 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, molecular structure, electrochemistry and DFT study of a ferrocenyl-substituted 4-quinazolinone and related heterocycles†
Jiří
Tauchman
a,
Kateřina
Hladíková
a,
Filip
Uhlík
b,
Ivana
Císařová
a and
Petr
Štěpnička
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 12840 Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz
bDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 12840 Prague, Czech Republic
First published on 26th March 2013
Abstract
2-Ferrocenyl-4(3H)-quinazolinone (2) was obtained by acylation of 2-aminobenzoic amide with ferrocenecarbonyl chloride (FcCOCl) and subsequent base-catalysed cyclisation of the intermediate 2-(ferrocenecarboxamido)benzamide (1). The related benzoxazin-4-one (4) was obtained analogously (though in one step) from FcCOCl and 2-aminobenzoic acid. Compounds 2 and 4 were thionated with P2S5 to give the corresponding ferrocenylated 4(3H)-quinazoline thione (3), and a mixture of benzothiazine-4-thione (5) and benzothiazine-4-one (6, major), respectively. Attempts to prepare 3-amino-2-ferrocenyl-4(3H)-quinazolinone from 4 and hydrazine hydrate failed. The reaction gave only 2-(ferrocenecarboxamido)benzohydrazide (7), which reacted with 2-chloro-1,3-dimethylimidazolidinium chloride to afford a mixture of cyclic and open (major) condensation products, both bearing the 1,3-dimethylimidazolidin-2-ylidene terminal group (8 and 9). All compounds were characterised by spectroscopic methods and the molecular structures of 1–7 and 9 were determined by X-ray diffraction analysis. Cyclic voltammetric measurements revealed that the compounds undergo a single one-electron oxidation, localised presumably at the ferrocene unit. DFT computations for 2 as a representative example performed in the gas phase and crystal state, reproduced well the experimental geometry and further supported the assignment of the redox processes.
Introduction
Studies dealing with the preparation of ferrocenylated heterocycles1 emerged shortly after the discovery of ferrocene,2 very likely in response to the important role which the uniquely large and diverse family of the conventional (i.e., organic) heterocyclic compounds has always played in chemistry.3 Nowadays, the studies into ferrocenyl-substituted heterocycles are motivated mostly by the search for new heterocyclic compounds and ligands4 and, further, by prospective applications of the specific electrochemical properties5 and biological activity of these compounds.6Although the structurally related ferrocenyl quinolines,7 quinolinones,8 flavones9 and quinoxalines10 have been already described, there is, up to the best of our knowledge, no report available in the literature concerning the structurally related, ferrocenyl-substituted quinazolinones and similar compounds, which all represent the basis of various biologically active substances.11 This prompted us to prepare and structurally characterise the directly ferrocenyl-substituted quinazolinone, viz. 2-ferrocenyl-4(3H)-quinazolinone (2), its corresponding 3,1-benzoxazin-4-one and their thionated analogues. Details of our investigations are presented in this contribution.
Results and discussion
Synthesis of 2-ferrocenyl-4(3H)-quinazolinone and the related compounds
2-Ferrocenyl-4-quinazolinone (2) was synthesised in the conventional manner12 as shown in Scheme 1. In the first step, 2-aminobenzoic amide was acylated with ferrocenecarbonyl chloride to give bis-amide 1. Upon treatment with NaOH in a methanol–water mixture, intermediate 1 underwent smooth heterocyclisation to afford quinazolinone 2 as an orange solid (76% isolated yield over the two steps). Compound 2 was further thionated with phosphorus pentasulphide13 in the presence of triethylamine to give the respective quinazoline thione 3 in a nice 75% yield after chromatographic purification.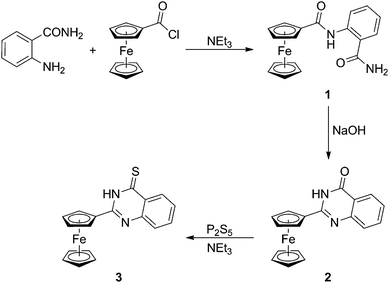 | ||
| Scheme 1 Synthesis of quinazolinone 2 and the corresponding thione 3. | ||
The structurally related benzoxazin-4-one 4 (Scheme 2) was obtained in one step by the reaction of ferrocenecarbonyl chloride with anthranilic amide in pyridine. Thionation of 4 with P2S5 provided a chromatographically separable mixture containing unreacted 4 (recovered: 17%) and two new compounds: the fully thionated benzothiazine-4-thione (5, 5%) and benzothiazine-4-one (6, 65%), in which only the in-ring oxygen atom was replaced with sulphur. It is noteworthy that the reactivity observed for 4 contrasts with the literature reports13 suggesting that the reaction of 2-substituted 3,1-benzoxazin-4-ones with P2S5/NEt3 proceeds under the replacement of both oxygen atoms to afford exclusively the corresponding benzothiazin-4-thiones.
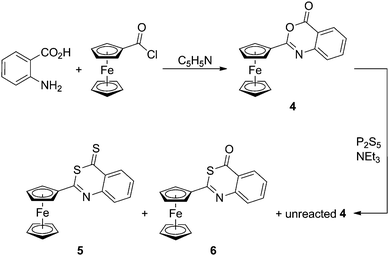 | ||
| Scheme 2 Preparation and thionation of benzoxazinone 4. | ||
Attempts to prepare 3-amino-2-ferrocenyl-4-quinazolinone by hydrazinolysis14 of 4 led only to an opened amide-hydrazide 7 as a product of formal hydrolysis. Likewise, attempted cyclisation of 7 in the presence of 2-chloro-1,3-dimethylimidazolidinium chloride failed.15 Instead of the anticipated cyclic product, the reaction afforded a mixture of two rather unexpected condensation products, viz. the cyclic 1,3-dimethylimidazolidin-2-ylidene hydrazide 8 and its opened zwitterionic analogue 9 in 14% and 75% isolated yields, respectively (Scheme 3).16
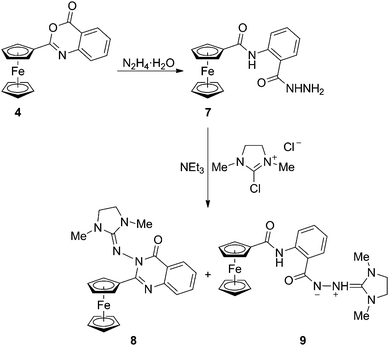 | ||
| Scheme 3 Opening of benzoxazinone 4 with hydrazine and the reaction of the resulting amido-hydrazide 7 with 2-chloro-1,3-dimethylimidazolidinium chloride. | ||
All compounds were characterised by 1H and 13C{1H} NMR, and IR spectroscopy, conventional and high-resolution electrospray ionisation (ESI) mass spectrometry, and by elemental analysis. Besides, the molecular structures of 1–7 and 9 were determined by single-crystal X-ray diffraction analysis.
Description of the molecular structures
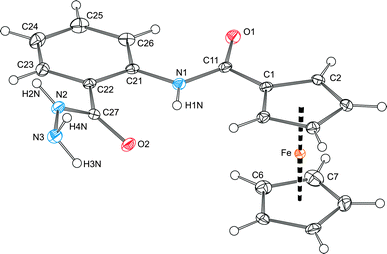
The molecular structure of the starting bis-amide 1, which crystallizes as a defined hemihydrate, 1·½H2O,17 is shown in Fig. 1. The molecular geometry of 1 is similar to that reported for simple ferrocene-based amides such as FcCONHPh18 and fc(CONHPh)219 (Fc = ferrocenyl, fc = ferrocene-1,1′-diyl), and for 2-(acetylamino)benzamide.20 The amide plane comprising atoms C11, O1 and N1 in 1 is rotated with respect to its parent cyclopentadienyl ring C(1–5) by 14.7(2)°. Mutual rotation of the benzene ring and its amide pendant (C27, N2, O2) is considerably larger, the respective dihedral angle being 29.3(2)°. These rotations, possibly destabilising the structure due to a decreased conjugation, seem to result from intra- and intermolecular hydrogen bonding interactions.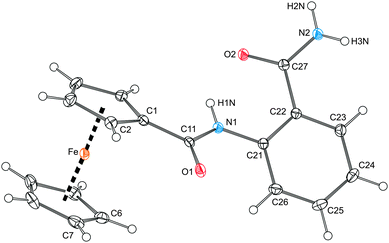 | ||
| Fig. 1 View of the molecule of amide 1 in the crystal structure of 1·½H2O showing displacement ellipsoids at the 30% probability level and the labelling scheme. Selected distances and angles (in Å and deg): C11–O1 1.234(2), C11–N1 1.361(2), C21–N1 1.408(2), C27–O2 1.248(2), C27–N2 1.328(2), O1–C11–N1 123.9(2), O2–C27–N2 122.3(2), C1–C11–N1–C21 177.6(2); Fe–Cg1 1.6463(9), Fe–Cg2 1.654(1), ∠Cp1, Cp2 2.6(1). Note: Cp1 and Cp2 are cyclopentadienyl rings C(1–5) and C(6–10), respectively; Cg1 and Cg2 denote their respective centroids. | ||
In fact, the individual molecules of amide 1 fully exploit their H-bonding ability. In the crystal, they assemble into dimers lying around the inversion centres via N–H⋯O interactions (A in Fig. 2) and further into infinite columnar stacks via the carboxyl-like double hydrogen bridges (B). An additional intramolecular N–H⋯O contact though with a rather acute angle at the H atom (C) is detected between the FcCONH proton and the CONH2 oxygen. The solvating water molecules (not shown in Fig. 2) reside on the crystallographic two-fold axes and between the columnar stacks, which they interconnect into a three-dimensional array via O1W–H1W⋯O1 hydrogen bonds towards the FcCO oxygens (O1W⋯O1 = 2.823(2), angle at H1W = 152°).
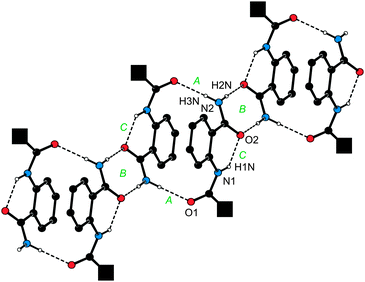 | ||
| Fig. 2 View of a single columnar stack built up from molecules of 1 in the crystals of 1·½H2O. For clarity, the bulky ferrocenyl groups were replaced with filled black squares and the CH protons were omitted. H-bond parameters are as follows. A: N2–H3N⋯O1, N2⋯O1 = 2.975(2) Å, angle at H3N = 176°; B: N2–H2N⋯O2, N2⋯O2 = 2.883(2) Å, angle at H2N = 176°; C: N1–H1N⋯O2, N1⋯O2 = 2.690(2) Å, angle at H1N = 136°. Symmetry codes: A: ½ − x, ½ − y, −z; B: −x, 1 − y, −z; C: intramolecular. | ||
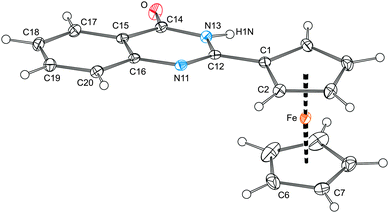
The overall molecular structure of hydrazide 7 (Fig. 3) compares well with that of the analogous amide 1. Similarly to 1, the ‘amide’ planes in 7 are rotated with respect to their parent aromatic rings [cf. the dihedral angles 9.0(2)° for planes (C11, O1, N1) and C(1–5), and 30.1(1)° for planes (C27, O2, N2) and C(21–26)]. The molecules of 7 assemble into ‘dimers’ via N–H⋯O hydrogen bonds (Fig. 4). However, the cross-linking of these dimers is achieved through N–H⋯O contacts involving the hydrazide NH2 (terminal) moieties, which thus take over the role played by the solvent molecules in the structure of 1·½H2O. As the result, the individual molecules of 7 associate into wave-like layers, which further assemble into a complicated three-dimensional network.
 | ||
| Fig. 3 View of the molecular structure of 7 showing displacement ellipsoids at the 30% probability level and the atomic labelling scheme. Selected distances and angles (in Å and deg): C11–O1 1.235(2), C11–N1 1.358(2), C21–N1 1.405(2), C27–O2 1.245(2), C27–N2 1.327(2), N2–N3 1.415(2), O1–C11–N1 124.1(1), O2–C27–N2 121.9(1), C27–N2–N3 122.6(1), C1–C11–N1–C21–172.6(1), O2–C27–N2–N3 3.7(2); Fe–Cg1 1.6408(6), Fe–Cg2 1.6463(7), ∠Cp1, Cp2 2.15(8). The ring planes are defined as for the analogous bis-amide 1 (cf.Fig. 1). | ||
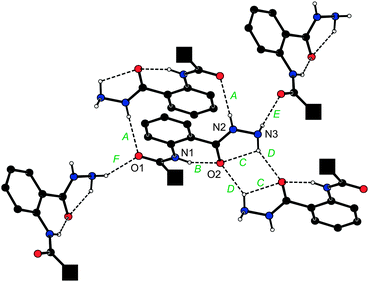 | ||
| Fig. 4 Different N–H⋯O hydrogen bonds (denoted as A–F) formed by a single molecule of 7 in the crystal. For clarity, the bulky ferrocenyl groups were replaced with filled black squares and all CH protons were omitted. H-bond parameters are as follows. A: N2–H2N⋯O1, N2⋯O1 = 3.073(2) Å, angle at H2N = 153°; B: N1–H1N⋯O2, N1⋯O2 = 2.696(2) Å, angle at H1N = 142°; C: N3–H3N⋯O2, N3⋯O2 = 2.751(1) Å, angle at H3N = 109°; D: N3–H3N⋯O2, N3⋯O2 = 3.040(2) Å, angle at H3N = 145°; E ≡ F: N3–H4N⋯O1, N3⋯O1 = 3.066(2) Å, angle at H4N = 151°. Symmetry codes: A: 1 − x, −y, 1 − z; B and C: intramolecular; D: ½ − x, ½ − y, 1 − z; E: x, −y, ½ + z, F: x, −y, z – ½. | ||
The molecular structures of compounds 2–6 are presented in Fig. 5 and 6, respectively. Structural data for these ferrocenyl-substituted heterocycles, which formally differ only by the type of the heteroatoms and their positions within the six-membered ring, are summarised in Table 1.
 | ||
| Fig. 5 View of the molecular structure of 2 showing the displacement ellipsoids at the 30% probability level. | ||
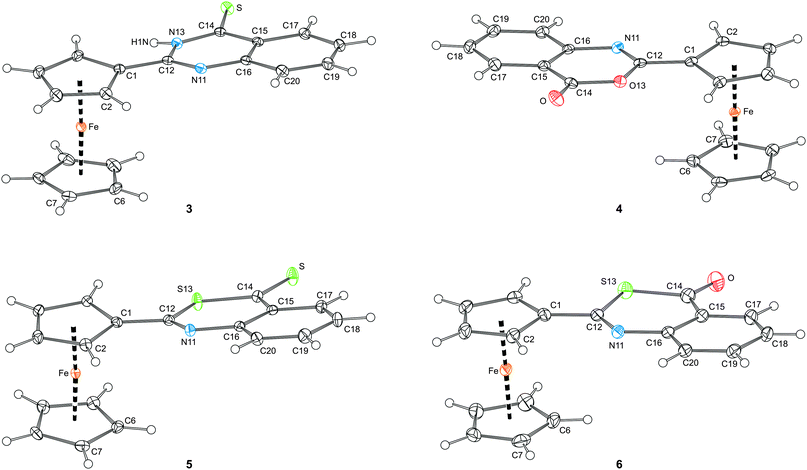 | ||
| Fig. 6 Views of the molecular structures of compounds 3–6 showing the atom labelling scheme and displacement ellipsoids at the 30% probability level. | ||
| Parametera | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| a Definitions: ψ is the dihedral angle subtended by the Cp1 plane and the least-squares plane of the entire heterocyclic moiety (N11, C12, Y13, C14, and C15–C20); cyclopentadienyl rings: Cp1 = C(1–5), Cp2 = C(6–10), Cg1/Cg2 are the respective ring centroids. | |||||
| Y13/Z | N13/O | N13/S | O13/O | S13/S | S13/O |
| N11–C12 | 1.298(2) | 1.302(2) | 1.279(2) | 1.289(3) | 1.277(3) |
| C12–Y13 | 1.382(2) | 1.382(2) | 1.379(2) | 1.762(2) | 1.767(3) |
| Y13–C14 | 1.371(2) | 1.361(2) | 1.390(2) | 1.737(2) | 1.780(3) |
| C14-Z | 1.238(2) | 1.672(1) | 1.198(2) | 1.655(2) | 1.218(4) |
| C14–C15 | 1.452(2) | 1.447(2) | 1.460(2) | 1.455(3) | 1.455(4) |
| C15–C16 | 1.406(2) | 1.412(2) | 1.401(2) | 1.410(3) | 1.416(4) |
| C16–N11 | 1.388(2) | 1.382(2) | 1.395(2) | 1.393(2) | 1.393(4) |
| C16–N11–C12 | 117.1(1) | 117.3(1) | 117.5(1) | 121.9(2) | 122.6(2) |
| N11–C12–Y13 | 123.3(1) | 122.4(1) | 125.5(1) | 125.5(2) | 126.7(2) |
| C12–Y13–C14 | 123.5(1) | 124.3(1) | 121.1(1) | 104.9(1) | 103.4(1) |
| Y13–C14–C15 | 114.7(1) | 114.7(1) | 115.2(1) | 126.5(2) | 118.4(2) |
| ψ | 12.40(7) | 8.95(6) | 7.06(7) | 14.7(1) | 4.0(1) |
| Fe–Cg1 | 1.6391(7) | 1.6457(6) | 1.6451(7) | 1.644(1) | 1.640(1) |
| Fe–Cg2 | 1.6506(9) | 1.6597(7) | 1.6497(8) | 1.652(1) | 1.649(2) |
| ∠Cp1, Cp2 | 0.3(1) | 2.91(9) | 0.34(9) | 3.3(1) | 0.6(2) |
The crystal structures reveal practically planar arrangements for the entire heterocyclic units (10 atoms) and their attached C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CS bonds. The maximum observed displacement from the mean planes of the bicyclic units of 0.042(1) and 0.045(1) Å are seen for N13 atoms in 2 and 3, respectively. These compounds also show the largest departure of the exo-cyclic O/S atoms from the plane of their parent heterocyclic moiety, the perpendicular distances from the mean ring plane being 0.082(1) Å for O in 2, and 0.071(1) Å from S in 3. The in-ring distances and angles (Table 1) clearly illustrate a distortion of the six-membered heterocyclic ring upon replacement of the oxygen and nitrogen atoms by the relatively larger sulphur.
O/CS bonds. The maximum observed displacement from the mean planes of the bicyclic units of 0.042(1) and 0.045(1) Å are seen for N13 atoms in 2 and 3, respectively. These compounds also show the largest departure of the exo-cyclic O/S atoms from the plane of their parent heterocyclic moiety, the perpendicular distances from the mean ring plane being 0.082(1) Å for O in 2, and 0.071(1) Å from S in 3. The in-ring distances and angles (Table 1) clearly illustrate a distortion of the six-membered heterocyclic ring upon replacement of the oxygen and nitrogen atoms by the relatively larger sulphur.
The heterocyclic units and their bonding cyclopentadienyl ring in 2–6 are rotated from a mutually coplanar arrangement. The rotation varies both in the magnitude (see ψ in Table 1) and also in the sense. Whereas the CO bond in 4 is directed inwards the ferrocene moiety (i.e., towards the Fe atom), the CO and CS bonds in all other structurally characterised compounds point away from the ferrocene unit. These rotations not exceeding ca. 15° do not seem to destabilise the structures much by, e.g., a decreased conjugation (see DFT computations below), and can be accounted for by the steric factors at molecular level and also the crystal packing effects.
Finally, the ferrocene units in 2–6 are regular, showing only negligible tilting. The observed shortening of the Fe–Cg1 distances as compared to the matched Fe–Cg2 distances, which is relatively small (0.3–0.9%) but systematic and statistically significant, can be explained by a strengthening of the Fe ← Cp1 donation due to an electron density transfer from the ferrocene unit to the electron-withdrawing heterocyclic unit.
Only compounds 2 and 3 possess both NH protons as the typical H-bond donors and suitable H-bond acceptors. Not surprisingly, therefore, the molecules in their crystal structures associate via N–H⋯Z (Z = O, S) interactions giving rise to dimers around the crystallographic inversion centres (Fig. 7a). Probably due to its spatial proximity, the adjacent CH proton (H5) becomes involved in a similar interaction (C5–H5⋯O), too. The molecules further associate via offset π⋯π stacking interactions of the ferrocene C5H5 ring and the C6H4 ring within the heterocyclic moiety (Fig. 7b). The distances of the ring centroids and the interplanar angles are 3.839(1) Å and 11.08(9)° for 2, and 3.8559(9) Å and 5.93(8)° for 3. Since these π-interactions involve molecules related by elemental translation along the b-axis, they interconnect the H-bonded dimers into infinite columnar stacks.
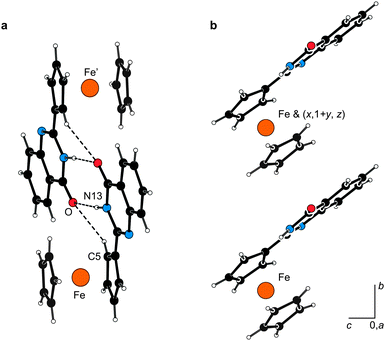 | ||
| Fig. 7 (a) View of the hydrogen-bonded dimer in the structure of 2 showing the hydrogen bonds as dashed lines. The H-bond parameters are as follows: N13–H1N⋯O, N13⋯O = 2.836(2) Å, angle at H1N = 173°; C5–H5⋯O, C5⋯O = 3.225(2) Å, angle at H5 = 136°. Compound 3 forms a similar dimer, the respective parameters being: N13–H1N⋯S, N13⋯S = 3.361(1) Å, angle at H1N = 161°; C5–H5⋯S, C5⋯S = 3.525(2) Å, angle at H5 = 132°. (b) π⋯π Stacking interactions of molecules of 2 leading to propagation of the molecular array along the crystallographic b-axis. | ||
In contrast, the molecules of 4–6, lacking the NH groups, associate predominantly via π⋯π interactions. In the case of 4 and 6, these interactions are formed between the C5H5 rings and the heterocyclic moieties in molecules related by elemental translation along the b-axis. A similar interaction dominates in the crystal assembly of 5, involving molecules related by the crystallographic inversion.
