 Open Access Article
Open Access ArticleNatural product isolation – how to get from biological material to pure compounds
Franz Bucar*a, Abraham Wubea and Martin Schmidb
aInstitute of Pharmaceutical Sciences, Department of Pharmacognosy, University of Graz, Universitätsplatz 4, 8010 Graz, Austria. E-mail: franz.bucar@uni-graz.at; Fax: +43 316 380 9860; Tel: +43 316 380 5531
bInstitute of Pharmaceutical Sciences, Department of Pharmaceutical Chemistry, University of Graz, Graz, Austria
First published on 8th February 2013
Abstract
Covering: 2008 to 2012
Since the last comprehensive review by Otto Sticher on natural product isolation in NPR (O. Sticher, Nat. Prod. Rep., 2008, 25, 517), a plethora of new reports on isolation of secondary compounds from higher plants, marine organisms and microorganisms has been published. Although methods described earlier like the liquid-solid chromatographic techniques (VLC, FC, MPLC, HPLC) or partition chromatographic methods are still the major tools for isolating pure compounds, some developments like hydrophilic interaction chromatography (HILIC) have not been fully covered in previous reviews. Furthermore, examples of using different preparative solid-phase extraction (SPE) columns including molecular imprinting technology have been included. Special attention is given to chiral stationary phases in isolation of natural products. Methods for proper identification of plant material, problems of post-harvest changes in plant material, extraction methods including application of ionic liquids, de-replication procedures during natural product isolation are further issues to be discussed by the review. Selected work published between 2008 and mid-2012 is covered.
 Franz Bucar | Franz Bucar studied pharmacy at the University of Graz, Austria, where he also received his doctoral degree in natural sciences. He performed postdoctoral studies at the School of Pharmacy, University of London, studying alkaloids and flavonoids, and at Uppsala University for anti-inflammatory bioassays. Currently, he is associate professor in pharmacognosy at the Institute of Pharmaceutical Sciences, University of Graz. His main research activities focus on analysis of traditional medicinal plants using bioassay-guided research strategies including antibacterial plant constituents as well as plant natural products as modulators of bacterial resistance. |
 Abraham Wube | Abraham Wube is a senior post-doc researcher at the Department of Pharmacognosy, University of Graz. He obtained his B.Sc. degree in chemistry from Asmara University, Eritrea, his M.Sc. degree in chemistry from Addis Ababa University, Ethiopia, and his Ph.D in natural sciences from University of Graz, studying Ethiopian traditional medicinal plants for anti-inflammatory and antimicrobial compounds. His research interests include investigation of diverse natural products such as alkaloids, quinones, flavonoids and terpenoids from a wide range of higher plant species for their antibacterial, antioxidant, anti-inflammatory, antimalarial and cytotoxic properties as well as synthesis of antimycobacterial quinolones. |
 Martin Schmid | Martin Schmid studied chemistry at the University of Graz, Austria, and received his doctoral degree in natural sciences in terms of pharmaceutical chemistry-drug analysis. He performed his postdoctoral studies at the Universities of Innsbruck, Greifswald, Pécs and at Uppsala University for development of chiral phases. Currently, he is associate professor in pharmaceutical chemistry at the Institute of Pharmaceutical Sciences, University of Graz. His research activities deal with development of new phases and methods for enantioseparation by chromatographic and electrophoretic techniques. Furthermore, his scientific interest is devoted to characterisation and separation of new drugs of abuse. |
1 Introduction
Despite of substantial developments of extraction and separation techniques, isolation of natural products (NPs) from plants, marine organisms or microorganisms is still a challenging task. Undoubtedly hybrid methods like LC-NMR or LC-MS made on-line structure elucidation possible and provided impressive examples of NP identification without prior isolation,2 however, in many cases the necessity to get the purified compounds in hand is still a fact. Full chemical structures including stereochemistry of new NPs most likely need isolated, highly purified compounds, however the amounts needed have significantly decreased and 2D NMR spectra of small molecules now can be obtained with less than 100 μg within a reasonable time. Furthermore, testing for biological activity in vitro and in vivo has to be done after a purification process in order to exclude interference with accompanying compounds. Last but not least, reference standards for quality control of herbal medicinal plants and herbal medicines largely depend on isolated compounds with documented purity. In recent years, NPs have experienced a renaissance in drug-discovery programmes, mainly due to their superior chemical diversity over synthetic compound libraries3 and their drug-like properties.4 The classical way of isolation of NPs starts with identification, collection and preparation of the biological material, usually by drying. Extraction with different solvents from low to higher polarity follows. Prior to isolation of pure compounds, often by (semi-)preparative HPLC or liquid-liquid chromatographic techniques, several purification steps are necessary to remove most of the unwanted matrix. This review will go through these essential steps except liquid-liquid chromatographic techniques and illustrate developments in these areas during the time since 2008 by selected examples of secondary metabolites, i.e. the review is focusing on small molecule NPs (Mr < 2000). In recent years, a trend towards isolation strategies driven by biological or pharmacological activity can be recognized. However, by following this approach extensively, our picture of the vast chemical diversity of plants, microbes or marine organisms will be narrowed and explorative work on the chemistry of living organisms and interactive changes of their metabolic profiles should be encouraged.Bioassay-guided isolation strategies connecting information on the chemical profiles of extracts and fractions with their activity data in in vitro bioassays performed at micro-scale significantly reduced the time for hit discovery. In principle it seems to be a straightforward procedure to get from a plant to an active compound, however there are some critical steps which have to be kept in mind, such as correct plant identification, consideration of transformations during preparation and extraction of the material, or de-replication of already known compounds at the earliest stage of the fractionation procedure. These issues will be discussed briefly in this review. The majority of studies aiming at isolation of significant amounts (mg to g quantities) of pure NPs still use the wide range of liquid chromatographic methods like VLC, MPLC and HPLC, taking advantage of improved separation capacities due to smaller particle size and different selectivity (e.g. HILIC stationary phases). Solid-phase extraction, originally established as a purification method prior to HPLC or GC analysis, is increasingly recognized as a method for rapid fractionation of crude plant extracts or for trapping pure compounds eluted after HPLC separation transferred to capillary NMR (capNMR) analysis for de novo structure elucidation.
Since the comprehensive review by Sticher1 on NP isolation which still represents a valuable overview of currently available methods, the topic has been covered by the recently published 3rd edition of Natural Products Isolation5 which outlines a selection of methods including protocols for extraction and application of chromatographic techniques for NP isolation. Detection, isolation and bioactivity testing of NPs is also covered by the book edited by Colegate and Molyneux.6 A review by Beek et al.7 covers methods for rapid analysis of plant constituents including miniaturized liquid-liquid extraction techniques. Aside from analytical methods, different modes of sample preparation are covered by a review on Chinese plants used for medicinal and food purposes.8
2 Authentication and preparation of plant material/marine organisms
Unequivocal identification of the investigated biological material is without doubt the key to all following steps in NP isolation. In a comprehensive review on marine organisms by Blunt et al.,9 major concerns regarding the recent trend to publish compounds isolated from unidentified microorganisms from sources that are not clearly defined is expressed, illustrating this important issue. In the medicinal plants area, the awareness of the necessity of authentication of biological material has been given a boost by the significantly increasing emergence of herbal drugs from traditional Chinese medicine and products derived thereof on the European market.10 As a result, in an international research programme, GP-TCM (www.gp-tcm.org), authentication projects involving the Chinese Medicinal Plant Authentication Centre at RBG Kew have been established for economically important plant species.11 Characteristics of Chinese medicinal plants and their corresponding herbal drugs have been recently illustrated.12 When collecting plant material, selection criteria might be based on ethnomedicinal data, chemosystematic relationships or ecological observations. Legal and ethical issues like the convention on biodiversity (CBD, http://www.cbd.int/) have to be respected.4,13,14In the following section, major tools which are used in plant authentication will be discussed. A wide range of methods is available for identification of biological materials (plants, marine organisms, microorganisms) which are applicable to different degrees for authentication of unknown material. A combination of several methods might be necessary for unequivocal authentication. In any case, a voucher specimen of authenticated reference material is an indispensable prerequisite. In order to keep track of investigated material of each study a voucher specimen should be kept locally and also be stored in a major herbarium.
2.1 Morphological/anatomical analysis
The primary way of authentication is by morphological as well as anatomical analysis. Both methods need profound expertise and training. One major concern for the authors is the disappearance of classical pharmacognosy, including training in morphological and anatomical analysis of herbal drugs, from curricula, and thus a lack of expertise in this area can be expected in the future. However, if strong anatomical characters like trichomes or calcium oxalate crystals are present, the classical light microscopic analysis of plant material is still a valuable and inexpensive method. Recently, microscopic characteristics of medicinal plants have been published by Upton et al.15 and Rahfeld.16 Morphological characterization of microorganisms usually is combined with genetic markers for identification.17–192.2 TLC/HPTLC analysis
For rapid comparison of a series of samples with reference material, fingerprint analysis by TLC or in the more sophisticated version by HPTLC is an option. In order to make results comparable between different laboratories and literature references, a number of parameters like saturation of the TLC chamber, mobile-phase composition, water content of the silica stationary phase, etc. have to be controlled. Meanwhile HPTLC can be regarded as an established method with application in GMP-compliant quality control of herbal drugs and preparations thereof.20–23 As exemplifying applications of HPTLC, the detection of 5% adulterations of black cohosh (Cimicifuga racemosa) with other Cimicifuga species,24 the identification of Hoodia gordonii25 or the differentiation between Arctostaphylos uva-ursi and A. pungens26 might serve. Coupling TLC/HPTLC with mass spectrometry either by compound extraction with specific interfaces or by ambient mass spectrometry significantly increased the spectral information on selected compounds.7,27,28 Recently, using a TLC-MS extraction interface and coupling to NMR, rutin, caffeic acid and chlorogenic acid could be identified and quantitatively determined.292.3 HPLC analysis
Hyphenation of HPLC separation with different spectroscopic detection methods like PDA, MS or NMR offers two ways of identification of plant material. On the one hand, specific marker compounds can be used for chemotaxonomic applications, e.g. 3-hydroxy-3-methylglutaric acid in flavonol acyl glycosides in the genus Rosa;30 on the other hand, HPLC fingerprints in combination with pattern recognition analysis can be applied for identification of the plant of origin in extracts.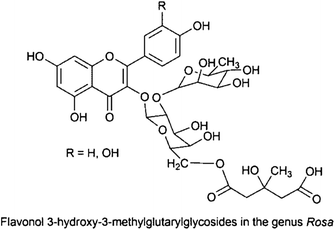
In a chemosystematic study of Taxus spp., LC-PDA-MS fingerprint chromatograms were analysed by hierarchical cluster analysis (HCA) and principal component analysis (PCA) leading to differentiation of eight investigated species to six well-supported groups and correct assignment of most species.31 Combination of PCA of two fingerprints of LC and 1H NMR with a pharmacological fingerprint was used for comprehensive characterization of commercial willow (Salix spp.) bark extracts.32 Differentiation of six Ganoderma species, fungi used in traditional Chinese medicine, was possible by combination of HILIC (see 4.2.5) and reversed-phase columns.33 HPLC analysis still plays a major role in phytochemical analysis including identification of crude plant extracts.8,23,34
Aside from identification purposes directed to organisms, a major application of HPLC methods is de-replication, i.e. the identification of known metabolites in extracts ideally at an early stage of the fractionation process. This is largely done by hyphenated techniques such as LC-NMR, LC-MS, LC-PDA35 and combinations thereof. Using a UHPLC-PDA-TOF-MS setup in Lippia spp. 14 compounds could be unambiguously and further 28 compounds tentatively identified.36 For analytical purposes UHPLC (UPLC), i.e. the application of stationary phases of sub-2 μm particle size combined with high speed elution and instrumentation capable of coping with high backpressures, has resulted in remarkable improvements of analysis of complex mixtures like plant extracts, as clearly outlined in a review by Eugster et al.37
A significant increase in sensitivity of NMR analysis could be gained by using micro-coil NMR which made successful recording of two dimensional NMR spectra (HMBC, HSQC) of 100 μg NP samples (Mrca. 500) in overnight runs possible, as outlined in a recent review on LC-NMR methods by Sturm and Seger.2
2.4 GC analysis
In case of analysing biological material containing volatile constituents like essential oils, GC-MS analysis still represents the method of choice, taking advantage of the unsurpassed peak capacity of capillary GC columns. Headspace solid-phase micro-extraction or steam distillation extraction can be used to collect the volatile fractions from small amounts of plant material.38 Comprehensive two dimensional GC (GC × GC) and multidimensional GC (MDGC),39,40 combining different GC instruments, columns and detectors and selective transfer of individual peaks in combination with multivariate data analysis (MVDA), made fingerprint analysis of volatiles even more informative.38,41,42 Identification of bacteria by GC analyses of bacterial fatty acid methyl esters is still a frequently used authentication technique.432.5 Spectroscopic methods: NMR, MS, NIR, FT-IR
Advances in data analysis of complex signal patterns enabled application of spectroscopic techniques to crude plant extracts for metabolic fingerprinting without prior HPLC separation.35,44,45 By using 1H-NMR metabolic fingerprinting in combination with PCA five different Verbascum species were divided in two groups, group A (Verbascum phlomoides and Verbascum densiflorum) and group B (Verbascum xanthophoeniceum, Verbascum nigrum and Verbascum phoeniceum).46 A similar approach of 1H-NMR-based metabolic profiling was used for discrimination of Ilex species and varieties.47NIR direct measurements of fresh and dry samples without prior extraction is possible, but samples may also include hydrodistillates and extracts. For quality control of the fruiting bodies of Ganoderma lucidum, NIR diffuse reflectance spectroscopy could be used in combination with chemometric techniques to discriminate the samples according to their cultivation area.48 Exploration of different IR techniques for identification of Epilobium spp. and Hypericum spp. from whole leaf samples showed that the morphological properties of the plant material have to be taken into consideration when developing the appropriate IR-based identification method.49 A review by Alvarez-Ordonez et al. covers the potential of FT-IR-based methods as rapid and non-invasive techniques for assessment of membrane composition and changes due to environmental and other stress factors in food-borne bacteria.50 Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) has revolutionized in situ identification of microorganisms by analysing them in a short time from colonies grown on culture plates.51,52
2.6 Molecular biological methods
Omics techniques have gained increasing importance in authentication of biological material during the last decades.10 DNA-based approaches to authenticate plant materials include comparison of internal transcribed spacer (ITS) sequences, random amplified polymorphic DNA (RAPD) markers, the use of sequence characterised amplified region (SCAR) markers or high resolution melting analysis (HRM). In a study by Ruzicka et al.53 on the problematic genus Verbena, which includes about 40 species with frequently occurring natural hybrids, it was possible to differentiate Verbena officinalis by SCAR markers from all species except the closest V. hastata while HRM even enabled discrimination from the latter species.In commercialized plant material, admixtures with different plant species represent a serious problem. By HRM, Mader et al.54 were able to detect the adulteration in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 with unknown plant species and a ratio of 1:200000 of added Veratrum nigrum. As a major drawback for the detection of unknown adulterations, the authors suggested that since universal primers might not react with all species it is important to design assays for specific contaminants or at least for higher level taxa (e.g. plant families).
:1000 with unknown plant species and a ratio of 1:200000 of added Veratrum nigrum. As a major drawback for the detection of unknown adulterations, the authors suggested that since universal primers might not react with all species it is important to design assays for specific contaminants or at least for higher level taxa (e.g. plant families).
SCAR markers were also applied for identification of important Indian medicinal Phyllanthus species, namely P. amarus, P. fraternus, P. debilis and P. urinaria.55 The issue of post-mortem alteration of DNA in herbarium material has been investigated. Although DNA modifications, most likely due to hydrolytic deamination of cytosine during long-term herbarium storage, were observed, herbarium specimens are considered a valuable source of reliable sequence data.56 Molecular identification methods of microorganisms include amplified and non-amplified nucleic acid probes and have been reviewed several times.57–59
2.7 Post-harvest changes in plant material
Post-harvest alteration of plant metabolites has to be taken into consideration as it can lead to significant changes due to plant immanent enzymes like hydrolases (glycosidases), peroxidases or polyphenol oxidases (PPO).60 Early studies by Janecke and Henning61 could identify a number of enzymes in dried plant material which can be reactivated after extraction with aqueous solvents even if lower percentages of ethanol or methanol are present.62 Especially caffeic acid derivatives seem to be subject to oxidative changes. Cichoric acid (2R,3R-O-dicaffeoyltartaric acid), a marker compound in Echinacea purpurea products, was shown to be highly susceptible to degradation by PPO.63 Salvianolic acid B was found only as minor component in fresh roots of Savia miltiorrhiza but significantly increased during drying.64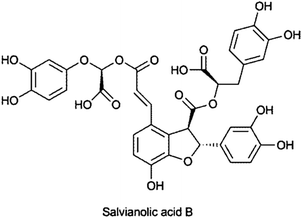
Similar processes were observed in rhizomes of Ligusticum chuanxiong when studying the influence of post-harvest drying and processing methods on nine major components. By drying at 60° or under the sun, the contents of senkyunolide A, coniferylferulate and Z-ligustilide significantly decreased while the content of corresponding compounds increased.65 Detailed studies of post-harvest changes of St. John's wort (Hypericum perforatum), marjoram (Majorana hortensis) and peppermint (Mentha x piperita) have been performed by the group of Boettcher et al.66–68 In addition, perishing of plant material by microbes or fungi has to be scrutinized. Not only can enzymatic degradation be caused by microbial enzymes, secondary metabolites can be induced if plant material was contaminated during life-time, as known for isoflavonoid phytoalexins in legumes.69
The problem of artefact formation during the isolation procedure was also discussed by Jones and Kinghorn.13
3 Extraction methods
Extracting the compounds of interest from the non-soluble matrix in which they are embedded needs several issues to be taken into account. These include the polarity and stability of the extractives and the solvent, the toxicity, volatility, viscosity and purity of the extraction solvent, the probability of artefact formation during the extraction process, and the amount of bulk material to be extracted. The issue of artefact formation due to solvents has been reviewed recently.70 In plant material secondary metabolites usually are found inside cells, thus grinding of the raw material and breaking tissue and cell integrity before extraction increases extraction yield. In the following section the most important methods for extraction of secondary metabolites from biological material applied in laboratory scale will be discussed.3.1 Classical solvent extraction procedures
The majority of isolation procedures still utilize simple extraction procedures with organic solvents of different polarity, water and their mixtures.1,71,72 The methods include maceration, percolation, Soxhlet extraction, ultrasound-assisted extraction and turbo-extraction. Maceration is carried out at room temperature by soaking the material with the solvent with eventual stirring. It has the advantage of moderate extraction conditions but suffers from high solvent consumption, long extraction times and low extraction yields. Extraction yield is improved by percolation, i.e. packing the pre-soaked plant material in a container which allows the constantly controlled removal of the extract via a valve at the bottom and adding fresh solvent from the top. Soxhlet extraction is a popular method for extraction due to its reduced solvent consumption; however, thermo-labile compounds might be degraded during the extraction process. For liquid samples extraction by organic solvents or heterogeneous solvent mixtures can be done, either simply in a separating funnel or similar to a Soxhlet apparatus in a perforator. On a smaller scale, extraction of the liquid sample absorbed on a porous matrix (like diatomaceous earth) packed in a column with non-miscible solvents is an option (e.g. Extrelut® columns).3.2 Ultrasound-assisted extraction (UAE)
In UAE the plant material, usually in a glass container, is covered by the extraction solvent and put into an ultrasonic bath. It decreases extraction time and improves extraction yields due to mechanical stress which induces cavitations and cellular breakdown, and has gained increasing popularity. Examples of NPs extracted by UAE include anthocyanidins, flavonols and phenolic acids from Delonix regia,73 capsaicinoids from Capsicum frutescens in lab and pilot-plant scale,74 cyanidin-3-rutinosid from Litchi chinensis75 or essential oils from laurel, rosemary, thyme, oregano and tuberose.76 In the latter study by Roldan-Gutierrez et al.,76 dynamic UAE, i.e. where the solvent (in this case ethanol) is pumped through the plant material which is placed in an extraction tube in a temperature-controlled water bath connected to an ultrasound probe, showed superior extraction efficiency compared to steam distillation or superheated water extraction.3.3 Microwave-assisted extraction (MAE)
Nowadays extraction employing either diffused microwaves in closed systems or focused microwaves in open systems are established methods. Principles of these technologies, their pros and cons as well as extraction protocols have been outlined in detail by Sticher1 and by Delazar et al.88 MAE has been modified in different ways leading to vacuum microwave-assisted extraction (VMAE), nitrogen-protected microwave-assisted extraction (NPMAE), ultrasonic microwave-assisted extraction (UMAE) or dynamic microwave-assisted extraction (DMAE), which are discussed in a review by Chan et al.89 Some recent examples of application of MAE to NP isolation employing ionic liquids are mentioned below (section 3.4).3.4 Extraction with ionic liquids
In recent years, application of ionic liquids (ILs) for UAE, MAE or simple batch extraction of plant metabolites at room temperature or elevated temperature has gained increasing attention and has been recently reviewed extensively.90 These ILs, also designated as “designer solvents”, are organic salts in the liquid state consisting of an organic cation and an organic or inorganic anion. ILs are able to dissolve a wide range of polar to non-polar compounds, have a low vapour pressure, show a high thermal stability and low combustibility, and some of them are biodegradable. Table 1 presents applications of ionic liquids with different extraction technologies like liquid-liquid extraction (LLE), UAE, MAE or liquid-phase micro-extraction (LPME). An exemplifying study was performed for extraction of artemisin by IL, N,N-dimethylethanolammonium octanoate (DMEA oct) and bis(2-methoxyethyl)ammonium bis(trifluoromethylsulfonyl)imide (BMOEA bst) showing the best performance.79 Artemisin was recovered from the extract after addition of water and crystallisation in 82% yield compared to the total extracted amount. The purity of artemisin crystals was 95% as determined by NMR. Meanwhile a number of studies have been performed mainly with the aim of enriching extracts for analysis by HPLC. Immobilized ILs for solid-phase extraction is discussed in section 3.7. Application of ILs as new solid-phase micro-extraction (SPME) stationary phases caused problems due to contamination of the GC injector when directly inserted into the system.90N,N-dimethylammonium N′,N′-dimethylcarbamate (DIMCARB) proved to be a distillable IL, and could be more easily removed from the extract compared to the majority of ILs which are minimally volatile.77 Another feature of ILs which is still insufficiently investigated is their biodegradability and impact on the environment if used at industrial scale,100 and this needs future attention. In eco-toxicological studies using a Vibrio fischeri bioluminescence quenching assay, longer side-chains, non-aromatic head groups and the anion BF4 showed the highest toxicological risk,101 but the potential to design more hydrophobic ILs with lower toxicity by avoiding aromatic substructures was indicated.102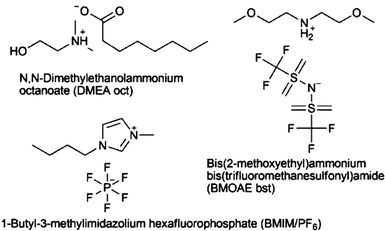
| Plant | Compound | Extraction methoda | ILb | Reference |
|---|---|---|---|---|
| a LSE: liquid-solid extraction; MAE: microwave-assisted extraction; SPE: solid-phase extraction; UAE: ultrasound-assisted extraction; UMAE: ultrasound/microwave-assisted extraction.b a.o.: and other anions; BMIM/BF4: 1-butyl-3-methylimidazolium bortetrafluoride; BMOEA bst: bis(2-methoxyethyl)ammonium bis(trifluoromethylsulfonyl)imide; CnMIM/Cl; Br; Sac; Ace: 1-alkyl-3-methylimidazolium chloride, bromide, saccharinate, acesulfamate; DIMCARB: N,N-dimethylammonium N′,N′-dimethylcarbamate; DMEA oct: N,N-dimethylethanolammonium octanoate; OMIM/Cl 1-octyl-3-methylimidazolium chloride; OMIM/PF6 1-octyl-3-methylimidazolium hexafluorophosphate. | ||||
| Acacia catechu | Hydrolysable tannins | LSE | DIMCARB, removable from extract by distillation | 77 |
| Apocynum venetum | Hyperoside, isoquercitrin | MAE | BMIM/BF4 | 78 |
| Artemisia annua | Artemisinin | LSE | DMEA oct; BMOEA bst | 79 |
| Cynanachum bungei | Acetophenones | UAE | BMIM/BF4 | 80 |
| Glaucium flavum | Alkaloids | LSE | CnMIM/Cl; Br; Sac; Ace | 81 |
| Nelumbo nucifera | Phenolic alkaloids | MAE | CnMIM/Cl; Br; BF4 | 82 |
| Polygonum cuspidatum | trans-Resveratrol | MAE | BMIM/Br | 83 |
| Psidium guajava | Gallic acid, ellagic acid, quercetin | MAE | CnMIM/Cl; Br; a.o. | 84 |
| Rheum spp. (rhubarb) | Anthraquinones | UMAE | CnMIM/Cl; Br; BF4 | 85 |
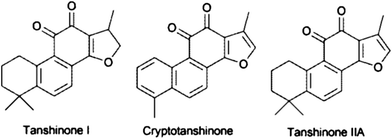
| Salvia miltiorrhiza | Cryptotanshinone, tanshinone I, tanshinone II A | UAE | Aqueous OMIM/Cl; analytes concentrated by anion metathesis to OMIM/PF6 | 86 |
| Smilax china | trans-Resveratrol, quercetin | MAE | CnMIM/Cl; Br; a.o. | 84 |
| Sophora flavescens | Oxymatrine | 1 .LSE; 2. SPE | 1. Silica-confined IL; 2. MeOH | 87 |
| Terminalia chebuja | Hydrolysable tannins | LSE | DIMCARB | 77 |
3.5 Accelerated (pressurized) solvent extraction (ASE)
In comparison to most other extraction systems which need an additional step for separation of the remaining non-soluble matter from the liquid extract, on-line filtration within the automatized extraction process of accelerated (or pressurized) solvent extraction (ASE, a patented system by Thermo/Dionex®) is included. The methodology is applied to solid and semisolid samples in 1–100 g scale using common solvents at elevated temperature and pressure.103 Up to 24 samples can be extracted automatically. In a study on altitudinal variation of phenolic compounds in Calluna vulgaris, Vaccinium myrtillus and Sambucus nigra, 205 samples of dried and ground material mixed 1:1 with diatomaceous earth (DE) or sea sand were extracted with 80% MeOH for their flavonoids and phenolic acids, illustrating the necessity of serial extraction under controlled conditions when doing comparative studies.104 In ASE, sequential extraction with solvents of different polarity and mixing of solvents is possible, as illustrated by Cicek et al.105 Consecutive extraction of subaereal parts of Actea racemosa with petroleum ether for defatting followed by dichloromethane led to isolation of 2.2 g enriched triterpene saponin fraction from 50 g of plant material. Although ASE usually is mainly used as a sample preparation method for analytical purposes,106–110 preparative scale application of ASE was performed with Hypericum perforatum to obtain the phloroglucinols adhyperforin and hyperforin as well as three caffeoyl quinic acid derivatives.111 Due to increased capacities of extraction cells in the latest version of ASE instrumentation, this type of application is likely to increase in the future. ASE or similar instrumentation can also be used for subcritical water extraction (SWE) employing temperatures of 100–280 °C. Subcritical water (superheated water, pressurized hot water) is heated to a temperature between the boiling point at atmospheric pressure (100 °C) and the critical temperature (374 °C) under pressure, thereby increasing its solution properties for organic lipophilic compounds. In the NPs field SWE has been employed to extract phenolic compounds from pomegranate (Punica granatum) seed residues,112 gallic acid and ellagitannins from Terminalia chebula,113 the flavonol quercetin from onion (Allium cepa) skin,114 phenolic compounds from potato (Solanum tuberosum) peels115 or essential oil from Cinnamomum ceylanicum.116 For phenolic type of compounds, SWE seems to be an attractive alternative to organic solvent extraction, however, artefact formation and degradation has to be scrutinized, as shown by Plaza et al. who observed formation of degradation products due to Maillard reaction, caramelization and thermo-oxidation when SWE was applied to extraction of different organic matter including microalgae, algae and plants.1173.6 Supercritical fluid extraction (SFE)
Replacing extraction with organic solvents by extraction technologies which are less detrimental to environment and meet the increasing regulatory requirements certainly can be considered as a driving force for the increasing application of supercritical fluid extraction, above all using supercritical CO2. An overview of methodology including extraction protocols and applications in NP isolation and extraction is given by Nahar and Sarker118 as well as Sticher.1 Mathematical models which represent the mass transfer mechanisms and the extraction process in order to design the SFE application properly have been reviewed by Huang et al.119 Recent reports on SFE for extraction of NPs and modelling include phloroglucinol and benzophenone derivatives from Hypericum carinatum,120 essential oils,121 gallic acid, quercetin and essential oil from the flowers of Achyrocline satureioides122 or phenolics including anthocyanidins from grape peels (Vitis labrusca).123The utilization of organic solvents as modifiers for supercritical CO2 to increase its solvating capabilities to medium-polar and polar compounds has broadened the spectrum of NP compound classes accessible to SFE, accepting the ecological problems related to organic solvent extractions which increase to a small extent.
3.7 Extraction on solid phases
Extraction processes which take advantage of adsorption of the analytes or unwanted impurities on a solid phase have gained a dominant role in purification of NP extracts, not least due to its integration into automated sample preparation and isolation systems. Most applications utilize solid-phase extraction (SPE) which employs a wide range of stationary phases with diverse chemistry like silica gel, reversed-phase material, ion-exchange resins or mixed-mode material and HILIC stationary phases in pre-packed glass or plastic columns. For HILIC, hydrophilic interaction chromatography, see section 4.2.5. Usually a forced flow technique using a vacuum manifold is applied. Several strategies can be used in SPE. Either unwanted impurities (like chlorophylls) are removed by adsorption on the stationary phase or the analytes of interest are adsorbed on the stationary phase, whereas impurities are eluted. In the latter version, a second step of elution will remove the concentrated analytes from the column. Elution of the compounds of interest might be done stepwise by applying a gradient with increasing eluting power, i.e. the procedure is then related to VLC (vacuum liquid chromatography). An exciting development of recent years was the design of molecularly imprinted polymers (MIP) to be used in SPE applications for selective enrichment of various compounds. Either ionic liquid-imprinted silica particles or copolymers of acrylamide and ethylene glycol dimethacrylate with the respective template compounds are used to create material which will have a high affinity to the template structures. In a first elution step the unwanted material is removed from the SPE column whereas target compounds bound to the solid phase are obtained in a concentrated solution usually upon elution with organic solvents like methanol, though additional purification steps might be necessary. Recent reports on isolation of NPs with MIP-SPE are summarized in Table 2.| Compound (template) | Plant | MIPa | Polymerization | SPE eluent | Ref. |
|---|---|---|---|---|---|
| a AA: acrylamide; ACN: acetonitrile; AEIB: 1-allyl-3-ethylimidazolium bromide; AIBN: 2,2′-azo-bis-isobutyronitrile; CHCl3: chloroform; Cl: cross linker; DMSO: dimethylsulfoxide; EDMA: ethylene glycol dimethacrylate; Fm: functional monomer; IL: ionic liquid; In: initiator; MAA: methacrylic acid; MeOH: methanol; n-BuOH: n-butanol; Pg: porogene; THF: tetrahydrofuran. | |||||
| Podophyllotoxin | Dysosma versipellis; Sinopodophyllum hexandrum; Diphylleia sinensis | Fm: AA | Microwave heating initiated precipitation polymerization, 60 °C | MeOH; MeOH/acetic acid (9:1, v/v) | 91 |
| Cl: EDMA + divinylbenzene | |||||
| Pg: AcCN | |||||
| In: AIBN | |||||
| Andrographolide | Andrographis paniculata | Fm: AA | Precipitation polymerization, 60 °C | MeOH/water (3:2, v/v); MeOH | 92 |
| Cl: EDMA | |||||
| Pg: ACN–toluene (3:1, v/v) | |||||
| In: AIBN | |||||
| Quercetin | Cacumen platycladi (Platycladus orientalis) | Fm: AA | Batch polymerization; 60 °C | MeOH; MeOH/acetic acid (9:1, v/v) | 93 |
| Cl: EDMA | |||||
| Pg: 1,4-dioxane, THF, acetone, ACN | |||||
| In: AIBN | |||||
| Kirenol | Siegesbeckia pubescens | Fm: AA | Batch polymerization; 60 °C | MeOH/acetic acid (9:1, v/v) | 94 |
| Cl: EDMA | |||||
| Pg: THF | |||||
| In: AIBN | |||||
| Berberine | Phellodendron wilsonii | Fm: AA | Batch polymerization, 60 °C | MeOH-CHCl3 (1:60, v/v) | 95 |
| Cl: EDMA | |||||
| Pg: CHCl3, DMSO, MeOH | |||||
| In: AIBN | |||||
| Protocatechuic acid | Homalomena occulta | Fm: AA | Precipitation polymerization, 60 °C | MeOH/acetic acid (9:1, v/v) | 96 |
| Cl: EDMA | |||||
| Pg: ACN | |||||
| In: AIBN | |||||
| 18β-glycyrrhetinic acid | Glycyrrhiza glabra | Fm: MAA | Batch polymerization, 60 °C | MeOH | 97 |
| Cl: EDMA | |||||
| Pg: CHCl3 | |||||
| In: AIBN | |||||
| Protocatechuic acid; caffeic acid; ferulic acid | Salicornea herbacea | Fm: IL monomer (AEIB) | Batch polymerization, 60 °C | Aqueous HCl (0.5 mol L−1) | 98 |
| Cl: EDMA | |||||
| Pg: n-BuOH/H2O (9:1, v/v) | |||||
| In: AIBN | |||||
| Cryptotanshinone, tanshinone I, tanshinone IIA, template: 9,10-phenanthrenequinone | Salvia miltiorrhiza | IL: 3-aminopropyl-trimethoxysilane + 3-chloropropionylchloride + imidazole; immobilized on silica | — | n-hexane (washing step) MeOH (elution) | 99 |
Aside from SPE as sample purification before LC or GC analysis, trapping compounds on SPE columns for off-line LC-NMR coupling has gained increasing importance for structure elucidation, metabolic profiling and de-replication strategies.2,124–126 As part of automated isolation systems SPE is combined with preparative HPLC like in the Sepbox® instrument127 or as proposed by Tu et al.128 A sophisticated combination of SPE columns representing strong anion and cation exchangers, a mixed-mode polymeric RP-anion exchanger with a poly(divinylbenzen-co-vinylpyrrolidone) backbone and a size exclusion column of a hydroxypropylated dextran gel (Sephadex LH-20®) were used for explorative fractionation of extracts from microorganisms.129 SPE might also be carried out by adding spatially separated anion and cation exchange resins in sachets to organic extract solutions for separating acidic, basic and neutral compounds.130 For micro-scale isolation, variants of SPE like SPME or stir-bar sorptive extraction (SBSE) can be used. For isolation of the volatile fraction of herbal teas SPE was used in comparison to hydro distillation,131,132 but headspace-SPME and SBSE are attractive alternatives for this type of application, as reviewed recently.133
3.8 Distillation methods
Volatiles such as essential oils are still obtained mainly by distillation techniques, although working at elevated temperatures can lead to chemical changes, most obvious in essential oils of chamomile (blue chamazulene originating from colourless matricin) or other proazulene-containing plants (e.g. yarrow, Achillea spp.). Recent developments in distillation methodology include microwave steam distillation (synonym: microwave steam diffusion) which applies microwaves to increase disruption of glands and cells while steam is passing through the plant material and carrying the essential oil.134,135 In a comparative study of the essential oil isolated from Salvia rosifolia136 by microwave-assisted hydro-distillation in 45 min an essential oil of similar yield and composition as the one obtained after 180 min of conventional hydro-distillation (HD) was obtained. Microdistillation was preferable for isolation of the most volatile fraction of monoterpene hydrocarbons.136 For characterisation of representative chamomile volatiles in the vapour upon inhalation, a combination of HD and RP18-SPE in a circulating apparatus (SD-SPE) was applied and compared to simultaneous distillation extraction (collecting the volatiles in a water non-soluble solvent) and HD. It could be shown that actually a much higher percentage of the more polar en-in-dicycloethers and bisabolol, important ingredients for the anti-inflammatory activity of chamomile oil, could be obtained by SD-SPE.1374 Isolation by liquid-solid chromatography techniques
A wide range of liquid chromatographic methods with solid stationary phases either as planar or column chromatography is available for further fractionation and final purification of NPs. The choice largely depends on the stage of purity of the extract or fraction, and the final purpose of the isolated NP. High sample capacity combined with relatively low costs made low pressure liquid chromatography (LPLC), vacuum liquid chromatography (VLC) or flash chromatography (FC) popular for fractionation of crude extracts, and in rare cases even pure compounds could be obtained by these single fractionation steps. However, in many cases medium-pressure liquid chromatography (MPLC) or semi-preparative and preparative HPLC with higher peak resolution power had to be applied for final purification.4.1 Preparative planar chromatography (PPC)
Due to its simplicity in use and relatively low costs for isolation of small molecule NPs, PPC is still a frequently used technique although the number of applications is lower than those of column chromatography. An attractive feature of PPC is the wide range of chemical detection methods characteristic for compound classes which can be carried out on a narrow section of the plate leaving most of the compound unchanged and available for isolation. In bio-assay-guided isolation strategies, planar chromatography has the advantage of direct application of bioassays on TLC plates, making the rapid localisation of bioactive compound zones possible. So far, bioautographic methods include antifungal and antibacterial activity, acetyl cholinesterase (AChE) inhibition, α- and β-glucosidase inhibition and radical scavenging or antioxidant activity, as reviewed recently by Marston.138 The search for AChE inhibitors by TLC bioautography can be illustrated by studies of the genus Peganum identifying harmine and harmaline as potent compounds.139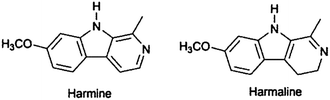
In addition to the optimization of growth media for bioautographic detection of antimicrobial activity of Cordia giletti, the ability to quench the bioluminescence of Vibrio fischeri, indicating toxicity, was checked in another TLC bioautographic approach.140 A review by Sherma141 on developments in planar chromatography between 2009 and 2011 presents some illustrative examples too.
To overcome the disadvantage of classical TLC of uncontrolled flow rates of the mobile phase, forced-flow techniques such as centrifugal planar chromatography or over-pressured layer chromatography have been developed enabling elution and on-line detection of compounds.142,143 A comprehensive outline of the application of PPC to isolation of NPs has been provided by Gibbons recently.144
4.2 Column chromatographic methods
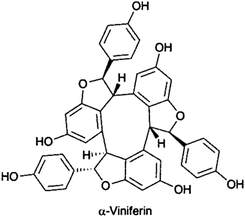
Recently, VLC was part of the isolation procedure of α-viniferin and hopeaphenol, trimeric and tetrameric stilbenes from Shorea ovalis,146,147 anthraquinones, naphthalenes and naphthoquinones from Asphodeline lutea,148 alantolactone and isoalantolactone from Inula helenium,149 the antifungal sakurasosaponin from Jacquinia flammea,150 and antimalarial diterpene formamides from the marine sponge Cymbastela hooperi.151
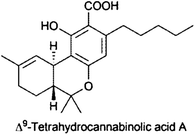
Two independent flash chromatography systems on normal phase and reversed phase were developed for the rapid isolation of Δ9-tetrahydrocannabinolic acid A (THCA) from Cannabis sativa.161 By normal-phase FC and gradient elution with cyclohexane and acetone 1.8 g, crude cannabis extract yielded 0.6 g THCA, whereas using an RP-18 phase with an isocratic elution with MeOH–formic acid (0.554%, pH 2.3), 85:15 v/v, 0.3 g extract resulted in 51 mg THCA; purity of THCA with both methods was >98.8%.161
Another example of the separation power of FC was provided by Uckoo et al.162 isolating four structurally similar polymethoxyflavones, i.e. tangeretin, nobiletin, tetramethoxy-flavone and sinensitin, from peels of Citrus reshni and C. sinensis by FC on silica with a hexane–acetone gradient. A mixture of diterpenes from the mollusc Thuridilla splendens, thuridillins D–F, was obtained by silica FC but could be finally separated by preparative TLC on AgNO3-impregnated silica gel plates.163
:23) using an RP-18 column at a flow rate of 100 ml min−1 afforded pure compounds of peoniflorin and albiflorin.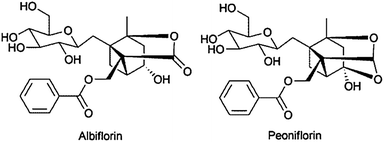
Silver nitrate-impregnated silica gel was employed for successful separation of the sesquiterpenes (Z)-α- and (Z)-β-santalol with >96% purities from the white sandalwood, Santalum album.170 These two sesquiterpene alcohols, which together constitute over 80% of the heartwood oil of matured trees, are responsible for the antifungal, anti-inflammatory, antidepressant and organoleptic properties of white sandalwood essential oil.170
:5 to 60:40). The triazole-bonded silica HILIC column employed by Morikawa et al.180 provided better separation for sesquiterpene glycosides from the Thai medicinal plant Sapindus rarak compared to a RP-30 column due to the positively charged triazole stationary phase. A polyamine-II column that possesses secondary and tertiary amine groups bonded to porous silica particles was used for the separation of triterpene glycosides from Physena sessiliflora in HILIC mode.181 Van Wagoner et al.182 isolated sulphonated karlotoxins from the microalgae Karlodinium veneficum using the reverse-phase Develosil TM-UG-5® C1 phase with a basic eluent. Cyano packing allowed efficient purification of the phytotoxic oleanane saponins of the leaves of Bellis sylvestris that differ greatly in hydrophobicity, without the need to use gradient elution.183
| Compounds | Source | Column | Mobile phase | Ref. | ||
|---|---|---|---|---|---|---|
| Stationary phasea | Dimension (mm) | PDb (μm) | ||||
| a C1: trimethylsilan chemically bonded to porous silica particle; β-CD: β-cyclodextrin bonded to porous silica particle; Click-CD: β-cyclodextrin bonded to porous silica particle by click chemistry; Diol: dihydroxypropane groups chemically bonded to porous silica particles; HILIC: hydrophilic interaction chromatography; Partisil 10: amino and cyano groups chemically bonded to porous silica particle; Polyamine II: secondary and tertiary amine groups bonded to porous silica particle.b PD: particle diameter. | ||||||
| Terpenoids | ||||||
| Sesquiterpenes | Acorus calamus | Silica gel-Diol | 10 × 250 | 10 | Isocratic hexane-2-propanol (97:39) | 190 |
| Silica gel C18 | 30 × 150 | 5 | Gradient H2O-MeOH (50:50 to 0:100) | |||
| Sesquiterpenes | Rolandra fruticosa | Silica gel-C18 | 10 × 150, 19 × 150 | 5 | Isocratic H2O-MeOH (50:50, 55:45) | 191 |
| Sesquiterpenes | Artemisia persica | Silica gel-C18 | 10 × 150 | 5 | Gradient H2O-MeCN (80:20 to 0:100); H2O-MeOH (70:30 to 0:100) | 192 |
| Diterpenoids | Leonotis leonurus | Silica gel-C18 | 21.2 × 150 | 7 | 100% MeOH | 193 |
| Partisil 10 | 4.6 × 250 | 10 | Isocratic MeOH-CH2Cl2 (1:99) | |||
| Diterpenoids | Ajuga bracteose | Silica gel-C18 | 2.1 × 100 | 1.7 | Gradient H2O/0.1% HCO2H-MeCN (70:30 to 5:95) | 194 |
| Triterpenoids | Lycopodium phlegmaria | Silica gel-C18 | 20 × 250 | 5 | Isocratic H2O-MeOH (15:85) | 195 |
| Silica gel-C18 | 19 × 250 | 5 | Isocratic H2O-MeOH (15:85) | |||
| Triterpenoids | Cogniauxia podolaena | Silica gel-C18 | 19 × 150 | 5 | Gradient H2O-MeCN (90:10 to 0:100) | 196 |
| Triterpenoid saponins | Aesculus glabra | Silica gel-C18 | 4.6 × 250 | 3.5 | Isocratic H2O/0.5% HOAc-MeCN (63:37, 60:40) | 197 |
| Silica gel-C18 | 22 × 250 | 10 | Isocratic H2O/0.5% AcOH-MeCN (60:40, 52:48, 45:55, 35:65) | |||
| Triterpene glycosides | Physena sessiliflora | Silica gel-C18 | 20 × 100 | 5 | Isocratic H2O-MeCN (70:30, 63:37) | 181 |
| Silical gel-Polyamine-II | 20 × 150 | 5 | Isocratic H2O-MeCN (17.5:82.5, 22.5:77.5) | |||
| Triterpenoid oligoglycosides | Sapindus rarak | Silica gel-C30 | 4.6 × 250 | 5 | Isocratic H2O-MeCN/1% AcOH (50:50) | 180 |
| Silica gel-Triazole (HILIC) | 20 × 250 | 5 | Isocratic H2O-MeCN (5:95) | |||
| Terpenoids, phenethyl glucosides | Hyssopus cuspidatus | Silica gel-Phenyl | 20 × 250 | 5 | Isocratic H2O-MeOH (25:75, 20:80, 60:40) | 198 |
| Silica gel-C18-Phenyl | 10 × 250 | 5 | Isocratic H2O-MeOH (10:90, 15:85, 25:75) | |||
| Sesquiterpenoids, macrolide and diterpenoid | Cyphostemma greveana | Silica gel-C18 | 10 × 250 | 5 | Isocratic H2O-MeOH (35:65) | 199 |
| Silica gel-Phenyl | 10 × 250 | 5 | Isocratic H2O-MeCN (55:45) | |||
| Oleanane saponins | Bellis sylvestris | Silica gel-C18 | 10 × 250 | 10 | Isocratic H2O-MeCN-MeOH (50:20:30) | 183 |
| Silica gel-CN | 10 × 250 | 5 | ||||
| Alkaloids | ||||||
| Cyclic diterpene alkaloids | Agelas mauritiana | Silica gel-C18 | 10 × 250 | 5 | Isocratic H2O-MeCN (46:54, 70:30, 75:25) | 200 |
| Quinoline alkaloids | Drummondita calida | Silica gel-C18 | 21.2 × 150 | 5 | Gradient H2O/0.1%TFA-MeOH/0.1% (90:10 to 0:100) | 201 |
| Silica gel-Diol | 20 × 150 | 5 | Gradient CH2Cl2-MeOH (90:10 to 0:100) | |||
| Stemona alkaloids | Stemona sp. | Silica gel-C18 | 4.6 × 250 | 5 | Gradient H2O in 10mM NH4OAc-MeOH (45:55 to 10:90, 19 min; 10:90 to 0:100, 1 min; 0:100, 10 min) | 202 |
| Eupolauridine alkaloids | Ambavia gerrardii | Silica gel-Phenyl | 10 × 250 | 5 | Isocratic H2O-MeOH (40:60) | 185 |
| Flavonoids | ||||||
| Anthocyanins | Asparagus officinalis | Silica gel-C18 | 20 × 250 | 5 | Gradient H2O/10%HCO2H 40%MeCN/50%H2O/10%HCO2H(75:25 to 50:50, 23 min) | 203 |
| Anthocyanins | Arabidopsis thaliana | Silica gel-C18 | 20 × 250 | 5 | Isocratic H2O/0.5% AcOH-MeOH (60:40) | 204 |
| Flavonoid glucuronides, chromone | Stratiotes aloides | Silica gel-phenyl | 10 × 250 | 7 | Gradient H2O/0.01%TFA-MeCN 84%/H2O 16% (100:0 to 80:20, 10 min; 80:20 to 60:40, 30 min; 60:40 to 50:50, 10 min); Gradient H2O/0.01% TFA-MeOH/H2O (84:16) (100:0, 60 min; 100:0 to 0:100, 20min) | 166 |
| Silica gel-CN | 25 × 250 | 5 | ||||
| Flavonoid glycosides | Citrus bergamia | Silica gel-C18 | 21.2 × 100 | 10 | Isocratic H2O/0.1%HCO2H-MeCN (55:45, 12 min; 77:23, 15 min | 205 |
| Flavones | Mimosa diplotricha | Silica gel-C18 | 20 × 250 | 5 | Isocratic H2O-MeOH (40:60) | 206 |
| Flavonoids, triterpene saponins | Glycyrrhiza sp. | Silica gel-C18 | 19 × 100 | 5 | Gradient H2O/0.1% HCO2H-MeCN (85:15, 5 min: 85:15 to 65:35, 55 min; 65:35 to 5:95,60 min | 176 |
| β-CD (HILIC)d | 30 × 150 | 5 | Gradient H2O-MeCN/0.1% HCO2H (5:95 to 10:90, 30 min; 10:90, 30 min) | |||
| Flavonolignans | Calamus quiquesetinerivius | Silica gel C18 | 10 × 250 | 5 | Isocratic H2O-MeOH (51:49, 65:35) | 207 |
| Neoflavonoids and Benzofurans | Pterocarpus santalinus | Silica gel-C18 | 10 × 250 | 5 | Isocratic H2O-MeOH (43:57) | 208 |
| Steroids | ||||||
| Bufadienolides | Bufo bufo gargarizans | Click-CD (HILIC) | 4.6 × 150 | 5 | Gradient H2O-MeCN/0.1%HCO2H (5:95 to 40:60) | 179 |
| Silica gel-C18 | 4.6 × 150 | 3 | Gradient H2O-MeCN (95:5 to 35:65, 0–60 min; 35:65 to 5:95, 60–70 min) | |||
| Lignans | ||||||
| Polyhenolic lignans | Capparis flavicana, Vitax glabrata | Silica gel-Phenyl | 22 × 250 | 5 | Isocratic H2O-MeCN (85:15, 87.5:12.5, 90:10, 95:5) | 184 |
| Silica gel-C18 | 20 × 250 | 5 | Isocratic H2O-MeCN (95:5), H2O-MeOH (90:10) | |||
| Lignan glucosides, flavanones | Macaranga tanarius | Silica gel-C18 | 6 × 250 | 3 | Isocratic H2O-MeCN (90:10, 19:1, 40:10, 41::9, 83:17, 40:10) | 209 |
| Tannins | ||||||
| Gallotannins | Eugenia jambolana | Silica gel-C18 | 10 × 250 | 5 | Isocratic H2O-MeOH (76:24, 70:30, 67:33, 65:35) | 210 |
| Peptides | ||||||
| Cyclopeptides | Annona montana | Silica gel-C18 | 4.6 × 250 | 5 | Isocratic H2O-MeCN (25:75) | 211 |
| Silica gel-C30 | 20 × 250 | 5 | Isocratic H2O-MeCN/0.5%TFA (25:75) | |||
| Cyclodepsipeptides | Lyngbya confervoides | Silica gel-C18 | 21.2 × 100 | 10 | Gradient H2O-MeOH (70:30 to 0:100, 40 min, 0:100, 10 min) | 212 |
| Silica gel C18 | 10 × 250 | 5 | H2O-MeOH/0.05% TFA (40:60 to 10:90, 25 min; 10:90 to 0:100, 10 min) | |||
| Lipopeptides | Nocardia sp. | Silica gel-C18 | 10 × 250 | 5 | Gradient H2O-MeCN/CH2Cl2 (98:2 to 50:50) | 213 |
| Others | ||||||
| Polyketides | Botryosphaeria rhodina | Silica gel-C18 | 16 × 250 | 5 | Gradient H2O-MeCN (75:25 to 0:100) | 214 |
| Cyanopyridone glucosides | Acalypha indica | Silica gel-C8 | 21.2 × 250 | 5 | Gradient H2O-MeOH (100:0, 20 min; 80:20, 30 min; 0:100, 40 min) | 168 |
| Acetophenone | Acronychia pedunculata | Silica gel-C8 | 10 × 250 | 5 | Gradient H2O-MeOH (30:70 to 0:100) | 215 |
| Karlotoxins | Karlodinium veneficum | Silica gel-C18 | 4.6 × 150 | 3.5 | Isocratic H2O-MeCN (62:38) | 182 |
| Silica gel-C1 | 4.6 × 250 | 5 | Isocratic 2 mM NH4Ac-MeCN (64:36) | |||
| Picolinic acid derivative | Fusarium fujikuroi sp. Tlau3 | Silica gel-C8 | 19 × 250 | 5 | Isocratic H2O/TFA-MeOH/TFA (45/0.1: 55/0.1) | 216 |
| Stilbenoids, phenanthraquinone | Oncidium microchilum, O. isthmi, Myrmecophila humboldtii | Silica gel-C18 | 21.2 × 100 | 5 | Gradient H2O/0.05% TFA-MeCN (40:60 to 15:85) | 217 |
| Silica gel-C18 | 10 × 250 | 5 | Gradient H2O/0.1%TFA-MeCN (various proportions) | |||
| Polycylic fatty acids | Beilschmiedia sp. | Silica gel-C18 | 10 × 250 | 5 | Isocratic H2O/0.05% TFA-MeCN (42:58, 45:55) | 218 |
A semi-preparative CN-phase HPLC column was employed to isolate six free amino acids from the aquatic macrophyte Stratiotes aloides, the European water soldier.166 In addition, a luteolin glycoside was purified from S. aloides using a phenyl-bonded silica column. As compared to the aliphatic straight-chain reversed phases such as C18 and C8, the π-electrons of the phenyl group can interact with aromatic residues of an analyte molecule in addition to hydrophobic interaction to increase retention relative to non-aromatic compounds. Thus, phenyl-modified silica gel columns were also employed to isolate lignans from the aerial parts of the Thai medicinal plant Capparis flavicans184 and antiproliferative eupolauridine alkaloids from the roots of Ambavia gerrardii.185
In recent years, a clear trend towards miniaturization of bioassay-guided setups like HPLC-based activity profiling in order to quickly identify metabolites of significant biological activity in crude plant extracts could be recognized.186,187 In this respect, a microfractionation strategy combined with activity testing in a zebrafish bioassay in combination with UHPLC-TOF-MS and microfluidic NMR was proposed for rapid detection of pharmacologically active natural products.188
5 Chiral chromatographic methods in natural products isolation
After isolation of chiral compounds of NPs, often a method to determine absolute configuration is needed. Different models for the requirements of chiral recognition have been discussed. The best known model is the three-point interaction model by Dalgliesh,189 which postulates that three interactions have to take effect and at least one of them has to be stereoselective. For enantioseparation at an analytical scale, high-performance separation techniques such as HPLC, GC, CE or SFC have widely been used, however HPLC is applied in most cases. This separation technique allows separating enantiomers either indirectly with chiral derivatization reagents or directly with chiral stationary phases or chiral mobile-phase additives. There are advantages and disadvantages for each of these techniques. Indirect separation is based on derivatization by chiral derivatization reagents to form diastereomeric derivatives. They differ in their chemical and physical behavior and therefore are resolved on achiral stationary phases, such as a reversed-phase column. This approach avoids the need for expensive columns with chiral stationary phases, however derivatization has to be regarded as an additional step which can have side reactions, formation of decomposition products and racemization as undesirable side effects. Furthermore, the chiral derivatization reagent has to be of high enantiomeric purity; also, derivatizable groups in the analyte have to be available. Direct enantioseparation using columns with chiral stationary phases is more convenient and also applicable for separations on preparative scale. On the other hand, a collection of expensive columns is required. Finally, the approach to add a chiral selector to the mobile phase can be regarded as a simple and flexible alternative, however applicability is limited. Since mobile phases containing a chiral selector cannot be reused, this technique should not be applied with expensive chiral additives.219 For detection, mostly UV-VIS is used, although polarimetric detectors are advantageous, since they produce a negative peak for (−)-enantiomers. For direct chiral separations, a variety of chiral separation principles is available: the most often used principle is based on enantioselective complexation in cavities of a chiral selector.220 As secondary interactions, hydrogen-bonding, dipole interactions and hydrophobic interactions can be taken into account. For example, cyclodextrin (CD) derivatives of α-CD, β-CD or γ-CD or synthesized chiral crown ethers are suitable. Also, macrocyclic antibiotics such as the glycopeptides vancomycin, ristocetin or teicoplanin are available. The latter compound contains 18 chiral centers and three chiral cavities bridged by 5 aromatic ring structures. As interactions, hydrogen donor and acceptor sites are readily available close to the ring structures. All these selectors can be either fixed on the silica support of a column or can be used as chiral additives to the mobile phase along with an achiral column. Gutierrez et al.221 isolated tanikolide seco-acid and tanikolide dimer from the Madagascar marine cyanobacterium Lyngbya majuscule. They used a chiral HPLC column based on the macrocyclic antibiotic teicoplanin along with mixtures of ethanol/water as mobile phase. Moreover, chiral stationary phases based on polysaccharides are commercially available. They showed a very broad applicability to different compound classes. Since the chiral cavities of native amylose and cellulose are too small, they are not available for interaction and have to be altered by derivatization. These columns have found a wide range of applicability. Besides columns bearing the polysaccharide covalently attached to the silica support, there are also coated polysaccharide CSPs available, however, the latter ones are limited with respect to the solvents that can be used in the mobile phase. Antonov et al.222 report on a new procedure for separation of highly polar glycoside fractions by a Chiralpak IC HPLC column consisting of cellulose tris(3,5-dichlorophenylcarbamate). Batista et al.223 elucidated the structure and absolute stereochemistry of isomeric monoterpene chromane esters by means of a Chiralcel OD-H HPLC column. In this case cellulose is derivatized by tris(3,5-methylphenylcarbamate). The same selector is also provided by other vendors: a new tyrosine-derived metabolite, namely aspergillusol A, was isolated as well as a methyl ester of 4-hydroxyphenylpyruvic acid oxime and secalonic acid A from the marine-derived fungus Aspergillus aculeatus CRI323-04. For chiral HPLC, a Phenomenex Lux Cellulose-1® was used.224A further chiral separation principle represents ligand-exchange chromatography, which was one of the first successful separation principles in chiral chromatography. In this case, chiral recognition is based on the formation of ternary mixed metal complexes between the selector and analyte ligand. As can be seen from Table 4, this separation principle was used most frequently. Immobilized amino acids, such as D-penicillamine or amino acid derivatives are complexed by the mobile phase containing Cu(II) for enantio-resolution.225,227–230,232–234,236–239
| Compounds | Source | CSPa | Chiral stationary phaseb | Mobile phase | Ref. |
|---|---|---|---|---|---|
| a CSP: Chiral separation principle; CIC: chiral inclusion complexation; LE: ligand-exchange; PT: Pirkle type.b D-PA: D-penicillamine. | |||||
| Malevamide E | Symploca laete-viridis | LE | Chirex; D-PA on silica | 1.7 mM Cu(II) in acetonitrile/water (14:86); mobile phase II: 1.9 mM Cu(II) in acetonitrile/water (5:95) | 225 |
| [8-Ethyl]-chlorophyll c3 | Emiliania huxleyi | CIC | Chiralpak IC; cellulose tris (3,5-dichlorophenylcarbamate) on silica | 1:2:2 (v/v/v) methanol–acetonitrile–100 mM aqueous ammonium acetate | 226 |
| Monoterpene chromane esters | Peperomia obtusifolia | CIC | Chiralcel OD-H; cellulose tris (3,5-dimethylphenylcarbamate) | n-hexane | 223 |
| Cordyheptapeptides C–E | Acremonium persicinum | LE | MCIGEL CRS10W; N,N-dioctyl-L(or D)-alanine | 2 mM Cu(II) | 227 |
| Lyngbyastatins 1 and 3, acyl proline derivatives, tumonoic acids D–I, tumonoic acid A | Blennothrix cantharidosmum | LE | Chirex 3126; D-PA on silica | 2 mM Cu(II) | 228 |
| Molassamide | Dichothrix utahensis | LE | Chirex 3126; D-PA on silica | 2 mM Cu(II) with acetonitrile | 229 |
| Carriebowmide | Lyngbya polychroa | LE | Chirex 3126; D-PA on silica | 2 mM Cu(II) | 230 |
| Tanikolide dimer, tanikolide seco-acid | Lyngbya majuscula | CIC | Chirobiotic T; teicoplanin on silica | 40:60 water/ethanol | 221 |
| Aspergillusol | Aspergillus aculeatus | CIC | Lux Cellulose-1, cellulose tris(3,5-dimethylphenylcarbamate) on silica | 2-propanol/hexane (20:80) | 224 |
| Quinadoline B | Aspergillus sp. FKI-1746 | PT | Sumichiral OA-3100; N-(3,5-dinitrophenylaminocarbonyl)-L-valine | methanol/acetonitrile (95:5) containing 1 mM citric acid | 231 |
| 3-Amino-6-hydroxy-2-piperidone | Lyngbya confervoides | LE | Chirex 3126; D-PA on silica | 2 mM Cu(II) or 2 mM Cu(II)/acetonitrile (95:5) | 232 |
| Coibamide A | Leptolyngbya sp. | LE | Chirex 3126; D-PA on silica | 2 mM Cu(II) or 2 mM Cu(II)/acetonitrile (95:5) | 233 |
| Pitipeptolides C–F | Lyngbya majuscula | LE | Chiralpak MA (+); amino acid derivatives on silica | acetonitrile/2 mM Cu(II) (10:90) | 234 |
| Diarylheptanoids | Alpinia katsumadai | CIC | Daicel Chiralpak IB; cellulose 3,5-dimethylphenylcarbamate on silica | n-Hexane/2-propanol (7:3) | 235 |
| Kempopeptins A, B | Lyngbya sp. | LE | Chirex 3126; D-PA on silica | 2 mM Cu(II) or 2 mM Cu(II)/acetonitrile (95:5) | 236 |
| Hexamollamide | Didemnum molle | LE | Chiralpak MA (+); amino acid derivatives on silica | 2 mM Cu(II)/acetonitrile (80:20) | 237 |
| Hantupeptin A | Lyngbya majuscula | LE | Chiralpak MA (+); amino acid derivatives on silica | 2 mM Cu(II)/acetonitrile (85:15); | 238 |
| Eudistomides A, B | Eudistoma sp. | LE | Chirex 3126; D-PA on silica | 1 mM Cu(II)/acetonitrile (95:5) | 239 |
Adams et al.225 isolated malevamide E, a dolastatin 14 analogue, from the marine cyanobacterium Symploca laete-viridis. They used aqueous Cu(II) solutions with acetonitrile as mobile phase. In another approach, Clark et al.228 discovered 6 new acyl proline derivatives and tumonoic acids D–I. Stereostructures were elucidated by chiral HPLC using a Phenomenex Chirex 3126® column consisting of D-penicillamine bonded on silica backbone. An aqueous solution of 2 mM copper(II) sulfate served as mobile phase. This column showed wide applicability for determination of absolute configuration.225,228–230,232,233,236,239 Teruya and coworkers applied another ligand-exchange column, namely a Daicel Chiralpak MA (+), for the determination of a hexapeptide, hexamollamide after bioassay-guided fractionation of the Okinawan ascidian Didemnum molle.237
Another approach for enantioseparation by HPLC represents the use of a so called Pirkle-column or brush-type phase. These columns provide various selectors for ionic or covalent bonding. The chiral selector consists of an optically pure amino acid bonded to γ-aminopropylsilanized silica. A linking of a π-electron group to the stereogenic center of the selector provides π-electron interactions and one point of chiral recognition. Koyama reports the elucidation of relative and absolute stereochemistry of quinadoline B, an inhibitor of lipid droplet synthesis in macrophages.231 For chiral HPLC, a Sumichiral OA-3100 column with covalently bonded (S)-valine as chiral selector and a mixture of methanol/acetonitrile (95:5) containing 1 mM citric acid was used. Further examples for the successful use of chiral HPLC columns can be found in Table 4.
Besides HPLC, GC and CE can be used for determination of stereostructure as well. Generally, the chiral selectors provided for HPLC are also applicable in GC and CE. For example, malyngolide dimer was isolated by Gutierrez et al. after the extract of the marine cyanobacterium Lyngbya majuscula was fractionated.240 The absolute configuration was determined by chiral GC-MS after chemical degradation and results were compared with an authentic sample. Pinto et al.241 reported the isolation of a new triquinane sesquiterpene, (−)-epi-presilphiperfolan-1-ol, from the essential oil of Anemia tomentosa var. anthriscifolia. They elucidated chiral configuration by bi-dimensional GC using 2,3-di-O-ethyl-6-O-tert-butyldimethyl-silyl-β-cyclodextrin as the chiral stationary phase.241 There is a variety of chiral capillaries for GC commercially available. First development of a chiral GC capillary was done by Gil-Av's group.242 An amino acid derivative served as chiral selector for enantioseparation of N-trifluoroacetyl amino acids. Chiral recognition on these phases is based on the formation of multiple hydrogen bonds. Moreover, columns based on the chiral separation principle of metal complexes, cyclodextrins, cyclocholates, calixarenes are used.219
6 Isolation by preparative gas chromatography (PGC)
For isolation of volatiles, PGC is an attractive option. Usually packed columns with higher sample capacity but lower peak resolution are employed,243,244 however there are an increasing number of successful applications of thick-phase film wide-bore capillaries with capillary GC instrumentation during the last years. PGC was reviewed recently giving also some practical advice to achieve satisfying results.245 Menthol and menthone from peppermint oil (Mentha x piperita) have been isolated using a 15 m × 0.32 mm i.d. DB-5 column (1 μm film thickness) and an external cryotrap. Flow switching between the cryotrap and the detector (FID) was gained by an Deans switch device.246 A multidimensional PGC consisting of three GC systems equipped with three Deans switch transfer devices was used for isolation of carotol, an oxygenated sesquiterpene from carrot seed oil (Daucus carota).247 By combining 5% diphenyl-polyethylene glycol-ionic liquid stationary phases with diverse selectivity in the preparative MDGC setup, 2.22 mg of carotol were collected in about 230 min.247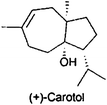
Compounds selected in a MDGC setup on the first GC column by microfluidic heart-cut could be enriched from multiple runs by an internal cryogenically cooled trap before transferring to the second column.248 For fractionation of volatiles emitted by Spodoptera-infested maize seedlings, which were most attractive to females of the parasitoid Cotesia marginiventris, even micro-bore capillary columns were used.249 (E,E)-2,4-Undecadienal was identified as the most deodorizing compound in the odor of coriander leaves (Coriandrum sativum) with aid of PGC on a 60 m × 0.75 mm column with a polyethylene glycol stationary phase.250
7 Conclusions
In recent years several major developments have been recognized in the field of NP isolation. An increasing number of methods have been developed by hyphenation of chromatographic and spectroscopic or spectrometric techniques with the aim to elucidate structures of known as well as novel compounds without the need for isolation. In the same direction goes coupling of LC with SPE trapping and transfer to capillary NMR illustrating the trend to downscale isolation procedures. Microwave and ultrasonic-assisted extraction procedures as well as accelerated solvent extraction seem to be established as methods increasing extraction efficacy and shortening extraction time. IL as extraction solvents are also an upcoming field in the natural products area and maybe will result in a more selective enrichment of compounds of interest already in crude extracts. SPE widened its application towards fractionation similar to VLC. However, the most exciting development in SPE seems to be the selective isolation of target compounds by molecularly imprinted stationary phases.Chiral separations are increasingly also applied at preparative scale, taking the chiral character of many NPs into account. Although the chromatographic principle was known for many years, HILIC is currently experiencing a significant increase of applications in NP isolation and analysis, providing an additional mechanism of separation compared to normal and reversed-phase chromatography. Although isolation of pure compounds from difficult matrices like organic matter is still challenging and we are far from isolation procedures in one step, the application of more selective methods from extraction to fractionation and purification will speed up the time from collection of biological material to final purified compound.
8 References
- O. Sticher, Nat. Prod. Rep., 2008, 25, 517–554 RSC.
- S. Sturm and C. Seger, J. Chromatogr., A, 2012, 1259, 50–61 Search PubMed.
- J. Rosen, J. Gottfries, S. Muresan, A. Backlund and T. I. Oprea, J. Med. Chem., 2009, 52, 1953–1962 CrossRef CAS.
- D. G. I. Kingston, J. Nat. Prod., 2011, 74, 496–511 CrossRef CAS.
- Natural Products Isolation. Methods and Protocols, 3rd edn, ed. S. D. Sarker and L. Nahar, Humana Press, New York, 2012 Search PubMed.
- Bioactive Natural Products, 2nd edn, ed. S. M. Colegate and R. J. Molyneux, CRC Press Inc., Boca Raton, 2007 Search PubMed.
- T. A. Beek, K. K. R. Tetala, I. I. Koleva, A. Dapkevicius, V. Exarchou, S. M. F. Jeurissen, F. W. Claassen and E. J. C. Klift, Phytochem. Rev., 2009, 8, 387–399 Search PubMed.
- J. Zhao, G.-P. Lv, Y.-W. Chen and S.-P. Li, J. Chromatogr., A, 2011, 1218, 7453–7475 CrossRef CAS.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2010, 27, 165–237 RSC.
- A. Buriani, M. L. Garcia-Bermejo, E. Bosisio, Q. Xu, H. Li, X. Dong, M. S. J. Simmonds, M. Carrara, N. Tejedor, J. Lucio-Cazana and P. J. Hylands, J. Ethnopharmacol., 2012, 140, 535–544 Search PubMed.
- K. Chan, D. Shaw, M. S. J. Simmonds, C. J. Leon, Q. Xu, A. Lu, I. Sutherland, S. Ignatova, Y.-P. Zhu, R. Verpoorte, E. M. Williamson and P. Duez, J. Ethnopharmacol., 2012, 140, 469–475 Search PubMed.
- A colored identification atlas of Chinese materia medica and plants as specified in the pharmacopoeia of the People's Republic of China, ed. S. Chen, Y. Lin, Z. Qian and C. Leon, People's Medical Publishing House, Beijing, 2010 Search PubMed.
- W. P. Jones and A. D. Kinghorn, Methods Mol. Biol., 2012, 864, 341–366 Search PubMed.
- D. D. Soejarto, C. Gyllenhaal, H. H. S. Fong, L. T. Xuan, N. T. Hiep, N. V. Hung, T. Q. Bich, B. Southavong, K. Sydara and J. M. Pezzuto, J. Nat. Prod., 2004, 67, 294–299 Search PubMed.
- American Herbal Pharmacopoeia. Botanical pharmacognosy-microscopic characterization of botanical medicines, ed. R. Upton, A. Graff, G. Jolliffe, R. Länger and E. M. Williamson, American Herbal Pharmacopoeia/CRC Press, Boca Raton, 2011 Search PubMed.
- B. Rahfeld, Mikroskopischer Farbatlas pflanzlicher Drogen, Spektrum, Akad. Verl., Heidelberg, 2009 Search PubMed.
- K. B. Sanon, A. M. Ba, C. Delaruelle, R. Duponnois and F. Martin, Mycorrhiza, 2009, 19, 571–584 Search PubMed.
- S. Monchy, J.-D. Grattepanche, E. Breton, D. Meloni, G. Sanciu, M. Chabe, L. Delhaes, E. Viscogliosi, T. Sime-Ngando and U. Christaki, PLoS One, 2012, 7, e39924 Search PubMed.
- R. L. Simister, P. Deines, E. S. Botte, N. S. Webster and M. W. Taylor, Environ. Microbiol., 2012, 14, 517–524 Search PubMed.
- E. Reich, A. Schibli and A. Schibli, High-performance thin-layer chromatography for the analaysis of medicinal plants//High-performance thin-layer chromatography for the analysis of medicinal plants, Thieme, Stuttgart, 2007 Search PubMed.
- S. Sudberg, E. M. Sudberg, J. Terrazas, S. Sudberg, K. Patel, J. Pineda and B. Fine, J. AOAC Int., 2010, 93, 1367–1375 Search PubMed.
- B. Meier and D. Spriano, J. AOAC Int., 2010, 93, 1399–1409 CAS.
- Chromatographic fingerprint analysis of herbal medicines. Thin-layer and high performance liquid chromatography of Chinese drugs, 2nd edn, ed. H. Wagner, R. Bauer, D. Melchart, P.-G. Xiao and A. Staudinger, Springer, Wien, New York, 2011 Search PubMed.
- A. Ankli, E. Reich and M. Steiner, J. AOAC Int., 2008, 91, 1257–1264 Search PubMed.
- V. Widmer, E. Reich and A. DeBatt, J. Planar Chromatogr.–Mod. TLC, 2008, 21, 21–26 Search PubMed.
- F. R. Gallo, G. Multari, G. Pagliuca, A. Panusa, G. Palazzino, M. Giambenedetti, V. Petitto and M. Nicoletti, Nat. Prod. Res. DOI:10.1080/14786419.2012.696253.
- J. Sherma, J. AOAC Int., 2012, 95, 992–1009 CrossRef CAS.
- J. Zhang, Z. Zhou, J. Yang, W. Zhang, Y. Bai and H. Liu, Anal. Chem., 2012, 84, 1496–1503 Search PubMed.
- A. Gossi, U. Scherer and G. Schlotterbeck, Chimia, 2012, 66, 347–349 Search PubMed.
- E. A. Porter, d. B. A. A. van, G. C. Kite, N. C. Veitch and M. S. J. Simmonds, Phytochemistry, 2012, 81, 90–96 Search PubMed.
- G.-B. Ge, Y.-Y. Zhang, D.-C. Hao, Y. Hu, H.-W. Luan, X.-B. Liu, Y.-Q. He, Z.-T. Wang and L. Yang, Planta Med., 2008, 74, 773–779 Search PubMed.
- S. Agnolet, S. Wiese, R. Verpoorte and D. Staerk, J. Chromatogr., A, 2012, 1262, 130–137 Search PubMed.
- Y. Chen, W. Bicker, J. Y. Wu, M. Y. Xie and W. Lindner, J. Chromatogr., A, 2010, 1217, 1255–1265 Search PubMed.
- High performance liquid chromatography in phytochemical analysis, M. Waksmundzka-Hajnos and J. Sherma, eds., CRC Press, Boca Raton, 2011 Search PubMed.
- J.-L. Wolfender, Planta Med., 2009, 75, 719–734 CrossRef CAS.
- C. S. Funari, P. J. Eugster, S. Martel, P.-A. Carrupt, J.-L. Wolfender and D. H. S. Silva, J. Chromatogr., A, 2012, 1259, 167–178 Search PubMed.
- P. J. Eugster, D. Guillarme, S. Rudaz, J.-L. Veuthey, P.-A. Carrupt and J.-L. Wolfender, J. AOAC Int., 2011, 94, 51–70 Search PubMed.
- E. Mateus, R. C. Barata, J. Zrostlikova, d. S. M. D. R. Gomes and M. R. Paiva, J. Chromatogr., A, 2010, 1217, 1845–55 Search PubMed.
- P. J. Marriott, G. T. Eyres and J.-P. Dufour, J. Agric. Food Chem., 2009, 57, 9962–9971 Search PubMed.
- L. Mondello, P. Q. Tranchida, P. Dugo and G. Dugo, Mass Spectrom. Rev., 2008, 27, 101–124 CrossRef CAS.
- Y. Qiu, X. Lu, T. Pang, C. Ma, X. Li and G. Xu, J. Sep. Sci., 2008, 31, 3451–3457 CrossRef CAS.
- J. Vial, H. Nocairi, P. Sassiat, S. Mallipatu, G. Cognon, D. Thiebaut, B. Teillet and D. N. Rutledge, J. Chromatogr., A, 2009, 1216, 2866–2872 Search PubMed.
- B. Slabbinck, B. de Baets, P. Dawyndt and P. de Vos, Syst. Appl. Microbiol., 2009, 32, 163–176 Search PubMed.
- F. van der Kooy, F. Maltese, Y. H. Choi, H. K. Kim and R. Verpoorte, Planta Med., 2009, 75, 763–775 CrossRef CAS.
- H. K. Kim, Y. H. Choi and R. Verpoorte, Nat. Protoc., 2010, 5, 536–549 Search PubMed.
- M. I. Georgiev, K. Ali, K. Alipieva, R. Verpoorte and Y. H. Choi, Phytochemistry, 2011, 72, 2045–2051 Search PubMed.
- H. K. Kim, Saifullah, S. Khan, E. G. Wilson, S. D. P. Kricun, A. Meissner, S. Goraler, A. M. Deelder, Y. H. Choi and R. Verpoorte, Phytochemistry, 2010, 71, 773–784 CrossRef CAS.
- Y. Chen, M.-Y. Xie, Y. Yan, S.-B. Zhu, S.-P. Nie, C. Li, Y.-X. Wang and X.-F. Gong, Anal. Chim. Acta, 2008, 618, 121–130 CrossRef CAS.
- M. Kokalj, J. Kolar, T. Trafela and S. Kreft, Planta Med., 2011, 77, PA38 Search PubMed.
- A. Alvarez-Ordóñez, D. J. M. Mouwen, M. López and M. Prieto, J. Microbiol. Methods, 2011, 84, 369–378 Search PubMed.
- A. Wieser, L. Schneider, J. Jung and S. Schubert, Appl. Microbiol. Biotechnol., 2012, 93, 965–974 CrossRef CAS.
- Y.-P. Ho and P. M. Reddy, Mass Spectrom. Rev., 2011, 30, 1203–1224 CrossRef CAS.
- J. Ruzicka, B. Lukas, L. Merza, I. Göhler, G. Abel, M. Popp and J. Novak, Planta Med., 2009, 75, 1271–1276 Search PubMed.
- E. Mader, J. Ruzicka, C. Schmiderer and J. Novak, Anal. Biochem., 2011, 409, 153–155 CrossRef CAS.
- N. Jain, A. Shasany, S. Singh, S. Khanuja and S. Kumar, Planta Med., 2008, 74, 296–301 Search PubMed.
- M. Staats, A. Cuenca, J. E. Richardson, G. R. Vrielink-van, G. Petersen, O. Seberg and F. T. Bakker, PLoS One, 2011, 6, e28448 Search PubMed.
- F. S. Nolte and A. M. Caliendo, Molecular detection and identification of microorganisms, in Man. Clin. Microbiol., 9th ed., American Society for Microbiology, 2007, vol. 1, pp. 218–244 Search PubMed.
- P. Cullen, H. Funke, H.-G. Klein, T. Langmann and M. Neumaier, Laboratoriumsmedizin, 2008, 32, 317–320 Search PubMed.
- M. Saker, C. Moreira, J. Martins, B. Neilan and V. M. Vasconcelos, Appl. Microbiol. Biotechnol., 2009, 85, 237–252 Search PubMed.
- W. Kreis, Enzyme bei der Gewinnung von Drogen und der Herstellung von Phytopharmaka, in Pharmakognosie - Phytopharmazie, ed. R. Hänsel and O. Sticher, Springer, Heidelberg, 2007, pp. 285–291 Search PubMed.
- H. Janecke and W. Hennig, Planta Med., 1959, 7, 41–55 Search PubMed.
- H. Janecke and W. Hennig, Mitt. Dtsch. Pharm. Ges., 1960, 30, 136–42 Search PubMed.
- B. Nuesslein, M. Kurzmann, R. Bauer and W. Kreis, J. Nat. Prod., 2000, 63, 1615–1618 Search PubMed.
- X.-B. Li, W. Wang, G.-J. Zhou, Y. Li, X.-M. Xie and T.-S. Zhou, Molecules, 2012, 17, 2388–2407 Search PubMed.
- S.-L. Li, R. Yan, Y.-K. Tam and G. Lin, Chem. Pharm. Bull., 2007, 55, 140–144 Search PubMed.
- H. Boettcher, I. Guenther and R. Franke, Gartenbauwissenschaft, 2002, 67, 243–254 Search PubMed.
- H. Boettcher, I. Gunther and U. Bauermann, Postharvest Biol. Technol., 1999, 15, 41–52 Search PubMed.
- H. Boettcher, I. Gunther and L. Kabelitz, Postharvest Biol. Technol., 2003, 29, 343–351 Search PubMed.
- F. Bucar, Phytoestrogens in plants: with special reference to isoflavones, in Isoflavones, Chemistry, Analysis, Function and Effects, ed. V. Preedy, RSC Publishing, Cambridge, 2013, pp. 14–27 Search PubMed.
- F. Maltese, F. van der Kooy and R. Verpoorte, Nat. Prod. Commun., 2009, 4, 447–454 Search PubMed.
- V. Seidel, Methods Mol. Biol., 2012, 864, 27–41 Search PubMed.
- E. Ghisalberti, Detection and Isolation of Bioactive Natural Products, in Bioactive Natural Products, ed. J. R. Molyneux and S. M. Colegate, CRC Press, Boca Raton, 2007, pp. 11–76 Search PubMed.
- F. Adje, Y. F. Lozano, P. Lozano, A. Adima, F. Chemat and E. M. Gaydou, Ind. Crops Prod., 2010, 32, 439–444 Search PubMed.
- S. Boonkird, C. Phisalaphong and M. Phisalaphong, Ultrason. Sonochem., 2008, 15, 1075–1079 Search PubMed.
- G. Rao, Anal. Methods, 2010, 2, 1166–1170 RSC.
- J. M. Roldan-Gutierrez, J. Ruiz-Jimenez and d. C. M. D. Luque, Talanta, 2008, 75, 1369–1375 CrossRef CAS.
- S. A. Chowdhury, R. Vijayaraghavan and D. R. MacFarlane, Green Chem., 2010, 12, 1023–1028 RSC.
- X. Lin, Y. Wang, X. Liu, S. Huang and Q. Zeng, Analyst, 2012, 137, 4076–4085 RSC.
- A. A. Lapkin, P. K. Plucinski and M. Cutler, J. Nat. Prod., 2006, 69, 1653–1664 CrossRef CAS.
- Y. Sun, Z. Liu, J. Wang, S. Yang, B. Li and N. Xu, Ultrason. Sonochem., 2013, 20, 180–186 Search PubMed.
- M. G. Bogdanov, I. Svinyarov, R. Keremedchieva and A. Sidjimov, Sep. Purif. Technol., 2012, 97, 221–227 Search PubMed.
- Y. Lu, W. Ma, R. Hu, X. Dai and Y. Pan, J. Chromatogr., A, 2008, 1208, 42–46 CrossRef CAS.
- F.-Y. Du, X.-H. Xiao and G.-K. Li, J. Chromatogr., A, 2007, 1140, 56–62 CrossRef CAS.
- F.-Y. Du, X.-H. Xiao, X.-J. Luo and G.-K. Li, Talanta, 2009, 78, 1177–1184 CrossRef CAS.
- C. Lu, H. Wang, W. Lv, C. Ma, P. Xu, J. Zhu, J. Xie, B. Liu and Q. Zhou, Chromatographia, 2011, 74, 139–144 CrossRef CAS.
- W. Bi, M. Tian and K. H. Row, Talanta, 2011, 85, 701–706 CrossRef CAS.
- W. Bi, M. Tian and K. H. Row, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2012, 880, 108–113 Search PubMed.
- A. Delazar, L. Nahar, S. Hamedeyazdan and S. D. Sarker, Methods Mol. Biol., 2012, 864, 89–115 Search PubMed.
- C.-H. Chan, R. Yusoff, G.-C. Ngoh and F. W.-L. Kung, J. Chromatogr., A, 2011, 1218, 6213–6225 CrossRef CAS.
- B. Tang, W. Bi, M. Tian and K. H. Row, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2012, 904, 1–21 CrossRef CAS.
- Y. Yuan, Y.-Z. Wang, M.-D. Huang, R. Xu, H. Zeng, C. Nie and J.-H. Kong, Anal. Chim. Acta, 2011, 695, 63–72 Search PubMed.
- X. Yin, Q. Liu, Y. Jiang and Y. Luo, Spectrochim. Acta, Part A, 2011, 79, 191–196 CrossRef CAS.
- X. Song, J. Li, J. Wang and L. Chen, Talanta, 2009, 80, 694–702 CrossRef.
- F.-F. Chen, R. Wang and Y.-P. Shi, Talanta, 2012, 89, 505–512 CrossRef CAS.
- C.-Y. Chen, C.-H. Wang and A.-H. Chen, Talanta, 2011, 84, 1038–1046 Search PubMed.
- F.-F. Chen, G.-Y. Wang and Y.-P. Shi, J. Sep. Sci., 2011, 34, 2602–2610 Search PubMed.
- B. Claude, P. Morin, M. Lafosse, A.-S. Belmont and K. Haupt, Talanta, 2008, 75, 344–350 CAS.
- W. Bi, M. Tian and K. H. Row, J. Chromatogr., A, 2012, 1232, 37–42 CrossRef CAS.
- M. Tian and K. H. Row, Chromatographia, 2011, 73, 25–31 Search PubMed.
- M. Markiewicz, C. Jungnickel, A. Markowska, U. Szczepaniak, M. Paszkiewicz and J. Hupka, Molecules, 2009, 14, 4396–4405 Search PubMed.
- P. C. A. G. Pinto, S. P. F. Costa, J. L. F. C. Lima and M. L. M. F. S. Saraiva, Ecotoxicol. Environ. Saf., 2012, 80, 97–102 Search PubMed.
- S. P. M. Ventura, A. M. M. Goncalves, T. Sintra, J. L. Pereira, F. Goncalves and J. A. P. Coutinho, Ecotoxicology, 2012 Search PubMed.
- M. A. Mottaleb and S. D. Sarker, Methods Mol. Biol., 2012, 864, 75–87 Search PubMed.
- G. Rieger, M. Mueller, H. Guttenberger and F. Bucar, J. Agric. Food Chem., 2008, 56, 9080–9086 Search PubMed.
- S. S. Cicek, S. Schwaiger, E. P. Ellmerer and H. Stuppner, Planta Med., 2010, 76, 467–473 Search PubMed.
- J. Chen, F. Wang, J. Liu, F. S.-C. Lee, X. Wang and H. Yang, Anal. Chim. Acta, 2008, 613, 184–195 CrossRef CAS.
- Z. Han, Y. Ren, J. Zhu, Z. Cai, Y. Chen, L. Luan and Y. Wu, J. Agric. Food Chem., 2012, 60, 8233–8247 CrossRef CAS.
- S. Fuchs, E. Gruenauer, G. Reich and G. Sontag, Ernaehrung, 2012, 36, 299–307 Search PubMed.
- Q. G. Liao, R. L. Li and L. G. Luo, Chromatographia, 2012, 75, 931–935 CrossRef CAS.
- J. Fojtova, L. Lojkova and V. Kuban, J. Sep. Sci., 2008, 31, 162–168 Search PubMed.
- Y. Zhang, C. Liu, M. Yu, Z. Zhang, Y. Qi, J. Wang, G. Wu, S. Li, J. Yu and Y. Hu, J. Chromatogr., A, 2011, 1218, 2827–2834 Search PubMed.
- L. He, X. Zhang, H. Xu, C. Xu, F. Yuan, Ž. Knez, Z. Novak and Y. Gao, Food Bioprod. Process., 2012, 90, 215–223 Search PubMed.
- P. Rangsriwong, N. Rangkadilok, J. Satayavivad, M. Goto and A. Shotipruk, Sep. Purif. Technol., 2009, 66, 51–56 Search PubMed.
- M.-J. Ko, C.-I. Cheigh, S.-W. Cho and M.-S. Chung, J. Food Eng., 2011, 102, 327–333 Search PubMed.
- P. P. Singh and M. D. A. Saldaña, Food Res. Int., 2011, 44, 2452–2458 Search PubMed.
- B. Jayawardena and R. M. Smith, Phytochem. Anal., 2010, 21, 470–472 Search PubMed.
- M. Plaza, M. Amigo-Benavent, M. D. del Castillo, E. Ibáñez and M. Herrero, Food Res. Int., 2010, 43, 2341–2348 Search PubMed.
- L. Nahar and S. D. Sarker, Methods Mol. Biol., 2012, 864, 43–74 Search PubMed.
- Z. Huang, X.-H. Shi and W.-J. Jiang, J. Chromatogr., A, 2012, 1250, 2–26 CrossRef CAS.
- F. M. C. Barros, F. C. Silva, J. M. Nunes, R. M. F. Vargas, E. Cassel and P. G. L. von, J. Sep. Sci., 2011, 34, 3107–3113 Search PubMed.
- J. P. Coelho, A. F. Cristino, P. G. Matos, A. P. Rauter, B. P. Nobre, R. L. Mendes, J. G. Barroso, A. Mainar, J. S. Urieta, J. M. N. A. Fareleira, H. Sovova and A. F. Palavra, Molecules, 2012, 17, 10550–10573 Search PubMed.
- T. Hatami, R. N. Cavalcanti, T. M. Takeuchi and M. A. A. Meireles, J. Supercrit. Fluids, 2012, 65, 71–77 Search PubMed.
- K. Ghafoor, J. Park and Y.-H. Choi, Innovative Food Sci. Emerging Technol., 2010, 11, 485–490 Search PubMed.
- J.-L. Wolfender, G. Marti and E. F. Queiroz, Curr. Org. Chem., 2010, 14, 1808–1832 CrossRef CAS.
- J.-L. Wolfender, Chromatogr. Sci. Ser., 2011, 102, 287–329 Search PubMed.
- K. T. Johansen, S. G. Wubshet, N. T. Nyberg and J. W. Jaroszewski, J. Nat. Prod., 2011, 74, 2454–2461 Search PubMed.
- M. Bhandari, A. Bhandari and A. Bhandari, J. Young Pharm., 2011, 3, 226–231 Search PubMed.
- Y. Tu, C. Jeffries, H. Ruan, C. Nelson, D. Smithson, A. A. Shelat, K. M. Brown, X.-C. Li, J. P. Hester, T. Smillie, I. A. Khan, L. Walker, K. Guy and B. Yan, J. Nat. Prod., 2010, 73, 751–754 CrossRef CAS.
- M. Maansson, R. K. Phipps, L. Gram, M. H. G. Munro, T. O. Larsen and K. F. Nielsen, J. Nat. Prod., 2010, 73, 1126–1132 CrossRef.
- J. J. Araya, G. Montenegro, L. A. Mitscher and B. N. Timmermann, J. Nat. Prod., 2010, 73, 1568–1572 CrossRef CAS.
- C. Tschiggerl and F. Bucar, Fitoterapia, 2011, 82, 903–910 Search PubMed.
- C. Tschiggerl and F. Bucar, Plant Foods Hum. Nutr., 2012, 67, 129–135 Search PubMed.
- C. Tschiggerl and F. Bucar, Phytochem. Rev. DOI:10.1007/s11101-012-9244-6.
- N. Sahraoui, M. A. Vian, I. Bornard, C. Boutekedjiret and F. Chemat, J. Chromatogr., A, 2008, 1210, 229–233 Search PubMed.
- A. Farhat, C. Ginies, M. Romdhane and F. Chemat, J. Chromatogr., A, 2009, 1216, 5077–5085 Search PubMed.
- G. Oezek, F. Demirci, T. Oezek, N. Tabanca, D. E. Wedge, S. I. Khan, K. H. C. Baser, A. Duran and E. Hamzaoglu, J. Chromatogr., A, 2010, 1217, 741–748 CrossRef.
- H. Krueger, Planta Med., 2010, 76, 843–846 Search PubMed.
- A. Marston, J. Chromatogr., A, 2011, 1218, 2676–2683 Search PubMed.
- X.-Y. Zheng, L. Zhang, X.-M. Cheng, Z.-J. Zhang, C.-H. Wang and Z.-T. Wang, J. Planar Chromatogr.–Mod. TLC, 2011, 24, 470–474 Search PubMed.
- P. N. Okusa, C. Stevigny, M. Devleeschouwer and P. Duez, J. Planar Chromatogr.–Mod. TLC, 2010, 23, 245–249 Search PubMed.
- J. Sherma, J. AOAC Int., 2012, 95, 992–1009 CrossRef CAS.
- E. Tyihak and E. Mincsovics, J. Planar Chromatogr.–Mod. TLC, 2010, 23, 382–395 Search PubMed.
- E. Mincsovics and E. Tyihak, Nat. Prod. Commun., 2011, 6, 719–732 Search PubMed.
- S. Gibbons, Methods Mol. Biol., 2012, 864, 117–153 Search PubMed.
- R. G. Reid and S. D. Sarker, Methods Mol. Biol., 2012, 864, 155–87 Search PubMed.
- S. Hadi and Noviany, Adv. Nat. Appl. Sci., 2009, 3, 107–112 Search PubMed.
- Noviany and S. Hadi, Mod. Appl. Sci., 2009, 3, 45–51 Search PubMed.
- G. Todorova, I. Lazarova, B. Mikhova and I. Kostova, Chem. Nat. Compd., 2010, 46, 322–323 Search PubMed.
- J. Y. Seo, S. S. Lim, J. R. Kim, J.-S. Lim, Y. R. Ha, I. A. Lee, E. J. Kim, J. H. Y. Park and J.-S. Kim, Phytother. Res., 2008, 22, 1500–1505 Search PubMed.
- K. Garcia-Sosa, A. Sanchez-Medina, S. L. Alvarez, S. Zacchino, N. C. Veitch, P. Sima-Polanco and L. M. Pena-Rodriguez, Nat. Prod. Res., 2011, 25, 1185–1189 Search PubMed.
- A. D. Wright and N. Lang-Unnasch, J. Nat. Prod., 2009, 72, 492–495 CrossRef CAS.
- L. Miller and M. Mahoney, J. Chromatogr., A, 2012, 1250, 264–273 Search PubMed.
- J. D. Fair and C. M. Kormos, J. Chromatogr., A, 2008, 1211, 49–54 CrossRef CAS.
- J. Sherma, Flash chromatography: TLC for method development and purity testing of fractions, in Encycl. Chromatogr. (3rd Ed.), CRC Press, 2010, vol. 2, pp. 874–877 Search PubMed.
- P. Weber, M. Hamburger, N. Schafroth and O. Potterat, Fitoterapia, 2011, 82, 155–161 Search PubMed.
- A. P. Breksa and K. Dragull, Food Chem., 2009, 113, 1308–1313 Search PubMed.
- S. Schmidt, G. Jurgenliemk, H. Skaltsa and J. Heilmann, Phytochemistry, 2012, 77, 218–225 Search PubMed.
- R. Graziose, T. Rathinasabapathy, C. Lategan, A. Poulev, P. J. Smith, M. Grace, M. A. Lila and I. Raskin, J. Ethnopharmacol., 2011, 133, 26–30 Search PubMed.
- F. Mattivi, U. Vrhovsek, G. Malacarne, D. Masuero, L. Zulini, M. Stefanini, C. Moser, R. Velasco and G. Guella, J. Agric. Food Chem., 2011, 59, 5364–5375 Search PubMed.
- P. W. Yang, M. G. Li, J. Y. Zhao, M. Z. Zhu, H. Shang, J. R. Li, X. L. Cui, R. Huang and M. L. Wen, Folia Microbiol., 2010, 55, 10–16 Search PubMed.
- A. Wohlfarth, H. Mahler and V. Auwaerter, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3059–3064 Search PubMed.
- R. M. Uckoo, G. K. Jayaprakasha and B. S. Patil, Sep. Purif. Technol., 2011, 81, 151–158 Search PubMed.
- M. J. Somerville, P. L. Katavic, L. K. Lambert, G. K. Pierens, J. T. Blanchfield, G. Cimino, E. Mollo, M. Gavagnin, M. G. Banwell and M. J. Garson, J. Nat. Prod., 2012, 75, 1618–1624 Search PubMed.
- H. Henke, Preparative Gel Chromatography on Sephadex LH-20, Huethig, Heidelberg, 1996, pp. 276–280 Search PubMed.
- Y. Cheng, Q. Liang, P. Hu, Y. Wang, F. W. Jun and G. Luo, Sep. Purif. Technol., 2010, 73, 397–402 CrossRef CAS.
- J. Conrad, B. Förster-Fromme, M.-A. Constantin, V. Ondrus, S. Mika, F. Mert-Balci, I. Klaiber, J. Pfannstiel, W. Möller, H. Rösner, K. Förster-Fromme and U. Beifuss, J. Nat. Prod., 2009, 72, 835–840 CrossRef CAS.
- J. Yang, H. Ye, H. Lai, S. Li, S. He, S. Zhong, L. Chen and A. Peng, J. Sep. Sci., 2012, 35, 256–262 Search PubMed.
- M. Hungeling, M. Lechtenberg, F. R. Fronczek and A. Nahrstedt, Phytochemistry, 2009, 70, 270–277 Search PubMed.
- R. Wang, X. Peng, L. Wang, B. Tan, J. Liu, Y. Feng and S. Yang, J. Sep. Sci., 2012, 35, 1985–1992 Search PubMed.
- P. P. Daramwar, P. L. Srivastava, B. Priyadarshini and H. V. Thulasiram, Analyst, 2012, 137, 4564–4570 RSC.
- A. J. Alpert, J. Chromatogr., A, 1990, 499, 177–196 CrossRef CAS.
- Y. Guo and S. Gaiki, J. Chromatogr., A, 2011, 1218, 5920–5938 Search PubMed.
- P. Jandera, Anal. Chim. Acta, 2011, 692, 1–25 CrossRef CAS.
- J. Bernal, A. M. Ares, J. Pol and S. K. Wiedmer, J. Chromatogr., A, 2011, 1218, 7438–7452 CrossRef CAS.
- M. R. Gama, R. G. da Costa Silva, C. H. Collins and C. B. G. Bottoli, TrAC, Trends Anal. Chem., 2012, 37, 48–60 CrossRef.
- H. Zhang, Z. Guo, W. Li, J. Feng, Y. Xiao, F. Zhang, X. Xue and X. Liang, J. Sep. Sci., 2009, 32, 526–535 CrossRef CAS.
- M. Karonen, J. Liimatainen and J. Sinkkonen, J. Sep. Sci., 2011, 34, 3158–3165 Search PubMed.
- T. Tan, Z.-G. Su, M. Gu, J. Xu and J.-C. Janson, Biotechnol. J., 2010, 5, 505–510 Search PubMed.
- Y. Liu, J. Feng, Y. Xiao, Z. Guo, J. Zhang, X. Xue, J. Ding, X. Zhang and X. Liang, J. Sep. Sci., 2010, 33, 1487–1494 Search PubMed.
- T. Morikawa, Y. Xie, Y. Asao, M. Okamoto, C. Yamashita, O. Muraoka, H. Matsuda, Y. Pongpiriyadacha, D. Yuan and M. Yoshikawa, Phytochemistry, 2009, 70, 1166–1172 CrossRef CAS.
- M. Inoue, K. Ohtani, R. Kasai, M. Okukubo, M. Andriantsiferana, K. Yamasaki and T. Koike, Phytochemistry, 2009, 70, 1195–1202 CrossRef CAS.
- R. M. van Wagoner, J. R. Deeds, A. O. Tatters, A. R. Place, C. R. Tomas and J. L. C. Wright, J. Nat. Prod., 2010, 73, 1360–1365 CrossRef CAS.
- M. Scognamiglio, B. D'Abrosca, V. Fiumano, A. Chambery, V. Severino, N. Tsafantakis, S. Pacifico, A. Esposito and A. Fiorentino, Phytochemistry, 2012, 84, 125–134 Search PubMed.
- P. Luecha, K. Umehara, T. Miyase and H. Noguchi, J. Nat. Prod., 2009, 72, 1954–1959 Search PubMed.
- E. Pan, S. Cao, P. J. Brodie, M. W. Callmander, R. Randrianaivo, S. Rakotonandrasana, E. Rakotobe, V. E. Rasamison, K. TenDyke, Y. Shen, E. M. Suh and D. G. I. Kingston, J. Nat. Prod., 2011, 74, 1169–1174 CrossRef CAS.
- P. Grabher, E. Durieu, E. Kouloura, M. Halabalaki, L. A. Skaltsounis, L. Meijer, M. Hamburger and O. Potterat, Planta Med., 2012, 78, 951–956 Search PubMed.
- H. J. Kim, I. Baburin, J. Zaugg, S. N. Ebrahimi, S. Hering and M. Hamburger, Planta Med., 2012, 78, 440–447 Search PubMed.
- S. Challal, N. Bohni, O. E. Buenafe, C. V. Esguerra, W. P. A. M. de, J.-L. Wolfender and A. D. Crawford, Chimia, 2012, 66, 229–232 Search PubMed.
- C. E. Dalgliesh, J. Chem. Soc., 1952, 3940–3942 RSC.
- J. Zaugg, E. Eickmeier, S. N. Ebrahimi, I. Baburin, S. Hering and M. Hamburger, J. Nat. Prod., 2011, 74, 1437–1443 CrossRef CAS.
- L. Pan, D. D. Lantvit, S. Riswan, L. B. S. Kardono, H.-B. Chai, E. J. Carcache Blanco, N. R. Farnsworth, D. D. Soejarto, S. M. Swanson and A. D. Kinghorn, Phytochemistry, 2010, 71, 635–640 CrossRef CAS.
- F. Moradi-Afrapoli, S. N. Ebrahimi, M. Smiesko, M. Raith, S. Zimmermann, F. Nadjafi, R. Brun and M. Hamburger, Phytochemistry, 2013, 85, 143–152 Search PubMed.
- F. He, C. Lindqvist and W. W. Harding, Phytochemistry, 2012, 83, 168–172 Search PubMed.
- A. Castro, J. Coll and M. Arfan, J. Nat. Prod., 2011, 74, 1036–1041 CrossRef CAS.
- S. Wittayalai, S. Sathalalai, S. Thorroad, P. Worawittayanon, S. Ruchirawat and N. Thasana, Phytochemistry, 2012, 76, 117–123 Search PubMed.
- J. T. Banzouzi, P. N. Soh, B. Mbatchi, A. Cave, S. Ramos, P. Retailleau, O. Rakotonandrasana, A. Berry and F. Benoit-Vical, Planta Med., 2008, 74, 1453–1456 Search PubMed.
- W. Yuan, P. Wang, G. Deng and S. Li, Phytochemistry, 2012, 75, 67–77 Search PubMed.
- M. Furukawa, M. Makino, E. Ohkoshi, T. Uchiyama and Y. Fujimoto, Phytochemistry, 2011, 72, 2244–2252 CrossRef CAS.
- S. Cao, Y. Hou, P. Brodie, J. S. Miller, R. Randrianaivo, E. Rakotobe, V. E. Rasamison and D. G. I. Kingston, Chem. Biodiversity, 2011, 8, 643–650 CrossRef CAS.
- F. Yang, M. T. Hamann, Y. Zou, M.-Y. Zhang, X.-B. Gong, J.-R. Xiao, W.-S. Chen and H.-W. Lin, J. Nat. Prod., 2012, 75, 774–778 Search PubMed.
- X. Yang, Y. Feng, S. Duffy, V. M. Avery, D. Camp, R. J. Quinn and R. A. Davis, Planta Med., 2011, 77, 1644–1647 Search PubMed.
- S. Kongkiatpaiboon, J. Schinnerl, S. Felsinger, V. Keeratinijakal, S. Vajrodaya, W. Gritsanapan, L. Brecker and H. Greger, J. Nat. Prod., 2011, 74, 1931–1938 Search PubMed.
- Y. Sakaguchi, Y. Ozaki, I. Miyajima, M. Yamaguchi, Y. Fukui, K. Iwasa, S. Motoki, T. Suzuki and H. Okubo, Phytochemistry, 2008, 69, 1763–1766 CrossRef CAS.
- R. Nakabayashi, M. Kusano, M. Kobayashi, T. Tohge, K. Yonekura-Sakakibara, N. Kogure, M. Yamazaki, M. Kitajima, K. Saito and H. Takayama, Phytochemistry, 2009, 70, 1017–1029 CrossRef.
- L. Di Donna, G. Luca, F. Mazzotti, A. Napoli, R. Salerno, D. Taverna and G. Sindona, J. Nat. Prod., 2009, 72, 1352–1354 CrossRef CAS.
- L.-C. Lin, C.-T. Chiou and J.-J. Cheng, J. Nat. Prod., 2011, 74, 2001–2004 Search PubMed.
- C.-L. Chang, G.-J. Wang, L.-J. Zhang, W.-J. Tsai, R.-Y. Chen, Y.-C. Wu and Y.-H. Kuo, Phytochemistry, 2010, 71, 271–279 Search PubMed.
- S.-F. Wu, F.-R. Chang, S.-Y. Wang, T.-L. Hwang, C.-L. Lee, S.-L. Chen, C.-C. Wu and Y.-C. Wu, J. Nat. Prod., 2011, 74, 989–996 CrossRef CAS.
- K. Matsunami, H. Otsuka, K. Kondo, T. Shinzato, M. Kawahata, K. Yamaguchi and Y. Takeda, Phytochemistry, 2009, 70, 1277–1285 CrossRef CAS.
- R. Omar, L. Li, T. Yuan and N. P. Seeram, J. Nat. Prod., 2012, 75, 1505–1509 Search PubMed.
- P.-H. Chuang, P.-W. Hsieh, Y.-L. Yang, K.-F. Hua, F.-R. Chang, J. Shiea, S.-H. Wu and Y.-C. Wu, J. Nat. Prod., 2008, 71, 1365–1370 CrossRef.
- S. Matthew, V. J. Paul and H. Luesch, Planta Med., 2009, 75, 528–533 CrossRef CAS.
- T. P. Wyche, Y. Hou, E. Vazquez-Rivera, D. Braun and T. S. Bugni, J. Nat. Prod., 2012, 75, 735–740 Search PubMed.
- R. Abdou, K. Scherlach, H.-M. Dahse, I. Sattler and C. Hertweck, Phytochemistry, 2010, 71, 110–116 Search PubMed.
- E. Kouloura, M. Halabalaki, M.-C. Lallemand, S. Nam, R. Jove, M. Litaudon, K. Awang, H. A. Hadi and A.-L. Skaltsounis, J. Nat. Prod., 2012, 75, 1270–1276 Search PubMed.
- N. Boonman, S. Prachya, A. Boonmee, P. Kittakoop, S. Wiyakrutta, N. Sriubolmas, S. Warit and C. A. Dharmkrong-At, Planta Med., 2012, 78, 1562–1567 Search PubMed.
- R. B. Williams, S. M. Martin, J.-F. Hu, E. Garo, S. M. Rice, V. L. Norman, J. A. Lawrence, G. W. Hough, M. G. Goering, M. O'Neil-Johnson, G. R. Eldridge and C. M. Starks, Planta Med., 2012, 78, 160–165 Search PubMed.
- R. B. Williams, S. M. Martin, J.-F. Hu, V. L. Norman, M. G. Goering, S. Loss, M. O'Neil-Johnson, G. R. Eldridge and C. M. Starks, J. Nat. Prod., 2012, 75, 1319–1325 Search PubMed.
- G. Guebitz and M. G. Schmid, Mol. Biotechnol., 2006, 32, 159–179 Search PubMed.
- G. Gubitz and M. G. Schmid, Biopharm. Drug Dispos., 2001, 22, 291–336 CrossRef CAS.
- M. Gutierrez, E. H. Andrianasolo, W. K. Shin, D. E. Goeger, A. Yokochi, J. Schemies, M. Jung, D. France, S. Cornell-Kennon, E. Lee and W. H. Gerwick, J. Org. Chem., 2009, 74, 5267–5275 CrossRef CAS.
- A. S. Antonov, S. A. Avilov, A. I. Kalinovsky, S. D. Anastyuk, P. S. Dmitrenok, E. V. Evtushenko, V. I. Kalinin, A. V. Smirnov, S. Taboada, M. Ballesteros, C. Avila and V. A. Stonik, J. Nat. Prod., 2008, 71, 1677–1685 CrossRef CAS.
- J. M. Batista, Jr., A. N. L. Batista, J. S. Mota, Q. B. Cass, M. J. Kato, V. S. Bolzani, T. B. Freedman, S. N. Lopez, M. Furlan and L. A. Nafie, J. Org. Chem., 2011, 76, 2603–2612 CrossRef.
- N. Ingavat, J. Dobereiner, S. Wiyakrutta, C. Mahidol, S. Ruchirawat and P. Kittakoop, J. Nat. Prod., 2009, 72, 2049–2052 CrossRef CAS.
- B. Adams, P. Poerzgen, E. Pittman, W. Y. Yoshida, H. E. Westenburg and F. D. Horgen, J. Nat. Prod., 2008, 71, 750–754 CrossRef CAS.
- S. Alvarez, M. Zapata, J. L. Garrido and B. Vaz, Chem. Commun., 2012, 48, 5500–5502 RSC.
- Z. Chen, Y. Song, Y. Chen, H. Huang, W. Zhang and J. Ju, J. Nat. Prod., 2012, 75, 1215–1219 Search PubMed.
- B. R. Clark, N. Engene, M. E. Teasdale, D. C. Rowley, T. Matainaho, F. A. Valeriote and W. H. Gerwick, J. Nat. Prod., 2008, 71, 1530–1537 CrossRef CAS.
- S. P. Gunasekera, M. W. Miller, J. C. Kwan, H. Luesch and V. J. Paul, J. Nat. Prod., 2010, 73, 459–462 CrossRef CAS.
- S. P. Gunasekera, R. Ritson-Williams and V. J. Paul, J. Nat. Prod., 2008, 71, 2060–2063 CrossRef CAS.
- N. Koyama, Y. Inoue, M. Sekine, Y. Hayakawa, H. Homma, S. Oinmura and H. Tomoda, Org. Lett., 2008, 10, 5273–5276 CrossRef CAS.
- S. Matthew, C. Ross, V. J. Paul and H. Luesch, Tetrahedron, 2008, 64, 4081–4089 CrossRef CAS.
- R. A. Medina, D. E. Goeger, P. Hills, S. L. Mooberry, N. Huang, L. I. Romero, E. Ortega-Barria, W. H. Gerwick and K. L. McPhail, J. Am. Chem. Soc., 2008, 130, 6324–6325 CrossRef CAS.
- R. Montaser, V. J. Paul and H. Luesch, Phytochemistry, 2011, 72, 2068–2074 Search PubMed.
- J.-W. Nam, G.-Y. Kang, A.-R. Han, D. Lee, Y.-S. Lee and E.-K. Seo, J. Nat. Prod., 2011, 74, 2109–2115 Search PubMed.
- K. Taori, V. J. Paul and H. Luesch, J. Nat. Prod., 2008, 71, 1625–1629 CrossRef CAS.
- T. Teruya, H. Sasaki and K. Suenaga, Tetrahedron Lett., 2008, 49, 5297–5299 CrossRef CAS.
- A. Tripathi, J. Puddick, M. R. Prinsep, P. P. F. Lee and L. T. Tan, J. Nat. Prod., 2009, 72, 29–32 CrossRef CAS.
- E. L. Whitson, A. S. Ratnayake, T. S. Bugni, M. K. Harper and C. M. Ireland, J. Org. Chem., 2009, 74, 1156–1162 CrossRef CAS.
- M. Gutierrez, K. Tidgewell, T. L. Capson, N. Engene, A. Almanza, J. Schemies, M. Jung and W. H. Gerwick, J. Nat. Prod., 2010, 73, 709–711 CrossRef CAS.
- S. C. Pinto, G. G. Leitao, H. R. Bizzo, N. Martinez, E. Dellacassa, d. S. F. Martins, F. L. P. Costa, d. A. M. Barbosa and S. G. Leitao, Tetrahedron Lett., 2009, 50, 4785–4787 CrossRef CAS.
- E. Gil-av, B. Feibush and R. Charles-Siger, Tetrahedron Lett., 1966, 8, 1009–1015 Search PubMed.
- H. L. Zuo, F. Q. Yang, X. M. Zhang and Z. N. Xia, J. Anal. Methods Chem., 2012, 402081, DOI:10.1155/2012/402081.
- F. Q. Yang, H. K. Wang, H. Chen, J. D. Chen and Z. N. Xia, J. Anal. Methods Chem., 2011, 942467, DOI:10.1155/2011/942467.
- T. Ozek and F. Demirci, Methods Mol. Biol., 2012, 864, 275–300 Search PubMed.
- H. E. Park, S.-O. Yang, S.-H. Hyun, S. J. Park, H.-K. Choi and P. J. Marriott, J. Sep. Sci., 2012, 35, 416–423 Search PubMed.
- D. Sciarrone, S. Panto, C. Ragonese, P. Q. Tranchida, P. Dugo and L. Mondello, Anal. Chem., 2012, 84, 7092–7098 Search PubMed.
- S.-T. Chin, B. Maikhunthod and P. J. Marriott, Anal. Chem., 2011, 83, 6485–6492 Search PubMed.
- M. D'Alessandro, V. Brunner, G. von Merey and T. C. J. Turlings, J. Chem. Ecol., 2009, 35, 999–1008 Search PubMed.
- H. Ikeura, K. Kohara, X.-X. Li, F. Kobayashi and Y. Hayata, J. Agric. Food Chem., 2010, 58, 11014–11017 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |