 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN,N′-Dibutylbarbituric acid as an acceptor moiety in push–pull chromophores†
Milan
Klikar
a,
Filip
Bureš
*a,
Oldřich
Pytela
a,
Tomáš
Mikysek
b,
Zdeňka
Padělková
c,
Alberto
Barsella
d,
Kokou
Dorkenoo
d and
Sylvain
Achelle
e
aInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic. E-mail: filip.bures@upce.cz; Fax: +420 46 603 7068; Tel: +420 46 603 7099
bDepartment of Analytical Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic
cDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic
dDépartement d'Optique ultrarapide et de Nanophotonique, IPCMS-CNRS, 23 Rue du Loess, BP 43, Strasbourg Cedex 2, 67034, France
eInstitut des Sciences Chimiques de Rennes UMR CNRS 6226, IUT de Lannion, rue Eduard Branly, BP 30219, Lannion Cedex, F22302, France
First published on 4th October 2013
Abstract
Twelve novel D–π–A chromophores with the N,N′-dibutylbarbituric acid acceptor, the N,N-dimethylamino donor and a systematically extended π-linker were synthesized. The extent of intramolecular charge-transfer, structure–property relationships and nonlinear optical properties were further investigated by X-ray analysis, electrochemistry, UV/Vis absorption spectra, calculations and EFISH experiments.
Introduction
Design, synthesis and application of novel π-conjugated molecules in materials science have been the subject of considerable research interest.1 In a π-conjugated molecule having an electron-donor (D) and an electron-acceptor (A), the so-called D–π–A or push–pull chromophore, intramolecular charge-transfer (ICT) from the donor to the acceptor occurs. The ICT can easily be illustrated by two limiting resonance forms (aromatic and quinoid/zwitterionic arrangement).2 The D–A interaction and simultaneous generation of a new low-energy molecular orbital determine the unique properties of push–pull chromophores such as intensive colour, dipolar character, electrochemical behaviour, crystallinity, intermolecular interactions (π-stacking) as well as nonlinear optical (NLO) properties. Within the last 30 years or so, it has been realized that prepolarization of an organic molecule in D–π–A arrangement significantly enhances its NLO response. Hence, various π-conjugated backbones and electron donating and withdrawing groups and moieties have been utilized in the construction of NLO-active molecules (NLOphores) to date.3 Since the first synthesis by Adolf von Baeyer in 1864, barbituric acid (BA) and its derivatives have found widespread application across many branches.4 The uses of C5-(di)alkylated BAs (barbitals) as hypnotic-sedative, anticonvulsant, antimicrobial, spasmolytic, antiinflammatory, antitumoral and fat-reducing agents are apparently the most widely known pharmacological applications.5 Besides this, BA-derived anionic dyes (Oxonols, DiSBACs) may act as fast voltage FRET probes monitoring changes in the cell membrane potential.6However, the pseudoaromatic pyrimidine-2,4,6-trione ring in BA can also be employed as an electron-withdrawing moiety in push–pull chromophores. This class of BA-derived compounds can be represented by merocyanine dyes, especially by the well-known merocyanine 540.7 To date, (thio)barbituric acid ((T)BA) and its push–pull derivatives have found an admirable number of applications, especially in materials science. Either N-alkylated or N-unsubstituted (T)BA derivatives showed very strong and distinct solvatochromism and were investigated as powerful probes (i) to study solvent effects, solvent/medium polarity and pH-sensitivity; (ii) to distinguish between the specific and non-specific solvent interactions and (iii) to assess the hydrogen bonding ability.8 The latter property of hydrogen-bonding was further utilized in supramolecular assemblies of (T)BA derivatives exhibiting photoelectrochemical, photorefractive, semiconducting and distinct absorption and fluorescent properties.9 Yagai et al. showed several impressive hydrogen-bonded nanorings, nanofibres and rosettes,10 while Bassani et al. demonstrated application of such hierarchical self-assemblies in organic photovoltaic devices.11
Push–pull chromophores featuring a (T)BA acceptor unit were also widely studied as efficient NLOphores. Hence, C5 methylidene substituted (T)BA push–pull derivatives proved to be highly prepolarized and showed high nonlinear optical coefficients β of up to 300 × 10−30 esu.12 Considering the number of (T)BA push–pull chromophores synthesized to date, it is somewhat curious that only few efforts were made to systematically study the influence of the structure and length of the π-linker connecting (T)BA acceptor and a given donor.13 Hence, in this work we have focused on push–pull chromophores 1–3 with the π-conjugated system consisting of multiple bonds and 1,4-phenylene moieties end-capped with N,N′-dibutylbarbituric acid and the N,N-dimethylamino group (Fig. 1). These groups have been chosen as the electron acceptor and donor, while the butyl substituents assure the chromophore sufficient solubility and simultaneously retain the possibility of its crystallization and study by X-ray analysis.
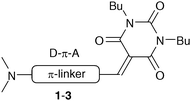 | ||
| Fig. 1 General structure of target push–pull chromophores 1–3. | ||
According to the N,N′-dibutylbarbituric acid connection to the central π-linker, three series of compounds 1–3 were synthesized. Each series contains four compounds a–d that differ in the length and structures of the π-linker (a – 1,4-phenylene, b – 4,4′-biphenylene, c – (E)-phenylethenylphenyl and d – phenylethynylphenyl). These model push–pull chromophores were further investigated by X-ray analysis, electrochemistry, UV/Vis spectra, calculations and EFISH experiments to evaluate the extent of the ICT and fundamental structure–property relationships.
Results and discussion
Synthesis
The obvious retrosynthetic strategy leading to target chromophores 1–3 involves the synthesis of N,N′-dibutylbarbituric acid and its Knoevenagel reaction14 with extended aldehydes. The barbituric acid was synthesized from N,N′-dibutylurea, generated from butyl isocyanate and butylamine in 99% yield15 and its subsequent treatment with malonic acid (91% yield).16 Extended benzaldehydes, cinnamaldehydes and propargyl aldehydes required for the final Knoevenagel condensation were prepared in a modular manner as shown in Scheme 1. 1,4-Diiodobenzene 4 was used as a suitable starting compound. Its formylation afforded 4-iodobenzaldehyde 6 in 44% yield. Transformation into (E)-4-iodocinnamaldehyde 7 was accomplished via Wittig reaction of 6 with (triphenylphosphoranylidene)acetaldehyde and subsequent isomerization in the overall yield of 59%. Sonogashira cross-coupling on 4 and 4-iodo-N,N-dimethylaniline 7 with propargyl alcohol afforded alcohols 8 (51%) and 9 (67%).17 Their oxidation with Dess–Martin periodinane provided extended propargyl aldehydes 10 and 11 in the yields of 94 and 76%, respectively.18 The π-systems of iodo-substituted aldehydes 6, 7 and 10 were further extended by Suzuki–Miyaura (Method A), Heck (Method B) and Sonogashira (Method C) reactions with boronic acid pinacol ester 12, styrene 13 and terminal acetylene 14, respectively. These reactions yielded biphenyl aldehydes 15–17, stilbene aldehydes 18–20 and phenylethynylphenyl aldehydes 21–23 in the indicated yields. For the synthetic details and the characterization of aldehydes 15–23 see the ESI.†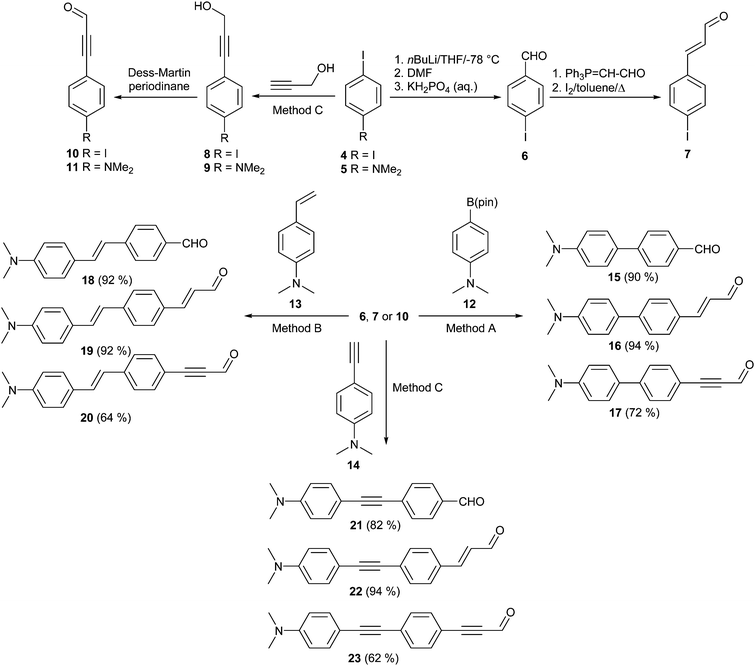 | ||
| Scheme 1 Synthetic approach to extended aldehydes 15–23. | ||
Despite the recent progress made in the catalytic Knoevenagel condensation,19 we have adopted in our strategy very mild and efficient methodology of Al2O3-catalyzed reaction developed by Foucaud20 and further applied by Diederich et al. for the construction of the dicyanovinyl acceptor moiety in cyanoethynylethenes (CEEs).21 This methodology proved to be also very useful for the condensation of aldehydes and N,N′-dibutylbarbituric acid. Commercially available 4-N,N-dimethylamino-substituted benzaldehyde 24 and cinnamaldehyde 25 as well as extended aldehydes 11 and 15–23 were converted into target push–pull chromophores 1a–d, 2a–d and 3a–d in the yields of 49–81, 76–91 and 69–82% (Scheme 2). Whereas the products of Knoevenagel reaction 1a, 2a–d and 3a–d could be purified by simple column chromatography, chromophores 1b–d decomposed already during the TLC analysis. It has recently been shown that the exocyclic double bond in BA benzylidene derivatives possesses highly electrophilic character and undergoes facile reaction with various nucleophiles.22 Hence, a retro Knoevenagel reaction with traces of water or acid media most likely takes place during the purification of 1b–d on silica. Therefore, the equilibrium condensation of extended benzaldehydes 15, 18 and 21 was carried out with 2 equivalents of N,N′-dibutylbarbituric acid and products 1b–d were purified by repeated precipitation from hot hexane.
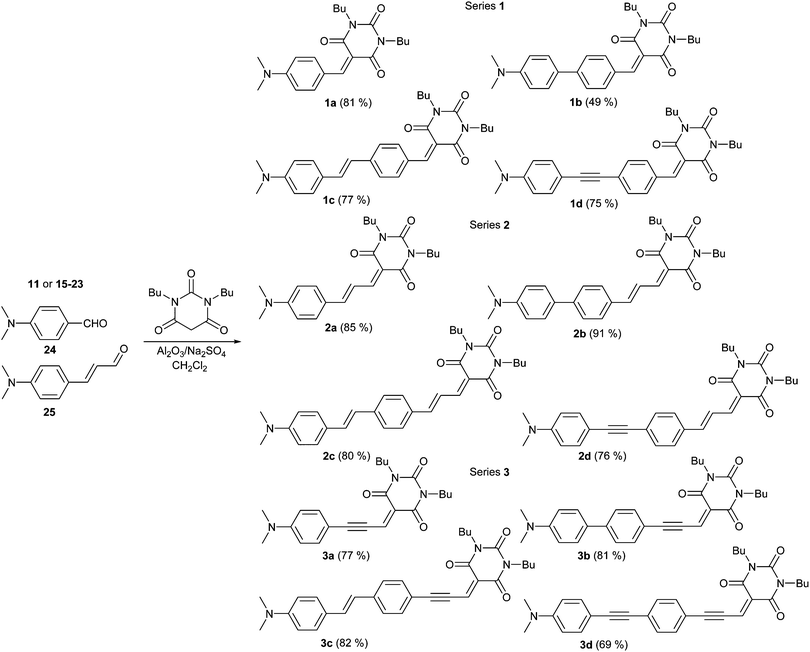 | ||
| Scheme 2 Knoevenagel condensation and structures of target chromophores 1–3. | ||
Having difficulties with the synthesis of 1b–d, we have attempted an alternative synthetic route as shown in Scheme S1 (see the ESI†). This strategy involves Knoevenagel condensation of iodo-substituted aldehydes 6, 7 and 10 yielding intermediates 26–28 and their subsequent modification via cross-coupling reactions (Methods A–C). However, these procedures led only to isolation of chromophores 2a–b in the yields of 25–70%. The cross-coupling reactions on 26 provided intensively coloured solutions, in which target compounds 1b–d were detected by TLC and HR-MALDI-MS. Nevertheless, isolation of pure chromophores was not possible. On the other hand, cross-coupling reactions on 28 provided directly a mixture of several products of degradation. Most probably, a conjugated nucleophilic addition on the electrophilic triple bond in 28 takes place under the conditions of cross-coupling reactions and caused degradation.21d
X-ray analysis
All target compounds 1–3 are thermally stable and intensely coloured solids readily soluble in chlorinated solvents. Target push–pull chromophores 1a and 2a were crystallized by slow diffusion of hexane into their solutions in dichloromethane. Fig. 2 shows the ORTEP plots and confirms the molecular structures assigned to 1a and 2a. Both front and side views are provided. As can be seen, both butyl residues stick oppositely and almost perpendicularly to the π-system mean plane. Average torsion angles between the barbituric acid ring and the adjacent 1,4-phenylene moieties are 4.5° (C2–C3–C6–C7) and 0.4° (C2–C3–C8–C9) for 1a and 2b, respectively. Such a planar arrangement of the π-systems in 1a and 2a would allow efficient D–A interaction. The extent of the ICT and molecule polarization can easily be assessed by evaluating quinoid character δr and bond length alternation of the π-system used. Average quinoid character of the 1,4-phenylene moieties in 1a and 2a are 0.05 and 0.04, respectively. For benzene, δr = 0, while in fully quinoid rings δr is equal to 0.08–0.1. Hence, the measured values indicate a considerable π-system polarization and rival those measured for highly polarized CEEs, which exhibited δr within the range of 0.03–0.07.21 Quinoid characters of the propen-1,3-diyl (C3–C5–C6) and pent-1,3-dien-1,5-diyl (C3–C5–C6–C7–C8) π-linker segments in 1a and 2a are 0.04 and 0.06. Fig. S1 and S2 in the ESI† show the crystal packing of 1a and 2a. In the solid state, chromophore 1a doesn't adopt a D to A molecular arrangement typical for the crystallization of dipolar compounds. This is most likely due to the less extended π-system and significant influence of the butyl residues (interlayer distance of about 3.6 Å). In contrast, the crystal packing of chromophore 2a shows typical D to A intermolecular interactions and π-stacks with the interlayer distance less than 3.4 Å.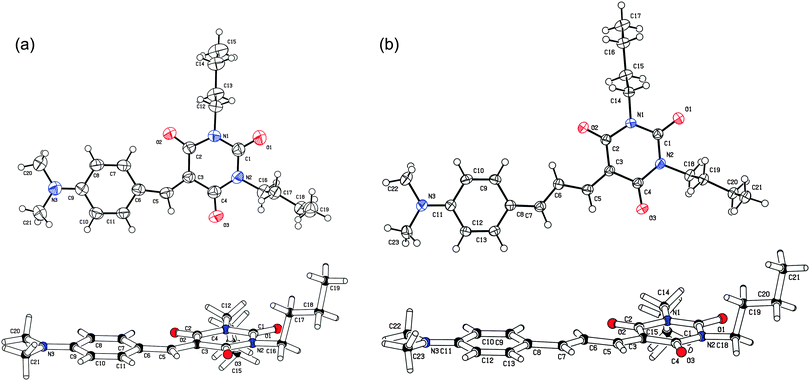 | ||
| Fig. 2 ORTEP representation of chromophores 1a (a) and 2a (b) measured by X-ray analysis at 150 K (50% probability level). | ||
Electrochemistry
Electrochemical behaviour of chromophores 1–3 was investigated by cyclic voltammetry (CV) and rotating-disc voltammetry (RDV). The measurements were carried out in acetonitrile that contained 0.1 M Bu4NPF6 as the electrolyte in a three-electrode cell. A saturated calomel electrode (SCE) separated by a bridge filled with supporting electrolyte and Pt wire were used as the reference and auxiliary electrodes, respectively. The acquired data are summarized in Table 1. Table S1 and Fig. S3–S6 in the ESI† contain full electrochemical data and representative CV curves for compounds 1a, 2b, 2c and 3d. Whereas the first oxidation was likely localized on the N,N-dimethylamino donor, the first reduction involved the barbituric acid acceptor moiety and the adjacent part of the π-linker. The half-wave potentials E1/2 of the first oxidation and reduction were further recalculated to afford energies EHOMO and ELUMO,23 that can be compared with the calculated HOMO and LUMO levels.| Comp. | E 1/2(ox1) [V] | E 1/2(red1) [V] | ΔEb [V] | E HOMO [eV] | E LUMO [eV] |
|---|---|---|---|---|---|
| a E 1/2(ox1) and E1/2(red1) are the half-wave potentials of the first oxidation and reduction, respectively, as measured by RDV. b ΔE = E1/2(ox1) − E1/2(red1); the potentials are given versus SCE. c E absHOMO/LUMO = E1/2(ox1/red1) + 4.43. | |||||
| 1a | +0.99 | −1.26 | 2.25 | −5.42 | −3.17 |
| 1b | +0.77 | −1.02 | 1.79 | −5.20 | −3.41 |
| 1c | +0.58 | −1.01 | 1.59 | −5.01 | −3.42 |
| 1d | +0.79 | −0.95 | 1.74 | −5.22 | −3.48 |
| 2a | +0.81 | −1.04 | 1.85 | −5.24 | −3.39 |
| 2b | +0.76 | −0.92 | 1.68 | −5.19 | −3.51 |
| 2c | +0.59 | −0.93 | 1.52 | −5.02 | −3.50 |
| 2d | +0.78 | −0.83 | 1.61 | −5.21 | −3.60 |
| 3a | +0.94 | −0.97 | 1.91 | −5.37 | −3.46 |
| 3b | +0.77 | −0.87 | 1.64 | −5.20 | −3.56 |
| 3c | +0.58 | −0.87 | 1.45 | −5.01 | −3.56 |
| 3d | +0.80 | −0.79 | 1.59 | −5.23 | −3.64 |
All target chromophores showed one or more one-electron oxidation processes that are mostly reversible. The first oxidation potential is within the range of +0.99 to +0.58 V. Within the particular series of compounds 1–3, the first oxidation potentials decreased in the following order: a > d > b > c. With the same donor and acceptor moieties and type of the BA connection to the π-linker, this observation reflects the composition and spatial arrangement of the π-linker. Whereas the planar stilbene π-linkers in chromophores c allowed very efficient D–A interaction (E1/2(ox1) = +0.58 to +0.59 V), the N,N-dimethylamino donors in chromophores a and b with short 1,4-phenylene (E1/2(ox1) = +0.81 to +0.99 V) and twisted 4,4′-biphenylene π-linkers (E1/2(ox1) = +0.76 to +0.77 V) were engaged in less ICT. In contrast to stilbene π-linkers in c, the planar phenylethynylphenyl π-linkers in chromophores d shifted the first oxidation potential anodically by about 200 mV up to +0.80 V. This clearly shows the difference between the electronegative and insulating acetylenic and polarizable olefinic units.2,21d A comparison of structural analogues 1b/2b/3b, 1c/2c/3c and 1d/2d/3d revealed almost the same values of the first oxidation potentials +0.76 to +0.77, +0.58 to +0.59 and +0.78 to +0.80 V. This indicates that N,N-dimethylamino donors are in these sets of compounds similarly involved in the ICT. However, compounds 1a/2a/3a with the shortest 1,4-phenylene π-linkers showed the expected trend for the first oxidation potentials (+0.99/+0.81/+0.94 V). The first oxidation potential decreased upon increasing the overall π-system length and polarizability.