The molecular structure of compound 9 is presented in Fig. 8 together with selected geometric data. According to a search in the Cambridge Structural Database,21 compound 9 represents the only structurally characterised compound possessing an extended (acyclic) (C3N2)⋯N⋯N–C(O) linkage, where ⋯ stands for any bond. An additional search in Chemical Abstracts for a similar fragment bearing the 1,3-dimethyl-2-imidazolidinylidene moiety resulted in only a few compounds (<30)22 that were all formulated conservatively as the tautomeric 2-(1,3-dimethyl-2-imidazolidinylidene)hydrazides, (1,3-Me2C3N2)N–NHC(O)R.
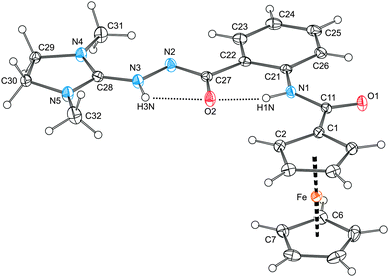 | ||
| Fig. 8 View of the molecular structure of 9 showing the hydrogen bonds as dotted lines. The displacement ellipsoids correspond to 30% probability. Selected distances and angles (in Å and deg): C1–C11 1.481(2), C11–O1 1.229(2), C11–N1 1.357(2), C22–C27 1.504(2), C27–O2 1.280(2), C27–N2 1.314(2), N2–N3 1.402(2), N3–C28 1.317(2), C28–N4 1.330(2), C28–N5 1.344(2), Fe–Cg1 1.6457(7), Fe–Cg2 1.6491(8), ∠Cp1, Cp2 1.6(1); O1–C11–N1 124.7(1), C22–C27–O2 121.4(1), C22–C27–N2 114.4(1), O2–C27–N2 124.1(1), C27–N2–N3 108.7(1), N2–N3–C28 123.5(1), N4–C28–N5 110.9(1). Note: The ring planes are defined as for 1. H-bond parameters: N1–H1N⋯O2, N1⋯O2 = 2.582(2) Å, angle at H1N = 147°; N3–H3N⋯O2, N3⋯O2 = 2.496(2) Å, angle at H3N = 110°. | ||
In our case, the good-quality diffraction data allowed to locate unambiguously the NH protons and thus to formulate compound 9 as a zwitterion (1,3-Me2C3N2)NH(+)–N(−)–C(O)RFc (RFc = 2-(ferrocenylcarboxamido)phenyl). Parameters describing the geometry of the C11(O1)N11 unit in 9 are very similar to those found in 1 and the precursor 7. On the other hand, the other amide moiety, N2(−)–C27O2, gains a partial N2(δ−)C27–O2(δ−) character, very likely due to an electron density transfer from the negatively charged N2, which in turn results in an elongation of the C27O2 bond and shortening of the C27–N2 bond. The C28N3(+) and N2–N3 bond lengths in 9 are similar to the respective distances in 4-chloro-N-(imidazolidin-2-ylidene)-1H-indazol-1-amine (N–N = 1.402(2) Å, NNC = 1.307(2) Å),23 whereas the N2–N3 separation in 9 is significantly longer than the N–N distances reported for 1,2-dibenzoylhydrazine (1.385(3) Å)24 and benzaldehyde benzoylhydrazone (1.368(7) Å).25
The extensive electronic delocalisation in the molecule of 9 corresponds with the unique all-planar geometry this compound assumes in the solid state. Taking the amide linker {C11, O1, N1} as the reference plane, the bonding cyclopentadienyl ring is found rotated at the dihedral angle of 8.2(2)°, while the entire extended pendant comprising as much as 18 atoms [N(1–5), O2 and C(21–32)], which are coplanar within ca. 0.07 Å, is rotated by 6.9(2)°. In addition to conjugation, the planar geometry and perhaps also the particular tautomeric form seem to be stabilised by intramolecular N–H⋯O contacts that can be detected in the structure albeit with unfavourably acute angles at the hydrogen atoms (see Fig. 8).
Electrochemistry
The electrochemical behaviour of compounds 1–7 was studied by cyclic voltammetry on a stationary platinum disc electrode using 1,2-dichloroethane solutions and Bu4N[PF6] as the supporting electrolyte. The representative cyclic voltammograms are shown in Fig. 9 and the pertinent data are summarised in Table 2.![Cyclic voltammograms of compounds 2 and 4 as recorded on a Pt disc electrode in 1,2-dichloroethane containing 0.1 M Bu4N[PF6] as the supporting electrolyte. Scan rate: 200 mV s−1.](/image/article/2013/NJ/c3nj00182b/c3nj00182b-f9.gif) | ||
| Fig. 9 Cyclic voltammograms of compounds 2 and 4 as recorded on a Pt disc electrode in 1,2-dichloroethane containing 0.1 M Bu4N[PF6] as the supporting electrolyte. Scan rate: 200 mV s−1. | ||
| Compound | E°′ [V] | Compound | E°′ [V] |
|---|---|---|---|
| a The potentials were determined as an average of the peak potentials in cyclic voltammetry and are given relative to ferrocene/ferrocenium reference. Conditions: Pt-disc electrode, ca. 0.5 mM solutions in dry 1,2-dichloroethane containing 0.1 M Bu4N[PF6] as the supporting electrolyte. Scan rate: 200 mV s−1. | |||
| 1 | 0.21 | 5 | 0.27 |
| 2 | 0.22 | 6 | 0.26 |
| 3 | 0.26 | 7 | 0.21 |
| 4 | 0.25 | ||
All compounds display one electrochemically reversible redox transition in the anodic region, which is controlled by diffusion as indicated by the peak potentials increasing linearly with the square root of the scan rate (ip ∝ ν½). Only in the case of thione 3 the primary redox change is complicated by some follow-up processes affording other redox-active species and is associated with a pre-wave (see Fig. S1, ESI†). The observed redox changes, which can be attributed to the ferrocene/ferrocenium couple, occur in a rather narrow range of ca. 60 mV for the whole series and at potentials more positive than the ferrocene/ferrocenium reference (Table 2). The anodic shift corresponds with the overall electron-withdrawing character of the amide and heterocyclic moieties that can be expected to make any electron removal more difficult (cf. the Hammett’s σp constants for CONH2: 0.36, and 2-pyrimidinyl: 0.53).26
Theoretical computations
In order to rationalise the observed redox behaviour and geometric structure determined by X-ray diffraction analysis, we studied quinazolinone 2 as the representative theoretically by density functional theory (DFT) calculations. The geometry obtained by DFT computations (B3LYP/6-31G*) for an isolated molecule matched quite well the experimental one. An average relative error of 0.6% was achieved for the bond distances and angles involving the non-hydrogen atoms, while the maximum error of about 2% was observed for the N13–C14 distance (see Table S1, ESI†). The quinazolinone moiety and its bonding cyclopentadienyl ring was found to be almost coplanar with the torsion angle N11–C12–C1–C2 (φ) being ca. −5°, which is considerably less that the value from the crystal structure data, φ = 15.5(2)°.The discrepancy between φ calculated for an isolated molecule and the value determined from X-ray diffraction data was tentatively attributed to crystal packing effects. Indeed, the geometry obtained from DFT calculations (PBE/6-31G*) performed for a periodic structure with four molecules in the unit cell gave φ = 15°, which is in a perfect agreement with the experimental value. In this case, however, a slightly worse average relative error for the bond length and angles was obtained (ca. 1% for the bond lengths and 0.9% for the angles), which is in line with the well known, slightly poorer performance of the PBE functional (that was used to make the calculations of the crystal structure more economical) as compared with B3LYP.
Next, starting from the geometry optimised for an isolated molecule, we carried out a fully relaxed potential energy surface scan (DFT/B3LYP) for this torsion angle and found a second minimum at φ = –172° (Fig. 10). The potential energy difference between the minima is negligible (about 0.02 kJ mol−1) with respect to thermal energy kT at ambient conditions (ca. 2.5 kJ mol−1). The energy barriers separating these minima are about 16 and 22 kJ mol−1 high. Similar calculations using PBE functional gave practically identical results. The energy minima were found at φ ca. −4° and –174°, and the calculated energy barriers were ca. 17 and 24 kJ mol−1 (for a comparison of the two energy profiles, see Fig. S2, ESI†). Considering the height of the energy barriers, one can expect that the ferrocenyl and quinazolinone moieties undergo a fast rotation at room temperature in solution. This is in accordance with the 1H and 13C{1H} NMR spectra showing degenerate signals for the CH groups in α and β positions of the substituted cyclopentadienyl ring. It is also noteworthy that structures corresponding to the minima are very nearly mirror images of each other except for a rotation of the unsubstituted cyclopentadienyl ring.
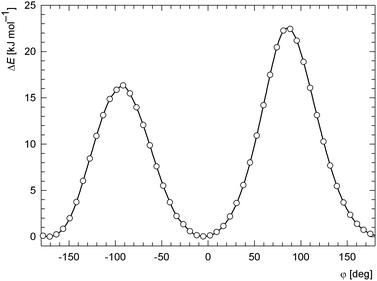 | ||
| Fig. 10 The dependence of the relative DFT (B3LYP/6-31G*) potential energy on the torsion angle N11–C12–C1–C2 (φ) for an isolated molecule. | ||
Furthermore, we investigated the electronic structure of 2. The highest occupied molecular orbital (HOMO; Fig. 11) was found to be contributed mainly from d-orbitals centred at the iron atom (68%). Since this contribution represents the only major entry (>10%) from one particular atom to HOMO, one may expect that the initial oxidation will occur there, which is in accordance with the observed electrochemical behaviour (vide supra). On the other hand, the lowest unoccupied molecular orbital (LUMO; Fig. 11) is more delocalized with major contributions from p-orbitals of the carbon atoms forming the quinazolinone ring (23% for the pivotal atom C18 and 11% for C12; for numbering, see Fig. 5), and further from d-orbitals of the iron atom (15%).
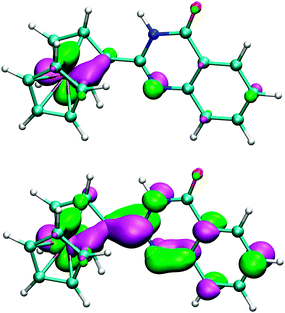 | ||
| Fig. 11 B3LYP/6-31G* HOMO (top) and LUMO (bottom) orbitals of quinazolinone 2 shown at the ±0.05 a.u. level. | ||
The extensive conjugation in the molecule of 2 is clearly manifested in UV-vis spectra (Fig. 12). For instance, the low-energy absorption (d–d transitions) of 2 appears shifted to lower energies and is more intense (2: λmax = 458 nm, ε ≈ 220 M−1 cm−1) than for unsubstituted ferrocene (λmax ≈ 440 nm)27 and even ferrocenecarboxamide (FcCONH2; λmax ≈ 445 nm).
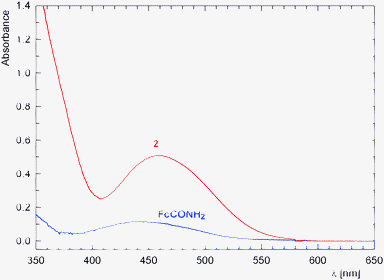 | ||
| Fig. 12 UV-vis spectra of 2 (red) and ferrocenecarboxamide (blue) as recorded for ca. 5 × 10−4 M solutions in methanol (optical path: 1 cm). | ||
The calculated electrostatic potential (ESP) is shown in Fig. 13 with values coded by colours on a contour of total electron density drawn at the 0.002 a.u. level that roughly corresponds to the Van der Waals envelope.28 The domain with the most negative ESP (most attractive for a positive test charge) is located on the NH group of quinazolinone, whilst the domain with the most positive ESP is localized at the CO moiety. This corresponds with the solid-state packing based on an assembly of dimers formed via the N–H⋯OC hydrogen bonds. A supportive C–H⋯O interaction also detected in the crystal structure (C5–H5⋯O see Fig. 5) probably reflects a polarisation of this CH group by the negatively charged CO oxygen from the other molecule, which is indicated by an increased ESP at this H atom as compared to α-CH close to the heterocyclic N atom.
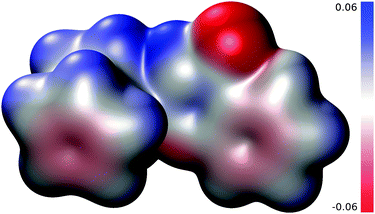 | ||
| Fig. 13 Electrostatic potential (with magnitude <0.06 a.u.) mapped on a B3LYP/6-31G* electron density contour corresponding to 0.002 a.u. | ||
Conclusions
2-Ferrocenyl-4(3H)-quinazolinone and the related heterocycles differing by the type and position of the heteroatoms (N → O, S) are accessible by the general methods devised for the preparation of their simple organic counterparts though not always in exactly the same manner. As revealed by electrochemical measurements and DFT computations, the directly connected heterocyclic and ferrocene moieties are conjugated. This conjugation affects both the molecular geometry (the molecular parts tend to be close to co-planar) and the electrochemical response (the electron-withdrawing heterocyclic pendant renders the ferrocene oxidation more difficult).Experimental
Materials and methods
All syntheses were performed under argon atmosphere with an exclusion of the direct day light. Ferrocenecarbonyl chloride29 and ferrocenecarboxamide30 were prepared according to the literature procedures. Solvents used in the syntheses were dried by standing over appropriate drying agents and distilled under argon [dichloromethane (K2CO3), pyridine (KOH), dioxane and triethylamine (sodium metal)]. Methanol was distilled from sodium methoxide. Anhydrous acetonitrile and ethanol were purchased from Sigma-Aldrich. Other chemicals (Sigma-Aldrich and Alfa-Aesar) and solvents used for crystallizations and in chromatography (Lachner) were used as received.NMR spectra were recorded with a Varian UNITY Inova 400 MHz spectrometer (1H, 399.95 MHz; 13C, 100.58 MHz) at 298 K. Chemical shifts (δ/ppm) are given relative to internal tetramethylsilane. Infrared spectra were recorded in Nujol mulls with a Nicolet 6700 FTIR spectrometer in the range 400–4000 cm−1. UV-vis spectra were recorded with Unicam UV300 spectrometer using methanolic solution and 1 cm quartz cells. Electrospray ionization (ESI) mass spectra were obtained with an Esquire 3000 (Bruker; low resolution) or an LTQ Orbitrap XL instrument (Thermo Fisher Scientific; high resolution).
Electrochemical measurements were performed with a computer-controlled potentiostat μAUTOLAB III (Eco Chemie, Netherlands) at room temperature (ca. 23 °C) using a standard Metrohm three-electrode cell equipped by a platinum disc working electrode (2 mm diameter), platinum sheet auxiliary electrode, and a double-junction Ag/AgCl (3 M KCl) reference electrode. The compounds were dissolved in dry 1,2-dichloroethane (Sigma-Aldrich, absolute) to give solutions containing ca. 0.5 mM of the analyte and 0.1 M Bu4N[PF6] (Fluka, puriss. for electrochemistry) as the supporting electrolyte. The solutions were deaerated with argon before the measurement and then kept under an argon blanket. The redox potentials (accuracy ca. 5 mV) are given relative to a ferrocene/ferrocenium reference (E°′ = 0.46 V under the experiment conditions).
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the eluent. A single orange band was collected and evaporated to afford analytically pure 1 as a yellow-orange solid (306 mg, 88%).
:1 v/v) as the eluent. A single orange band was collected and evaporated to afford analytically pure 1 as a yellow-orange solid (306 mg, 88%).
M.p. 161–163 °C (toluene). 1H NMR (CDCl3): δ 4.25 (s, 5H, C5H5), 4.43 (virtual t, J = 2.0 Hz, 2H, C5H4), 4.43 (virtual t, J = 1.9 Hz, 2H, C5H4), 5.70 (br s, 1H, NH2), 6.32 (br s, 1H, NH2), 7.07 (ddd, J = 1.2, 7.4, 7.9 Hz, 1H, C6H4), 7.53 (ddd, J = 0.4, 1.6, 7.4 Hz, 1H, C6H4), 7.57 (ddd, J = 0.3, 1.5, 7.9 Hz, 1H, C6H4), 8.78 (dd, J = 1.1, 8.6 Hz, 1H, C6H4), 11.72 (br s, 1H, NH). 13C{1H} NMR (CDCl3): δ 68.63 (CH C5H4), 69.97 (CH C5H5), 71.06 (CH C5H4), 76.48 (Cipso C5H4), 117.72 (CCipso C6H4), 121.23 (CH C6H4), 122.07 (CH C6H4), 127.36 (CH C6H4), 133.60 (CH C6H4), 140.95 (NCipso C6H4), 169.75 (CONH2), 171.50 (CONH). IR (Nujol): νmax 3380 m, 3320 m, 3169 m, amide I 1655 vs, 1624 s, 1595 s, 1579 s, amide II 1514 vs, 1448 vs, 1400 vs, 1308 vs, 1270 s, 1217 w, 1171 w, 1142 m, 1124 w, 1107 m, 1083 w, 1043 w, 1029 m, 1002 w, 950 w, 886 w, 848 m, 825 s, 760 s, 682 w, 641 m, 527 m, 496 m, 482 m, 464 w cm−1. MS (ESI+): m/z 371 ([M+Na]+). HR MS (ESI+) calc. For C18H16O2N2FeNa ([M+Na]+) 371.0453, found 371.0453. Anal. Calcd for C18H16N2O2Fe·0.15 CH2Cl2: C 60.40, H 4.55, N 7.76. Found: C 60.37, H 4.63, N 7.54.
:1 (v/v) as the eluent afforded a single orange band, which was evaporated to give 2 as an orange solid (141 mg, 86%).
M.p. 190 °C dec. (chloroform). 1H NMR (CDCl3): δ 4.25 (s, 5H, C5H5), 4.58 (virtual t, J = 1.9 Hz, 2H, C5H4), 5.24 (virtual t, J = 1.8 Hz, 2H, C5H4), 7.50 (ddd, J = 1.5, 6.8, 8.1 Hz, 1H, C6H4), 7.73 (ddd, J = 0.6, 1.5, 8.2 Hz, 1H, C6H4), 7.78 (ddd, J = 1.5, 6.7, 8.2 Hz, 1H, C6H4), 8.36 (ddd, J = 0.5, 1.5, 7.9 Hz, 1H, C6H4), 11.20 (br s, 1H, CONH). 13C{1H} NMR (CDCl3): δ 68.13 (CH C5H4), 70.03 (CH C5H5), 71.45 (CH C5H4), 76.06 (Cipso C5H4), 120.53 (CCipso C6H4), 125.92 (CH C6H4), 126.38 (CH C6H4), 127.35 (CH C6H4), 134.81 (CH C6H4), 149.92 (NCipso C6H4), 155.14 (NCN), 163.49 (CO). IR (Nujol): νmax 3178 w, νNH 3122 m, 3053 w, amide I 1660 vs, 1596 vs, amide II 1564 s, 1507 m, 1440 s, 1413 w, 1339 m, 1291 s, 1265 w, 1246 w, 1219 w, 1153 m, 1107 m, 1099 w, 1063 w, 1041 w, 1030 w, 1020 w, 1003 w, 957 m, 892 m, 872 w, 836 w, 818 s, 782 s, 729 w, 690 w, 623 w, 609 w, 551 m, 494 s, 484 s cm−1. MS (ESI+): m/z 331 ([M+H]+), 353 ([M+Na]+). Anal. Calcd for C18H14N2OFe·0.04 CHCl3: C 64.69, H 4.23, N 8.37. Found: C 64.71, H 4.14, N 8.22.
M.p. 170–172 °C (toluene). 1H NMR (CDCl3): δ 4.24 (s, 5H, C5H5), 4.58 (virtual t, J = 2.0 Hz, 2H, C5H4), 5.00 (virtual t, J = 1.9 Hz, 2H, C5H4), 7.48 (ddd, J = 1.3, 7.0, 8.2 Hz, 1H, C6H4), 7.69 (ddd, J = 0.5, 1.3, 8.2 Hz, 1H, C6H4), 7.78 (ddd, J = 1.5, 7.0, 8.3 Hz, 1H, C6H4), 8.68 (ddd, J = 0.5, 1.5, 8.1 Hz, 1H, C6H4), 10.38 (br s, 1H, CONH). 13C{1H} NMR (CDCl3): δ 67.82 (CH C5H4), 70.20 (CH C5H5), 71.76 (CH C5H4), 74.96 (Cipso C5H4), 127.36 (CH C6H4), 127.97 (CH C6H4), 127.97 (CCipso C6H4), 130.49 (CH C6H4), 135.72 (CH C6H4), 145.41 (NCipso C6H4), 153.28 (NCN), 186.29 (CS). IR (Nujol): νmax 3200 w, 3121 w, 1582 vs, 1566 vs, 1499 vs, 1481 vs, 1432 s, 1410 w, 1346 m, 1318 w, 1293 w, 1256 w, 1241 w, 1225 vs, 1205 vs, 1153 m, 1138 w, 1107 m, 1056 w, 1023 m, 1006 w, 925 m, 866 m, 859 w, 842 w, 824 m, 806 w, 769 w, 774 s, 767 s, 742 w, 724 w, 706 w, 592 w, 559 w, 483 s, 438 m cm−1. MS (ESI+): m/z 369 ([M+Na]+). Anal. Calcd for C18H14N2SFe: C 62.44, H 4.08, N 8.09. Found: C 62.28, H 4.08, N 8.04.
M.p. 162–164 °C (heptane). 1H NMR (CDCl3): δ 4.23 (s, 5H, C5H5), 4.55 (virtual t, J = 1.9 Hz, 2H, C5H4), 5.08 (virtual t, J = 1.9 Hz, 2H, C5H4), 7.46 (ddd, J = 1.2, 7.3, 7.8 Hz, 1H, C6H4), 7.57 (ddd, J = 0.6, 1.2, 8.2 Hz, 1H, C6H4), 7.77 (ddd, J = 1.6, 7.3, 8.1 Hz, 1H, C6H4), 8.20 (ddd, J = 0.6, 1.6, 7.9 Hz, 1H, C6H4). 13C{1H} NMR (CDCl3): δ 69.26 (CH C5H4), 70.06 (CH C5H5), 71.86 (CH C5H4), 73.12 (Cipso C5H4), 116.47 (CCipso C6H4), 126.39 (CH C6H4), 127.20 (CH C6H4), 128.64 (CH C6H4), 136.52 (CH C6H4), 147.60 (NCipso C6H4), 159.97 (NCO), 162.46 (OCO). IR (Nujol): νmax 1760 vs, 1708 w, 1620 vs, 1599 vs, 1570 s, 1474 vs, 1447 vs, 1409 m, 1395 w, 1351 w, 1329 w, 1318 m, 1307 m, 1262 vs, 1224 w, 1205 w, 1162 w, 1108 w, 1095 s, 1056 w, 1045 w, 1036 m, 1024 m, 1008 vs, 968 w, 936 m, 892 m, 842 w, 816 m, 794 w, 779 vs, 735 w, 726 w, 690 m, 629 m, 500 s, 485 s cm−1. MS (ESI+): m/z 332 ([M+H]+), 354 ([M+Na]+). Anal. Calcd for C18H13NO2Fe: C 65.28, H 3.96, N 4.23. Found: C 65.11, H 4.00, N 4.11.
M.p. 173–175 °C (chloroform). 1H NMR (CDCl3): δ 4.26 (s, 5H, C5H5), 4.44 (virtual t, J = 1.8 Hz, 2H, C5H4), 4.90 (virtual t, J = 1.9 Hz, 2H, C5H4), 7.07 (ddd, J = 1.2, 7.4, 7.9 Hz, 1H, C6H4), 7.47 (ddd, J = 0.4, 1.6, 7.9 Hz, 1H, C6H4), 7.52 (ddd, J = 0.4, 1.6, 7.4 Hz, 1H, C6H4), 7.56 (br s, 1H, NH), 8.74 (dd, J = 1.2, 8.4 Hz, 1H, C6H4); signals due to hydrazide protons were not found. 13C{1H} NMR (CDCl3): δ 68.61 (CH C5H4), 69.98 (CH C5H5), 71.09 (CH C5H4), 76.41 (Cipso C5H4), 117.70 (CCipso C6H4), 121.33 (CH C6H4), 122.36 (CH C6H4), 126.23 (CH C6H4), 133.28 (CH C6H4), 140.20 (NCipso C6H4), 169.61 (CONH), 170.04 (CONH). IR (Nujol): νmax 3393 m, 3319 s, 3206 w, 3111 w, 3075 w, amide I 1637 vs, 1615 m, 1594 vs, amide II 1522 vs, 1445 vs, 1339 m, 1315 s, 1299 s, 1268 m, 1240 w, 1220 w, 1163 w, 1148 m, 1106 w, 1078 w, 1045 w, 1033 w, 1006 w, 945 s, 830 s, 756 s, 736 w, 669 w, 558 w, 534 m, 500 s, 486 m cm−1. MS (ESI+): m/z 386 ([M+Na]+). Anal. Calc. For C18H17N3O2Fe: C 59.52, H 4.72, N 11.57. Found: C 59.23, H 4.73, N 11.32.
:1 (v/v) as the eluent. After the first yellow band was collected, the eluent was changed for dichloromethane/methanol 10:1 (v/v) affording a second yellow band, which was also collected. The collected fractions were evaporated and dried under vacuum to provide analytically pure 8 (first band; 18 mg, 14%) and 9 (second band; 100 mg, 75%) as yellow-orange solids.
X-ray crystallography
Single-crystals suitable for X-ray diffraction analysis were grown by liquid-phase diffusion of hexane into a solution in ethyl acetate (1: orange plate, 0.15 × 0.36 × 0.45 mm3) or, similarly from chloroform/hexane (2: red prism, 0.39 × 0.44 × 0.61 mm3; 7: red prism, 0.24 × 0.37 × 0.43 mm3), dichloromethane/hexane (9: red prism, 0.30 × 0.34 × 0.52 mm3), and from toluene/hexane (3: red prism, 0.45 × 0.46 × 0.58 mm3). Crystals of 4 (red prism, 0.13 × 0.18 × 0.49 mm3), 5 (red plate, 0.06 × 0.13 × 0.32 mm3) and 6 (red prism, 0.22 × 0.26 × 0.31 mm3) were obtained from a concentrated heptane solution upon slow cooling.The diffraction data (±h ±k ±l; θmax range 27.0–27.5°, data completeness ≥99.8%) were collected with an Apex II CCD diffractometer (Bruker) equipped with a Cryostream Cooler (Oxford Cryosystems) using graphite monochromated MoKα radiation (λ = 0.71073 Å). The data were corrected for absorption using the methods incorporated in the diffractometer software.
The structures were solved by the direct methods (SHELXS9731) and refined by full-matrix least squares based on F2 (SHELXL9731). All non-hydrogen atoms were refined with anisotropic displacement parameters. The NH and OH hydrogens were identified on the difference density maps and refined as riding atoms with Uiso assigned to 1.2 Ueq of their bonding atoms. Other hydrogen atoms were included in their calculated positions and refined similarly (riding model). Relevant crystallographic data and structure refinement parameters are available as (Table S2, ESI†).
The geometric data and structural drawings were obtained with a recent version of the PLATON program.32 The numerical values were rounded with respect to their estimated deviations (ESDs) given to one decimal place. Parameters relating to atoms located in constrained positions are given without ESDs.
DFT calculations
The geometry optimization, starting from the experimentally determined solid-state structure of 2, was performed using the density-functional theory (DFT) with Becke's three parameter functional33 with the non-local Lee–Yang–Parr correlation functional (B3LYP)34 and the standard 6-31G* basis set with analytically constructed energy gradient as implemented in the Gaussian 09 program package.35 For the stationary points at the potential energy surface (PES), the harmonic vibrational analysis was carried out by using the analytically calculated force-constant matrix to show that the points are a minima.Similar calculations were done also for the crystal structure using periodic boundary conditions (PBC). As the unit cell contains four molecules, a computationally less intensive method was used, namely a functional developed by Perdew, Burke and Ernzerhof (PBE)36 with standard 6-31G* basis set. The calculated geometric data (distances and angles of the non-hydrogen atoms) are available as (Table S1, ESI†).
Acknowledgements
This work is a part of the long-term research plan of the Faculty of Science, Charles University in Prague supported by the Ministry of Education, Youth and Sports of the Czech Republic (project no. MSM0021620857). An access to the National Grid Infrastructure MetaCentrum (project LM2010005) and CERIT-SC (project CZ.1.05/3.2.00/08.0144) are also gratefully acknowledged.Notes and references
- M.-G. A. Shvekhgeimer, Russ. Chem. Rev., 1996, 65, 41 CrossRef.
- (a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039 CrossRef CAS; (b) S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632 RSC; (c) G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125 CrossRef CAS; (d) E. O. Fischer and W. Pfab, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1952, 7, 377 Search PubMed.
- See, for instance: (a) Chem. Heterocycl. Compd, 1–64 DOI:10.1002/SERIES1079, Wiley (an on-line book series); (b) J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, Chichester, 5th edn, 2010 Search PubMed.
- For recent examples, see: (a) J. R. Wright, K. J. Shaffer, C. J. McAdam and J. D. Crowley, Polyhedron, 2012, 36, 73 CrossRef CAS; (b) E. I. Klimova, M. Flores-Alamo, S. C. Maya, M. E. Martínez, L. Ortiz-Frade and T. Klimova, Molecules, 2012, 17, 10079 CrossRef CAS; (c) P. Štépnička, H. Solařová and I. Císařová, J. Organomet. Chem., 2011, 696, 3727 CrossRef.
- (a) A. Hildebrandt, D. Schaarschmidt, L. van As, J. C. Swarts and H. Lang, Inorg. Chim. Acta, 2011, 374, 112 CrossRef CAS; (b) A. Hildebrandt, D. Schaarschmidt and H. Lang, Organometallics, 2011, 30, 556 CrossRef CAS; (c) A. Hildebrandt and H. Lang, Dalton Trans., 2011, 40, 11831 RSC; (d) J. M. Speck, R. Claus, A. Hildebrandt, T. Rüffer, E. Erasmus, L. van As, J. C. Swarts and H. Lang, Organometallics, 2012, 31, 6373 CrossRef CAS; (e) U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Hahn, S. Liebing, J. Kortus and H. Lang, Organometallics, 2012, 31, 6761 CrossRef CAS; (f) A. Hildebrandt, D. Schaarschmidt, R. Claus and H. Lang, Inorg. Chem., 2011, 50, 10623 CrossRef CAS; (g) P. V. Solntsev, S. V. Dudkin, J. R. Sabin and V. N. Nemykin, Organometallics, 2011, 30, 3037 CrossRef CAS; (h) D. Plazuk, B. Rychlik, A. Blauz and S. Domagala, J. Organomet. Chem., 2012, 715, 102 CrossRef CAS; (i) J. Tauchman, J. Trnka, I. Císařová and P. Štépnička, Collect. Czech. Chem. Commun., 2010, 75, 1023 CrossRef CAS.
- C. Ornelas, New J. Chem., 2011, 35, 1973 RSC.
- M. Zora and Ö. Velioglu, J. Organomet. Chem., 2008, 693, 2159 CrossRef CAS.
- A. Patti, S. Pedotti, T. Grassi, A. Idolo, M. Guido and A. De Donno, J. Organomet. Chem., 2012, 716, 216 CrossRef CAS.
- (a) J.-P. Monserrat, G. G. Chabot, L. Hamon, L. Quentin, D. Scherman, G. Jaouen and E. A. Hillard, Chem. Commun., 2010, 46, 5145 RSC; (b) K. N. Tiwari, J.-P. Monserrat, F. de Montigny, G. Jaouen, M.-N. Rager and E. A. Hillard, Organometallics, 2011, 30, 5424 CrossRef CAS. See also: (c) A. Ghose and J. N. Srivastava, Indian J. Heterocycl. Chem., 1992, 1, 241 ( Chem. Abstr. , 1992 , 117 , 192009 ) CAS.
- P. Zanello, M. Fontani, S. Z. Ahmed and C. Glidewell, Polyhedron, 1998, 17, 4155 CrossRef CAS.
- (a) T. P. Selvam and P. V. Kumar, Res. Pharm., 2011, 1, 1 Search PubMed; (b) R. Rajput and A. P. Mishra, Int. J. Pharm. Pharm. Sci., 2012, 4, 66 CAS.
- D. J. Connolly, D. Cusack, T. P. O’Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153 CrossRef CAS.
- B. Dash, E. K. Dora and C. S. Panda, Heterocycles, 1982, 19, 2093 CrossRef CAS.
- This procedure, which failed in our case, is well established in the literature. For selected recent examples, see: (a) M. A. El-Hashash and Y. A. El-Badr, Helv. Chim. Acta, 2011, 94, 389 CrossRef CAS; (b) K. S. Kumar, S. Ganguly, R. E. Veerasamy and E. De Clerq, Eur. J. Med. Chem., 2010, 45, 5474 CrossRef CAS; (c) M. T. H. Khan, R. Khan, Y. Wuxiuer, M. Arfan, M. Ahmed and I. Sylte, Bioorg. Med. Chem., 2010, 18, 4317 CrossRef CAS.
- This reaction was attempted in analogy to the known cyclisation of N-acylanthranilic acid: T. Isobe and T. Ishikawa, J. Org. Chem., 1999, 64, 6989 CrossRef CAS.
- The formation of 1,3-dimethylimidazolidin-2-ylidene derivatives (N-acylguanidines) from 2-chloro-1,3-dimethylimidazolidinium chloride and primary amides has been reported: T. Isobe and T. Ishikawa, J. Org. Chem., 1999, 64, 6984 CrossRef CAS.
- The crystal structure of 1·½H2O has been determined before but was not discussed in the literature: R. Janner, R. Gompper and K. Polborn, Private communication to CCDC (2003).
- N. Malek-Saied, R. El Aissi, S. Ladeira and E. Benoist, Appl. Organomet. Chem., 2011, 25, 680 CAS.
- M. Oberhoff, L. Duda, J. Karl, R. Mohr, G. Erker, R. Fröhlich and M. Grehl, Organometallics, 1996, 15, 4005 CrossRef CAS.
- L. A. Errede, M. C. Etter, R. C. Williams and S. M. Darnauer, J. Chem. Soc., Perkin Trans. 2, 1981, 233 RSC.
- Cambridge Structural Database, version 5.33 of November 2011 with updates of November, 2011, and February, May and August 2012 Search PubMed.
- The majority of compounds were claimed in patents. Original literature reports are: (a) S. Hünig and F. Müller, Justus Liebigs Ann. Chem., 1962, 651, 89 CrossRef; (b) F. L. Scott, J. K. O’Halloran, J. O’Driscoll and A. F. Hegarty, J. Chem. Soc., Perkin Trans. 1, 1972, 2224 RSC.
- F. Saczewski, A. Kornicka, A. Rybczynska, A. L. Hudson, S. S. Miao, M. Gdaniec, K. Boblewski and A. Lehmann, J. Med. Chem., 2008, 51, 3599 CrossRef CAS.
- S. S. S. Raj, B. M. Yamin, A. M. A. Boshaala, M. T. H. Tarafder, K. A. Crouse and H.-K. Fun, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 1011 Search PubMed.
- I. A. Litvinov, O. N. Kataeva, L. V. Ermolaeva, G. A. Vagina, T. V. Troepolskaya and V. A. Naumov, Izv. Akad. Nauk SSSR, Ser. Khim., 1991, 75 CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- Y. S. Sohn, D. N. Hendrickson and H. B. Gray, J. Am. Chem. Soc., 1971, 93, 3603 CrossRef.
- J.-P. Doucet and J. Weber, Computer-Aided Molecular Design: Theory and Applications, Academic Press, London, 1996 Search PubMed.
- T. Kojima, D. Noguchi, T. Nakayama, Y. Inagaki, Y. Shiota, K. Yoshizawa, K. Ohkubo and S. Fukuzumi, Inorg. Chem., 2008, 47, 886 CrossRef CAS.
- P. Štépnička, I. Císařová, D. Nižňanský and S. Bakardjieva, Polyhedron, 2010, 29, 134 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc, Wallingford CT, 2010 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammogram of 3 (Fig. S1), a comparison of calculated and experimentally determined bond lengths and angles for 2 (Table S1), comparison of energy profiles calculated for 2 at two different levels of theory (Fig. S2), and summary of the crystallographic data (Table S2). CCDC 922133–922140. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3nj00182b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |