Outer sphere hydrogenation catalysis†
Odile Eisenstein*a and Robert H. Crabtree*b
aInstitut Charles Gerhardt UMR 5253 CNRS, Université Montpellier 2 cc 1501, Place E. Bataillon, 34095 Montpellier, France. E-mail: Odile.Eisenstein@univ-montp2.fr
bDepartment of Chemistry, Yale University, 225 Prospect St., New Haven, CT 06520-8107, USA. E-mail: robert.crabtree@yale.edu
First published on 20th September 2012
Abstract
In the title catalysts, the substrate, typically a ketone, imine or N-heterocycle, remains in the outer sphere (OS). The catalyst transfers hydride and a proton to the unbound substrate either by a concerted or by a stepwise path. These include catalysts not always considered together, such as Bullock's ionic hydrogenation catalysts, bifunctional catalysts in the tradition of Shvo and Noyori and Stephan's frustrated Lewis pair catalysts. By omitting the oxidative addition, insertion and reductive elimination pathways of conventional inner sphere (IS) catalysts, these OS pathways are in principle equally open to inexpensive metals and even nonmetal catalysts. These OS pathways lead to useful selectivity properties, particularly Noyori's asymmetric catalysis, but much more remains to be done in this rapidly developing field.
 Odile Eisenstein | Odile Eisenstein is ‘Directeur de Recherche’ in the CNRS. First at the Paris-Sud University, Orsay till 1996 and then at Montpellier University, her goal is to show the power of computational chemistry to analyse experimental questions mostly associated with organometallic reactivity. She won the CNRS Silver medal, the Langevin award from the French Academy of Science, the SFC Le Bel award, the RSC Frankland award, the ACS Organometallic Award and she is a member of IAQMS. She is DSc(Hons) of Indiana University and Université Laval. |
 Robert H. Crabtree | At Yale since 1977, Bob Crabtree is now Whitehead Professor. He has been ACS and RSC organometallic chemistry awardee, Baylor Medallist, Mond lecturer, Kosolapoff awardee, Stauffer Lecturer, has chaired the ACS Inorganic Division and is the author of an organometallic textbook. Early work on catalytic alkane C–H activation and functionalization was followed by work on H2 complexes, dihydrogen bonding, and catalysis for green and energy chemistry. He is an ACS Fellow and a Fellow of the American Academy of Arts& & Sciences. |
Introduction
The majority of homogeneous hydrogenation catalysts operate by an inner sphere (IS) mechanism.1 In this case, an unsaturated organic substrate binds directly to the catalyst, typically by π-bonding between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X double bond and the metal (X = C, N, O). Subsequent insertion, elimination and H2 oxidative addition steps complete the cycle (eqn (1)). In outer sphere (OS) catalysts, in contrast, the substrate does not bind to the metal but H+ and H− are instead transferred, either together or stepwise, from the catalyst to the unbound substrate. Eqn (2) shows a common case of OS catalysis where an adjacent NH acts as the proton donor. The implications of OS catalysis, currently of rising importance, are discussed in this review.