N,N′-Dibutylbarbituric acid can be denoted as the main reduction centre of chromophores 1–3. The observed first reductions were one-electron irreversible processes with E1/2(red1) ranging from −1.26 to −0.79 V. The measured first reduction potentials were again dependent on the structure and length of the π-linker. Within the particular series of compounds 1–3, the values of E1/2(red1) were shifted to more positive values in the following order: d > c ≥ b > a. This observation is in accordance with the previous discussion on their oxidations. Thus, the most positively shifted first reduction potentials −0.79 to −0.83 V were measured for planar chromophores d with phenylethynylphenyl π-linker. A combination of two electronegative acetylenic units as in 3d caused a shift of E1/2(red1) up to −0.79 V. Whereas the first reduction potentials of chromophores b and c were almost identical, the most negative first reduction potentials were measured for chromophores a. A comparison across three series 1–3 showed that the first reductions are facilitated upon increasing the length and planarity of the π-linker as well as its connection to the acceptor.
The difference between the first oxidation and reduction potentials ΔE (electrochemical gap) assesses the D–A interaction and the extent of the ICT in the best way. The data and energy level diagram in Table 1 and Fig. 3 show a clear trend for decreasing ΔE in the order 1 > 2 ≥ 3 and within each series in the order a > b > d > c. Chromophores 1c (ΔE = 1.59 V), 2c (ΔE = 1.52 V) and 3c (ΔE = 1.45 V) showed the lowest electrochemical gaps, mainly due to their raised HOMO level.
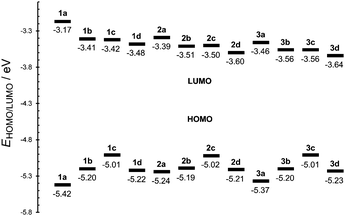 | ||
| Fig. 3 Energy level diagram of chromophores 1–3. | ||
UV/Vis spectra
All target chromophores 1–3 are coloured compounds. Their electronic absorption spectra were measured in three solvents with different properties – cyclohexane (nonpolar, protic), dichloromethane (nonpolar, aprotic, polarizable) and acetonitrile (dipolar, aprotic, nonpolarizable) at a concentration of 2 × 10−5 M. All spectra are shown in Fig. 4 and Fig. S7 and S8 (the ESI†). The optical properties are summarized in Table 2. The measured spectra in all solvents are dominated by intensive longest-wavelength absorption CT-bands with λmax appearing between 439–495 (cyclohexane), 462–527 (dichloromethane) and 438–516 nm (acetonitrile). The molar absorption coefficients ε range from 3.5–64.8 × 103 mol−1 dm3 cm−1 (Table 2). These values are consistent with those measured for previous N,N-dialkylamino substituted BA derivatives.12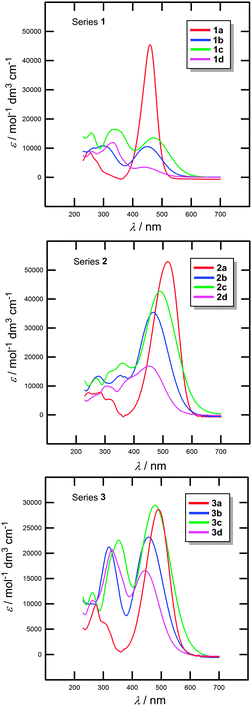 | ||
| Fig. 4 UV/Vis absorption spectra of chromophores in series 1–3 measured in acetonitrile (c = 2 × 10−5 M). | ||
| λmax [nm (eV)]/ε(×10−3) [mol−1 dm3 cm−1] | |||
|---|---|---|---|
| Comp. | Cyclohexane | Dichloromethane | Acetonitrile |
| a Indistinguishable shoulder. b Shoulder at λ = 476 nm (ε = 41.6 × 103 mol−1 dm3 cm−1). c Shoulder at λ = 454 nm (ε = 26.6 × 103 mol−1 dm3 cm−1). | |||
| 1a | 439(2.82)/63.7a | 462(2.68)/64.8 | 459(2.70)/45.7 |
| 1b | 444(2.79)/25.1a | 467(2.66)/23.7 | 451(2.75)/10.5 |
| 1c | 476(2.61)/35.8a | 493(2.52)/31.0 | 472(2.63)/13.5 |
| 1d | 450(2.76)/16.1a | 460(2.70)/13.9 | 438(2.83)/3.50 |
| 2a | 495(2.51)/39.6b | 527(2.35)/49.4 | 516(2.40)/53.0 |
| 2b | 460(2.70)/26.2 | 481(2.58)/34.4 | 467(2.66)/35.5 |
| 2c | 487(2.55)/36.8 | 508(2.44)/38.3 | 490(2.53)/42.8 |
| 2d | 454(2.73)/22.4 | 470(2.64)/32.4 | 449(2.76)/16.9 |
| 3a | 481(2.58)/34.6c | 503(2.47)/26.6 | 491(2.53)/28.8 |
| 3b | 458(2.71)/34.3a | 476(2.61)/23.4 | 458(2.71)/23.2 |
| 3c | 486(2.55)/25.5a | 501(2.48)/30.9 | 479(2.59)/29.5 |
| 3d | 456(2.72)/27.3a | 467(2.66)/21.7 | 444(2.79)/16.6 |
Except for chromophores 1a and 1b, the λmax values of chromophores 1–3 measured in cyclohexane and dichloromethane are linearly dependent. The slope calculated by orthogonal linear fit is 1.305 (Fig. S9, the ESI†). A similar but less tight dependence can be derived from the correlation between the λmax values measured in cyclohexane and acetonitrile (slope = 1.518; Fig. S10, the ESI†). These slope values imply that a bathochromic shift of the longest-wavelength absorption maxima is being observed when going from nonpolar aprotic cyclohexane to polarizable dichloromethane or dipolar acetonitrile. This can be explained as a higher solvation stabilization of the excited than the ground state. This further implies that the excited state is more polarizable and polar (most probably the dominant feature) than the ground state.
The following structure–property relationships were deduced from the spectra measured in acetonitrile (the solvent used for electrochemical measurements). Chromophores 1a (λmax = 459 nm), 2a (λmax = 516 nm) and 3a (λmax = 491 nm) featuring the shortest π-linkers showed remarkably bathochromically shifted CT-bands. However, in comparison with 1a the positions of the longest-wavelength absorption maxima of 2a and 3a shifted bathochromically as a result of their extended π-linkers. A comparison of structurally similar chromophores 1c/1d, 2c/2d and 3c/3d reveals bathochromically shifted CT-bands (Δλmax = 34–41 nm) for chromophores in series c that feature the olefinic subunit (stilbene). This observation is in agreement with the previous findings made by electrochemistry. Cinnamaldehyde-derived chromophores 2a–d showed the most bathochromically shifted CT-bands (λmax = 449–516 nm) across three series 1–3, which reflects their planar and most polarizable connection of the π-linker to the BA acceptor via a but-1,3-diene spacer.
Calculations and EFISH experiments
The calculations were performed with PM3 and PM7 semi-empirical methods implemented in programs ArgusLab24 and MOPAC201225 and visualization in OPchem.26 Energies of HOMO and LUMO, their difference ΔE, dipole moment μ and first hyperpolarizabilities β were calculated (Table 3). Although the PM7 calculated values of EHOMO and ELUMO are in absolute values different from those measured by CV and RDV, they showed similar trends. Namely, the lowest calculated HOMO–LUMO differences within the series of compounds 1–3 were calculated for chromophores 1c (ΔE = −6.76 eV), 2c (ΔE = −6.40 eV) and 3c (ΔE = −6.50 eV) with a planar stilbene π-linker.| Comp. | E HOMO [eV] | E LUMO [eV] | ΔEa [eV] | μ [D] | β [1029 esu] | μβ (2ω)b [1048 esu] |
|---|---|---|---|---|---|---|
| a The data were calculated by PM7 (MOPAC2012). b Measured at 1907 nm in CHCl3, c was within the range 10−3 to 10−2 M; μβ ± 10%. c Underestimated due to aggregation. | ||||||
| 1a | −8.52 | −1.23 | −7.29 | 7.47 | 2.97 | 270 |
| 1b | −8.26 | −1.36 | −6.90 | 5.54 | 3.04 | 1080 |
| 1c | −8.13 | −1.37 | −6.76 | 5.92 | 4.33 | 1390 |
| 1d | −8.14 | −1.29 | −6.85 | 5.97 | 4.09 | 750 |
| 2a | −8.36 | −1.49 | −6.87 | 7.93 | 6.35 | 1000 |
| 2b | −8.25 | −1.65 | −6.60 | 6.56 | 5.99 | 1040 |
| 2c | −8.08 | −1.68 | −6.40 | 7.73 | 10.6 | 1880 |
| 2d | −8.10 | −1.64 | −6.46 | 7.66 | 8.97 | 1270 |
| 3a | −8.55 | −1.42 | −7.13 | 7.03 | 3.22 | 900 |
| 3b | −8.32 | −1.56 | −6.76 | 6.68 | 4.13 | 940 |
| 3c | −8.10 | −1.60 | −6.50 | 8.12 | 8.15 | 1270 |
| 3d | −8.37 | −1.61 | −6.76 | 5.94 | 5.08 | 870c |
As can be seen from these selected values, a connection of the BA acceptor to the π-system in 2 through a but-1,3-diene spacer allowed the most efficient D–A interaction. Hence, these cinnamaldehyde-derived chromophores generally showed the lowest HOMO–LUMO gap. Localization and gradual separation of the HOMO and the LUMO can be visualized on compounds 2a–d (Fig. 5). For complete listing of the HOMO–LUMO localizations see Fig. S11–S22 in the ESI.† As expected, the HOMO is localized on the N,N-dimethylanilino donor moiety, whilst the LUMO is spread over the barbituric acid C5 and adjacent part of the π-linker. This is in agreement with the aforementioned electrochemical conclusions.
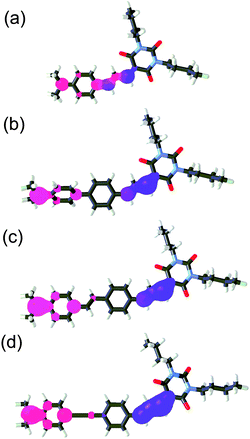 | ||
| Fig. 5 Optimized geometries and HOMO (red) and LUMO (blue) localizations in chromophores 2a (a), 2b (b), 2c (c) and 2d (d). | ||
Second-order non-linear optical properties were studied in CHCl3 solution by the electric-field-induced second-harmonic generation technique (EFISH) which provides information about the scalar product μβ(2ω) of the vector component of the first hyperpolarisability tensor β and the dipole moment vector.27 This product is derived according to eqn (1) and considering the third-order term γ0(−2ω,ω,ω,0) as negligible for the push–pull compounds under study. This approximation is usually used for push–pull organic and organometallic molecules.
| γEFISH = μβ/5kT + γ0(−2ω,ω,ω,0) | (1) |
Measurements were performed at 1907 nm, obtained from a Raman-shifted Nd:YAG+ laser source, which allowed us to work far from the resonance peaks of 1–3. It should be noted that the sign and values of μβ depend on the “direction” of the transition implied in the NLO phenomena and on the direction of the ground-state dipole moment. When β and μ were parallel (antiparallel), positive (negative), maximal μβ values were reached. All the μβ values of compounds 1–3 (Table 3) are positive which indicates more polarized excited than ground states (μe > μg). In addition, this implies that the ground and excited states are polarized in the same direction. The μβ values observed are relatively high compared to Disperse Red 1 used as the NLO standard.28
A comparison of measured μβ values with the calculated first hyperpolarizabilities β in Table 2 revealed that both quantities correspond to general trends. Namely, the highest β/μβ coefficients were calculated/measured for NLOphores in series 2. Both coefficients increased within the particular series in the order of a > b ≥ d > c. This observed variation in nonlinear optical properties mimics the trends seen in the values of ΔE and λmax. The highest μβ value (1880 × 10−48 esu) was measured for NLOphore 2c with the planar and polarizable stilbene π-linker, while NLOphore 1a showed the weakest NLO response (270 × 10−48 esu). Thus, an insertion of one 1,4-phenylene and two olefinic subunits into the π-linker (2cvs.1a) enhanced the NLO response seven times.
Conclusions
N,N-Dimethylamino donor-substituted benzaldehydes, cinnamaldehydes and propargyl aldehydes with systematically extended π-systems were prepared and efficiently combined with the N,N′-dibutylbarbituric acid acceptor via Al2O3-catalyzed Knoevenagel condensation. Twelve new push–pull chromophores were synthesized and the extent of the ICT was further evaluated by X-ray analysis, electrochemistry, absorption spectra, calculation and EFISH experiments. With the given electron donor and acceptor, the electrochemical, optical and NLO behaviour of charge-transfer chromophores 1–3 can be finely tuned by extension, composition and spatial arrangement of the π-linker used. Push–pull chromophore 2c showed well balanced properties such as electrochemical gap ΔE = 1.52 V, the most bathochromically shifted CT-band appearing at 490 nm (2.53 eV), calculated HOMO–LUMO difference of −6.40 eV, dipole moment μ = 7.73 D and thermal stability (m.p. 228–232 °C). Considering the highest first hyperpolarizability β attained for (T)BA derivatives (∼300 × 10−30 esu) known to date and a relatively small π-system of 2c, its calculated (β = 10.6 × 10−29 esu) and measured NLO responses (μβ = 1880 × 10−48 esu) are remarkable.In conclusion, N,N′-dibutylbarbituric acid proved to be a very efficient electron acceptor moiety in push–pull chromophores assuring their large dipolar character and simultaneously maintaining their sufficient solubility in common organic solvents. In view of the current interest in new organic materials for optoelectronics and ease of its synthesis, N,N′-dibutylbarbituric acid can be regarded as an alternative to former BA derivatives.
Experimental
For more synthetic details and instrumentation used see the ESI.† All cross-coupling reactions were carried out in Schlenk flasks dried under vacuum and filled with argon.General method for Suzuki–Miyaura reaction (Method A)
An iodo derivative (1.0 mmol) and boron ester 12 (259 mg, 1.05 mmol) were dissolved in the mixture of THF–H2O (20 ml, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Argon was bubbled through the solution for 15 min whereupon [PdCl2(PPh3)2] (28 mg, 0.04 mmol) and Na2CO3 (159 mg, 1.5 mmol) were added and the reaction mixture was stirred at 60 °C for 12 h. The reaction was diluted with aq. NH4Cl (50 ml) and extracted with CH2Cl2 (2 × 50 ml). The combined organic extracts were dried (Na2SO4), the solvents were evaporated in vacuo and the crude product was purified by column chromatography (SiO2; CH2Cl2).
:1). Argon was bubbled through the solution for 15 min whereupon [PdCl2(PPh3)2] (28 mg, 0.04 mmol) and Na2CO3 (159 mg, 1.5 mmol) were added and the reaction mixture was stirred at 60 °C for 12 h. The reaction was diluted with aq. NH4Cl (50 ml) and extracted with CH2Cl2 (2 × 50 ml). The combined organic extracts were dried (Na2SO4), the solvents were evaporated in vacuo and the crude product was purified by column chromatography (SiO2; CH2Cl2).
General method for Heck reaction (Method B)
Iodo derivative (1.0 mmol) and styrene 13 (155 mg, 1.05 mmol) were dissolved in toluene (30 ml) and iPr2NH (0.21 ml, 1.5 mmol) was added. Argon was bubbled through the solution for 15 min whereupon [Pd(P-tBu3)2] (28 mg, 0.04 mmol) was added and the reaction mixture was stirred at 80 °C for 12 h. The reaction was diluted with aq. NH4Cl (50 ml) and extracted with CH2Cl2 (2 × 50 ml). The combined organic extracts were dried (Na2SO4), the solvents were evaporated in vacuo and the crude product was purified by column chromatography (SiO2; CH2Cl2).General method for Sonogashira reaction (Method C)
Iodo derivative (1.0 mmol) and acetylene 14 (152 mg, 1.05 mmol) were dissolved in THF (20 ml) and Et3N (5 ml). Argon was bubbled through the solution for 15 min whereupon [PdCl2(PPh3)2] (21 mg, 0.03 mmol) and CuI (6 mg, 0.03 mmol) were added and the reaction mixture was stirred at 25 °C for 12 h. The reaction was diluted with aq. NH4Cl (50 ml) and extracted with CH2Cl2 (2 × 50 ml). The combined organic extracts were dried (Na2SO4), the solvents were evaporated in vacuo and the crude product was purified by column chromatography (SiO2; CH2Cl2).General method for Knoevenagel reaction
A mixture of N,N′-dibutylbarbituric acid (240/480 mg, 1.0/2.0 mmol), aldehyde (1.0 mmol), Al2O3 (510 mg, 5.0 mmol, activity II–III) and Na2SO4 (710 mg, 5.0 mmol) in CH2Cl2 (10 ml) was stirred at 25 °C for 12 h. The suspension was filtered and the solvent was evaporated in vacuo. The crude product was purified by column chromatography (SiO2; CH2Cl2) or by washing with hexane.Chromophore 1a
Compound 1a was synthesized from commercially available aldehyde 24 (149 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 1a was isolated as an orange-red solid (300 mg, 81%). M.p. 156–161 °C. Rf = 0.67 (SiO2; CH2Cl2). Found: C, 67.47; H, 7.92; N, 11.21. C21H29N3O3 requires C, 67.90; H, 7.87; N, 11.31%. IR (HATR): νmax/cm−1 2921, 2359, 1712, 1645, 1362, 1090, 1017, 787. 1H NMR (400 MHz; CDCl3): δH 0.92–0.96 (6H, m, 2×CH3), 1.34–1.40 (4H, m, 2×CH2), 1.60–1.64 (4H, m, 2×CH2), 3.13 (6H, s, N(CH3)2), 3.94–3.98 (4H, m, 2×NCH2), 6.69 (2H, d, J = 9.2, CHar), 8.37 (2H, d, J = 9.2, CHar) and 8.42 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 14.01, 14.04, 20.41, 20.47, 30.43, 30.46, 40.26, 41.69, 42.28, 110.32, 111.20, 121.32, 139.57, 151.53, 154.47, 158.86, 161.56 and 163.96. HR-FT-MALDI-MS (DHB) m/z: 372.2281 ([M + H]+), C21H30N3O3+ requires 372.2282.Chromophore 1b
Compound 1b was synthesized from aldehyde 15 (225 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (480 mg, 2.0 mmol). The crude product was purified by repeated washing with hexane. Compound 1b was isolated as a red-violet solid (219 mg, 49%). M.p. 166–172 °C. Rf = 0.65 (SiO2; CH2Cl2). Found: C, 71.90; H, 7.38; N, 9.46. C27H33N3O3 requires C, 72.46; H, 7.43; N, 9.39%. IR (HATR): νmax/cm−1 2922, 2360, 1712, 1654, 1363, 806.1H NMR (400 MHz; CDCl3): δH 0.93–0.98 (6H, m, 2×CH3), 1.40–1.44 (4H, m, 2×CH2), 1.59–1.68 (4H, m, 2×CH2), 3.02 (6H, s, N(CH3)2), 3.94–3.99 (4H, m, 2×NCH2), 6.78 (2H, d, J = 8.8, CHar), 7.60 (2H, d, J = 8.8, CHar), 7.67 (2H, d, J = 8.8, CHar), 8.22 (2H, d, J = 8.4, CHar) and 8.54 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 13.99, 20.39, 20.43, 30.36, 40.52, 41.97, 42.55, 112.68, 116.20, 125.56, 127.01, 128.19, 130.39, 135.58, 146.32, 150.94, 151.12, 159.02, 160.75 and 162.90. HR-FT-MALDI-MS (DHB) m/z: 448.2588 ([M + H]+), C27H34N3O3+ requires 448.2595.Chromophore 1c
Compound 1c was synthesized from aldehyde 18 (251 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (480 mg, 2.0 mmol). The crude product was purified by repeated washing with hexane. Compound 1c was isolated as a violet solid (364 mg, 77%). M.p. 172–177 °C. Rf = 0.63 (SiO2; CH2Cl2). Found: C, 72.78; H, 7.34; N, 8.75. C29H35N3O3 requires C, 73.54; H, 7.45; N, 8.87%. IR (HATR): νmax/cm−1 2921, 2360, 1714, 1657, 1363, 789. 1H NMR (400 MHz; CDCl3): δH 0.93–0.98 (6H, m, 2×CH3), 1.34–1.42 (4H, m, 2×CH2), 1.59–1.68 (4H, m, 2×CH2), 3.00 (6H, s, N(CH3)2), 3.94–3.99 (4H, m, 2×NCH2), 6.70 (2H, d, J = 8.8, CHar), 6.92 (1H, d, J = 16.0, CH), 7.22 (1H, d, J = 16.0, CH), 7.44 (2H, d, J = 8.8, CHar), 7.53 (2H, d, J = 8.4, CHar), 8.17 (2H, d, J = 8.4, CHar) and 8.49 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 14.01, 20.38, 20.43, 30.35, 40.51, 41.96, 42.54, 112.40, 116.06, 123.10, 124.95, 125.82, 128.82, 131.02, 132.97, 135.57, 144.01, 150.88, 151.11, 158.67, 160.73 and 162.90. HR-FT-MALDI-MS (DHB) m/z: 474.2752 ([M + H]+), C29H36N3O3+ requires 474.2751.Chromophore 1d
Compound 1d was synthesized from aldehyde 21 (249 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (480 mg, 2.0 mmol). The crude product was purified by repeated washing with hexane. Compound 1d was isolated as a red-violet solid (353 mg, 75%). M.p. 139–143 °C. Rf = 0.63 (SiO2; CH2Cl2). Found: C, 74.05; H, 7.12; N, 8.86. C29H33N3O3 requires C, 73.86; H, 7.05; N, 8.91%. IR (HATR): νmax/cm−1 2957, 2360, 1712, 1667, 1362, 1221, 1093. 1H NMR (400 MHz; CDCl3): δH 0.93–0.98 (6H, m, 2×CH3), 1.37–1.41 (4H, m, 2×CH2), 1.58–1.64 (4H, m, 2×CH2), 3.01 (6H, s, N(CH3)2), 3.93–3.99 (4H, m, 2×NCH2), 6.66 (2H, d, J = 8.8, CHar), 7.42 (2H, d, J = 8.8, CHar), 7.53 (2H, d, J = 8.4, CHar), 8.11 (2H, d, J = 8.4, CHar) and 8.49 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 14.00, 20.38, 20.42, 30.35, 40.37, 42.04, 42.62, 88.19, 96.11, 109.37, 111.95, 117.30, 129.70, 131.01, 131.57, 133.37, 134.34, 150.71, 151.02, 158.19, 160.54 and 162.62. HR-FT-MALDI-MS (DHB) m/z: 472.2583 ([M + H]+), C29H34N3O3+ requires 472.2595.Chromophore 2a
Compound 2a was synthesized from commercially available aldehyde 25 (175 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 2a was isolated as a dark violet solid (338 mg, 85%). M.p. 169–171 °C. Rf = 0.46 (SiO2; CH2Cl2). Found: C, 69.34; H, 7.85; N, 10.39. C23H31N3O3 requires C, 69.49; H, 7.86; N, 10.57%. IR (HATR): νmax/cm−1 2923, 2360, 1713, 1648, 1533, 1366, 1223, 1164, 809. 1H NMR (400 MHz; CDCl3): δH 0.93–0.97 (6H, m, 2×CH3), 1.33–1.41 (4H, m, 2×CH2), 1.59–1.64 (4H, m, 2×CH2), 3.08 (6H, s, N(CH3)2), 3.91–3.95 (4H, m, 2×NCH2), 6.65 (2H, d, J = 8.8, CHar), 7.37 (1H, d, J = 14.8, CH), 7.58 (2H, d, J = 8.8, CHar), 8.16 (1H, d, J = 12.4, CH) and 8.41 (1H, dd, J1 = 12.4, J2 = 14.8, CH). 13C NMR (100 MHz, CDCl3): δC 14.01, 14.03, 20.39, 20.49, 30.43, 30.50, 40.33, 41.41, 41.92, 110.53, 112.01, 120.82, 123.66, 132.30, 151.53, 153.14, 157.09, 158.69, 162.35 and 162.94. HR-FT-MALDI-MS (DHB) m/z: 398.2436 ([M + H]+), C23H32N3O3+ requires 398.2438.Chromophore 2b
Compound 2b was synthesized from aldehyde 16 (251 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 2b was isolated as a dark violet solid (431 mg, 91%). M.p. 199–204 °C. Rf = 0.18 (SiO2; CH2Cl2–Hex 1:1). Found: C, 73.34; H, 7.57; N, 8.69. C29H35N3O3 requires C, 73.54; H, 7.45; N, 8.87%. IR (HATR): νmax/cm−1 2924, 2360, 1713, 1653, 1550, 1361, 1222, 809. 1H NMR (400 MHz; CDCl3): δH = 0.93–0.98 (6H, m, 2×CH3), 1.35–1.42 (4H, m, 2×CH2), 1.59–1.65 (4H, m, 2×CH2), 3.01 (6H, s, N(CH3)2), 3.92–3.96 (4H, m, 2×NCH2), 6.78 (2H, d, J = 7.2, CHar), 7.42 (1H, d, J = 15.2, CH), 7.56 (2H, d, J = 7.2, CHar), 7.61 (2H, d, J = 8.4, CHar), 7.69 (2H, d, J = 8.4, CHar), 8.19 (1H, d, J = 12.0, CH) and 8.61 (1H, dd, J1 = 15.2, J2 = 12.0, CH). 13C NMR (100 MHz, CDCl3): δC 14.00, 14.02, 20.38, 20.47, 30.37, 30.43, 40.58, 41.63, 42.13, 112.74, 113.98, 124.50, 126.50, 127.33, 127.95, 130.12, 133.12, 144.60, 150.72, 151.25, 154.65, 157.70, 161.87 and 162.43. HR-FT-MALDI-MS (DHB) m/z: 474.2727 ([M + H]+), C29H36N3O3+ requires 474.2725.
Chromophore 2c
Compound 2c was synthesized from aldehyde 19 (277 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 2c was isolated as a dark violet solid (400 mg, 80%). M.p. 228–232 °C. Rf = 0.73 (SiO2; CH2Cl2). Found: C, 74.42; H, 7.57; N, 8.37. C31H37N3O3 requires C, 74.52; H, 7.46; N, 8.41%. IR (HATR): νmax/cm−1 2923, 2360, 1715, 1656, 1543, 1396, 1361, 1221, 821. 1H NMR (400 MHz; CDCl3): δH 0.93–0.98 (6H, m, 2×CH3), 1.36–1.40 (4H, m, 2×CH2), 1.59–1.65 (4H, m, 2×CH2), 2.99 (6H, s, N(CH3)2), 3.92–3.96 (4H, m, 2×CH2), 6.69 (2H, d, J = 8.8, CHar), 6.89 (1H, d, J = 16.0, CH), 7.15 (1H, d, J = 16.0, CH), 7.37 (1H, d, J = 15.2, CH), 7.42 (2H, d, J = 8.8, CHar), 7.48 (2H, d, J = 8.4 Hz, CHar), 7.62 (2H, d, J = 8.4, CHar), 8.18 (1H, d, J = 12.0, CH) and 8.55 (1H, dd, J1 = 15.2, J2 = 12.0, CH). 13C NMR (100 MHz, CDCl3): δC 13.99, 14.02, 20.38, 20.47, 30.37, 30.43, 40.54, 41.63, 42.13, 112.45, 113.93, 123.21, 124.54, 125.15, 126.67, 128.30, 130.03, 131.65, 133.76, 142.14, 150.70, 151.24, 154.45, 157.61, 161.88 and 162.42. HR-FT-MALDI-MS (DHB) m/z: 500.2893 ([M + H]+), C31H38N3O3+ requires 500.2908.Chromophore 2d
Compound 2d was synthesized from aldehyde 22 (275 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 2d was isolated as a dark violet solid (378 mg, 76%). M.p. 194–199 °C. Rf = 0.85 (SiO2; CH2Cl2). Found: C, 74.28; H, 7.08; N, 8.36. C31H35N3O3 requires C, 74.82; H, 7.09; N, 8.44%. IR (HATR): νmax/cm−1 2922, 2360, 1713, 1653, 1558, 1397, 1362, 1222, 816. 1H NMR (400 MHz; CDCl3): δH 0.93–0.98 (6H, m, 2×CH3), 1.32–1.42 (4H, m, 2×CH2), 1.57–1.65 (4H, m, 2×CH2), 3.00 (6H, s, N(CH3)2), 3.92–3.96 (4H, m, 2×NCH2), 6.65 (2H, d, J = 8.8, CHar), 7.37 (1H, d, J = 15.6, CH), 7.41 (2H, d, J = 8.8, CHar), 7.50 (2H, d, J = 8.0, CHar), 7.62 (2H, d, J = 8.0, CHar), 8.18 (1H, d, J = 12.0, CH) and 8.59 (1H, dd, J1 = 15.6, J2 = 12.0, CH). 13C NMR (100 MHz, CDCl3): δC 13.99, 14.01, 20.37, 20.46, 30.36, 30.42, 40.37, 41.69, 42.18, 87.96, 95.17, 109.48, 111.96, 114.77, 125.50, 127.80, 129.26, 131.89, 133.20, 134.32, 150.58, 151.17, 153.46, 157.14, 161.77 and 162.30. HR-FT-MALDI-MS (DHB) m/z: 498.2741 ([M + H]+), C31H36N3O3+ requires 498.2751.Chromophore 3a
Compound 3a was synthesized from aldehyde 11 (173 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 3a was isolated as a dark violet solid (304 mg, 77%). M.p. 144–148 °C. Rf = 0.70 (SiO2; CH2Cl2). Found: C, 69.75; H, 7.41; N, 10.54. C23H29N3O3 requires C, 69.85; H, 7.39; N, 10.62%. IR (HATR): νmax/cm−1 2923, 2360, 1713, 1362, 1258, 1222, 1090, 1014, 794. 1H NMR (400 MHz; CDCl3): δH 0.92–0.97 (6H, m, 2×CH3), 1.33–1.41 (4H, m, 2×CH2), 1.58–1.65 (4H, m, 2×CH2), 3.07 (6H, s, N(CH3)2), 3.91–3.95 (4H, m, 2×NCH2), 6.65 (2H, d, J = 8.8, CHar), 7.58 (2H, d, J = 8.8, CHar) and 7.81 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 13.96, 13.98, 20.36, 20.41, 30.34, 40.24, 41.59, 42.12, 94.03, 107.99, 111.77, 121.56, 125.55, 136.29, 137.35, 151.17, 152.53, 159.92 and 161.90. HR-FT-MALDI-MS (DHB) m/z: 396.2275 ([M + H]+), C23H30N3O3+ requires 396.2282.Chromophore 3b
Compound 3b was synthesized from aldehyde 17 (249 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 3b was isolated as a dark violet solid (382 mg, 81%). M.p. 179–184 °C. Rf = 0.77 (SiO2; CH2Cl2). Found: C, 73.66; H, 7.02; N, 8.90. C21H33N3O3 requires C, 73.86; H, 7.05; N, 8.91%. IR (HATR): νmax/cm−1 2928, 2360, 1717, 1658, 1567, 1556, 1398, 1363, 1291, 808. 1H NMR (400 MHz; CDCl3): δH 0.93–0.98 (6H, m, 2×CH3), 1.32–1.44 (4H, m, 2×CH2), 1.58–1.68 (4H, m, 2×CH2), 3.01 (6H, s, N(CH3)2), 3.92–3.97 (4H, m, 2×NCH2), 6.78 (2H, d, J = 8.8, CHar), 7.55 (2H, d, J = 8.4, CHar), 7.61 (2H, d, J = 8.4, CHar), 7.70 (2H, d, J = 8.8, CHar) and 7.80 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 13.99, 14.01, 20.38, 20.43, 29.93, 30.33, 40.57, 41.82, 42.33, 91.41, 112.74, 119.02, 119.51, 124.77, 126.16, 127.24, 128.02, 134.38, 136.73, 144.29, 150.77, 150.92, 159.44 and 161.40. HR-FT-MALDI-MS (DHB) m/z: 472.2589 ([M + H]+), C29H34N3O3+ requires 472.2595.Chromophore 3c
Compound 3c was synthesized from aldehyde 20 (275 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 3c was isolated as a dark violet solid (408 mg, 82%). M.p. 207–212 °C. Rf = 0.83 (SiO2; CH2Cl2). Found: C, 74.69; H, 7.16; N, 8.36. C31H35N3O3 requires C, 74.82; H, 7.09; N, 8.44%. IR (HATR): νmax/cm−1 2922, 2360, 1713, 1659, 1565, 1400, 1362, 1222, 825. 1H NMR (400 MHz; CDCl3): δH 0.93–0.98 (6H, m, 2×CH3), 1.34–1.42 (4H, m, 2×CH2), 1.59–1.66 (4H, m, 2×CH2), 2.98 (6H, s, N(CH3)2), 3.90–3.96 (4H, m, 2×NCH2), 6.68 (2H, d, J = 8.8, CHar), 6.87 (1H, d, J = 16.0, CH), 7.13 (1H, d, J = 16.0, CH), 7.40 (2H, d, J = 8.8, CHar), 7.47 (2H, d, J = 8.4, CHar), 7.62 (2H, d, J = 8.4, CHar) and 7.78 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 13.92, 13.96, 20.31, 20.37, 30.25, 40.46, 41.74, 42.24, 91.76, 112.37, 119.41, 119.50, 123.03, 124.59, 124.96, 126.21, 128.29, 131.97, 134.24, 136.52, 141.72, 150.66, 150.84, 159.38 and 161.32. HR-FT-MALDI-MS (DHB) m/z: 498.2740 ([M + H]+), C31H36N3O3+ requires 498.2751.Chromophore 3d
Compound 3d was synthesized from aldehyde 23 (273 mg, 1.0 mmol) and N,N′-dibutylbarbituric acid (240 mg, 1.0 mmol). Compound 3d was isolated as a dark violet solid (342 mg, 69%). M.p. 190–195 °C. Rf = 0.71 (SiO2; CH2Cl2). Found: C, 75.10; H, 6.80; N, 8.49. C31H33N3O3 requires C, 75.13; H, 6.71; N, 8.48%. IR (HATR): νmax/cm−1 2922, 2360, 1715, 1675, 1654, 1567, 1400, 1360, 1094, 789. 1H NMR (400 MHz; CDCl3): δH 0.93–0.97 (6H, m, 2×CH3), 1.32–1.43 (4H, m, 2×CH2), 1.57–1.67 (4H, m, 2×CH2), 3.00 (6H, s, N(CH3)2), 3.92–3.96 (4H, m, 2×NCH2), 6.65 (2H, d, J = 8.8, CHar), 7.41 (2H, d, J = 8.8, CHar), 7.50 (2H, d, J = 8.4, CHar), 7.63 (2H, d, J = 8.4, CHar) and 7.78 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δC 13.95, 13.98, 20.35, 20.40, 29.91, 30.30, 40.35, 41.85, 42.35, 87.71, 91.58, 95.49, 109.36, 111.96, 117.71, 120.45, 125.45, 127.65, 131.50, 133.21, 133.58, 136.26, 150.64, 150.85, 159.34 and 161.26. HR-FT-MALDI-MS (DHB) m/z: 496.2597 ([M + H]+), C31H34N3O3+ requires 496.2595.Acknowledgements
This work was supported by the Czech Science Foundation (13-01061S) and Technology Agency of the Czech Republic (TE01020022, Flexprint).Notes and references
- (a) Special issue on “Organic electronics and Optoelectronics”, ed. S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923–1386; (b) Special issue on “Materials for electronics”, ed. R. D. Miller and E. A. Chandross, Chem. Rev., 2010, 110, 1–574; (c) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS PubMed; (d) Y. Ohmori, Laser Photonics Rev., 2009, 4, 300 CrossRef.
- J. Kulhánek, F. Bureš, J. Opršal, W. Kuznik, T. Mikysek and A. Růžička, Asian J. Org. Chem., 2013, 2, 422 CrossRef.
- (a) S. R. Marder and J. W. Perry, Adv. Mater., 1993, 5, 804 CrossRef CAS; (b) D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195 CrossRef CAS; (c) L. R. Dalton, J. Phys.: Condens. Matter, 2003, 15, R897 CrossRef CAS; (d) M. G. Kuzyk, J. Mater. Chem., 2009, 19, 7444 RSC.
- (a) M. K. Carter, J. Chem. Educ., 1951, 28, 524 CrossRef CAS; (b) R. Y. Levina and F. K. Velichko, Russ. Chem. Rev., 1960, 29, 437 CrossRef; (c) R. G. Sans and M. G. Chozas, Crit. Rev. Food Sci. Nutr., 1998, 38, 315 CrossRef PubMed.
- (a) R. G. Sans and M. G. Chozas, Pharmazie, 1988, 43, 827 CAS; (b) L. Ma, C. Xie, Y. Ran, X. Liang, L. Huang, H. Pei, J. Chen, J. Liu, Y. Sang, H. Lai, A. Peng, M. Xiang, Y. Wei and L. Chen, J. Med. Chem., 2012, 55, 9958 CrossRef CAS PubMed.
- (a) J. E. Gonzáles and R. Y. Tsien, Chem. Biol., 1997, 4, 269 CrossRef; (b) J. E. Gonzáles and R. Y. Tsien, Biophys. J., 1995, 69, 1272 CrossRef; (c) C. Wolff, B. Fuks and P. Chatelain, J. Biomol. Screening, 2003, 8, 533 CrossRef CAS PubMed.
- (a) F. Sieber, J. Photochem. Photobiol., A, 1987, 46, 1035 CrossRef CAS; (b) A. C. Benniston, A. Harriman and K. S. Gulliya, J. Chem. Soc., Faraday Trans., 1994, 90, 953 RSC; (c) A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973 CrossRef CAS.
- (a) M. Bauer, A. Rollberg, A. Barth and S. Spange, Eur. J. Org. Chem., 2008, 4475 CrossRef CAS; (b) M. C. Rezende, P. Compodonico, E. Abuin and J. Kossanyi, Spectrochim. Acta, Part A, 2001, 57, 1183 CrossRef CAS; (c) J. J. Fox and D. Shugar, Bull. Soc. Chim. Belg., 1952, 61, 44 CrossRef CAS; (d) L. Lu, R. J. Lachicotte, T. L. Penner, J. Perlstein and D. G. Whitten, J. Am. Chem. Soc., 1999, 121, 8146 CrossRef CAS; (e) J. Schmidt, R. Schmidt and F. Würthner, J. Org. Chem., 2008, 73, 6355 CrossRef CAS PubMed; (f) S. Seifert, A. Seifert, G. Brunklaus, K. Hofmann, T. Rüffer, H. Lang and S. Spange, New J. Chem., 2012, 36, 674 RSC.
- (a) F. Würthner and S. Yao, J. Org. Chem., 2003, 68, 8943 CrossRef PubMed; (b) A. Ciesielski, G. Schaeffer, A. Petitjean, J.-M. Lehn and P. Samorì, Angew. Chem., Int. Ed., 2009, 48, 2039 CrossRef CAS PubMed; (c) K. Motesharei and D. C. Myles, J. Am. Chem. Soc., 1998, 120, 7328 CrossRef CAS; (d) R. Baron, C.-H. Huang, D. M. Bassani, A. Onopriyenko, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2005, 44, 4010 CrossRef CAS PubMed; (e) P. Zhu, H. Kang, A. Facchetti, G. Evmenenko, P. Dutta and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 11496 CrossRef CAS; (f) H. Saadeh, L. Wang and L. Yu, J. Am. Chem. Soc., 2000, 122, 546 CrossRef CAS; (g) W. S. Jahng, D. Yoo, H. Moon, I. K. Moon, D.-H. Shin, M. Joo, E. Lee and N. Kim, Mol. Cryst. Liq. Cryst., 2000, 349, 1 CrossRef CAS.
- (a) S. Yagai, Y. Goto, T. Karatsu, A. Kitamura and Y. Kikkawa, Chem.–Eur. J., 2011, 17, 13657 CrossRef CAS PubMed; (b) S. Yagai, T. Nakajima, T. Karatsu, K. Saitow and A. Kitamura, J. Am. Chem. Soc., 2004, 126, 11500 CrossRef CAS PubMed; (c) S. Yagai, M. Ishii, T. Karatsu and A. Kitamura, Angew. Chem., Int. Ed., 2007, 46, 8005 CrossRef CAS PubMed.
- (a) C.-H. Huang, N. D. McClenaghan, A. Kuhn, G. Bravic and D. M. Bassani, Tetrahedron, 2006, 62, 2050 CrossRef CAS; (b) C.-H. Huang, N. D. McClenaghan, A. Kuhn, J. W. Hofstraat and D. M. Bassani, Org. Lett., 2005, 7, 3409 CrossRef CAS PubMed.
- (a) C. R. Moylan, R. J. Twieg, V. Y. Lee, S. A. Swanson, K. M. Betterton and R. D. Miller, J. Am. Chem. Soc., 1993, 115, 12599 CrossRef CAS; (b) H. Ikeda, Y. Kawabe, T. Sakai and K. Kawasaki, Chem. Lett., 1989, 1803 CrossRef CAS; (c) H. Ikeda, T. Sakai and K. Kawasaki, Chem. Phys. Lett., 1991, 179, 551 CrossRef CAS; (d) Y. Kawabe, H. Ikeda, T. Sakai and K. Kawasaki, J. Mater. Chem., 1992, 2, 1025 RSC; (e) Y.-W. Cao, X.-D. Chai, S.-G. Chen, Y.-S. Jiang, W.-S. Yang, R. Lu, Y.-Z. Ren, M. Blanchard-Desce, T.-J. Li and J.-M. Lehn, Synth. Met., 1995, 71, 1733 CrossRef CAS; (f) Y.-W. Cao, X.-D. Chai, W.-S. Yang, R. Lu, Y.-S. Jiang, T.-J. Li, M. Blanchard-Desce and J.-M. Lehn, Thin Solid Films, 1996, 284–285, 859 CrossRef CAS; (g) S. Barlow, H. E. Bunting, C. Ringham, J. C. Green, G. U. Bublitz, S. G. Boxer, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 1999, 121, 3715 CrossRef CAS; (h) S. Das, M. George, T. Mathew and C. V. Akosan, J. Chem. Soc., Perkin Trans. 2, 1996, 731 RSC; (i) J. Garín, J. Orduna, J. I. Rupérez, R. Alcalá, B. Villacampa, C. Sánchez, N. Martín, J. L. Segura and M. González, Tetrahedron Lett., 1998, 39, 3577 CrossRef; (j) E. Stankovic, S. Toma, R. V. Boxel, I. Asselberghs and A. Persoons, J. Organomet. Chem., 2001, 637–639, 426 CrossRef CAS; (k) X. Zhou, A.-M. Ren, J.-K. Feng, X.-J. Liu and C.-C. Shu, Chin. J. Chem., 2004, 22, 39 Search PubMed; (l) B. R. Cho, K. N. Son, S. J. Lee, T. I. Kang, M. S. Han and S. J. Jeon, Tetrahedron Lett., 1998, 39, 3167 CrossRef CAS; (m) G. Koeckelberghs, S. Sioncke, T. Verbiest, I. V. Severen, I. Pickard, A. Persoons and C. Samyn, Macromolecules, 2003, 36, 9736 CrossRef CAS; (n) S. M. Lee, W. S. Jahng, J. H. Lee, B. K. Rhee and K. H. Park, Chem. Phys. Lett., 2005, 411, 496 CrossRef CAS; (o) A. V. Kulinich, N. A. Derevyanko and A. A. Ishchenko, Russ. J. Gen. Chem., 2006, 76, 1441 CrossRef CAS.
- (a) G. Bourhill, J.-L. Brédas, L.-T. Cheng, S. R. Marder, F. Meyers, J. W. Perry and B. G. Tiemann, J. Am. Chem. Soc., 1994, 116, 2619 CrossRef CAS; (b) M. Blanchard-Desce, V. Alain, P. V. Bedworth, S. R. Marder, A. Fort, C. Runser, M. Barzoukas, S. Lebus and R. Wortmann, Chem.–Eur. J., 1997, 3, 1091 CrossRef CAS; (c) C. Maertnes, C. Detrembleur, P. Dubois, R. Jérôme, C. Boutton, A. Persoons, T. Kogej and J. L. Brédas, Chem.–Eur. J., 1999, 5, 369 CrossRef; (d) E. Genin, V. Hugues, G. Clermont, C. Herbivo, M. C. R. Castro, A. Comel, M. M. M. Raposo and M. Blanchard-Desce, Photochem. Photobiol. Sci., 2012, 11, 1756 RSC.
- E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1894, 27, 2345 CrossRef.
- M. Roman, B. Andrioletti, M. Lemaire, J.-M. Bernard, J. Schwartz and P. Barbeau, Tetrahedron, 2011, 67, 1506 CrossRef CAS.
- A. M. Gilbert, S. Caltabiano, F. E. Koehn, Z.-I. Chen, G. D. Francisco, J. W. Ellingboe, Y. Kharode, A. M. Mangine, R. Francis, M. TrailSmith and D. Gralnick, J. Med. Chem., 2002, 45, 2342 CrossRef CAS.
- (a) J. J. La Clair, J. Am. Chem. Soc., 1997, 119, 7676 CrossRef CAS; (b) A. Auffrant, B. Jaun, P. D. Jarowski, K. N. Houk and F. Diederich, Chem.–Eur. J., 2004, 10, 2906 CrossRef CAS PubMed.
- (a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS; (b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed.
- (a) K. M. Khan, M. Ali, M. Khan, M. Taha and S. Perveen, Lett. Org. Chem., 2011, 8, 28 CrossRef CAS; (b) J. M. Khurana and K. Vij, Catal. Lett., 2010, 138, 104 CrossRef CAS; (c) B. S. Jursic, J. Heterocycl. Chem., 2001, 38, 655 CrossRef CAS; (d) M. L. Deb and P. J. Bhuyan, Tetrahedron Lett., 2005, 46, 6453 CrossRef CAS; (e) T. Szotkowski, F. Bureš, O. Pytela, J. Kulhánek and Z. Trávníček, J. Heterocycl. Chem., 2006, 43, 1583 CrossRef CAS; (f) J. S. Yadav, B. V. S. Reddy, A. K. Basak, B. Visali, A. V. Narsaiah and K. Nagaiah, Eur. J. Org. Chem., 2004, 546 CrossRef CAS.
- F. Texier-Boullet and A. Foucaud, Tetrahedron Lett., 1982, 23, 4927 CrossRef CAS.
- (a) N. N. P. Moonen, R. Gist, C. Boudon, J.-P. Gisselbrecht, P. Seiler, T. Kawai, A. Kishioka, M. Gross, M. Irie and F. Diederich, Org. Biomol. Chem., 2003, 1, 2032 RSC; (b) N. N. P. Moonen, W. C. Pomerantz, R. Gist, C. Boudon, J.-P. Gisselbrecht, T. Kawai, A. Kishioka, M. Gross, M. Irie and F. Diederich, Chem.–Eur. J., 2005, 11, 3325 CrossRef CAS PubMed; (c) J. C. May, J. H. Lim, I. Biaggio, N. N. P. Moonen, T. Michinobu and F. Diederich, Opt. Lett., 2005, 30, 3057 CrossRef CAS PubMed; (d) F. Bureš, W. B. Schweizer, J. C. May, C. Boudon, J.-P. Gisselbrecht, M. Gross, I. Biaggio and F. Diederich, Chem.–Eur. J., 2007, 13, 5378 CrossRef PubMed.
- (a) J. D. Figueroa-Villar and A. A. Vieira, J. Mol. Struct., 2013, 1034, 310 CrossRef CAS; (b) M. C. Rezende and I. Almodovar, Magn. Reson. Chem., 2012, 50, 266 CrossRef CAS PubMed.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894 CrossRef CAS PubMed.
- ArgusLab, Mark Thompson and Planaria Software LLC, Version 4.01, webpage: http://www.arguslab.com.
- MOPAC2012, J. J. P. Stewart, Stewart Computational Chemistry, version 13.084W, webpage: http://OpenMOPAC.net.
- O. Pytela, OPchem, version 6.2, webpage: http://pytela.upce.cz/OPgm Search PubMed.
- (a) K. D. Singer and A. F. Garito, J. Phys. Chem., 1981, 75, 3572 CrossRef CAS; (b) B. F. Levine and C. G. Bethea, Appl. Phys. Lett., 1974, 24, 445 CrossRef CAS; (c) I. Ledoux and J. Zyss, Chem. Phys., 1982, 73, 203 CrossRef CAS; (d) T. Thami, P. Bassoul, M. A. Petit, J. Simon, A. Fort, M. Barzoukas and A. Villaeys, J. Am. Chem. Soc., 1992, 114, 915 CrossRef CAS.
- L.-T. Cheng, W. Tam, S. H. Stevenson, G. R. Meredith, G. Rikken and S. R. Marder, J. Phys. Chem., 1991, 95, 10631 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization of compounds 6–11, 15–23, 26–28, crystallographic data for 1a and 2a, full electrochemical data for 1–3, HOMO–LUMO localizations in 1–3, 1H and 13C NMR spectra of 1–3. CCDC 932394 and 932395. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00683b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |