Functional mesoporous materials for energy applications: solar cells, fuel cells, and batteries
Youngjin
Ye†
,
Changshin
Jo†
,
Inyoung
Jeong†
and
Jinwoo
Lee
*
Department of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Hyo-ja dong, Pohang, Kyungbuk 790-784, Korea. E-mail: jinwoo03@postech.ac.kr; Fax: +82 54 279 5528; Tel: +82 54 279 2395
First published on 13th March 2013
Abstract
This feature article presents recent progress made in the synthesis of functional ordered mesoporous materials and their application as high performance electrodes in dye-sensitized solar cells (DSCs) and quantum dot-sensitized solar cells (QDSCs), fuel cells, and Li-ion batteries. Ordered mesoporous materials have been mainly synthesized using two representative synthetic methods: the soft template and hard template methods. To overcome the limitations of these two methods, a new method called CASH was suggested. The CASH method combines the advantages of the soft and hard template methods by employing a diblock copolymer, PI-b-PEO, which contains a hydrophilic block and an sp2-hybridized-carbon-containing hydrophobic block as a structure-directing agent. After discussing general techniques used in the synthesis of mesoporous materials, this article presents recent applications of mesoporous materials as electrodes in DSCs and QDSCs, fuel cells, and Li-ion batteries. The role of material properties and mesostructures in device performance is discussed in each case. The developed soft and hard template methods, along with the CASH method, allow control of the pore size, wall composition, and pore structure, providing insight into material design and optimization for better electrode performances in these types of energy conversion devices. This paper concludes with an outlook on future research directions to enable breakthroughs and overcome current limitations in this field.
 Youngjin Ye | Youngjin Ye obtained his B.S. (2010) from the Department of Chemical Engineering of Pohang University of Science and Technology (POSTECH) in Korea. His current research focuses on the synthesis of Pt-based nanocrystals as electrocatalysts in fuel cells, under the supervision of Professor Jinwoo Lee. |
 Changshin Jo | Changshin Jo obtained a B.S. degree (2010) from the Department of Chemical Engineering of Pohang University of Science and Technology in Korea. He has worked on his doctoral thesis studying the synthesis of nanostructured electrode materials for energy storage applications under the supervision of Professor Jinwoo Lee. |
 Inyoung Jeong | Inyoung Jeong obtained his B.S. degree (2011) from the Department of Chemical Engineering of Pohang University of Science and Technology in Korea. He is currently a M.S. course student in the research group of Prof. Jinwoo Lee at POSTECH. His current research focuses on the synthesis and application of functional nanomaterials for dye sensitized solar cells. |
 Jinwoo Lee | Prof. Jinwoo Lee obtained his B.S. (1998) and Ph.D. (2003) from the Department of Chemical and Biological Engineering of Seoul National University, Korea. After his postdoctoral research at Seoul National University (with Prof. Taeghwan Hyeon, 2003–2005) and Cornell University (with Prof. Ulrich Wiesner, 2005–2008), he joined the faculty of the Department of Chemical Engineering at Pohang University of Science and Technology (POSTECH) in June, 2008. His research field includes synthesis and applications of ordered functional mesoporous materials and shape controlled nanocrystals in energy conversion and storage devices. He is also working on the development of nanostructured materials for high performance biosensors and biocatalysis. He has published more than 90 papers in prominent international journals and has had more than 4400 citations. |
Introduction
In recent decades, considerable research effort has been devoted globally to the development of new and renewable energy sources because of the fossil fuel depletion crisis and its hazardous environmental impact.1 Renewable energy does not produce harmful emissions, and hence, to preserve our environment, there is a high demand for replacing the currently used fossil fuels with new renewable energy sources and developing devices for efficiently utilizing them or storing the unused energy. It is expected that recent advances in the technology of solar cells, fuel cells, and rechargeable batteries will allow us to achieve a sustainable energy society with renewable energy sources in the near future. Solar cells convert solar radiation energy directly into electricity, with a current conversion efficiency of higher than 20% using silicon-based solar cells.2 However, the commercialization of silicon-based solar cells has been hampered to some extent by the high cost of silicon and the difficulty in manufacturing such cells. Among the various types of solar cells, dye-sensitized solar cells (DSCs) and quantum dot-sensitized solar cells (QDSCs) are of great interest as alternatives to silicon-based solar cells because of their low cost manufacturing processes and relatively high efficiency of 11%.3,4 Fuel cells transform the chemical energy stored in fuels such as hydrogen and small organic molecules directly into electricity, with a theoretical fuel efficiency that is significantly better than that of conventional heat engines.5,6 Hydrogen, which is a clean energy source, can be efficiently converted into electricity by utilizing high performance fuel cells.5,6 Renewable energy sources are variable over time, and their distributions are not even in space. In this sense, along with the development of highly efficient energy conversion devices, there is also a great need to develop highly efficient and low cost energy storage devices aiming at the efficient utilization of the energy generated by renewable energy sources. Among the various energy storage systems that could be utilized, electrochemical Li-ion rechargeable batteries have become the dominant power source for consumer electronic devices and hybrid electric vehicles (HEVs).7,8Recent advances in nanomaterial synthesis now allow the application of various nanostructured materials as electrode materials in solar cells, fuel cells, and batteries to improve the performance of such energy conversion and storage devices.9–14 Nanomaterials have attracted considerable attention because they generally provide the following features as electrode materials in energy conversion and storage devices: (i) a high surface area for electrochemical reactions in Li-ion batteries and fuel cells or for attaching numerous sensitized molecules (or inorganic sensitizers) in DSCs and QDSCs and (ii) fast diffusion of an electrolyte onto the surfaces of the electrochemically active materials in solar cells, fuel cells, and batteries, along with a shorter solid state diffusion path for the Li ions in batteries, which is beneficial for a high charging–discharging rate.
Among the various nanostructures, ordered mesoporous materials, which have interconnected pores with sizes ranging from 2 to 50 nm, can be good candidates as electrode materials because of their interconnected pores, controllable pore sizes and structures, and controllable pore wall chemical compositions.15–17 The structures and compositions of ordered mesoporous materials can be tailor-made for the electrode materials in energy conversion and storage applications through two representative synthetic methods: the hard template method18–21 and the soft template method.22,23 After the discovery of the first mesostructured aluminosilicates by Mobil researchers, the syntheses of mesostructured materials with various compositions have been sought for applications in catalysis, separations, including bioseparation, and electrode materials.24–29
In the soft template method, amphiphilic molecules, including surfactants and block copolymers, are self-assembled with inorganic precursors or nanoparticles into organized organic–inorganic hybrids. The organic materials used as templates are subsequently removed by heat-treatment under air, generating ordered mesoporous inorganic materials (see Fig. 1). Low molecular weight surfactants (e.g., cetyltrimethylammonium bromide) were first employed in this soft template method, and mesostructured aluminosilicates with amorphous structures have been successfully synthesized.16 In addition, mesostructured carbons with various pore sizes and pore structures have been synthesized using the soft-template method.30–34 However, in the case of transition metal oxides, the mesostructure is easily collapsed upon removal of the surfactants by calcination and solvent extraction.35,36 Poly(alkylene oxide)-based amphiphilic block copolymers have been used as structure-directing agents to fabricate a semicrystalline mesoporous metal oxide framework. The amorphous metal oxide/amphiphilic block copolymers have been heat-treated to 400 °C, and small crystals were found to nucleate and grow in the amorphous mesoporous walls. Although this approach allows the possibility of generating crystalline walls in mesostructured walls, it is still necessary to maintain the mesostructures at higher crystallization temperatures because some crystals are formed at a high temperature (e.g., lithium titanate is obtained at a temperature higher than 650 °C)37–39 and fully crystalline metal oxides are needed to enhance the electron mobility in n-type semiconductor oxides for dye-sensitized solar cells.40
 | ||
| Fig. 1 Schematic illustrations of (a) the hard template method and (b) the soft template method. | ||
In the hard template method, inorganic precursors are filled into pre-synthesized hard template mesoporous silica or colloidal spheres, which are subsequently removed by etching with HF or NaOH, or heat-treatment under air. By using this method, mesostructured carbons with different pore sizes and structures have been successfully synthesized.18,28,41,42 In addition, a hard template method is used to increase the crystalline temperature while maintaining the ordered mesostructure of mesoporous metal oxides.43–45 In this method, it is possible to heat-treat the mesostructured materials at an elevated temperature (∼700 °C, below the silica melting temperature), and the crystallinity can also be easily controlled by selecting the heating temperature. Using this method, Bruce group reported that either nanocrystals embedded in amorphous walls (∼500 °C) or fully crystalline walls (∼600 °C) could be selectively fabricated by controlling the heating temperature.46 Although the hard template method allows us to obtain thermally stable and highly crystalline mesostructured materials with various wall compositions, the synthetic approach is considerably long and cumbersome because of the following steps: (i) preparation of the organic template–silica composite, (ii) removal of the organic template by calcination, (iii) infiltration of the inorganic precursors into the mesoporous template, (iv) crystallization at the desired temperature, and (v) removal of the hard template.18–21
From these considerations, Lee et al. suggested a new method that combines the advantages of both the soft and hard template methods by employing a diblock copolymer, poly(isoprene-block-ethylene oxide) (PI-b-PEO), which contains a hydrophilic block and an sp2-hybridized-carbon-containing hydrophobic block as a structure-directing agent (Fig. 2).47 This method was referred to as combined assembly by soft and hard (CASH) chemistries. In this method, PI-b-PEO is combined with sols of metal oxides, which selectively swell the hydrophilic block PEO to form PI-b-PEO–metal oxide hybrids. When the resulting organic–inorganic hybrids are heat-treated under an inert atmosphere, part of the PI, containing sp2 carbons, is converted to an amorphous carbon, which acts as a rigid support for the mesostructured walls. Because the mesostructures are supported by the rigid carbons, the heating temperature can be easily elevated to a high temperature to synthesize thermally stable and highly crystalline mesoporous metal oxides. The carbon can be removed by heat treatment in air. This process provides a simple and viable approach to synthesize various types of mesoporous metal oxides (transition metal oxides), which need to be heat-treated at high temperatures for high performance in energy devices.
 | ||
| Fig. 2 Schematic representation of the CASH method.47 A hexagonally arranged polymer–metal oxide hybrid is heat-treated under argon and an in situ formed carbon acts as a rigid support for mesostructures during high temperature crystallization (∼700 °C). Subsequent air calcination leads to thermally stable and highly crystalline mesoporous metal oxides (reprinted with permission from ref. 47, Copyright Nature Publishing Group, 2008). | ||
In addition to CASH, it is worthwhile mentioning other promising approaches to synthesize crystalline mesoporous metal oxides. One effective approach is to use pre-formed nanocrystals as building blocks.40,48–50 Since this method does not require a high temperature for crystallization, mesoporous materials with high crystallinity can be easily obtained while retaining their original structures. In this system, the properties of nanocrystals such as ligand and size greatly affect the polymer–nanocrystal self-assembly.48 Therefore, better understanding of the self-assembly of polymers and nanocrystals is necessary to obtain precisely controlled mesostructures with high volumetric loading of nanocrystals. The other approach to the synthesis of highly crystalline metal oxides is the incorporation of laboratory-synthesized block copolymers as the structure directing agents.38,51–55 In the soft template method, relatively large repeat distances (>15 nm) with sufficient thick walls are required in order to crystallize mesoporous metal oxides without collapse of the structure. Unfortunately, commercially available Pluronic copolymers are not suitable for crystalline mesoporous oxides because they have relatively short hydrophobic poly(propylene oxide) blocks, yielding mesoporous structures with a pore wall thickness of 4–6 nm.52 Recent studies have shown that several types of block copolymers such as KLE (poly(ethylene-co-butylene)-b-poly(ethylene oxide)),53,56 poly(isobutylene-b-ethylene oxide),53 poly(styrene-b-ethylene oxide) (PS-b-PEO),54 and poly(ethylene oxide)-b-polybutadiene-b-poly(ethylene oxide)55 have been proven to produce crystalline mesostructured oxides with relatively large repeat distances and sufficiently thick walls. By using these systemically designed block copolymers, one-pot synthesis of crystalline metal oxides would be viable. It is expected that rapidly developing polymer technology would allow more precise control of the porous structure combined with the achievement of high crystallinity.
As previously described, synthetic methods for functional ordered mesoporous materials have been developed and optimized by many research groups, and various mesostructured materials can be tailor-made for use as high performance electrodes in solar cells, batteries, and fuel cells using such methods. In this feature article, we will discuss the recent advances made in the synthesis of functional ordered mesoporous materials and their applications in dye and quantum dot sensitized solar cells, fuel cells, and Li-ion batteries.
Mesoporous materials as electrodes for dye sensitized solar cells
Dye sensitized solar cells (DSCs), which have fascinating advantages such as cost effectiveness and an easy fabrication process, have emerged as promising alternatives to conventional silicon solar cells, showing a recorded efficiency of 12.3%.3,57 A DSC is a photoelectrochemical cell composed of three parts: the working electrode (WE), the electrolyte, and the counter electrode (CE) (Fig. 3). The WE is a dye sensitized metal oxide with a large band gap, typically TiO2, that transports photoexcited electrons injected from the lowest unoccupied molecular orbital (LUMO) of the dye to the conducting substrate. Then, the electrons move to the counter electrode, followed by successive redox reactions in the CE–electrolyte and oxidized dye–electrolyte interfaces to complete the circuit of the DSC. Each of these three parts plays an important role in the whole system, and there have been many developments for each of them to optimize the power conversion efficiency (PCE).58–60 In short, the developments are (i) application of various materials and nanostructures in the working electrode as well as counter electrode, (ii) design of novel dyes which can absorb light over a wide range of wavelengths with a high extinction coefficient and (iii) synthesis of suitable electrolytes to gain high open circuit voltage and stability (especially, solid state hole transport materials). Among them, as structural properties of the WE oxide film are powerful factors to determine and improve the PCE, research on functional nanostructures of WE has explosively studied and boosted the field of DSCs. In this article, our discussion is limited to the nanostructures of WE and CE, especially the templated mesoporous structure. | ||
| Fig. 3 Mesoporous working electrode and counter electrode in a dye sensitized solar cell (DSC). The novel physicochemical properties of a mesoporous structure (high surface area, uniform mesopores, and interconnected framework) can satisfy all the requirements of an ideal WE and CE. | ||
The most commonly used WE film is composed of 20 nm TiO2 nanoparticles with a large surface area for high dye loading. However, in addition to the low electron mobility of anatase TiO2, the many grain boundaries at the contacts between the nanoparticles and the three-dimensional (3-D) random walk pathway through the disordered nanoparticle network make the electron transport slow, which enhances recombination and inhibits charge collection.61 In the case of CE, even though platinum (Pt) shows a high catalytic activity and PCE, its scarcity (0.0037 ppm) and high price (US $50 per g) make it difficult to use for a large scale system.62 Moreover, Pt has also shown selective catalytic activity for some iodine free electrolytes.63–65 Therefore, in addition to the search for various other suitable materials for WEs and CEs, the application of nanostructures other than nanoparticles such as one-dimensional nanostructures (nanotubes,61,66 nanowires,67,68 nanorods,69etc.), hierarchical structures,70–73 hollow spheres,74,75 and mesoporous structures76–85 has attracted considerable attention because of the various advantages offered by these nanostructures, which can improve specific characteristics of a WE or CE. Among the nanostructures, ordered and interconnected mesoporous structural materials synthesized using various hard and soft template methods have shown promising and ideal features for both the WE and CE of DSCs, as discussed in detail in the following text.
Mesoporous materials as working electrodes
To increase the overall conversion efficiency in aspect of WE, the following factors are essential. First, a higher dye loading will produce a higher light harvest. A large internal surface area for the electrode is the chief requirement for high dye loading. Second, the injected electrons from the dye molecules need to be transported to the external circuit as efficiently as possible to decrease the back reaction with oxidized species in the electrolyte and holes in the dye. Third, sufficient porosity is needed for binding the dye molecules, as well as the diffusion of the electrolyte. Fourth, the scattering functions of the WE can lengthen the light pathway, which will improve the light harvesting.To satisfy the requirements for highly efficient DSCs and improve some characteristics of DSCs compared with conventional particulate systems, various functional nano-structures such as one-dimensional (1-D) nanostructures, hierarchical structures, and mesoporous structures for the WE film have been investigated. 1-D nanostructured materials such as nanowires, nanofibers, nanotubes, etc. have been actively applied to the WE of DSCs because the 1-D nanostructured WE film can provide direct path for electrons injected from dyes rather than the 3-D random walk pathway in the case of conventional 20 nm sized TiO2 nanoparticles.86–88 Law et al. reported that ZnO nanowires show several hundred times higher electron diffusion coefficient than that of the TiO2 nanoparticle and ZnO nanoparticle WE.67 However, most reported 1-D material based DSCs have shown lower overall cell efficiency than that of conventional DSCs using TiO2 nanoparticles due to insufficient surface area of the 1-D structure for high dye loading. Contrarily, a template derived mesoporous structure can provide a high surface area for high dye adsorption, ordered and interconnected continuous mesopores for efficient electron transport and electrolyte permeation with an additional light scattering effect caused by the sub-micrometer sized particles. In other words, the mesoporous structure can fulfill all the requirements of a WE for efficient DSCs, while overcoming the limitations of nanoparticles (disordered necking and electron trap sites between adjacent particles) and 1-D structures (insufficient surface area and dye loading). In this regard, much research on mesoporous metal oxides for WEs, mainly mesoporous TiO2 (hereafter, m-TiO2), has made extensive progress, from the synthesis of materials to the fabrication of films suitable for DSCs.
A mesoporous structure can be synthesized using either a soft template (block copolymers, ionic and nonionic surfactants as structure directing agents) or a hard template (mesoporous silica or carbon) method. Wei et al. used a screen printed an m-TiO2 film prepared using a triblock copolymer template (Pluronic P123, EO20PO70EO20) as the WE of a DSC, which showed superior photovoltaic performance with a high PCE of 10%.77 They found that the amount of dye loading for the m-TiO2 WE was about 1.5–2 times greater than that of a standard TiO2 particle WE (P25, ∼20 nm size), which was attributed to its higher surface area (195 m2 g−1). They also highlighted how the novel properties of m-TiO2 such as its uniform nanochannels and homogeneous nanocrystalline TiO2 arranged in a framework significantly influenced the photovoltaic performances of the DSCs. In a thicker WE film, photoexcited electrons should travel a longer distance, thereby increasing the recombination sites and dark current. They found that m-TiO2 WE based cells show a smaller decrease in open circuit voltage (Voc) with increasing film thickness compared with that of P25, indicating that electron transport is more efficient in a mesoporous structure due to the smooth necking region along the framework. Recently, Phadke et al. prepared DSCs based on an oil-in-water emulsion templated mesoporous TiO2 WE film and non-templated TiO2 WE. They studied the templating effect on electron transport energetic and kinetics of DSCs by Electrochemical Impedance Spectroscopy (EIS).89 They found that the templated WE has a higher electron diffusion coefficient (Deff) as compared to a TiO2 nanoparticulate (P25) film, because of the interconnected pore network of the m-TiO2 WE developed using an emulsion template.
Ahmed et al. investigated the properties of a mesoporous TiO2 electrode synthesized by supramolecular self-assembly using the cetyl-trimethylammonium bromide (CTAB) surfactant.90 The CTAB-templated mesoporous TiO2 electrode which has meso- and macro-pores showed a longer electron life time than that of a P25 electrode, as well as the light scattering effect, which were confirmed through Bode phase plots and diffuse reflectance spectra, respectively.
In addition to TiO2, there have also been other attempts to adopt mesoporous structures for other metal oxides. SnO2, which is a promising alternative material to TiO2 because of its chemical stability, three-order higher electron mobility (100 cm2 V−1 s−1vs. 0.1 cm2 V−1 s−1 for TiO2), and large band gap (3.6 eV),78 has been studied with various nanostructures such as multilayered hollow microspheres,75 hierarchical octahedral72 and 1-D structures.91 In addition, Ramasamy and Lee introduced an ordered mesoporous structure for SnO2 and applied in DSCs.78 They prepared mesoporous SnO2 (hereafter, m-SnO2) with the hard template method using KIT-6 silica, followed by the HF etching process. SnCl2·2H2O, as SnO2 precursors which are infiltrated into the pores of KIT-6 by melting the precursor at 85 °C, is converted into crystalline tin oxide by heating at 700 °C in air. After etching the silica template, the interwoven pore network is formed, replacing the silica framework. The m-SnO2 with a 3-D bicontinuous cubic mesostructure has a high BET surface area of 105 m2 g−1 and a pore volume of 0.27 cm3 g−1, with a bimodal pore system composed of 3 nm and ∼20 nm pores (see Fig. 4a and b). The interconnected 3 nm pores are generated by the removal of the silica template, whereas the 20 nm sized pores are generated from the selective filling of one of the two chiral channels of KIT-6 with SnO2. They coated the m-SnO2 with an ultra-thin TiO2 layer to suppress the charge recombination. Even though the thickness of the WE studied is 3 μm which is much thinner than that of other reported SnO2 based DSCs,72,75,92 the TiO2-coated m-SnO2 WE-based DSC showed a high current density (Jsc) of 10.42 mA cm−2, resulting in a PCE of 3.8%, which is 40% higher than its counterpart using a TiO2-coated commercial SnO2 nanoparticle WE (22–43 nm sized nanoparticle, Jsc: 6.60 mA cm−2). Interestingly, diffuse reflectance spectra results showed the light scattering ability of m-SnO2. This may be attributed to the sub-micrometer sized m-SnO2 particles with interconnected mesopores. The results confirmed that a mesoporous structure improves the Jsc and overall PCE of m-SnO2 by a higher dye-uptake and light scattering.
 | ||
| Fig. 4 (a and b) Synthesis scheme and TEM image of m-SnO2 prepared using a KIT-6 silica template (reprinted with permission from ref. 78, Copyright American Chemical Society, 2010). (c and d) Synthesis scheme and TEM image of Zn-doped m-SnO2 prepared by the double-replication method using SBA-15 silica and CMK-3 carbon (reprinted with permission from ref. 81). | ||
On the other hand, despite some advantages of SnO2, SnO2 WE based DSCs have shown low Voc due to more a positive conduction band level than that of TiO2. Therefore, approaches to increase the Voc of SnO2 have been investigated using core–shell or composite structures with wide band gap oxide.92,93 Recently, Ramasamy and Lee synthesized Zn-doped mesoporous SnO2 and used for the WE of DSCs to gain high Voc as well as high Jsc which can be obtained by Zn doping and mesoporous structures. Zn-doped m-SnO2 was synthesized by the double-replication method using SBA-15 silica and CMK-3 carbon as successive hard templates (Fig. 4c and d).81 Zn-doped m-SnO2 has a high surface area of 97 m2 g−1, with large pores of 11.5 nm. This study showed that the Zn doping of m-SnO2 leads to a negative shift in the flat band potential (Vfb) of SnO2 and increases the isoelectric point. These effects of Zn-doped m-SnO2 showed an increased Voc and higher dye uptake, resulting in a five-fold improvement (3.73% of PCE) compared to the undoped m-SnO2 WE-based DSCs. Recently, Dou et al. also reported Zn-doped SnO2 based DSCs which have a nanoflower structure.94 They showed a high electron mobility of a Zn-doped SnO2 nanoflower film through EIS measurement. Structurally, however, the Zn-doped SnO2 nanoflower has a smaller surface area (∼10 m2 g−1) than that of Zn-doped m-SnO2, resulting in lower dye uptake and relatively lower PCE of DSCs. The Zn-doped m-SnO2 WE, therefore, paved the way to develop various types of doped functional WEs combined with the advantages of a mesoporous structure for highly efficient DSCs.
As mentioned above, mesoporous structures that possess interconnected networks and high surface areas have shown advantages when used for WEs of DSCs, with improved characteristics such as electron transport, dye loading, electrolyte permeation, and light scattering when compared to those of typical nanoparticle electrodes. However, it should be noted that mesoporous structures do not provide ideal and positive effects unconditionally. An electrode with a mesoporous structure not only provides high surface area and efficient electrolyte diffusion but also increases the chance of electron recombination with an electrolyte simultaneously, resulting in a lower Voc for a mesoporous WE than a nanoparticulate WE.95,96 In addition, there are key factors controlling the optimum conditions of mesoporous electrodes suitable for DSCs.
First, the porosity and the pore size of electrodes are critical factors that can determine the performance of DSCs because they affect the usable surface area of the electrodes for dye adsorption and interfacial charge transfer with the electrolytes related to recovery of the excited dye and recombination. Ni et al. conducted an analytical study of the porosity effect of WEs on DSC performance.97 In the case of Voc, its value increases initially with increasing electrode porosity and continues to increase marginally at higher porosities. Although Jsc also increases initially as the porosity increases, it begins to drop at higher porosities. A WE with a porosity higher than the optimum value has a lower surface area for dye loading and increases the proportion of dead ends in the film, leading to negative effects on the photocurrent.98,99 However, these trends come from the nanoparticulate film system. Thus, templated mesoporous structures with highly interconnected networks can relieve the negative effect from dead ends. Pan et al.80 and Yun et al.96 have studied the pore size effect of mesoporous electrodes by synthesizing various materials with pore sizes ranging from 3 nm to 11 nm and analyzing the photovoltage transient measurements and EIS data. They found that even though m-TiO2 with smaller pores has a higher surface area, there is an optimum pore size to attain the highest PCE because undersized channels hamper the perfect dye adsorption and diffusion of the electrolyte. On the other hand, a WE with oversized open channels experiences severe recombination with redox shuttles. On the basis of these qualities, the soft template method employing synthetic block copolymers has more advantages compared to the hard template method to achieve the optimal pore size for high-performance DSCs. This is ascribed to the fact that the pore size can be easily controlled by changing the molecular weight of block copolymers. The pore connectivity can be changed by simply varying the amount of the copolymer or the ratio of the amount of inorganic precursor to polymer.100 In contrast, the pore size of a hard-templated material is typically limited to values below 10 nm.46,101
Second, the crystallinity of the mesoporous electrode is another crucial factor in DSC performance because semiconductor oxide WEs should transport electrons injected from dye molecules to the TCO substrate as soon as possible to reduce recombination with redox shuttles; moreover, numerous grain boundaries and junctions act as recombination sites. Electron transport becomes faster as the crystallinity increases. Typically, highly crystalline materials can be obtained with high-temperature annealing. Recently, Guldin et al. studied the influence of crystallization on conductivity and the electron transport properties of m-TiO2 WEs by varying the annealing temperature (400–700 °C).102 They used PI-b-PEO as a template to prevent collapse of the mesoporous structure at a high temperature of 700 °C and investigated the DSC performance with increasing m-TiO2 crystallinity for both liquid DSCs and solid-state DSCs (ssDSCs). In short, as shown in Fig. 5a and b, crystallization at high temperature yields larger crystallites and higher conductivity, while it decreases the surface area at the same time. This trade-off property differentiates the performance of liquid DSCs from that of ssDSCs. The liquid DSCs showed negative trends with increasing crystallization temperature; the ssDSCs exhibited PCEs that were three times higher when utilizing highly crystalline TiO2 electrodes annealed at 700 °C than the PCE of m-TiO2 heat-treated at 450 °C. This is attributed to the fact that while a liquid DSC is not influenced strongly by electron recombination, a ssDSC is more influenced by improved crystallinity and conductivity of TiO2 rather than decreased surface area.
 | ||
| Fig. 5 (a and b) Schematic representations of the crystallite size and orientation in a mesoporous TiO2 network derived using a block copolymer (PI-b-PEO) at different annealing temperatures of 400 °C and 650 °C, respectively (reprinted with permission from ref. 102). (c and d) TEM images of mesoporous TiO2 films which have different orderings derived by varying the amount of F127 copolymer (reprinted with permission from ref. 103, Copyright American Chemical Society, 2009). | ||
Third, the ordering of the mesoporous structure can have an influence on the properties of a WE. Zhang et al.103 prepared ordered body-centered orthorhombic (Fig. 5c) and disordered worm-hole-like m-TiO2 (Fig. 5d) in DSCs via the supramolecular template method using Pluronic F127 (EO106PO70EO106). When they studied the effect of morphology with different orderings on performance, they discovered that DSCs based on highly ordered m-TiO2 WEs showed higher Jsc and PCEs than the values corresponding to DSCs based on disordered m-TiO2 WEs. Meanwhile, ordered m-TiO2 electrodes have similar dye loading and trap densities as compared to those of their disordered counterparts, resulting in similar values of Voc in both electrodes. These results indicate that the higher Jsc of ordered m-TiO2 could only be explained by improved electron transport through a well-connected TiO2 framework.
As discussed above, the control of crystallinity, pore size, porosity, and ordering in the mesoporous electrode is crucial to obtaining optimum performance as an ideal WE in DSCs. The self-assembly of synthetic block copolymers allows the control of pore size and electronic properties of mesoporous oxides as well as crystallinity, with thermal stability at high temperature, when following the CASH method. The novel structure-directing agents PI-b-PEO and PS-b-PEO, used in the CASH methods demonstrated by Lee et al.47 can control the morphology (porosity and pore size) and crystallinity on the 10 nm scale. In addition, Docampo et al.104 reported that the control of the ratio of inorganic to organic components can make the density of sub-bandgap states favorable, resulting in improvement of electron transfer and charge separation.
The advantages of mesoporous structured working electrodes can be more remarkable when used for solid state DSCs (ssDSCs) because ordered, large open mesopores and good inter-connectivity enhance the solid-electrolyte pore filling and overall performances. Nedelcu et al. reported the use of m-TiO2 synthesized by PI-b-PEO via a non-hydrolytic sol–gel route for solid state electrolyte (spiro-OMeTAD)-based DSCs.79 They fine-tuned the pore sizes from 20 nm to 80 nm by changing the polymer molecular weight and TiO2 precursor content (see Fig. 6), as well as increasing the crystallization temperature up to 450 °C to 600 °C, without losing the porous structure. The solar cell performances were maximized when the films were heated to 550 °C, and the films had a highly open porous system composed of 30 nm pores. This study demonstrated the feasibility of using PI-b-PEO with the CASH method for the mesoporous WEs in solid state DSCs.
 | ||
| Fig. 6 Top view SEM images of various m-TiO2 films with different morphologies (pore diameter, pore volume and network) synthesized by varying the PI-b-PEO molecular weight and titania precursor content. Column header indicates the mass ratio of polymer to TiO2 precursor and Mn means the PI-b-PEO weight (reprinted with permission from ref. 79). | ||
Recently, Docampo et al. designed a new triblock-terpolymer-directed self-assembly for fully interconnected and bicontinuous mesoporous TiO2 films. They were the first group to report that mesoporous WEs outperformed conventional particulate DSCs, showing a PCE of 5% when performed with the hole-transport material spiro-OMeTAD.105 The triblock terpolymer PI-b-PS-b-PEO was designed to reside in an inorganic material (TiO2) in the minority phase, with a morphology of 3-D continuous and gyroid-like networks to reduce the volume contraction of the film during evaporation of the solvent and calcination (Fig. 7). They indicated that the improved PCE of m-TiO2 derived from the triblock terpolymer was mainly a result of the high density of sub-bandgap states induced by a large organic fraction, which led to improved transfer of electrons injected from dyes. Likewise, the mesoporous WEs are suitable for ssDSCs and can optimize the performance of ssDSCs via precise control of well-designed block-copolymer self-assembly.
 | ||
| Fig. 7 (a) Schematic representation of the chemical configuration of the triblock terpolymer PI(31%)-PS(53%)-PEO(16%) (each number in parenthesis means volume fraction) and illustrating the preferential residing of TiO2 sol (back particle) in the hydrophilic PEO part. (b) Schematic representation of a single alternating gyroid network of the PEO and TiO2 phase. (c) SEM image of the continuously connected and highly porous TiO2 film deposited onto a FTO (fluorine-doped tin oxide) substrate. (d) Magnified top view SEM image of mesoporous TiO2 after calcination at 500 °C (reprinted with permission from ref. 105, Copyright Wiley-VCH, 2012). | ||
Mesoporous materials as counter electrodes
A catalytic material coating on a counter electrode plays an important role, facilitating the reduction reaction of the oxidized species in an electrolyte with electrons to complete the circuit of a DSC. For an ideal counter electrode, the catalyst should possess high catalytic activity, as well as high electrical conductivity. In addition, low cost materials are greatly needed because the Pt used for a conventional CE is expensive and scarce, which prohibits the commercialization and mass production of DSCs. Many low cost alternative materials for Pt free CEs,106 including various types of carbonaceous materials,107–109 conducting polymers,62,110 and inorganic compounds,111–113 have been investigated and shown promising results comparable to a Pt CE. Structurally, to obtain a high catalytic activity in a CE, CE materials should possess large surface area as well as porous structure, for the efficient electrolyte permeation. In the case of carbon black, a layer thickness of ∼15 μm is required to obtain a PCE that is comparable to that of a Pt CE.114 However, a film of this thickness simultaneously causes fluidic resistance in the electrolyte and a mass transfer limitation.In this context, the introduction of a mesoporous structure into a CE material can be a powerful tool to improve the catalytic activities and interaction with the electrolyte because of its internal high surface area, ordered mesopores, and good connectivity. Ramasamy and Lee reported a mesoporous carbon (MC) CE and investigated its high catalytic activities.85 The mesoporous carbon CE (MSU-F-C)115 was synthesized by the hard template method using mesocellular silica (MSU-F silica).83 It has a high surface area of 894 m2 g−1 and large interconnected bimodal mesopores (20 nm, 4 nm) as shown in Fig. 8a. Interestingly, despite the use of a significantly thinner film (1.1 μm) than that reported for carbon CEs, the MSU-F-C CE showed a high PCE of 8.18%, corresponding to 92.4% for that of a platinized CE. Compared with a carbon black CE which can approach comparable performance of a Pt CE when its thickness is ∼15 μm, the high performance of a thin MSU-F-C CE is attributed to structural advantages such as high surface area and connected large pores. To elucidate the effect of the MSU-F-C mesostructure, conventional platinum and other carbon materials (Vulcan XC-72 and CMK-3) were employed as CEs. Vulcan XC-72 has 50% of the pore volume consisted of micropores and CMK-3 has small 3 nm pores. They showed larger charge transfer resistance (Rct) and low fill factor (ff) values than those of MSU-F-C. Although CMK-3 has a higher surface area than MSU-F-C, the DSC with CMK-3 CE exhibited lower PCEs than that of those with the MSU-F-C CEs. The better photovoltaic performance compared to the Vulcan and CMK-3 CEs can be attributed to the high internal surface area and large interconnected pores, which provide efficient redox electrolyte penetration, and thus a high catalytic active surface area for the reduction of I3− ions.
 | ||
| Fig. 8 (a) High resolution TEM image of microtomed MSU-F-C carbon synthesized using a MSU-F silica template. MSU-F-C has interconnected 20 nm large pores (reprinted with permission from ref. 85). (b) TEM image of ordered mesoporous carbon (OMC) synthesized by the soft template method using a F127 polymer. It shows a highly ordered 2-D hexagonal mesoporous structure with the well-connected framework (reprinted with permission from ref. 117, Copyright Elsevier, 2009). (c) Schematic representation illustrating the efficient electron transport and higher catalytic activity of a OM TiN–C CE compared with TiN nanoparticle CE, which are attributed to the embedded TiN crystals and OMC framework (reprinted with permission from ref. 120, Copyright American Chemical Society, 2012). | ||
The advantages of a mesostructure with large pores were further investigated for a quasi-solid state electrolyte using poly(vinylidenefluoride-co-hexafluropropylene) (PVDF-HFP). A quasi-solid DSC based on an MSU-F-C CE showed a 30% higher efficiency than CMK-3 and Vulcan CEs, which can be attributed to the efficient infiltration of the viscous polymer gel electrolyte through the ordered and large mesoporous channels. The MSU-F-C CE was also exploited in a quantum dot sensitized solar cell (QDSC) by Seol et al.116 In a QDSC, the commonly used polysulfide electrolyte induces a poisoning of the Pt, and QDSCs that use Pt CEs have shown poor PCEs and instability. Because of the corrosion resistance of carbon materials and the mesoporous structure effects, the MSU-F-C CE showed a superior catalytic activity (higher FF, smaller Rct) compared to Au and Pt CEs in ZnO nanowire WEs sensitized with CdS and CdSe and used for QDSCs, resulting in a PCE of 3.6%. In addition, the MSU-F-C CE showed better stability than the Au and Pt CEs.
Mesoporous carbon can also be synthesized using the soft template method by employing the triblock copolymer Pluronic F127 as a template. Ramasamy et al. reported an ordered mesoporous carbon CE prepared by using F127, where resol and tetraethyl orthosilicate were used as the polymer and inorganic precursor, respectively.117 The ordered mesoporous carbon (hereafter, OMC) possessing 6.1 nm mesopores generated by the decomposition of F127 and ∼2.5 nm pores generated by the removal of the silica source has a high BET surface area of 1575 m2 g−1 and a pore volume of 1.76 cm3 g−1. The TEM and SEM images of the OMC in Fig. 8b show the highly ordered 2-D hexagonal mesoporous structure with the well-connected framework. The large surface area and interconnected mesoporous structure maximize the electrolyte–CE interface, decreasing the Rct and consequently increasing the FF and PCE compared with Vulcan.
Moreover, the interconnected framework for the OMC particles of a few microns provides a more efficient electron pathway than the nano-sized Vulcan particles, which enhances the catalytic activities. Interestingly, although the OMC film was deposited on F-doped tin oxide (FTO) glass at a low temperature (125 °C), a cell based on the OMC CE showed a high efficiency of 7.46% because the OMC already had a connected framework generated by the soft template method, whereas the Vulcan CE showed a low efficiency of 4.30% because of the poor necking between the Vulcan nanoparticles by the low temperature heat treatment. This interesting feature paves the way for applying a flexible conducting plastic substrate such as indium tin oxide-coated polyethylene terephthalate (ITO-PET), which is thermally unstable at a high sintering temperature.
Although the triiodide/iodide (I3−/I−)-based electrolyte has been popularly used as a redox couple in the DSC electrolyte and the conversion efficiency (η) is higher than ∼10%, there are several disadvantages to using it, including the corrosion of Ag current collectors and the partial absorption of the visible light at approximately 430 nm by I3− ions. Very recently, to tackle the problems with an I3−/I− based electrolyte, considerable research effort has been devoted to the development of new alternative electrolytes and nontoxic and noncorrosive redox couples such as metal complexes (Fc+/Fc (Fc:ferrocene), Ni(IV)/(III) bis(dicarbollide))118,119 and the disulfide/thiolate (T2/T−) organic redox couple65 as alternatives to iodine-based redox couple electrolytes. Among these, metal free T2/T− based organic electrolytes have attracted considerable attention and exhibited high performance primarily because of their high transmittance for wavelengths lower than 450 nm and comparable dye regeneration rate by the T− redox species.65 However, the overall conversion efficiency was somewhat lower than expected as a result of the slow reduction of T2 to T− at the Pt counter electrode, showing a large charge transfer resistance.
To overcome this limitation, Ramasamy et al. synthesized an ordered mesoporous titanium nitride–carbon composite (hereafter, OM TiN–C) by soft-template simple synthesis and the resulting material was exploited as a CE material for highly efficient T2/T− electrolyte-based DSCs.120 As depicted in Fig. 8c, the small sized TiN nanocrystals (∼8 nm) embedded in the mesostructured walls efficiently catalyzed the redox reaction of T2 to T− and the interconnected mesostructure facilitated electrolyte and electron transport. Cells fabricated with OM TiN–C CE exhibited an energy conversion efficiency of 6.71% which is one of the highest efficiencies so far achieved with a Pt free CE and T2/T− electrolyte. It is higher than those fabricated with OMC (5.16%), TiN nanoparticles (1.73%), and a conventional Pt counter electrode (3.32%). In addition, the OM TiN–C CE also exhibited outstanding PCE which is comparable to that of Pt CEs, in a conventional I3−/I− redox couple (8.41% vs. 8.00%). This result suggests a high possibility of using OM TiN–C as a promising Pt-free counter electrode for other iodine-free electrolytes.
Catalytic materials in a CE which has a mesoporous structure have shown comparable performance to Pt electrodes and superior properties than bulk and non-structured materials. Recently, various inorganic compounds as CE catalysts have been extensively investigated, but most of them did not have a specific structure. By application of mesoporous structure into the promising materials, catalytic activities of them can be further improved.
Mesoporous materials as alternative catalyst supports in PEMFC
A proton exchange membrane fuel cell (PEMFC) has been considered to be one of the most promising alternative, clean power generation devices because of its high conversion efficiency and small amount of environmental pollution. The development of highly active electrocatalysts is a key issue for the commercialization of fuel cells, because the Pt catalyst layer alone would account for more than half of the stack costs.121,122 The most commonly used electrocatalyst for a PEMFC is 2–3 nm Pt nanoparticles supported on carbon black. In this article, we will focus on the recent developments of alternative support materials, because it has been proven that supports play a crucial role in PEMFCs, influencing the dispersion of Pt nanoparticles and the transport of reactants and products.123–125 Vulcan XC-72 carbon black is the most popular support material because of its relatively high BET surface area of ∼230 m2 g−1 and high electrical conductivity of ∼4 S cm−1.126 However, about 50% of the total pore volume of Vulcan XC-72 consists of micropores, which are not accessible to Pt nanoparticles and the Nafion electrolyte,125,127 reducing the formation of the triple phase boundary, which is a gas, electrolyte, and catalyst interface. To design support materials that facilitate the formation of the triple phase boundary, it is necessary to understand the structure of the Nafion electrolyte. Although the structure of the Nafion ionomer is still debated, from the original Gierke model that explains the geometry of the hydrophilic domains of Nafion as an interconnected network of 4 nm diameter spherical water clusters to the parallel 2.4 nm diameter cylindrical water channel model, it has been believed that the characteristic length scale of the hydrophilic domain is ∼2 to 5 nm.128–130 Considering the fact that the size of Pt nanoparticles is generally 2–3 nm, support materials should ideally have pores larger than 10 nm to facilitate the efficient diffusion of reactants and products, in addition to the high formation of the triple phase boundary (Fig. 9). | ||
| Fig. 9 Ideal structure of a support material. The pore size should ideally be larger than 10 nm for the penetration of Nafion electrolytes with a hydrophilic domain size of 2–5 nm, and spherical primary particles with a diameter smaller than one micrometer are beneficial for the fabrication of thinner electrodes. | ||
Fang et al. suggested a good way to maximize the triple phase boundary by replacing the carbon black support with an ordered hierarchical nanostructured carbon, comprised of large macropores (440 nm in diameter) interconnected with smaller macropores (160 nm in diameter), and carbon walls with 20 nm mesopores (Fig. 10).125 The presence of mesopores combined with micropores results in a relatively high BET surface area of 962 m2 g−1. This carbon structure makes possible a high dispersion of up to 60 wt% of Pt nanoparticles without a significant increase in particle size. Thus, the maximum power density of a membrane-electrode-assembly (MEA) using 60 wt% Pt nanoparticles dispersed on the hierarchical structured carbon was 53% higher than that using a commercial Pt/C catalyst with the same metal loading. It is clear that this increase in power density can be attributed to the structure of the hierarchical structured carbon, which allows the high utilization of the Pt nanoparticles and the achievement of a thin electrode, leading to a decrease in the mass transport loss.
 | ||
| Fig. 10 SEM image of hierarchically nanostructured carbon, in which large macropores (440 nm) are interconnected with smaller windows (160 nm), and 20 nm mesopores are located in carbon walls (reprinted with permission from ref. 125, Copyright American Chemical Society, 2009). | ||
Large-pore conductive mesoporous metal oxides as catalyst supports
Although carbon supports offer great properties such as a large surface area and high electrical conductivity for PEMFC applications, the carbon should eventually be replaced with conductive metal oxides that are stable over the long-term operation of a fuel cell, because carbon corrosion causes isolation of the catalyst particles, leading to the aggregation of nanoparticles and a decrease in gas permeability, which hinders gas transport.131 Although carbon corrosion has been known to be kinetically very slow, it could cause a significant decrease in fuel cell performance after long-term operation. In addition, because repeated start-up and shut-down operations facilitate carbon corrosion, the development of corrosion-resistant conductive metal oxides is very important.132,133 However, these usually lack sufficient surface area for the high dispersion of nanoparticles, which is a very important property of supporting materials. For example, Ebonex, which is a mixture of the TiOx phases and niobium-doped rutile oxide, has a BET surface area of ∼1 m2 g−1.134 The use of a mesoporous metal oxide could be a versatile way to improve the surface area and minimize the mass transport loss.Recently, Huang et al. reported the synthesis of mesoporous TiO2 which is a highly stable cathode support material for a PEMFC.135 In this work, TiO2 was obtained by the calcination of hydrolyzed titanium isopropoxide in the presence of a commercial triblock copolymer, Pluronic P123. The nitrogen sorption isotherm of the mesoporous TiO2 confirmed that the resulting material has a high surface area of 250 m2 g−1, which is comparable to that of Vulcan XC, along with a pore size of 4 nm. The high density of mesoporous TiO2, which makes it possible to achieve a catalyst layer that has one-third the thickness of Pt/C, plays a key role in reducing the mass transport loss. Therefore, although the Pt nanoparticles supported on m-TiO2 (6.2 nm) were 2.5 times larger than those of commercial Pt/C (2.5 nm) and larger than the m-TiO2 pores (4 nm), the current density at 0.6 V and the maximum power density of an MEA using Pt/m-TiO2 were both higher than those using Pt/Vulcan-XC. In addition, an accelerated durability test, which was performed by holding the potential at 1.2 V for 80 h, showed the significantly higher stability of Pt/TiO2 compared to Pt/C. After this durability test, Pt/TiO2 retained 80% of its initial ECSA, whereas Pt/Vulcan-XC lost 93% of its initial ECSA. This work has demonstrated that use of mesoporous metal oxides as supports could be a versatile method for improving stability while maintaining the catalytic activity of Pt-based catalysts. It should be noted that m-TiO2 used in these studies has relatively small pores (∼4 nm) and a low electrical conductivity of 1.47 μS cm−1.136 Although the desired formation of TPB phase and facile electron transfer would be unexpected from this material, it is highly remarkable that achievement of thin electrode combined with high corrosion resistance of the material greatly enhance the performance and stability of the MEA. Therefore, further enhancement of catalytic activity would be viable when corrosion-resistant large-pore metal oxides with high electrical conductivity are used as supports.
In this manner, Kang et al. synthesized conductive WO3−x using KIT-6 as a hard template.137 The use of KIT-6 silica,138 which possesses three-dimensionally well-interconnected pores with a diameter of 7.5 nm, results in ordered mesoporous tungsten sub-oxides with a relatively large BET surface area of 54.2 m2 g−1, thanks to the presence of smaller interconnected pores (3.5 nm), the result of the negative replicas of the KIT-6 template after the removal of the silica walls, and larger pores of 20 nm size, which are generated from the partial occupation of tungsten oxide filling in one of the two chiral channel systems of the KIT-6 template.139 It should be emphasized that the resulting mesoporous W18O49 showed a high conductivity of 1.76 S cm−1 at 300 K, which is comparable to that of a mesocellular carbon foam with large pores (3 S cm−1) and three times higher than that of mesoporous indium tin oxide, as reported by the Nazar group.140 The high electrical conductivity, which has been known to be difficult to obtain from mesoporous conductive metal oxides, was achieved because the protection of the structure by the rigid hard KIT-6 template allowed heat-treatment at a high temperature (600 °C), resulting in a highly crystalline mesoporous structure (Fig. 11). When the resulting oxide was tested as a catalyst support for fuel cells, Pt/W18O49 preserved 87% of its initial ECSA after an accelerated durability test involving cycling between 0.6 and 1.3 VNHE for 1000 cycles. In contrast, Pt/C only preserved 13% of its initial electrical surface area.
 | ||
| Fig. 11 (a and b) TEM image of mesoporous WO3−x showing (a) highly crystalline nature and (b) well-connected pore structure of mesoporous WO3−x (reprinted with permission from ref. 137). | ||
Intermetallics supported on mesoporous materials for formic acid oxidation
Direct formic acid fuel cells (DFAFCs) have attracted a great deal of attention as an energy source for portable electronic devices. The use of formic acid as fuel offers many advantages such as high power density and lower crossover rate.141,142 One of the main drawbacks is the development of active and durable electrocatalysts. Pd and Pt nanoparticles, which are currently used, do not fulfil the activity and durability required for commercialization, because Pt nanoparticles have relatively low catalytic activity and Pd nanoparticles show poor durability.142 Intermetallic PtPb and PtBi nanoparticles are promising substitutes for Pd and Pt catalysts in terms of low onset potential of formic acid oxidation, high current density, and high CO tolerance.143–145 Intermetallic nanoparticles, however, have generally been synthesized at relatively high temperatures (>600 °C), thus resulting in larger nanoparticles than desired.144,145 In order to achieve high mass activity, size reduction through fine dispersion of nanoparticles is necessary.Recently, X. Ji et al. employed surface-modified mesoporous carbon as a support to synthesize small-sized nanoparticles (<4 nm) embedded within the porous structure.146 In this work, they modified the surface of CMK-3 with sulfur to trap metal precursors inside the pores to inhibit further growth of the metal particles. They subsequently reduced the metal precursors by heating the materials at 360 °C in a forming gas (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :93 H2:N2). Sulfur was removed from the nanocomposites through evacuation at 300 °C for 12 h. After annealing at 600 °C under Ar atmosphere, they successfully prepared 3.5 nm intermetallic PtBi nanoparticles that were finely dispersed on the CMK-3 surface. The electrochemical results of materials under half-cell conditions demonstrated that PtBi/CMK-3 had both high mass activity and excellent long-term stability.
:93 H2:N2). Sulfur was removed from the nanocomposites through evacuation at 300 °C for 12 h. After annealing at 600 °C under Ar atmosphere, they successfully prepared 3.5 nm intermetallic PtBi nanoparticles that were finely dispersed on the CMK-3 surface. The electrochemical results of materials under half-cell conditions demonstrated that PtBi/CMK-3 had both high mass activity and excellent long-term stability.
As described earlier, the CASH method could be a versatile way to design a corrosion-resistant metal oxide support material because it allows us to synthesize a highly crystalline mesoporous metal oxide with a simple method. Noteworthy work adapting CASH was performed by Orilall et al., who suggested the one-pot synthesis of 10 nm PtPb nanoparticles selectively incorporated into the pore part of a mesoporous niobium–carbon composite with 30 nm pores.147 The use of a diblock copolymer, PI-b-PEO, makes possible the self-assembly of hydrophilic niobium oxide in the PEO block and hydrophobic Pt and Pb precursors in the PS block. Heat treatment at 700 °C leads to the synthesis of PtPb nanoparticles incorporated into a niobium oxide–carbon composite, by the simultaneous removal of the PEO part and the carbonization of the PS part. When the resulting material was tested as an electrocatalyst for formic acid oxidation, it showed an onset potential of −0.25 V vs. (3 M) Ag/AgCl and a mass activity of 3.3 mA μg−1, which are 60 mV more negative onset potential and 4 times higher mass activity than those of PtPb nanoparticles supported on carbon. When Pt nanoparticles incorporated into Nb2O5 were synthesized using the same method, Pt/Nb2O5 showed a one order of magnitude lower mass activity for the formic acid electro-oxidation.
Since the advancement of the design and fabrication of materials is expected to enhance the performance of catalysts, it is very helpful to test the quality of nanomaterials under more realistic (i.e., single cell) conditions. Lee and co-workers recently reported the one-pot synthesis of well-dispersed 10 nm intermetallic PtPb nanoparticles incorporated into the channels of large-pore (>30 nm) mesoporous carbon–silica composites.148 They employed a diblock copolymer, PS-b-PEO, as the structure directing agent. As shown in Fig. 12, PS-b-PEO co-assembled hydrophobic Pt and Pb precursors as well as hydrophobic carbon and silica precursors, yielding a nanocomposite with small intermetallic nanoparticles well-dispersed in ordered channels of OMCs. They found that PS-b-PEO played a critical role in achieving fine dispersion, by showing that the use of the commercial triblock copolymer F127 did not allow high dispersion, which resulted in larger intermetallics with a size of ∼30 nm. In the single cell tests at 50 °C, the resulting PtPb–OMCs showed higher maximum power density (239.8 mW mgPtPb−1) than that of Pt/C (156.63 mW mgPt−1). In addition, MEA durability tests were performed by applying 0.4 V for 6 h at 50 °C, confirming the enhanced durability of PtPb–OMCs when compared to that of Pt/C and Pd/C. The enhanced durability could be attributed to the high CO tolerance of PtPb intermetallic nanoparticles. These results demonstrated that PtPb–OMC catalysts have high potential for application in real fuel cell catalysts for DFAFCs. Because these one-pot methods employing lab-synthesized diblock copolymers generate highly ordered alloys or intermetallic nanoparticles incorporated into mesoporous carbon or crystalline metal oxides, they could open up great opportunities for the design of active and durable catalysts with the desired morphologies.
 | ||
| Fig. 12 Schematic figure showing the one-pot synthesis of intermetallic PtPb nanoparticles incorporated into ordered large-pore mesoporous carbon–silica composites (reprinted with permission from ref. 148, Copyright American Chemical Society, 2012). | ||
Ordered mesoporous materials for lithium ion battery applications
Lithium ion batteries (LIBs) have received considerable attention as one of the most promising power sources for mobile devices and electric vehicles (EVs) because of their high energy/power densities and long cycle life.8,149,150 Various approaches have been used in the battery field to increase the energy/power densities and decrease the weight and volume of such devices. Among the various parts of LIBs such as electrode, electrolyte, separator, and cell design, electrodes have been extensively studied and new electrode materials are proposed because the electrochemical performance strongly depends on the electrode active materials. However, the bulk form of the new active materials limits full utilization of all crystallites at high current density because of the low surface-to-volume ratio and the long diffusion length for lithium ions and electrons. As a result, nanoengineering approaches have received much attention because nanoscale materials have high electrode–electrolyte interface areas and short diffusion lengths for ions and electrons. Kinetically, the time constant (τ) for the solid-state diffusion of ions and electrons within the electrode material is associated with the diffusion length (L) and diffusion coefficient (D) (τ = L2/2D).151,152 Therefore, in order to reduce the diffusion length, many researchers have introduced nanoarchitecture into the active materials and have confirmed that the nanoactive materials exhibit a higher capacity and better rate performance than the same materials in the bulk form.153,154 However, it is natural that nanoscale materials also have demerits such as easy aggregation due to the high surface energy of nanomaterials, low coulombic efficiency from the formation of large-area solid electrolyte interface, and low volumetric energy density because of the low packing density. Among the representative solutions, a surface-coating method using carbon, SiO2, TiO2, MgO, Al2O3, and other materials has been introduced to prevent nanomaterial-related problems. The challenging issue of nanomaterials for LIB electrodes remains.155–159 Meanwhile, nanomaterials effectively relieve the stress caused by volume expansion during the lithium insertion and extraction cycles, resulting in a long cycle life. Nanostructured materials with various structures, including 0-D (nanoparticles), 1-D (nanowires, nanofibers, and nanotubes), 2-D (nanosheets), and 3-D (porous and hierarchical structure), have been applied as an electrode material in LIBs; studies on their applications are covered by many review articles.151,160–164 In this article, among many nanomaterials, we intensively discuss the utilization of ordered mesoporous structured materials as high performance electrodes in LIBs.Micrometer-size mesoporous materials with ordered nanopore arrays (2–50 nm) have many unique features152,165–167 (Fig. 13): (i) an electrolyte easily penetrates the 2-D or 3-D mesopores, which constitute a high electrolyte–electrode interface area. (ii) Mesopore walls that are a few nanometers thick (a small crystalline structure develops because of the nano-confinement effect) are more advantageous for the fast diffusion of lithium ions and electrons than the bulk-materials. (iii) The micrometer-size interconnected porous structures reveal a higher electrical conduction compared to separated nanoparticles. Therefore, less carbon additive is required to provide sufficient electric contacts in the case of a mesoporous active material. (iv) While micrometer-size mesoporous materials are effectively packed, nanometer-size materials suffer from a low packing density, which leads to a low volumetric capacity. (v) Active materials are more easily lost from the electrode in a separate nanoparticle format than in a mesoporous format during cycles. They can also pass through the separator. Moreover, as a host, mesoporous materials can retain dissolvable active materials such as polysulfides and organic radical polymers.
 | ||
| Fig. 13 Schematic representation of merits of mesoporous materials as electrodes. | ||
Because of these merits, mesoporous materials have been extensively studied for both anodes and cathodes to develop high performance LIBs. For example, attempts have been made to use the hard template method for various crystalline metal oxides. Bruce's group reported the synthesis and battery application of highly ordered mesoporous structured LiCoO2, β-MnO2, Li1.12Mn1.88O4, and TiO2 (anatase) by employing mesoporous silica (SBA-15 or KIT-6) as a template.166,168–170 Through a direct comparison with bulk materials, they demonstrated that the mesoporous structured materials exhibited higher capacities, faster rate performances, and enhanced cyclabilities compared to the bulk materials. Moreover, in some cases, a mesoporous active material reversibly accommodated a large amount of lithium ion intercalation, which was almost impossible in the bulk form of the material.169 In addition, many researchers have synthesized different types of mesoporous electrode materials171–174 and even intrinsically improved mesoporous materials through high temperature reduction or doping, along with the hard template method.175–177
However, although the hard template method seems to be a generic way to synthesize various mesoporous metal oxides, this method requires a long time and multiple steps. The removal of the silica template, using hydrofluoric acid (HF), is quite tedious and dangerous. In addition, the reaction between the lithium precursors and the silica template makes it difficult to synthesize normal cathode materials (lithium metal oxides) through a hard template approach. Therefore, additional treatment of mesoporous metal oxides (after silica etching) with lithium precursors is required.170,178 From a practical point of view, the soft template method using commercially available block copolymers is more useful for producing mesoporous electrode materials.179–184 Although there are many reports about the high performance mesoporous metal oxide electrode materials synthesized at low temperature (below 600 °C),39,54,185 the soft template approach has some limits, especially when the process requires high synthetic temperature for the formation of the fully crystalline metal oxides.179,180 For these reasons, the CASH method, which combines the strengths of the hard and soft template methods, has been suggested as an alternative method for the synthesis of mesoporous metal oxide active materials. This will be discussed later in the article.
Meanwhile, high capacitive electrode materials such as metals or metal oxides, sulfur, and polymers usually suffer from structural instability as a result of the aggregation, cracks, pulverization, dissolution, etc. that occur during the lithium insertion–extraction cycles. Therefore, in order to stabilize the active materials, ordered mesoporous structured carbon materials have been employed as host materials, providing not only ordered void pores for the volume change of guest materials, but also sufficient space to prevent the dissolution of active materials during cycling.182,186–188 This section also introduces the host–guest approach that uses highly conductive mesoporous carbon materials.
Ordered mesoporous active materials using the CASH method
As previously discussed, the CASH method is well suited for the synthesis of mesoporous active materials because a high temperature synthesis is possible with a very simple synthetic route. Because of the role of the sp2-hybridized carbon containing block, block copolymers allow the formation of crystallites without structural collapse at a high temperature (>700 °C). Moreover, the carbon coating (carbonization of the sp2-hybridized carbon block) on the surface of the metal oxide wall provides enough electrical conductivity and stability during the battery cycles. Hence, we expect that various mesoporous lithium metal oxides (or metal oxides), requiring high synthetic temperatures, can easily be synthesized using this method.As the first example of the use of the CASH method for the synthesis of electrode materials for LIBs, Wiesner and co-workers synthesized a highly crystalline TiO2 and carbon composite by using the copolymer PI-b-PEO as a structure directing agent.189 In order to increase the electrical conductivity of the composite material, carbon precursors (resol) were added to the block copolymer and titanium dioxide solution. The resol and TiO2 precursors interacted with the hydrophilic PEO part and formed a metal oxide–carbon wall after heat treatment at 700 °C. The pore sizes were tuned by changing the molecular weights using the precursors of TiO2 and carbon. As an anode, the mesoporous TiO2–carbon electrode showed better performance (lithium desertion of 0.55 Li/Ti) compared to pure mesoporous TiO2 (0.30 Li/Ti) or non-porous TiO2 (0.15 Li/Ti) samples. Moreover, the mesoporous TiO2–carbon electrode exhibited a stable cycle performance for 50 cycles. This stability was attributed to the nanoporous structure effect, which facilitated electrolyte diffusion and buffered volume changes during the cycles, along with enhanced electrical conductivity from the carbon wiring effect.
As another example, the CASH method was employed to synthesize an ordered mesostructured Li4Ti5O12–carbon nanocomposite, denoted as Meso-LTO–C, using the copolymer PI-b-PEO.190 The spinel Li4Ti5O12 has attracted considerable attention because it has several merits. For instance, its negligible volume expansion/contraction (<1%) and high voltage potential range (minimized side reaction) of ∼1.55 V (vs. Li/Li+) result in good safety and durability, making it applicable to electric vehicles (EVs). However, its major demerit is the electrically insulating (<10−13 S cm−1) nature of the LTO.
Recently, Feckl et al. synthesized an ultrasmall LTO with a porous structure via a solvothermal reaction in tert-butanol solution.39 In their study, the Pluronic polymer in the tert-butanol solution formed a porous structure and assisted the construction of the single-phase LTO. This LTO electrode was very impressive because it showed excellent rate capability: 175 mA h g−1 at C-rates of 1–50 C and 72% of capacity at a C-rate of 400 C. However, in a typical synthesis process, LTOs and other lithium metal oxides require a high temperature above 700 °C, making it infeasible to synthesize mesoporous LTOs at high temperatures using a soft template method. In order to overcome these drawbacks, a facile CASH method was applied. The in situ generated carbon from the PI chain covers the surface of the crystallized metal oxide and acts as a rigid support to prevent structural collapse at a high temperature (700 °C). This method was used to synthesize Meso-LTO–C with large uniform pores (>15 nm) and a high surface area (68.7 m2 g−1).
To study its anode performance, Meso-LTO–C/Li coin cells were tested at 1.0–2.5 V using two different electrode compositions (80:10:10 and 90:0:10, weight ratios of LTO–C powder, conductive additive (carbon black), and binder (PVDF)). As seen in Fig. 14, the Meso-LTO–C electrodes exhibited a higher capacity (144 mA h g−1) than micrometer-size LTO (bulk-LTO, 130 mA h g−1) at 0.2 C. Interestingly, in the absence of a conductive aid condition, a definite capacity difference was observed (Meso-LTO–C: 141 mA h g−1, bulk-LTO: 61 mA h g−1). Moreover, at a 10 C rate, the Meso-LTO–C with a carbon additive showed a high capacity of 115 mA h g−1 (69 mA h g−1 in the bulk-LTO electrode) and 90% of the initial capacity was maintained after 500 cycles.190 The high rate performance of Meso-LTO–C was also revealed by an electrochemical impedance spectroscopy (EIS) test using symmetric LTO/LTO cells. A highly improved electrical contact and reduced charge transfer resistance were observed in the Meso-LTO–C symmetric cell when compared with bulk-LTO. The results demonstrated that the electrical conductivity of the nanoporous structure is a key to high power performance. Therefore, due to the flexibility of this method, the simple CASH method can be regarded as a general route for the synthesis of mesoporous lithium metal oxides, requiring a high crystalline formation temperature and high electrical conductivity, to enhance the rate and cycle performance of LIB electrodes.
 | ||
| Fig. 14 Schematic illustration of preparation of Meso-LTO–C and cycle performance of (b) bulk-LTO/Li and (c) Meso-LTO–C/Li cells with C-rate provided on top axis (within the potential range of 1.0–2.5 V (vs. Li/Li+)) (reprinted with permission from ref. 190, Copyright Wiley-VCH, 2011). | ||
Ordered mesoporous carbon as a host material in a host–guest approach
The ordered mesoporous materials can act as host materials for the effective uptake of high-capacity guests. When used as anode materials, transition-metal oxides (with high capacities of up to 1000 mA h g−1) usually suffer from severe volume-change and aggregation problems, resulting in poor charge–discharge cycle performance. Moreover, most of the metal oxides have low electrical conductivity, requiring nanoengineering, surface coating, or composite structures. The mesoporous carbon materials can relieve these problems because nanosized transition-metal oxides are located inside the conductive carbon nanopores, and the pores prevent the aggregation of transition-metal oxides. In addition, mesopores provide an empty space for large volume changes in the transition-metal oxides, while there is little room for lithiated transition-metal oxides in the case of surface carbon-coated nanomaterials. Owing to these scenarios, various transition-metal oxides have been introduced into the mesoporous carbon pores.182,191 However, most mesoporous materials would not be appropriate hosts for transition-metal oxides because of their limited pore size (<10 nm).192 The small pores would limit the access of pre-synthesized guest materials. In addition, even after the loading of guest materials, the diffusion of the electrolyte into the active materials may be hindered within the small pores. Therefore, mesoporous carbon with large pore size is more pertinent as an effective host material than mesoporous carbon with small pore size.As one example, recently, Kang et al. synthesized Fe3O4 nanocrystals confined in a mesocellular carbon foam (MSU-F-C) using a host–guest approach and applied it as an anode material for LIBs.193 In this composite structure, MSU-F-C115 provides not only large pores (26 nm) that act as a buffer space for the volume change of Fe3O4 particles. Moreover, in this study, an ultrathin (<1 nm) conformal Al2O3 coating was applied using an atomic layer deposition (ALD) process in order to improve the durability and rate performance of composite materials. The Al2O3 ALD coating was carried out on an Fe3O4–MSU-F-C-61 (61 wt% Fe3O4) composite electrode by using trimethylaluminum (TMA) and H2O as vapor phase precursors. In addition, in order to evenly apply the Al2O3 layer on the inert basal plane of the carbon surface, a TMA/NO2 pretreatment was also performed (before the TMA–H2O ALD cycles). Fig. 15a and b show TEM images of the bare MSU-F-C and Fe3O4–MSU-F-C-61. As shown in Fig. 15b, the Fe3O4 (about 20 nm) nanoparticles are mainly located inside the large pores. This structure is beneficial for a battery electrode material because the electrolyte and Fe3O4 nanoparticles easily meet inside the pores, and the carbon host can absorb the volume expansion stress of the nanoparticles during the cycles, resulting in a high rate capability and high durability.
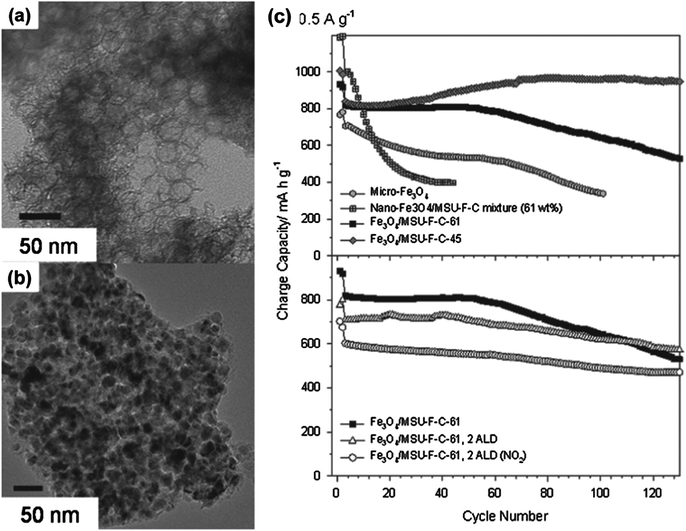 | ||
| Fig. 15 TEM images of (a) pure MSU-F-C and (b) Fe3O4–MSU-F-C-61. (c) Cycle performances of Fe3O4–MSU-F-Cs and ALD-coated Fe3O4–MSU-F-C-61 electrodes at 0.5 A g−1 (reprinted with permission from ref. 193, Copyright Wiley-VCH, 2011). | ||
The electrochemical performance of Fe3O4–MSU-F-C-61 exhibited a high first discharge (Li+ insertion) capacity of 1357 mA h g−1 at a 0.1 A g−1 current density, which is higher than the theoretical capacity of pure Fe3O4 (926 mA h g−1). This high discharge capacity of composite materials is partly attributed to MSU-F-C. The large charging capacity of MSU-F-C was also confirmed (983 mA h g−1).193 The irreversible capacity from the high surface area of the composite material was reduced by the ALD coating with and without the TMA/NO2 pretreatment. Fig. 15c shows the cycling performance of the Fe3O4–MSU-F-Cs and ALD-coated electrodes. The test was performed at 0.1 A g−1 for the first two cycles and 0.5 A g−1 for the following cycles in the range of 0.01–3.00 V (vs. Li/Li+). Unlike micro-Fe3O4 (<5 μm) and a mixture of Fe3O4 nanoparticles (∼13 nm) and MSU-F-C (61:39 weight ratio), the Fe3O4–MSU-F-C electrodes revealed excellent cycle durability. In addition, although lower capacities were obtained, the ALD-coated electrodes with and without TMA/NO2 pretreatment showed enhanced cycle performances. These results confirm that intimate contact between the nanoparticles and carbon surface in a nano-architecture lessens the volume change stress of Fe3O4 during cycling. Moreover, the Al2O3 surface coating with the ALD process protects the electrode by restraining undesirable side reactions.
Meanwhile, mesoporous materials have also been accepted as hosts for electrically insulating materials or easily dissolved active materials. In Li–S batteries, the polysulfides formed during cycling are easily dissolved from the positive electrode, and they migrate to the negative electrode through the separator, reducing the capacity. Moreover, the electrical conductivity of sulfur is also very low. In response to these problems, various mesoporous carbons have been introduced as sulfur host materials in Li–S batteries.194–199 Nazar and co-workers reported the application of a mesoporous carbon–sulfur nanocomposite in a Li–S cathode.200 As seen in Fig. 16, about 70 wt% of sulfur was trapped inside the 3 nm pores of CMK-3. The carbon host provided a highly interconnected pore structure for good electrical contact to the sulfur, as well as a large pore volume for sulfur trapping. In a coin cell test, a CMK-3–S electrode exhibited a high capacity of up to 1320 mA h g−1, whereas a mechanical mixture of CMK-3 and sulfur only had a value of 390 mA h g−1. In the case of the CMK-3–S electrode, decreased polarization was discovered when compared to the mechanical mixture because of the high electrical contact between sulfur and the carbon host. Moreover, a very high coulombic efficiency (99.94%) was shown in the first charge–discharge cycle, signifying the low dissolution of polysulfide in the carbon host. To prevent leaching of the guest in the Nazar study, external surface functionalization was performed on the CMK-3–S nanocomposite using a hydrophilic polyethylene glycol coating. The dissolution of polysulfide from the electrode was hindered by this surface treatment, resulting in a high cycling stability.
 | ||
| Fig. 16 Schematic diagrams of (a) a CMK-3–S nanocomposite and (b) synthesis and redox process of CMK-3–S (reprinted with permission from ref. 200, Copyright Nature Publishing Group, 2009). | ||
Spherical ordered mesoporous carbon nanoparticles are another example of a sulfur host. They were synthesized using inverse-opal silica as a template.196 By loading precursors such as resol (carbon precursor), tetraethylorthosilicate (SiO2 precursor), and Pluronic F127 block copolymer into the inverse opal template, spherical OMCs (diameter: 300 nm) were synthesized after the carbonization and silica-etching processes. This hierarchical mesoporous carbon had a high surface area (2445 m2 g−1) and inner pore volume (2.32 cm3 g−1) with bimodal pores (6 and 3.1 nm). As shown in Fig. 17a and b, the C–S (denoted as S-BMC–S-50, 50 wt% sulfur) electrode exhibited high reversible discharge capacity of 1200 mA h g−1 and 730 mA h g−1 after 100 cycles at a charge–discharge C-rate of 1 C. Despite increased sulfur content (up to 70 wt%), the electrochemical performance of the C–S samples was not affected, and the samples exhibited excellent cycle stability. Moreover, in order to relieve the overcharge capacity, two methods such as SC2 treatment and SiOx coating were introduced. These studies suggested that highly conductive mesoporous carbon can form high-performance sulfur electrodes because the carbon host provides sufficient electrical conduction and holds the soluble polysulfides inside the pores.
 | ||
| Fig. 17 (a) Cycle stability of 70 wt% sulfur in a spherical OMC, as prepared (S-BMC/S-70), washed (S-BMC/S-70-W) and SiOx coated (S-BMC-70-Si). (b) Initial charge–discharge profiles of electrodes (reprinted from permission from ref. 196, Copyright Wiley-VCH, 2012). | ||
Conclusions and outlook
Functional ordered mesoporous materials with various pore structures and wall compositions have been specifically designed to improve the performances of electrodes in solar cells, fuel cells, and batteries. The present feature article has focused on the recent developments in the preparation and further functionalization of ordered mesoporous materials as electrode materials in dye-sensitized solar cells, quantum-dot sensitized solar cells, polymer-membrane-electrolyte fuel cells, and Li-ion batteries. The developed soft and hard template methods, along with the CASH method, provide pore size control, controlled wall compositions, highly interconnected surface areas, and pore structure control, which allow the optimization of electrode performance in such electrochemical conversion and storage devices.In the near future, it is expected that additional research efforts will be devoted to the synthesis and post-functionalization of new functional ordered mesoporous materials and their applications to such energy fields. For example, some of the very recent studies on systemically designed block copolymers as structure directing agents have proven the concepts of designing mesoporous oxides with fully crystalline walls, mesoporous materials with functional nanocrystals on pores walls, multiple component mesoporous composites in a continuous structure, and hierarchical porous structures at multiple length scales.201 Rapid development of polymer synthesis technology, combined with a better understanding of the fundamental behavior of block copolymers and inorganic precursors in self-assembly would provide breakthroughs in the design of electrode materials.
Some challenges still remain in this field. Firstly, alternative high energy density devices should be further exploited, such as metal–air batteries as an alternative to lithium ion batteries.202 Secondly, a novel synthetic procedure for ordered mesoporous materials should be developed for practical applications. The synthetic procedure needs to meet criteria such as simplicity, mildness, and low cost. In addition, it should provide routes to more complex mesoscopic structures. For example, recent work by Vliet et al. showed that a mesostructured PtNi thin film, comprising large surface areas extended in 2-D thin films with mesoscale architectures, exhibited 20-fold enhanced oxygen reduction reaction activity than a commercial Pt/C catalyst.203 However, this structure is not yet amenable to high-volume manufacturing. From this point of view, a large-scale synthetic procedure for complex mesosopic structures is much in demand.
It is also expected that various functional mesoporous materials will be possible to synthesize and apply to other types of energy conversion such as biomass conversion and CO2 conversion, instead of being limited to solar cells, fuel cells, and batteries.
Acknowledgements
This work was supported by the Global Frontier R&D Program on Center for Multiscale Energy System funded by the National Research Foundation under the Ministry of Education. This work was further supported by National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2012R1A2A2A01002879), and by the second stage of the BK 21 program of Korea.References
- G. Boyle, Renewable Energy: Power for a Sustainable Future, Oxford, 2nd edn, 2004 Search PubMed.
- M. A. Green, IEEE Trans. Electron Devices, 1984, 31, 671–678 CrossRef.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CAS.
- J. Stumper and C. Stone, J. Power Sources, 2008, 176, 468–476 CrossRef CAS.
- R. P. O'Hayre, S. W. Cha, W. Colella and F. B. Prinz, Fuel Cell Fundamentals, John Wiley & Sons, New Jersey, 2006 Search PubMed.
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- F.-F. Cao, Y.-G. Guo and L.-J. Wan, Energy Environ. Sci., 2011, 4, 1634–1642 CAS.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294–1305 RSC.
- K.-Y. Chan, J. Ding, J. Ren, S. Cheng and K. Y. Tsang, J. Mater. Chem., 2004, 14, 505–516 RSC.
- Y. Qiao and C. M. Li, J. Mater. Chem., 2011, 21, 4027–4036 RSC.
- Y. Zhou, M. Eck and M. Kruger, Energy Environ. Sci., 2010, 3, 1851–1864 CAS.
- Q. Zhang, S. Yodyingyong, J. Xi, D. Myers and G. Cao, Nanoscale, 2012, 4, 1436–1445 RSC.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909–4927 RSC.
- J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. Bum Kim, Chem. Commun., 1999, 2177–2178 RSC.
- F. Jiao and P. G. Bruce, Angew. Chem., Int. Ed., 2004, 43, 5958–5961 CrossRef CAS.
- J. Lee, S. Yoon, S. M. Oh, C. H. Shin and T. Hyeon, Adv. Mater., 2000, 12, 359–362 CrossRef CAS.
- W.-C. Li, A.-H. Lu, C. Weidenthaler and F. Schüth, Chem. Mater., 2004, 16, 5676–5681 CrossRef CAS.
- D. M. Antonelli and J. Y. Ying, Angew. Chem., Int. Ed., 1996, 35, 426–430 CrossRef CAS.
- A. Sayari and Y. Yang, J. Phys. Chem. B, 2000, 104, 4835–4839 CrossRef CAS.
- K.-S. Kim, J.-H. Kim and G. Seo, Chem. Commun., 2003, 372–373 RSC.
- J. M. Thomas and R. Raja, Chem. Commun., 2001, 675–687 RSC.
- T. Sen, A. Sebastianelli and I. J. Bruce, J. Am. Chem. Soc., 2006, 128, 7130–7131 CrossRef CAS.
- J. Lee, D. Lee, E. Oh, J. Kim, Y.-P. Kim, S. Jin, H.-S. Kim, Y. Hwang, J. H. Kwak, J.-G. Park, C.-H. Shin, J. Kim and T. Hyeon, Angew. Chem., Int. Ed., 2005, 44, 7427–7432 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS.
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2005, 2125–2127 RSC.
- F. Zhang, Y. Meng, D. Gu, Y. Yan, C. Yu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef CAS.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS.
- Y. Huang, H. Cai, T. Yu, F. Zhang, F. Zhang, Y. Meng, D. Gu, Y. Wan, X. Sun, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2007, 46, 1089–1093 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schuth and G. D. Stucky, Nature, 1994, 368, 317–321 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, D. G. Demuth, P. Feng, T. E. Gier, P. Sieger, A. Firouzi and B. F. Chmelka, Chem. Mater., 1994, 6, 1176–1191 CrossRef CAS.
- Y. Li, G. L. Pan, J. W. Liu and X. P. Gao, J. Electrochem. Soc., 2009, 156, A495–A499 CrossRef CAS.
- J. Haetge, P. Hartmann, K. Brezesinski, J. Janek and T. Brezesinski, Chem. Mater., 2011, 23, 4384–4393 CrossRef CAS.
- J. M. Feckl, K. Fominykh, M. Döblinger, D. Fattakhova-Rohlfing and T. Bein, Angew. Chem., Int. Ed., 2012, 51, 7459–7463 CrossRef CAS.
- A. Corma, P. Atienzar, H. Garcia and J. Y. Chane-Ching, Nat. Mater., 2004, 3, 394–397 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS.
- K. Zhu, B. Yue, W. Zhou and H. He, Chem. Commun., 2003, 98–99 RSC.
- B. Tian, X. Liu, H. Yang, S. Xie, C. Yu, B. Tu and D. Zhao, Adv. Mater., 2003, 15, 1370–1374 CrossRef CAS.
- S. C. Laha and R. Ryoo, Chem. Commun., 2003, 2138–2139 RSC.
- F. Jiao, J.-C. Jumas, M. Womes, A. V. Chadwick, A. Harrison and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 12905–12909 CrossRef CAS.
- J. Lee, M. Christopher Orilall, S. C. Warren, M. Kamperman, F. J. DiSalvo and U. Wiesner, Nat. Mater., 2008, 7, 222–228 CrossRef CAS.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS.
- J. Ba, J. Polleux, M. Antonietti and M. Niederberger, Adv. Funct. Mater., 2005, 17, 2509–2512 CrossRef CAS.
- I. E. Rauda, R. Buonsanti, L. C. Saldarriaga-Lopez, K. Benjauthrit, L. T. Schelhas, M. Stefik, V. Augustyn, J. Ko, B. Dunn, U. Wiesner, D. J. Milliron and S. H. Tolbert, ACS Nano, 2012, 6, 6386–6399 CrossRef CAS.
- E. K. Richman, C. B. Kang, T. Brezesinski and S. H. Tolbert, Nano Lett., 2008, 8, 3075–3079 CrossRef CAS.
- Y. Wang, T. Brezesinski, M. Antonietti and B. Smarsly, ACS Nano, 2009, 3, 1373–1378 CrossRef CAS.
- C. Weidmann, K. Brezesinski, C. Suchomski, K. Tropp, N. Grosser, J. Haetge, B. M. Smarsly and T. Brezesinski, Chem. Mater., 2011, 24, 486–494 CrossRef.
- N. Krins, J. D. Bass, D. Grosso, C. Henrist, R. Delaigle, E. M. Gaigneaux, R. Cloots, B. n. d. Vertruyen and C. m. Sanchez, Chem. Mater., 2011, 23, 4124–4131 CrossRef CAS.
- E. Ortel, A. Fischer, L. Chuenchom, J. Polte, F. Emmerling, B. Smarsly and R. Kraehnert, Small, 2012, 8, 298–309 CrossRef CAS.
- A. Thomas, H. Schlaad, B. Smarsly and M. Antonietti, Langmuir, 2003, 19, 4455–4459 CrossRef CAS.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef.
- T. W. Hamann, R. A. Jensen, A. B. F. Martinson, H. Van Ryswyk and J. T. Hupp, Energy Environ. Sci., 2008, 1, 66–78 CAS.
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2006, 7, 69–74 CrossRef.
- S. Ahmad, J. H. Yum, H. J. Butt, M. K. Nazeeruddin and M. Grätzel, ChemPhysChem, 2010, 11, 2814–2819 CrossRef CAS.
- H. Wu, Z. Lv, Z. Chu, D. Wang, S. Hou and D. Zou, J. Mater. Chem., 2011, 21, 14815–14820 RSC.
- Y. Liu, J. R. Jennings, M. Parameswaran and Q. Wang, Energy Environ. Sci., 2011, 4, 564–571 CAS.
- M. Wang, N. Chamberland, L. Breau, J.-E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389 CrossRef CAS.
- O. K. Varghese, M. Paulose and C. A. Grimes, Nat. Nanotechnol., 2009, 4, 592–597 CrossRef CAS.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS.
- S. H. Ko, D. Lee, H. W. Kang, K. H. Nam, J. Y. Yeo, S. J. Hong, C. P. Grigoropoulos and H. J. Sung, Nano Lett., 2011, 11, 666–671 CrossRef CAS.
- S. H. Kang, S. H. Choi, M. S. Kang, J. Y. Kim, H. S. Kim, T. Hyeon and Y. E. Sung, Adv. Mater., 2008, 20, 54–58 CrossRef CAS.
- Q. Zhang, T. P. Chou, B. Russo, S. A. Jenekhe and G. Cao, Angew. Chem., Int. Ed., 2008, 47, 2402–2406 CrossRef CAS.
- D. Chen, F. Huang, Y.-B. Cheng and R. A. Caruso, Adv. Mater., 2009, 21, 2206–2210 CrossRef CAS.
- Y. F. Wang, J. W. Li, Y. F. Hou, X. Y. Yu, C. Y. Su and D. B. Kuang, Chem.–Eur. J., 2010, 16, 8620–8625 CrossRef CAS.
- J.-Y. Liao, B.-X. Lei, D.-B. Kuang and C.-Y. Su, Energy Environ. Sci., 2011, 4, 4079–4085 CAS.
- H. J. Koo, Y. J. Kim, Y. H. Lee, W. I. Lee, K. Kim and N. G. Park, Adv. Mater., 2008, 20, 195–199 CrossRef CAS.
- J. Qian, P. Liu, Y. Xiao, Y. Jiang, Y. Cao, X. Ai and H. Yang, Adv. Mater., 2009, 21, 3663–3667 CrossRef CAS.
- M. Zukalová, A. Zukal, L. Kavan, M. K. Nazeeruddin, P. Liska and M. Grätzel, Nano Lett., 2005, 5, 1789–1792 CrossRef.
- M. Wei, Y. Konishi, H. Zhou, M. Yanagida, H. Sugihara and H. Arakawa, J. Mater. Chem., 2006, 16, 1287–1293 RSC.
- E. Ramasamy and J. Lee, J. Phys. Chem. C, 2010, 114, 22032–22037 CAS.
- M. Nedelcu, J. Lee, E. J. W. Crossland, S. C. Warren, M. C. Orilall, S. Guldin, S. Huttner, C. Ducati, D. Eder, U. Wiesner, U. Steiner and H. J. Snaith, Soft Matter, 2009, 5, 134–139 RSC.
- K. Pan, W. Zhou, G. Tian, Q. Pan, C. Tian, T. Xie, Y. Dong, D. Wang and H. Fu, Eur. J. Inorg. Chem., 2011, 2011, 4730–4737 CrossRef CAS.
- E. Ramasamy and J. Lee, Energy Environ. Sci., 2011, 4, 2529–2536 CAS.
- S. H. Ahn, W. S. Chi, J. T. Park, J. K. Koh, D. K. Roh and J. H. Kim, Adv. Funct. Mater., 2012, 24, 519–522 CrossRef CAS.
- E. Ramasamy and J. Lee, Carbon, 2010, 48, 3715–3720 CrossRef CAS.
- J. H. Pan, X. S. Zhao and W. I. Lee, Chem. Eng. J., 2011, 170, 363–380 CrossRef CAS.
- E. Ramasamy and J. Lee, Chem. Commun., 2010, 46, 2136–2138 RSC.
- A. S. Nair, Z. Peining, V. J. Babu, Y. Shengyuan and S. Ramakrishna, Phys. Chem. Chem. Phys., 2011, 13, 21248–21261 RSC.
- P. Poudel and Q. Qiao, Nanoscale, 2012, 4, 2826–2838 RSC.
- P. Roy, D. Kim, K. Lee, E. Spiecker and P. Schmuki, Nanoscale, 2010, 2, 45–59 RSC.
- S. Phadke, A. Du Pasquier and D. P. Birnie, J. Phys. Chem. C, 2011, 115, 18342–18347 CAS.
- S. Ahmed, A. Du Pasquier, D. P. Birnie and T. Asefa, ACS Appl. Mater. Interfaces, 2011, 3, 3002–3010 CAS.
- E. N. Kumar, R. Jose, P. S. Archana, C. Vijila, M. M. Yusoff and S. Ramakrishna, Energy Environ. Sci., 2012, 5, 5401–5407 CAS.
- A. Kay and M. Grätzel, Chem. Mater., 2002, 14, 2930–2935 CrossRef CAS.
- C. Prasittichai and J. T. Hupp, J. Phys. Chem. Lett., 2010, 1, 1611–1615 CrossRef CAS.
- X. Dou, D. Sabba, N. Mathews, L. H. Wong, Y. M. Lam and S. Mhaisalkar, Chem. Mater., 2011, 23, 3938–3945 CrossRef CAS.
- P. K. Nayak, G. Garcia-Belmonte, A. Kahn, J. Bisquert and D. Cahen, Energy Environ. Sci., 2012, 5, 6022–6039 CAS.
- T. K. Yun, S. S. Park, D. Kim, Y.-K. Hwang, S. Huh, J. Y. Bae and Y. S. Won, J. Power Sources, 2011, 196, 3678–3682 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Sol. Energy Mater. Sol. Cells, 2006, 90, 1331–1344 CrossRef CAS.
- K. D. Benkstein, N. Kopidakis, J. van de Lagemaat and A. J. Frank, J. Phys. Chem. B, 2003, 107, 7759–7767 CrossRef CAS.
- J. Bisquert, Phys. Chem. Chem. Phys., 2008, 10, 49–72 RSC.
- M. Templin, A. Franck, A. Du Chesne, H. Leist, Y. Zhang, R. Ulrich, V. Schädler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS.
- F. Jiao, A. Harrison, J.-C. Jumas, A. V. Chadwick, W. Kockelmann and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 5468–5474 CrossRef CAS.
- S. Guldin, S. Hüttner, P. Tiwana, M. C. Orilall, B. Ülgüt, M. Stefik, P. Docampo, M. Kolle, G. Divitini, C. Ducati, S. A. T. Redfern, H. J. Snaith, U. Wiesner, D. Eder and U. Steiner, Energy Environ. Sci., 2011, 4, 225–233 CAS.
- Y. Zhang, Z. Xie and J. Wang, ACS Appl. Mater. Interfaces, 2009, 1, 2789–2795 CAS.
- P. Docampo, S. Guldin, M. Stefik, P. Tiwana, M. C. Orilall, S. Hüttner, H. Sai, U. Wiesner, U. Steiner and H. J. Snaith, Adv. Funct. Mater., 2010, 20, 1787–1796 CrossRef CAS.
- P. Docampo, M. Stefik, S. Guldin, R. Gunning, N. A. Yufa, N. Cai, P. Wang, U. Steiner, U. Wiesner and H. J. Snaith, Adv. Energy Mater., 2012, 2, 676–682 CrossRef CAS.
- M. Wu and T. Ma, ChemSusChem, 2012, 5, 1343–1357 CrossRef CAS.
- E. Ramasamy, W. J. Lee, D. Y. Lee and J. S. Song, Electrochem. Commun., 2008, 10, 1087–1089 CrossRef CAS.
- M. Wu, X. Lin, T. Wang, J. Qiu and T. Ma, Energy Environ. Sci., 2011, 4, 2308–2315 CAS.
- L. Kavan, J. H. Yum and M. Grätzel, Nano Lett., 2011, 11, 5501–5506 CrossRef CAS.
- Q. Tai, B. Chen, F. Guo, S. Xu, H. Hu, B. Sebo and X.-Z. Zhao, ACS Nano, 2011, 5, 3795–3799 CrossRef CAS.
- X. Xin, M. He, W. Han, J. Jung and Z. Lin, Angew. Chem., Int. Ed., 2011, 50, 11739–11742 CrossRef CAS.
- M. Wu, X. Lin, A. Hagfeldt and T. Ma, Angew. Chem., Int. Ed., 2011, 50, 3520–3524 CrossRef CAS.
- G. R. Li, J. Song, G. L. Pan and X. P. Gao, Energy Environ. Sci., 2011, 4, 1680–1683 CAS.
- T. N. Murakami, S. Ito, Q. Wang, M. K. Nazeeruddin, T. Bessho, I. Cesar, P. Liska, R. Humphry-Baker, P. Comte, P. Pechy and M. Grätzel, J. Electrochem. Soc., 2006, 153, A2255–A2261 CrossRef CAS.
- J. Lee, K. Sohn and T. Hyeon, Chem. Commun., 2002, 2674–2675 RSC.
- M. Seol, E. Ramasamy, J. Lee and K. Yong, J. Phys. Chem. C, 2011, 115, 22018–22024 CAS.
- E. Ramasamy, J. Chun and J. Lee, Carbon, 2010, 48, 4563–4565 CrossRef CAS.
- T. Daeneke, T.-H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211–215 CrossRef CAS.
- T. C. Li, A. M. Spokoyny, C. She, O. K. Farha, C. A. Mirkin, T. J. Marks and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 4580–4582 CrossRef CAS.
- E. Ramasamy, C. Jo, A. Anthonysamy, I. Jeong, J. K. Kim and J. Lee, Chem. Mater., 2012, 24, 1575–1582 CrossRef CAS.
- B. D. James and J. A. Kalinoski, U.S. Department of Energy-Hydrogen Program, 2008, Annual Progress Report, http://www.hydrogen.energy.gov/pdfs/progress08/v_a_2_james.pdf Search PubMed.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- G. S. Chai, I. S. Shin and J.-S. Yu, Adv. Mater., 2004, 16, 2057–2061 CrossRef CAS.
- H. Chang, S. H. Joo and C. Pak, J. Mater. Chem., 2007, 17, 3078–3088 RSC.
- B. Fang, J. H. Kim, M. Kim and J.-S. Yu, Chem. Mater., 2009, 21, 789–796 CrossRef CAS.
- D. Panthea, H. Darmstadt, S. Kaliaguine, L. Summchen and C. Roy, Carbon, 2001, 39, 1147–1158 CrossRef.
- K. W. Park, B. K. Kwon, J. H. Choi, I. S. Park, Y. M. Kim and Y. E. Sung, J. Power Sources, 2002, 109, 439–445 CrossRef CAS.
- T. Gierke, G. Munn and F. Wilson, J. Polym. Sci., Part B: Polym. Phys., 1981, 19, 1687–1704 CrossRef CAS.
- K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2008, 7, 75–83 CrossRef CAS.
- C. K. Knox and G. A. Voth, J. Phys. Chem. B, 2010, 114, 3205–3218 CrossRef CAS.
- Y. J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS.
- H. Tang, Z. Qi, M. Ramani and J. F. Elter, J. Power Sources, 2006, 158, 1306–1312 CrossRef CAS.
- K. H. Lim, H.-S. Oh, S.-E. Jang, Y.-J. Ko, H.-J. Kim and H. Kim, J. Power Sources, 2009, 193, 575–579 CrossRef CAS.
- G. Chen, S. R. Bare and T. E. Mallouk, J. Electrochem. Soc., 2002, 149, A1092–A1099 CrossRef CAS.
- S.-Y. Huang, P. Ganesan, S. Park and B. N. Popov, J. Am. Chem. Soc., 2009, 131, 13898–13899 CrossRef CAS.
- S.-Y. Huang, P. Ganesan and B. N. Popov, ACS Catal., 2012, 2, 825–831 CrossRef CAS.
- E. Kang, S. An, S. Yoon, J. K. Kim and J. Lee, J. Mater. Chem., 2010, 20, 7416–7421 RSC.
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- Y. Doi, A. Takai, Y. Sakamoto, O. Terasaki, Y. Yamauchi and K. Kuroda, Chem. Commun., 2010, 46, 6365–6367 RSC.
- T. T. Emons, J. Li and L. F. Nazar, J. Am. Chem. Soc., 2002, 124, 8516–8517 CrossRef CAS.
- C. Rice, S. Ha, R. I. Masel, P. Waszczuk, A. Wieckowski and T. Barnard, J. Power Sources, 2002, 111, 83–89 CrossRef CAS.
- X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS.
- C. Roychowdhury, F. Matsumoto, V. B. Zeldovich, S. C. Warren, P. F. Mutolo, M. Ballesteros, U. Wiesner, H. D. Abruña and F. J. DiSalvo, Chem. Mater., 2006, 18, 3365–3372 CrossRef CAS.
- E. Casado-Rivera, D. J. Volpe, L. Alden, C. Lind, C. Downie, T. Vázquez-Alvarez, A. C. D. Angelo, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2004, 126, 4043–4049 CrossRef CAS.
- Y. Liu, M. A. Lowe, F. J. DiSalvo and H. c. D. Abruña, J. Phys. Chem. C, 2010, 114, 14929–14938 CAS.
- X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286–293 CrossRef CAS.
- M. C. Orilall, F. Matsumoto, Q. Zhou, H. Sai, H. D. Abruña, F. J. Disalvo and U. Wiesner, J. Am. Chem. Soc., 2009, 131, 9389–9395 CrossRef CAS.
- J. Shim, J. Lee, Y. Ye, J. Hwang, S.-K. Kim, T.-H. Lim, U. Wiesner and J. Lee, ACS Nano, 2012, 6, 6870–6881 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS.
- Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- Y. Ren, A. R. Armstrong, F. Jiao and P. G. Bruce, J. Am. Chem. Soc., 2009, 132, 996–1004 CrossRef.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- W.-M. Zhang, X.-L. Wu, J.-S. Hu, Y.-G. Guo and L.-J. Wan, Adv. Funct. Mater., 2008, 18, 3941–3946 CrossRef CAS.
- D. Arumugam and G. Paruthimal Kalaignan, J. Electroanal. Chem., 2008, 624, 197–204 CrossRef CAS.
- K. A. Walz, C. S. Johnson, J. Genthe, L. C. Stoiber, W. A. Zeltner, M. A. Anderson and M. M. Thackeray, J. Power Sources, 2010, 195, 4943–4951 CrossRef CAS.
- Y. Yu, L. Gu, C. Zhu, S. Tsukimoto, P. A. van Aken and J. Maier, Adv. Mater., 2010, 22, 2247–2250 CrossRef CAS.
- L. J. Fu, H. Liu, C. Li, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Solid State Sci., 2006, 8, 113–128 CrossRef CAS.
- M. G. Kim and J. Cho, Adv. Funct. Mater., 2009, 19, 1497–1514 CrossRef CAS.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS.
- F. Cheng, Z. Tao, J. Liang and J. Chen, Chem. Mater., 2007, 20, 667–681 CrossRef.
- K. T. Lee and J. Cho, Nano Today, 2011, 6, 28–41 CrossRef CAS.
- H. Zhou, D. Li, M. Hibino and I. Honma, Angew. Chem., Int. Ed., 2005, 44, 797–802 CrossRef CAS.
- Y. Ren, L. J. Hardwick and P. G. Bruce, Angew. Chem., Int. Ed., 2010, 49, 2570–2574 CrossRef CAS.
- K. Saravanan, K. Ananthanarayanan and P. Balaya, Energy Environ. Sci., 2010, 3, 939–948 CAS.
- F. Jiao, K. M. Shaju and P. G. Bruce, Angew. Chem., Int. Ed., 2005, 44, 6550–6553 CrossRef CAS.
- F. Jiao and P. G. Bruce, Adv. Mater., 2007, 19, 657–660 CrossRef CAS.
- F. Jiao, J. Bao, A. H. Hill and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 9711–9716 CrossRef CAS.
- H. Zhou, S. Zhu, M. Hibino, I. Honma and M. Ichihara, Adv. Mater., 2003, 15, 2107–2111 CrossRef CAS.
- J.-Y. Luo, J.-J. Zhang and Y.-Y. Xia, Chem. Mater., 2006, 18, 5618–5623 CrossRef CAS.
- H. Kim and J. Cho, J. Mater. Chem., 2008, 18, 771–775 RSC.
- H. Liu, G. Wang, J. Liu, S. Qiao and H. Ahn, J. Mater. Chem., 2011, 21, 3046–3052 RSC.
- Y. Shi, B. Guo, S. A. Corr, Q. Shi, Y.-S. Hu, K. R. Heier, L. Chen, R. Seshadri and G. D. Stucky, Nano Lett., 2009, 9, 4215–4220 CrossRef CAS.
- S. Yoon, C. Jo, S. Y. Noh, C. W. Lee, J. H. Song and J. Lee, Phys. Chem. Chem. Phys., 2011, 13, 11060–11066 RSC.
- X. Fang, B. Guo, Y. Shi, B. Li, C. Hua, C. Yao, Y. Zhang, Y.-S. Hu, Z. Wang, G. D. Stucky and L. Chen, Nanoscale, 2012, 4, 1541–1544 RSC.
- J.-Y. Luo, Y.-G. Wang, H.-M. Xiong and Y.-Y. Xia, Chem. Mater., 2007, 19, 4791–4795 CrossRef CAS.
- Z. Shi, Y. Li, W. Ye and Y. Yang, Electrochem. Solid-State Lett., 2005, 8, A396–A399 CrossRef CAS.
- Z. C. Shi, A. Attia, W. L. Ye, Q. Wang, Y. X. Li and Y. Yang, Electrochim. Acta, 2008, 53, 2665–2673 CrossRef CAS.
- H.-Q. Li, R.-L. Liu, D.-Y. Zhao and Y.-Y. Xia, Carbon, 2007, 45, 2628–2635 CrossRef CAS.
- D. Wang, D. Choi, Z. Yang, V. V. Viswanathan, Z. Nie, C. Wang, Y. Song, J.-G. Zhang and J. Liu, Chem. Mater., 2008, 20, 3435–3442 CrossRef CAS.
- Y. Ishii, Y. Kanamori, T. Kawashita, I. Mukhopadhyay and S. Kawasaki, J. Phys. Chem. Solids, 2010, 71, 511–514 CrossRef CAS.
- Y. Zhou, Y. Kim, C. Jo, J. Lee, C. W. Lee and S. Yoon, Chem. Commun., 2011, 47, 4944–4946 RSC.
- J. Haetge, I. Djerdj and T. Brezesinski, Chem. Commun., 2012, 48, 6726–6728 RSC.
- F. Chen, Z. Shi and M. Liu, Chem. Commun., 2000, 2095–2096 RSC.
- I. Grigoriants, L. Sominski, H. Li, I. Ifargan, D. Aurbach and A. Gedanken, Chem. Commun., 2005, 921–923 RSC.
- Y. Kim, C. Jo, J. Lee, C. W. Lee and S. Yoon, J. Mater. Chem., 2012, 22, 1453–1458 RSC.
- J. Lee, Y. S. Jung, S. C. Warren, M. Kamperman, S. M. Oh, F. J. DiSalvo and U. Wiesner, Macromol. Chem. Phys., 2011, 212, 383–390 CAS.
- E. Kang, Y. S. Jung, G.-H. Kim, J. Chun, U. Wiesner, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 4349–4357 CrossRef CAS.
- M.-Y. Cheng and B.-J. Hwang, J. Power Sources, 2010, 195, 4977–4983 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- E. Kang, Y. S. Jung, A. S. Cavanagh, G.-H. Kim, S. M. George, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 2430–2438 CrossRef CAS.
- S.-R. Chen, Y.-P. Zhai, G.-L. Xu, Y.-X. Jiang, D.-Y. Zhao, J.-T. Li, L. Huang and S.-G. Sun, Electrochim. Acta, 2011, 56, 9549–9555 CrossRef CAS.
- X. Li, Y. Cao, W. Qi, L. V. Saraf, J. Xiao, Z. Nie, J. Mietek, J.-G. Zhang, B. Schwenzer and J. Liu, J. Mater. Chem., 2011, 21, 16603–16610 RSC.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS.
- C. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724–4730 CrossRef CAS.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531–1537 CAS.
- X. Ji, S. Evers, R. Black and L. F. Nazar, Nat. Commun., 2011, 2, 325 CrossRef.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- J.-S. Lee, S. Tai Kim, R. Cao, N.-S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- D. F. van der Vliet, C. Wang, D. Tripkovic, D. Strmcnik, X. F. Zhang, M. K. Debe, R. T. Atanasoski, N. M. Markovic and V. R. Stamenkovic, Nat. Mater., 2012, 11, 1051–1058 CAS.
Footnote |
| † These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2013 |