (d) P7[Au–Si] (64000× magnification). Because of the low scattering factor of Si, TEM cannot confirm the presence or absence of SiQDs within the polymer matrix. | ||
The TEM images of P3[Au–Si], P5[Au–Si], and P7[Au–Si] are depicted in Fig. 3b–d. It can clearly be seen that the Au–Si polymer composite nanoparticles possess a spherical shape with diameters approximately 100–200 nm. TEM measurements (Fig. 3) clearly show the encapsulation of AuNPs, however, because of the low scattering factor of Si, TEM cannot confirm the presence or absence of SiQDs within the polymer matrix. Sample P3[Au–Si] showed only a relatively small amount of encapsulated AuNPs with several empty polymeric nanoparticles observed (Fig. 3b). In contrast, the sample P5[Au–Si] (Fig. 3c), which is prepared with an increased mass ratios of PS-AuNPs to SiQDs, significant numbers of PS-AuNPs were incorporated into each polymer composite nanoparticle and no empty polymer particles were observed. P7[Au–Si] displays unusual morphology, as although it appears that some of the PS-AuNPs encapsulated successfully inside polymer matrix, most of the PS-AuNPs was observed outside the polymer shells in the form of PS-AuNPs clusters (Fig. 3d). This poor encapsulation of PS-AuNPs probably arises when the maximum possible mass of AuNPs was encapsulated within polymer nanoparticles, resulting in excess PS-AuNPs aggregating with themselves to form unencapsulated clusters. This observation correlated with UV-vis spectroscopy, which displayed a broad surface plasmon resonance (SPR) spectrum for this batch of polymer composite nanoparticles (Fig. 2). These results suggest an ‘upper-limit’ of encapsulation of PS-AuNPs within the polymer nanoparticles, and that when the amount of PS-AuNPs further increases beyond this limit, poor encapsulation is obtained in the miniemulsion polymerization.
Evaluation of fluorescence enhancement behaviour of Au–Si polymer composite nanoparticles by confocal microspectroscopy
Confocal microspectroscopy studies were performed to evaluate the enhancements in the luminescence spectra of Au–Si polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]) relative to the control samples in which either SiQDs (P1[Au]) or AuNPs (P2[Si]) are absent. The luminescence enhancement was investigated with the polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]), which possess a constant amount of SiQDs but differing amounts of PS-AuNPs. Each sample was scanned by an Ar ion laser at excitation wavelength of 488 nm, and the emitted/scattered light was dispersed on a grating of 150 lines per mm to exhibit both luminescence and Raman spectra. Fig. 4 shows typical dark field images (reflected images) and confocal luminescence images of Au–Si polymer composite nanoparticles (P3[Au–Si], P5[Au–Si] and P7[Au–Si]) drop-casted onto a glass cover slip. The bright regions observed in the confocal luminescence images belong to those polymer composite nanoparticles containing SiQDs, observations which are consistent with our previous work9 and suggest the successful encapsulation of SiQDs into the polymer matrix. In contrast, no luminescence was observed from the sample of polymer composite prepared in the absence of SiQDs (P1[Au]) (data not shown).![Reflected light image of (a) P3[Au–Si] (b) P5[Au–Si] and (c) P7[Au–Si] and confocal luminescence spectrum images of (d) P3[Au–Si] (e) P5[Au–Si] and (f) P7[Au–Si] polymer composite nanoparticles respectively. The polymer composite nanoparticles samples were purified by dialysis and drop cast on a glass cover slip. Scale bar on the reflected image = 10 μm and scan size of luminescence images = 50 × 50 μm.](/image/article/2013/NR/c3nr00421j/c3nr00421j-f4.gif) | ||
| Fig. 4 Reflected light image of (a) P3[Au–Si] (b) P5[Au–Si] and (c) P7[Au–Si] and confocal luminescence spectrum images of (d) P3[Au–Si] (e) P5[Au–Si] and (f) P7[Au–Si] polymer composite nanoparticles respectively. The polymer composite nanoparticles samples were purified by dialysis and drop cast on a glass cover slip. Scale bar on the reflected image = 10 μm and scan size of luminescence images = 50 × 50 μm. | ||
Fig. 5a shows the average luminescence/Raman spectra obtained for polymer composite nanoparticles P3[Au–Si]–P7[Au–Si] and the control specimen P2[Si]. These average spectra were obtained from the selection of bright regions collected from confocal images which were combined together to attain complete average luminescence/Raman spectra for the polymer composite nanoparticles. The large and broad signals present within the spectra correspond to the luminescence signals of SiQDs, which are incorporated inside or on the surface of the polymer nanocomposites. The Raman bands of PDVB appear around 3070–3000 cm−1 (aromatic C–H stretching) whilst the signal at approximately 2833–2886 cm−1 was attributed to aliphatic C–H stretching modes. Weak Raman signals were observed for the polymer composite nanoparticles P2[Si] and P7[Au–Si], where both of the polymer composite nanoparticles samples displayed small luminescence enhancements. However, the Raman bands of PDVB cannot be clearly identified from the average luminescence/Raman spectra in other samples because of the large broad luminescence peak which obscures the weak Raman signals.
![(a) Average luminescence/Raman spectra of Au–Si polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]) and control polymer composite nanoparticles (P2[Si]). (b) Total intensity and reflected light intensity of Au–Si polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]) and control polymer composite nanoparticles (P2[Si]). Total intensity was calculated as area underneath of the fluorescence peaks obtained from the averages over the particles identified in confocal images. The higher intensity peak at 0 cm−1 is the elastically scattered laser light. The excitation wavelength, λ = 488 nm.](/image/article/2013/NR/c3nr00421j/c3nr00421j-f5.gif) | ||
| Fig. 5 (a) Average luminescence/Raman spectra of Au–Si polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]) and control polymer composite nanoparticles (P2[Si]). (b) Total intensity and reflected light intensity of Au–Si polymer composite nanoparticles (P3[Au–Si]–P7[Au–Si]) and control polymer composite nanoparticles (P2[Si]). Total intensity was calculated as area underneath of the fluorescence peaks obtained from the averages over the particles identified in confocal images. The higher intensity peak at 0 cm−1 is the elastically scattered laser light. The excitation wavelength, λ = 488 nm. | ||
The luminescence/Raman spectra of P3[Au–Si]–P7[Au–Si] displayed significant improvements in the luminescence of the encapsulated SiQDs as the relative mass of encapsulated AuNPs increased (Fig. 5a). The fluorescence intensity of Au–Si polymer composite nanoparticles were enhanced about 7 and 9 times when 3 mg and 6 mg of AuNPs were used in the miniemulsion polymerization (P3[Au–Si] and P4[Au–Si]), respectively, relative to polymer composite nanoparticles which encapsulated only SiQDs. For Au–Si polymer composite nanoparticles possessing 12 mg and 18 mg of PS-AuNPs (P5[Au–Si] and P6[Au–Si]), a 15-fold increase in the fluorescence intensity was observed as compared to P2[Si]. These observations indicate that the co-incorporation of PS-AuNPs gives rise to a significant luminescence enhancement of the encapsulated SiQDs.
It is somewhat surprising that the Au–Si polymer composite nanoparticles sample P7[Au–Si], which was prepared with the largest mass of PS-AuNPs, displays a significant drop in fluorescence enhancement. A possible explanation for this observation may be that when the concentration of PS-AuNPs used in the miniemulsion polymerization is increased beyond a certain limit, the metal nanoparticles tend to self-aggregate and are consequently poorly encapsulated within the polymer matrix. This interpretation is consistent with the results of the UV-vis spectroscopy and the TEM study, where a broad plasmon resonance band was observed in sample P7[Au–Si], and further observations for the agglomeration of the AuNPs (Fig. 3d). Our observation is in agreement with the work reported by Chandra et al.31 on the luminescence enhancement of CdSe/ZnS QDs by AuNPs, showing that the degree of fluorescence enhancement depends upon the relative amount of AuNPs encapsulated. Importantly, in their work the emission intensity is also initially enhanced when the concentrations of AuNPs increased, however, further increases in the quantity of encapsulated AuNPs causes a reduction in emission fluorescence intensity.
The total luminescence intensity and the intensity of elastically scattered light for co-encapsulated AuNPs and SiQDs are shown in Fig. 5b. These intensities are effectively normalised to the number of particles by averaging the spectra of all the particles in the field of view and therefore correspond to the intrinsic properties of an average particle and are not simply determined by the number of particles in the field of view. From Fig. 5, it was noted that as the ratio of PS-AuNPs:SiQDs increased, the enhancement of total luminescence intensity increased until it reached a maximum (P5[Au–Si] and P6[Au–Si]). The optimum formulation in this study was obtained using 12 mg of PS-AuNPs (P5[Au–Si]), where it gives the greatest total intensity (2200000 counts per cm). With further increases in the mass of PS-AuNPs used in the formulation, a massive drop of total intensity was then observed (P7[Au–Si]). There are several possible explanations for this. In the 24 mg PS-AuNPs sample (P7[Au–Si]), we estimate a mole ratio of 3:1 PS-AuNP:SiQD from the particle sizes and masses used and therefore it is possible that the AuNPs actually reflect light at the polymer particle surface and that a kind of inner filter effect occurs, reducing the emission from each polymer particle. We tested this by plotting the intensity of elastically scattered light, but find that no increase in reflectivity occurs. Equally, the observation that the reflected light intensity does not decrease markedly also suggests that the low emission from P7[Au–Si] is not due to absorption of light by the AuNPs. In view of our observations of aggregation of the PS-AuNPs by TEM and absorption spectroscopy, it seems that the loss of intensity is related to this aggregation. If we use the size data to estimate the mole ratio of PS-AuNPs:SiQDs and the number of each inside every polymer particle we obtain about 40 SiQDs per polymer particle and about 120 AuNPs per polymer for P7[Au–Si]. The high number of PS-AuNPs seems to be too much for the polymer to encapsulate. We therefore suggest that the breakdown of encapsulation by overloading the polymer particles and the aggregation of the AuNPs leads to poor coupling of the excitation light at 488 nm to the red-shifted plasmon of the aggregate. Interestingly, there is also a significant blue-shift in the PL maximum of the samples which show enhancement, but this is absent in the P7[Au–Si] and the P2[Si] sample without Au. This is consistent with the explanation based on loss of the enhancement mechanism.
Inkjet printing application
To demonstrate the processability, and hence increased utility of the polymer composite nanoparticles, we inkjet printed a sample onto a glass substrate. A sample for printing was obtained from P6[Au–Si] suspended in cyclohexanone. A regular pattern of small spots (diameter between 20 and 25 μm) was printed on the glass slide and the dark field scattering and luminescence images were monitored by using luminescence microscopy (epifluorescence).Fig. 6a shows bright field optical image of a square lattice of small spots of P6[Au–Si] which has been printed uniformly upon the glass slide. The luminescence image (Fig. 6b) shows the existence of small right spots possessing a very similar regular pattern. The spots are annular rather than disc. The results of these preliminary ink-jet printing studies suggest that these composite polymer nanoparticles can be printed onto surfaces with a reasonable degree of fidelity, and hence are suitable of use as components within more complex materials fabricated by ink-jet printing techniques.
![Inkjet printed images of P6[Au–Si] of (a) bright field optical image and (b) the corresponding luminescence image at exposure time of 5000 ms measured in an epifluorescence microscopy (excitation provided by an Hg Lamp/bandpass filter 300–400 nm and the emission was filtered by a 420 nm longpass filter); scale bar = 100 μm.](/image/article/2013/NR/c3nr00421j/c3nr00421j-f6.gif) | ||
| Fig. 6 Inkjet printed images of P6[Au–Si] of (a) bright field optical image and (b) the corresponding luminescence image at exposure time of 5000 ms measured in an epifluorescence microscopy (excitation provided by an Hg Lamp/bandpass filter 300–400 nm and the emission was filtered by a 420 nm longpass filter); scale bar = 100 μm. | ||
Conclusions
Multicomponent polymer composite nanoparticles encapsulating two different inorganic nanoparticles, namely SiQDs and AuNPs, have been prepared using miniemulsion polymerization techniques. Co-encapsulation of AuNPs and SiQDs was found to enhance the luminescence of the SiQDs, which is observed because the excitation light is coupled to the plasmon band of the AuNPs and in turn the intense field at the AuNPs surface couples strongly to the SiQDs. Enhancement factors up to 15 times were observed. Our study also suggests that there exists an optimum loading of AuNPs within the polymer nanoparticles, beyond which the encapsulation fails and aggregation of Au-NPs occurred. To further demonstrate the utility of the polymer composite nanoparticles, we demonstrated their ink-jet printing onto a glass substrate. This work provides a synthetic route to prepare highly fluorescence SiQD probes through the generation of multimodal polymer composite nanoparticles.Acknowledgements
We thank the Ministry of Higher Education, Malaysia and Universiti Malaysia Terengganu, Malaysia for generous support and providing graduate funding. We also would like to acknowledge Prof Robin Harris and the Electron Microscopy Research Service, Newcastle University for the helps in TEM. The regional development agency One North East is also thanked for support.Notes and references
- T. H. Chen, K. W. Kuo, W. T. Kuo, H. Y. Huang and Y. Y. Huang, J. Bionanosci., 2008, 2, 109 CrossRef.
- A. Fu, W. Gu, C. Larabell and A. P. Ali visatos, Curr. Opin. Neurobiol., 2005, 15, 568 CrossRef CAS.
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Nat. Biotechnol., 2003, 21, 47 CrossRef CAS.
- R. Bakalo va, Z. Zhelev, H. Ohba and Y. Baba, J. Am. Chem. Soc., 2005, 127, 11328 CrossRef CAS.
- X. Gao, Y. Cui, R. M. Le venson, L. W. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969 CrossRef CAS.
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. De Grand, J. Lee, A. Nakayama, J. A. P arker, T. Mihaljevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93 CrossRef CAS.
- R. Sarkar, S. S. Narayanan, L. O. Palsson, F. Dias, A. Monkman and S. K. Pal, J. Phys. Chem. B, 2007, 111, 12294 CrossRef CAS.
- Y. Fu, J. Zhang and J. R. Lakowicz, Chem. Commun., 2009, 313–315 RSC.
- N. A. Harun, B. R. Horrocks and D. A. Fulton, Nanoscale, 2011, 3, 4733 RSC.
- J. S. Biteen, N. S. Lewis, H. A. Atwater, H. Mertens and A. Polman, Appl. Phys. Lett., 2006, 88, 131109 CrossRef.
- K. Zidek, I. Pelant, F. Trojanek, P. Maly, P. Gilliot, B. Honerlage, J. Oberle, L. Siller, R. Little and B. R. Horrocks, Phys. Lett. B, 2011, 84, 085321 Search PubMed.
- T. Fischer, V. Petrova-Koch, K. Shcheglov, M. S. Brandt and F. Koch, Thin Solid Films, 1996, 276, 100 CrossRef CAS.
- M. V. Wolkin, J. Jorne, P. M. Fauchet, G. Allan and C. Delerue, Phys. Rev. Lett., 1999, 82, 197 CrossRef CAS.
- N. H. Alsharif, C. E. M. Berger, S. S. Varanasi, Y. Chao, B. R. Horrocks and H. K. Datta, Small, 2009, 5(2), 221–228 CrossRef CAS.
- J. S. Biteen, D. Pacifici, N. S. Lewis and H. A. Atwater, Nano Lett., 2005, 5(9), 1768 CrossRef CAS.
- J. S. Biteen, L. A. Sweatlock, H. Mertens, N. S. Lewis, A. Polman and H. A. Atwater, J. Phys. Chem. C, 2007, 111, 13372–13377 CAS.
- L. M. Liz-Marzán, Mater. Today, 2004, 7(2), 26–31 CrossRef.
- P. Mulvaney, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- L. M. Liz-Marzán, Langmuir, 2006, 22, 32–41 CrossRef.
- M. A. Martins, S. Fateixa, A. V. Girão, S. S. Pereira and T. Trindada, Langmuir, 2010, 26(13), 11407–11412 CrossRef CAS.
- A. M. Schwartzberg and J. Z. Zhang, J. Phys. Chem. C, 2008, 112, 10323–10337 CAS.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824–830 CrossRef CAS.
- F. Le, D. W. Brandl, Y. A. Urzhumov, H. Wang, J. Kundu, N. J. Halas, J. Aizpurua and P. Nordlander, ACS Nano, 2008, 2(4), 707–718 CrossRef CAS.
- W. A. Murray and W. L. Barnes, Adv. Mater., 2007, 19(22), 3771–3782 CrossRef CAS.
- S. Lee, S. Kim, J. Choo, S. Y. Shin, Y. H. Lee, H. Y. Choi, S. Ha, K. Kang and C. H. Oh, Anal. Chem., 2007, 79, 916 CrossRef CAS.
- K. Y. Van Berkel, A. M. Piekarski, P. H. Kierstead, E. D. Pressly, P. C. Ray and C. J. Hawker, Macromolecules, 2009, 42(5), 1425–1427 CrossRef CAS.
- B. S. Kim and T. A. Taton, Langmuir, 2007, 23(4), 2198–2202 CrossRef CAS.
- T. D. Schladt, M. I. Shukoor, K. Schneider, M. N. Tahir, F. Natalio, I. Ament, J. Becker, F. D. Jochum, S. Weber, O. Köhler, P. Theato, L. M. Schreiber, C. Sönnichsen, H. C. Schröder, W. E. G. Müller and W. Tremel, Angew. Chem., Int. Ed., 2010, 49(23), 3976–3980 CrossRef CAS.
- K. Ray, M. H. Chowdhury, J. Zhang, Y. Fu, H. Szmacinski, K. Nowaczyk and J. R. Lakowicz, Adv. Biochem. Eng./Biotechnol., 2009, 116, 29–72 CAS.
- S. Roy, C. K. Dixit, R. Woolley, R. O'Kennedy and C. McDonagh, Nanotechnology, 2012, 23, 325603 CrossRef.
- S. Chandra, J. Doran, S. J. McCormack, M. Kennedy and A. J. Chatten, Sol. Energy Mater. Sol. Cells, 2012, 98, 385–390 CrossRef CAS.
- L. Lu, D. Chen, F. Sun, X. Ren, Z. Han and G. Guo, Chem. Phys. Lett., 2010, 492, 71–76 CrossRef CAS.
- J. Ramos and J. Forcada, Langmuir, 2011, 27, 7222 CrossRef CAS.
- K. Landfester and C. K. Weiss, Adv. Polym. Sci., 2010, 229, 1 CrossRef CAS.
- N. Bernardy, A. P. Romio, E. I. Barcelos, C. Dal Pizzol, C. L. Dora, E. Lemos-Senna, P. H. H. Araujo and C. J. Sayer, Biomed. Nanotechnol., 2010, 6, 181 CrossRef CAS.
- A. V. Fuchs and G. D. Will, Polymer, 2010, 51, 2119 CrossRef CAS.
- K. Landfester, A. Musyanovych and V. J. Maillander, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 493 CrossRef CAS.
- K. Y. van Berkel and C. J. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1594 CrossRef CAS.
- Y. Xu, H. Xu and H. J. Gu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2284 CrossRef CAS.
- R. Chen, F. Chu, L. C. Gauthier, I. Chaduc, E. Bourgeat-Lami and M. J. Lansalot, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2329 CrossRef CAS.
- B. Erdem, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4419–4430 CrossRef CAS.
- N. Joumaa, M. Lansalot, A. Theretz, A. Elaissari, A. Sukhanova and M. Artemyev, et al., Langmuir, 2006, 22(4), 1810–1816 CrossRef CAS.
- Y. Mori and H. Kawaguchi, Colloids Surf., B, 2007, 56, 246–254 CrossRef CAS.
- J. K. Oh, D. J. Siegwart and K. Matyjaszewski, Biomacromolecules, 2007, 8, 3326–3331 CrossRef CAS.
- U. Paiphansiri, P. Tangboriboonrat and K. Landfester, Macromol. Symp., 2007, 251, 54–62 CrossRef CAS.
- M. Takasu, T. Shiroya, K. Takeshita, M. Sakamoto and H. Kawaguchi, Colloid Polym. Sci., 2004, 282, 740–746 CAS.
- F. Tronc, M. A. Winnik, B. L. Kaul and J. C. Graciet, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1999–2009 CrossRef CAS.
- C. Tu, Y. Yang and M. Gao, Nanotechnology, 2008, 19 CrossRef CAS , 105601/1.
- S. D. Choudhury, R. Badugu, K. Ray and J. R. Lakowicz, J. Phys. Chem. C, 2012, 116, 5042 Search PubMed.
- N. Sui, V. Monnier, Y. Zakharko, Y. Chevolot, S. Alekseev, J. M. Bluet, V. Lysenko and E. Souteyrand, J. Nanopart. Res., 2012, 14, 1004 CrossRef.
- J. R. Lakowicz, Anal. Biochem., 2001, 298, 1–24 CrossRef CAS.
- K. Ray, R. Badugu and J. R. Lakowicz, J. Am. Chem. Soc., 2006, 128(28), 8998–8999 CrossRef CAS.
- F. Erogbogbo, C. W. Chang, J. May, P. N. Prasad and M. T. Swihart, Nanoscale, 2012, 4(16), 5163 RSC.
- J. H. Song, T. Atay, S. Shi, H. Urabe and A. V. Nurmikko, Nano Lett., 2005, 5, 1557–1561 CrossRef CAS.
- O. Kulakovich, N. Strekal, A. Yaroshevich, S. Maskevich, S. Gaponenko, I. Nabiev, U. Woggon and M. Artemyev, Nano Lett., 2002, 2, 1449–1452 CrossRef CAS.
- E. Hwang, I. I. Smolyaninov and C. C. Davis, Nano Lett., 2010, 10, 813–820 CrossRef CAS.
- K. Munechika, Y. Chen, A. F. Tillack, A. P. Kulkarni, I. J. L. Plante, A. M. Munro and D. S. Ginger, Nano Lett., 2010, 10, 2598–2603 CrossRef CAS.
- L. H. Lie, M. Duerdin, E. M. Tuite, A. Houlton and B. R. Horrocks, J. Electroanal. Chem., 2002, 183, 538 Search PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- B. Erdem, E. David Sudol, V. L. Dimonie and M. S. El-Aasser, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4431 CrossRef CAS.
- N. Zheng, J. Fan and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 6550 CrossRef CAS.
- M. M. Maye, W. Zheng, F. L. Leibowitz, N. K. Ly and C. J. Zhong, Langmuir, 2000, 16, 490–497 CrossRef CAS.
- X. M. Lin, C. M. Sorensen and K. J. Klabunde, J. Nanopart. Res., 2000, 2, 157–164 CrossRef CAS.
- X. M. Lin, H. M. Jaeger, C. M. Sorensen and K. J. Klabunde, J. Phys. Chem. B, 2001, 105, 3353–3357 CrossRef CAS.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen and K. J. Klabunde, Chem. Mater., 2003, 15, 935–942 CrossRef CAS.
- M. Kanehara, J. Sakurai, H. Sugimura and T. Teranishi, J. Am. Chem. Soc., 2009, 131, 1630–1631 CrossRef CAS.
- C. Li, K. L. Shuford, Q. H. Park, W. Cai, Y. Li, E. J. Lee and S. O. Cho, Angew. Chem., Int. Ed., 2007, 46, 3264–3268 CrossRef CAS.
- P. N. Njoki, I. S. Lim, D. Mott, H. Y. Park, B. Khan, S. Mishra, R. Sujakumar, J. Luo and C. J. Zhong, J. Phys. Chem. C, 2007, 111, 14664–14669 CAS.
- M. J. Yacamán, J. A. Ascencio, H. B. Liu and J. Gardea-Torresdey, J. Vac. Sci. Technol., B, 2001, 19, 1091–1103 Search PubMed.
- M. K. Corbierre, N. S. Cameron, M. Sutton, S. G. J. Mochrie, L. B. Lurio, A. Ruhm and R. B. Lennox, J. Am. Chem. Soc., 2001, 123, 10411–10412 CrossRef CAS.
- J. Shan and H. Tenhu, Chem. Commun., 2007, 4580–4598 RSC.
- M. K. Corbierre, N. S. Cameron and R. B. Lennox, Langmuir, 2004, 20, 2867–2873 CrossRef CAS.
- P. Alexandridis, Chem. Eng. Technol., 2011, 34(1), 15–28 CrossRef CAS.
- X. Wang, G. Li, T. Chen, M. Yang, Z. Zhang, T. Wu and H. Chen, Nano Lett., 2008, 8(9), 2643 CrossRef CAS.
- Z. Merican, T. L. Schiller, C. J. Hawker, P. M. Fredericks and I. Blakey, Langmuir, 2007, 23(21), 10539–10545 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. J. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347 CrossRef CAS.
- D. Crespy and K. Landfester, Soft Matter, 2011, 7, 11054–11064 RSC.
- M. S. El-Aasser, J. Coat. Technol. Res., 2004, 1, 1 CrossRef.
- Y. Chao, L. Šiller, S. Krishnamurthy, P. R. Coxon, U. Bangert, M. Gass, L. Kjeldgaard, S. N. Patole, L. H. Lie, N. O'Farrell, T. A. Alsop, A. Houlton and B. R. Horrocks, Nat. Nanotechnol., 2007, 2(8), 486–489 CrossRef.
- H. Y. Chen, S. Abraham, J. Mendenhall, S. C. Delamarre, K. Smith, I. Kim and C. A. Batt, ChemPhysChem, 2008, 9, 388 CrossRef CAS.
Footnote |
| † The particle size of citrate stabilized AuNPs slightly larger than PS-AuNPs because citrate stabilized AuNPs was measured in water, so it will promote hydration sphere around the particles and thus effect on the increasing of the particle sizes. |
| This journal is © The Royal Society of Chemistry 2013 |