X double bond and the metal (X = C, N, O). Subsequent insertion, elimination and H2 oxidative addition steps complete the cycle (eqn (1)). In outer sphere (OS) catalysts, in contrast, the substrate does not bind to the metal but H+ and H− are instead transferred, either together or stepwise, from the catalyst to the unbound substrate. Eqn (2) shows a common case of OS catalysis where an adjacent NH acts as the proton donor. The implications of OS catalysis, currently of rising importance, are discussed in this review. | (1) |
 | (2) |
 | ||
| Fig. 1 General reaction pattern. | ||
While all three classes are of mechanistic interest, Noyori catalysts have also been of great practical use for the catalytic asymmetric reduction of ketones,4 an important problem in organic synthesis. By the early 2000s, this had focused attention on the Ru catalysts rather than the ionic hydrogenation class. With the perspective afforded by the passage of time, we can now better appreciate the mechanistic analogies across the whole series. In each case, we have a frustrated proton–hydride pair, consisting of a proton donor A–H and a weak hydride donor H–B pair, where A is an electronegative element such as N or O and B is an electropositive element such as a d-block metal or boron. These are frustrated in the sense that they do not exclusively react to form H2, as CaH2 + H2O would do, for example. They tend instead to form a dihydrogen bond A–H⋯H–B, an attractive proton–hydride interaction with a bond strength comparable with that of traditional hydrogen bonds.10
Ionic hydrogenation
Ionic hydrogenation, initially stoichiometric,11,12 but later2 catalytic, requires a combination of a metal hydride and acid, such as CpW(CO)3H + HOTf, leading to initial formation of a protonated form that seems to be a dihydride12b but a dihydrogen complex [CpW(CO)3(H2)] OTf may be in equilibrium with it. Such complexes are excellent proton donors13,14 and, following proton transfer to the CX substrate, the carbocation center (+C–XH) of the resulting protonated substrate abstracts hydride from CpW(CO)3H to complete the hydrogenation. If it involves a single molecule of the complex, the process necessarily involves stepwise rather than concerted (H+ + H−) transfer. The protonated complex, whether [LnM(H2)]+ or [LnM(H)2]+ can only act as a proton donor and the hydride donor, neutral [LnMH], is only formed after proton transfer. Kinetic studies for the CpW(CO)3H case by Bullock, Norton and their coworkers, suggest a pre-equilibrium protonation of the complex is followed by proton transfer to substrate, then rate-determining hydride transfer.12cIrreversible adduct formation between the metal and the conjugate base of the acid is avoided by having an acid such as TfOH with a noncoordinating or reversibly coordinating anion. Magee and Norton15 introduced an enantioselective reduction of an iminium cation with CpRu(chiraphos)H as hydride donor and H2 as the reductant to regenerate the active hydride, and Bullock16 has recently extended his work to the acid-catalyzed dehydration of vicinal diols to give aldehydes that are subsequently hydrogenated to the monohydric alcohols. In the main group, combinations of R3SiH + acid give stoichiometric ionic hydrogenation.17
A recent ionic hydrogenation18 involves reduction of 2-methyl quinoline and acridine by catalyst 2 formed from 1 and H[B{3,5-(CF3)2C6H3}4]. In each case the heterocyclic ring but not the carbocyclic ring(s) are reduced. DFT calculations suggest that the intermediacy of dihydrogen complex 2 with stepwise proton transfer to the N lone pair of the heterocycle, followed by hydride transfer to the 2-position in the quinoline but to the 4-position for acridine (eqn (3)). The catalysts are very active, operating at near-ambient conditions with high TOF. Contrary to the usual case of having faster catalysis with lower steric hindrance, unsubstituted pyridine requires harsher conditions, for reasons discussed below.

 | (3) |
Shvo and Noyori catalysts
Moving to the second class,19–21 the Shvo3 and Noyori4 catalysts incorporate both AH and BH sites within the same complex. The proton donor site is typically a ligand OH or NH unit adjacent to the metal, and the hydride donor is a metal hydride, typically Ru–H. The two sites being adjacent and coupled electronically facilitates concerted (H+ + H−) transfer (Fig. 2). | ||
| Fig. 2 Noyori catalyst mechanism. | ||
The Shvo catalyst (3 in eqn (4)) has attracted detailed mechanistic study, notably by the groups of Bäckvall and of Casey.22–24 The key question was whether the reaction really is OS. Casey and coworkers saw significant H/D isotope effects for both RuH and OH sites, consistent with concerted (H+ + H−) transfer; DFT calculations also supported this conclusion with the OS mechanism being favored by at least 10 kcal mol−1.25 A mechanism involving η5- to η3-slip of the cyclopentadienyl and inner sphere coordination of the substrate CX bond was hard to exclude, however. An elegant trapping experiment by Casey's group probed the problem.26 On the IS hypothesis, the CN bond of the substrate imine in eqn (4) is reduced to amine while the imine nitrogen remains bound. In that case the coordinatively inert Ru should retain that same nitrogen to give 4 as the final product. If OS reduction occurs, then nothing should prevent binding at the alternative –NH2 binding site of the substrate from giving product 5 as well. In an experiment run below −20 °C in toluene, a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 kinetic mixture of 4 and 5 was found, consistent with the OS path. On warming to 25°, where kinetic lability has already set in, a thermodynamic 94:6 ratio was found (eqn (4)). Bäckvall22 argued that Ru migration across the arene ring could reconcile the results with the IS hypothesis and so some doubt still remained. Casey24 responded with a study of a saturated substrate with results again strongly supporting the OS mechanism. This important point now seems to be definitely established for imines and, until counter-examples are found, by inference for all the substrates and catalysts in this class.
:50 kinetic mixture of 4 and 5 was found, consistent with the OS path. On warming to 25°, where kinetic lability has already set in, a thermodynamic 94:6 ratio was found (eqn (4)). Bäckvall22 argued that Ru migration across the arene ring could reconcile the results with the IS hypothesis and so some doubt still remained. Casey24 responded with a study of a saturated substrate with results again strongly supporting the OS mechanism. This important point now seems to be definitely established for imines and, until counter-examples are found, by inference for all the substrates and catalysts in this class.
 | (4) |
O. A judicious ligand choice can give an extremely high level of asymmetric induction. Fig. 3 shows the hydrogenated form of typical catalysts. With the key NH sites methylated, however, all activity is lost.28a In one case,28b asymmetric OS and IS mechanisms were followed depending on the presence of acid or base. | ||
| Fig. 3 Some Noyori catalyst types. | ||
The hydrogenation step is reversible for many of the OS catalysts, so that transfer hydrogenation is an alternative means of recycling the catalyst, avoiding H2. Isopropanol is a classic (H+ + H−) donor because of its high driving force for production of acetone, a byproduct that is easily removed. Transfer hydrogenation has been successfully employed in reduction of ketones, although the reversibility of the process with i-PrOH/Me2CO militates against obtaining high enantiomeric excesses, since the undesired 50:50 thermodynamic enantiomer ratio is necessarily slowly approached under reversible conditions. Formic acid, as a 1:1 mixture with NEt3, is a better (H+ + H−) donor since the resulting loss of CO2 makes the process irreversible. For example, benzils, ArCOCOAr, were successfully reduced in this way to the dl diols, ArCH(OH)CH(OH)Ar, with up to 99% ee.20
Many other reactions have been carried out asymmetrically with Noyori catalysts.4 Chloromethyl ketones can be reduced to the asymmetric alcohols; base treatment then gives the chiral epoxides with retention of configuration at the carbinol carbon. If H2 is replaced by a nucleophile such as dimethyl malonate, asymmetric addition of the Nu–H takes place across the CC bond of a suitable Michael acceptor such as 2-cyclopentenone or a nitroalkene; ees in the 90–98% range were achieved. To show practical utility a gram scale synthesis was carried out with 95% ee and 94% yield involving dimethyl malonate and an aromatic nitroalkene to give an intermediate in the synthesis of the anti-inflammatory neuroprotective drug, Rolipram. The structure of the η6-arene in the arene ruthenium catalysts had a big effect on the ee obtained, C6Me6 being preferred. The malonate is believed to bind as Ru–CH(COOMe)2, with the accompanying proton binding at the adjacent N; an OS mechanism is envisaged for the subsequent reaction with the Michael acceptor.
Once the hydride transfers to the substrate from the metal in a Noyori or Shvo catalyst, the remaining metal center is left as an unsaturated 16e fragment. Any factors stabilizing this fragment help the hydride depart. This role is played by the deprotonated amine adjacent to M, which acts as a π-donor and partially compensates for the unsaturation (eqn (5)). Stabilization of coordinative unsaturation by an adjacent heteroatom lone pair along with relief of steric pressure is well documented.29
Hydrogen activation can convert the unsaturated form of the catalyst back to the 18e starting complexes (Fig. 3). This can happen by coordination of H2 to the unsaturated metal to give an H2 complex. These are good proton donors, and so the adjacent base can deprotonate the H2 to restore the original structure for the next cycle of catalysis (eqn (5)). The enhanced acidity of bound H2 arises from the contrast between the relatively weak binding in M–H2versus the very much stronger binding of the hydride left behind after proton transfer. This provides a high driving force for H+ transfer even to a weak base such as the ketones typical in ionic hydrogenation.
 | (5) |
Frustrated Lewis pair catalysts
In the third class, an FLP pair such as a borane of type BR3 and a Lewis base of type AR3′ (A = P or N) reacts with hydrogen to give a transient ion pair, [R3B–H][H–AR3′], that transfers (H+ + H−) to the substrate. The acid and base sites can either combine in the same molecule or remain separate.8,9 Spectroscopic and computational work shows the importance of inter and intramolecular phosphine borane interactions in FLPs.34 Concerted addition was shown to be possible by DFT calculations35 and heterolytic cleavage of an N-heterocyclic carbene (NHC) borane adduct to an FLP was also shown to have a low energy barrier.36 A frontier orbital analysis showed the importance of polarization of the H2 by the FLP in facilitating the H–H cleavage.37 This was emphasized by showing that H2 could be cleaved in a sufficiently strong electric field even without new bonds being formed to H+ and H−.38For catalysis, the acidic and basic sites of the FLP pair must avoid deactivation by irreversible adduct formation as would occur for NH3 and BH3 combining to give the catalytically inert H3N–BH3. Unless any such adduct formation is reversible, the Lewis acid–base pair will lose the ability to activate H2 and thus fail to act as a catalyst. Steric bulk is typically involved in preventing this deactivation, or at least promoting its reversibility.
In a somewhat related case, Miller, Labinger and Bercaw have proposed an FLP activation of H2 by a ligand-centered boron Lewis acid site in the stoichiometric hydrogenation of CO by a Re complex.39
Inexpensive metal catalysts
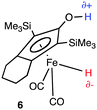
With increasing prices for precious metals, an urgent goal is their replacement by the inexpensive metals, Mo, W and the 1st row transition metals. Such catalysts are also less likely to leave toxic residues.2,40 The precious metals have provided extremely useful catalysts, many of which operate by IS mechanisms via oxidative addition and reductive elimination (OA/RE). As 2e processes, OA and RE are appropriate for precious metals because they have stable oxidation states two units apart (e.g., Ir(I), (III) and (V)). In contrast, first row transition metals tend to have stable oxidation states only one unit apart (M(I), (II) and (III) for Cu and Ni).OS hydrogenation requires no change of oxidation state during the cycle. In principle, this should open the field to first row transition metals and even to main group elements. Indeed the known main group FLP catalysts confirms the potential in this area; in addition, Shvo-type catalysts include one (6) based on a first row transition metal, iron.41,42 Morris et al.43 also have a series of Fe catalysts that adopt an OS mechanism. For the moment the efficiency of the first row and main group catalysts is limited, but the relative youth of the field gives hope for future progress. First row metals with their lower M–H bond strengths and greater tolerance for coordinative unsaturation may even be better suited than heavier metals to act as hydride donors. Bullock2 has emphasized the suitability of inexpensive metals for ionic hydrogenation.
One barrier to progress is the greater stress laid on high efficiency catalysts that have the potential for immediate application versus the progressive development of inexpensive catalysts of much lower initial efficiency. If turnover activity per unit cost were adopted as a ‘green’ metric in comparisons, however, inexpensive catalysts would look far more attractive.
Nature of the catalyst
The pKa data for the proton donor, AH, can aid in catalyst design and ligand tuning. The hydride donor function is quantified in terms of hydricity, both thermodynamic and kinetic.44,45 From equilibrium electrochemical and calorimetric data, Rakowski–Dubois, DuBois, and co-workers report a thermodynamic hydricity range of well over 30 kcal mol−1.46 As a broad guide, a hydride is expected to become more hydridic when located trans to a high trans effect ligand, a situation that also facilitates H− transfer since a vacancy is more easily accessed in such a site.Since the hydrogenated and dehydrogenated catalyst are both present together, formation of an adduct between them is in principle possible, as in the Shvo catalyst (eqn (6)). This dimerization is clearly reversible with H2 for catalysis to occur. Additional steric bulk may promote dissociation and no such dimerization is seen for the Noyori catalysts.
 | (6) |
The H+ and H− donors
A variety of proton donors is seen for OS hydrogenation catalysts. In ionic hydrogenation, these are dihydrogen complexes formed from strong acids. In Shvo and Noyori systems, the proton donors are ligand –OH or –NH groups and for FLP catalysts, the conjugate acids of the bulky base. The substrate is likely to pre-associate with these groups via CX⋯H–A hydrogen bonds (XO, N; AH = proton donor). The great majority of hydride donors are hydrides either of a transition metal or of a main group element, typically boron. For pre-association, a nonpolar solvent may favor the formation of hydrogen bonds and indeed toluene seems to be the most common solvent choice.Substrates
Organic carbonyls are the most common substrates. CO bonds are highly polar and very suitable to act as (H+ + H−) acceptors. More importantly, they and their reduction products have only a weak tendency to bind to the metal site. If binding were strong, H2 activation would be difficult, since H2 is a rather poor ligand.OS hydrogenation is therefore less often seen for imines and for N-heterocycles. The N lone pairs of these failed substrates do bind strongly to the metal and can suppress the catalysis. H2 is sterically small, so by introducing sufficient bulk in the system, we can block the substrate, but not H2, from the metal and permit catalysis. In a recent case,18 for example, quinoline blocked the metal site but the much more bulky 2-methyl quinoline did not do so and was successfully reduced by an OS route. Initial protonation of the N lone pair polarizes the CN bond, decreases the aromaticity of the heterocyclic ring and facilitates H− transfer to carbon (eqn (7)). On protonation, the substrate's affinity for H− greatly increases.
 | (7) |
An advantage of OS hydrogenation is its ability to reduce very hindered substrates. Another advantage is its selectivity for polar CX bonds over CC bonds.42 Noyori catalysts with adjacent AH and BH sites would be expected to favor 1,2-addition to double bonds. This would no longer apply for ionic and FLP catalysts where stepwise (H+ + H−) transfer would permit 1,4-addition as has been seen for acridine reduction.18
CO2 is not at all bulky but relatively hard to reduce. While the molecule as a whole is nonpolar, it contains polar CO bonds that lend themselves to OS reduction by H− transfer to the CO2 carbon.50 The mechanism then typically converts to inner sphere in the sense that the formate ion produced in the H− transfer is thought to subsequently bind to the metal.
An important advance by Nozaki et al.51 was the introduction of complex 7 as an extremely active CO2 reduction catalyst to give HCOOH with TOFs up to 1.5 × 105 h−1 and operating by an OS H− transfer. The structures of 1 and 7 are obviously related, but in the case of 7 the H− transfer occurs first, rather than protonation being needed as for 1.
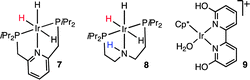
Hazari et al. introduced a similar catalyst, 8, for which DFT calculations suggested that the ligand NH proton is involved in polarizing the CO2 by an OS interaction.52 Fujita, Himeda et al. have recently described a water soluble catalyst, 9, in which H2 activation is believed to be accelerated by the pendant base effect of a deprotonated OH.53 Although these catalysts are not fully OS, since formate binds to the metal, yet they seem to be OS in the key H− transfer.50a
For esters and nitriles, Grey, Pez et al. saw hydrogenation with the anionic hydride salts, K[RuH2L2L′] (L = PPh3; L′ = cyclometalated L) and plausibly proposed H− transfer to the substrate with simultaneous polarization of the CX bond by the potassium counterion within an ion pair.54
Intermediate IS/OS cases
In strict IS hydrogenation, all the chemistry occurs at the metal and the CX substrate binds in the η2-form (eqn (1)). In the OS case, (H+ + H−) transfer occurs between the catalyst and the unbound substrate (eqn (2)). An intermediate situation applies in the case of the Meerwein–Ponndorf–Verley (MPV) mechanism of hydrogen transfer catalysis by catalysts such as Al(III). In this case a substrate CO binds in the η1-form via O and a H− transfer occurs from the CH bond of the alkoxide co-reactant, typically OiPr, remote from the metal in the intermediate sphere (eqn (8)). The MPV pathway has been proposed for several transition metal catalysts but is hard to distinguish experimentally from IS and OS routes, so calculations have taken a central place. For example, hydrogen-transfer from alcohols to ketones with Ir(cod) complexes of aminoalcohols as catalysts was proposed to go by the MPV mechanism on the basis of DFT calculations by Meijer and colleagues.49 In a highly effective asymmetric peptide catalyst series based on (arene)Ru complexes, an MPV-like mechanism involved the alkali metal cation deriving from the base needed for activity.55 An MPV pathway may easily escape detection because it seems to be considered much less often by workers in the field. Computational work is powerless if a potentially applicable mechanism is not considered. | (8) |
OS H atom transfer
Outer sphere hydrogenation goes back to the earliest days of homogeneous transition metal catalysis. In 1942, Iguchi looked at the reaction of cobalt cyanide solutions under an H2 atmosphere originally intended as an inert blanket.58 A fast reaction occurred that eventually led to the discovery that the solution catalyzed the reduction of a wide range of compounds, both organic and inorganic. [CoIII(CN)5H]3− was later identified as the main species in solution under H2,59 with an OS transfer of a hydrogen atom to the substrate as the mechanism of hydrogenation.60 Styrene, for example, is a good substrate because it can form a stabilized benzylic radical after H transfer. A second H atom transfer from another molecule of cobalt hydride then completes the reduction to ethylbenzene.Conclusion
Outer sphere hydrogenation provides important advantages not all of which have yet been exploited. In particular, a much wider variety of catalysts involving inexpensive metals should be viable. The analogy between FLP, Shvo–Noyori and ionic hydrogenation catalysts is likely to make for cross-fertilization between these fields, hitherto somewhat isolated from one another. The area has so far shown the most dramatic applications of these catalysts in the current rich development of the Noyori asymmetric hydrogenation, but much remains to be done in this rapidly developing field.Acknowledgements
This focus review developed from work supported as part of the Center for Electrocatalysis, Transport Phenomena, and Materials (CETM) for Innovative Energy Storage, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award no. DE-SC00001055 (energy storage applications) and DE-FG02-84ER13297 (synthesis). O.E. thanks the CNRS and the MESR for funding.Notes and references
- S. Bhaduri and D. Mukesh, Homogeneous Catalysis, J. Wiley, NY, 2000 Search PubMed.
- R. M. Bullock, Chem.–Eur. J., 2004, 10, 2366 CrossRef CAS.
- Y. Blum, D. Czarkie, Y. Rahamim and Y. Shvo, Organometallics, 1985, 4, 1459 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS; D. W. Stephan, Org. Biomol. Chem., 2012, 10, 5740 Search PubMed.
- A. L. Kenward and W. E. Piers, Angew. Chem., Int. Ed., 2008, 47, 38 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS.
- D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338 CrossRef CAS.
- G. Erker, Dalton Trans., 2011, 40, 7475 RSC.
- R. H. Crabtree, P. E. M. Siegbahn, O. Eisenstein and A. L. Rheingold, Acc. Chem. Res., 1996, 29, 348 CrossRef CAS.
- R. M. Bullock and B. J. Rappoli, Chem. Commun., 1989, 1447 RSC.
- (a) R. M. Bullock and M. H. Voges, J. Am. Chem. Soc., 2000, 122, 12594 CrossRef CAS; (b) R. M. Bullock, J.-S. Song and D. J. Szalda, Organometallics, 1996, 15, 2504 CrossRef CAS; (c) J. S. Song, D. J. Szalda, R. M. Bullock, C. J. C. Lawrie, M. A. Rodkin and J. R. Norton, Angew. Chem., Int. Ed. Engl., 1992, 31, 1233 CrossRef.
- P. G. Jessop and R. H. Morris, Coord. Chem. Rev., 1992, 121, 155 CrossRef CAS.
- R. H. Crabtree, Acc. Chem. Res., 1990, 23, 95 CrossRef CAS.
- M. P. Magee and J. R. Norton, J. Am. Chem. Soc., 2001, 123, 1778 CrossRef CAS.
- M. Schlaf, P. Ghosh, P. J. Fagan, E. Hauptman and R. M. Bullock, Adv. Synth. Catal., 2009, 351, 789 CrossRef CAS.
- D. N. Kursanov, Z. N. Parnes and N. M. Loim, Synthesis, 1974, 633 CrossRef CAS.
- G. Dobereiner, A. Nova, N. Schley, N. Hazari, S. Miller, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2011, 133, 7547 CrossRef CAS.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201 CrossRef CAS.
- T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393 CAS.
- J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC.
- J. S. M. Samec, A. H. Éll, J. B. Åberg, T. Privalov, L. Eriksson and J.-E. Bäckvall, J. Am. Chem. Soc., 2006, 128, 14293 CrossRef CAS.
- C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123, 1090 CrossRef CAS.
- C. P. Casey, T. B. Clark and I. A. Guzei, J. Am. Chem. Soc., 2007, 129, 11821 CrossRef CAS.
- (a) C. P. Casey, G. A. Bikzhanova, Q. Cui and I. A. Guzei, J. Am. Chem. Soc., 2005, 127, 14062 CrossRef CAS; (b) A. Comas-Vives, G. Ujaque and A. Lledós, Organometallics, 2007, 26, 4135 CrossRef CAS; (c) A. Comas-Vives, G. Ujaque and A. Lledós, Organometallics, 2008, 27, 4854 CrossRef CAS; (d) A. Comas-Vives, G. Ujaque and A. Lledós, THEOCHEM, 2009, 903, 123 CrossRef CAS; (e) A. Comas-Vives, G. Ujaque and A. Lledós, Adv. Inorg. Chem., 2010, 62, 231 CrossRef CAS.
- C. P. Casey, G. A. Bikzhanova and I. A. Guzei, J. Am. Chem. Soc., 2006, 128, 2286 CrossRef CAS.
- M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466 CrossRef CAS.
- (a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (b) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724 CrossRef CAS.
- K. G. Caulton, New J. Chem., 1994, 18, 25 Search PubMed; T. J. Johnson, K. Folting, W. E. Streib, J. D. Martin, J. C. Huffman, S. A. Jackson, O. Eisenstein and K. G. Caulton, Inorg. Chem., 1995, 34, 488 CrossRef CAS.
- W. N. O, A. J. Lough and R. H. Morris, Organometallics, 2012, 31, 2137 CrossRef CAS; W. N. O, A. J. Lough and R. H. Morris, Organometallics, 2012, 31, 2152 CrossRef.
- T. Li, I. Bergner, F. N. Haque, M. Zimmer-De Iuliis, D. Song and R. H. Morris, Organometallics, 2007, 26, 5940 CrossRef CAS.
- W. Baratta and P. Rigo, Eur. J. Inorg. Chem., 2008, 4041 CrossRef CAS.
- M. Martín, E. Sola, S. Tejero, J. L. Andrés and L. A. Oro, Chem.–Eur. J., 2006, 12, 4043 CrossRef.
- T. Wiegand, H. Eckert, O. Ekkert, R. Fröhlich, G. Kehr, G. Erker and S. Grimme, J. Am. Chem. Soc., 2012, 134, 4236 CrossRef CAS.
- (a) Y. Guo and S. H. Li, Inorg. Chem., 2008, 47, 6212 CrossRef CAS; (b) D. M. Camaioni, B. Ginovska-Pangovska, G. K. Schenter, S. Kathmann and T. Autrey, J. Phys. Chem. A, 2012, 116, 7228 CrossRef CAS.
- D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef CAS.
- A. Hamza, A. Stirling, T. A. Rokob and I. Pápai, Int. J. Quantum Chem., 2009, 109, 2416 CrossRef CAS.
- S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402 CrossRef CAS.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2010, 132, 3301 CrossRef CAS.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS.
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816 CrossRef CAS.
- R. M. Bullock, Angew. Chem., Int. Ed., 2007, 46, 7360 CrossRef CAS.
- (a) C. Sui-Seng, F. N. Haque, A. Hadzovic, A.-M. Pütz, V. Reuss, N. Meyer, A. J. Lough, M. Zimmer-De Iuliis and R. H. Morris, Inorg. Chem., 2009, 48, 735 CrossRef CAS; (b) A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266 CrossRef CAS.
- T.-Y. Cheng, B. S. Brunschwig and R. M. Bullock, J. Am. Chem. Soc., 1998, 120, 13121 CrossRef CAS.
- T.-Y. Cheng and R. M. Bullock, Organometallics, 1995, 14, 4031 CrossRef CAS.
- (a) D. E. Berning, B. C. Noll and D. L. DuBois, J. Am. Chem. Soc., 1999, 121, 11432 CrossRef CAS; (b) C. J. Curtis, A. Miedaner, W. W. Ellis and D. L. DuBois, J. Am. Chem. Soc., 2002, 124, 1918 CrossRef CAS; (c) W. W. Ellis, J. W. Raebiger, C. J. Curtis, J. W. Bruno and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 2738 CrossRef CAS; (d) W. W. Ellis, R. Ciancanelli, S. M. Miller, J. W. Raebiger, M. Rakowksi DuBois and D. L. DuBois, J. Am. Chem. Soc., 2003, 125, 12230 CrossRef CAS and refs cited.
- A. Nova, D. Balcells, N. D. Schley, G. E. Dobereiner, R. H. Crabtree and O. Eisenstein, Organometallics, 2010, 29, 6548 CrossRef CAS.
- S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884 RSC.
- J.-W. Handgraaf, J. N. H. Reek and E.-J. Meijer, Organometallics, 2003, 22, 3150 CrossRef CAS.
- (a) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef CAS; (b) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254 CrossRef CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168 CrossRef CAS.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274 CrossRef CAS.
- W.-H. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923 CAS.
- R. A. Grey, G. P. Pez and A. Wallo, J. Am. Chem. Soc., 1981, 103, 7536 CrossRef CAS.
- J. Wettergren, E. Buitrago, P. Ryberg and H. Adolfsson, Chem.–Eur. J., 2009, 15, 5709 CrossRef CAS.
- R. Radhakrishan, D. M. Do, S. Jaenicke, Y. Sasson and G.-K. Chuah, ACS Catal., 2011, 1, 1631 CrossRef CAS.
- G. K. Chuah, S. Jaenicke, Y. Z. Zhu and S. H. Liu, Curr. Org. Chem., 2006, 10, 1639 CrossRef CAS.
- M. Iguchi, J. Chem. Soc. Jpn., 1942, 63, 634 CrossRef CAS.
- R. G. S. Banks and J. M. Pratt, J. Chem. Soc. A, 1968, 854 RSC.
- J. Kwiatek, Catal. Rev., 1968, 1, 37 Search PubMed.
Footnote |
| † This article is included in the All Aboard 2013 themed issue. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |