 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral thiouronium salts: synthesis, characterisation and application in NMR enantio-discrimination of chiral oxoanions†‡
Magdalena B.
Foreiter
,
H. Q. Nimal
Gunaratne
*,
Peter
Nockemann
,
Kenneth R.
Seddon
,
Paul J.
Stevenson
and
David F.
Wassell
QUILL Research Centre, School of Chemistry and Chemical Engineering, The Queen's University of Belfast, Belfast BT9 5AG, Northern Ireland, UK. E-mail: quill@qub.ac.uk
First published on 15th November 2012
Abstract
Chiral thioureas and functionalised chiral thiouronium salts were synthesised starting from the relatively cheap and easily available chiral amines: (S)-methylbenzylamine and rosin-derived (+)-dehydroabietylamine. The introduction of a delocalised positive charge to the thiourea functionality, by an alkylation reaction at the sulfur atom, enables dynamic rotameric processes: hindered rotations about the delocalised CN and CS bonds. Hence, four different rotamers/isomers may be recognised: syn–syn, syn–anti, anti–syn and anti–anti. Extensive 1H and 13C NMR studies have shown that in hydrogen-bond acceptor solvents, such as perdeuteriated dimethyl sulfoxide, the syn–syn conformation is preferable. On the other hand, when using non-polar solvents, such as CDCl3, the mixture of syn–syn and syn–anti isomers is detectable, with an excess of the latter. Apart from this, in the case of S-butyl-N,N′-bis(dehydroabietyl)thiouronium ethanoate in CDCl3, the 1H NMR spectrum revealed that strong bifurcated hydrogen bonding between the anion and the cation causes global rigidity without signs of hindered rotamerism observable on the NMR time scale. This suggested that these new salts might be used as NMR discriminating agents for chiral oxoanions, and are indeed more effective than their archetypal guanidinium analogues or the neutral thioureas. The best results in recognition of a model substrate, mandelate, were obtained with S-butyl-N,N′-bis(dehydroabietyl)thiouronium bistriflamide. It was confirmed that the chiral recognition occurred not only for carboxylates but also for sulfonates and phosphonates. Further 1H NMR studies confirmed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 recognition mode between the chiral agent (host) and the substrate (guest); binding constants were determined by 1H NMR titrations in solutions of DMSO-d6 in CDCl3. It was also found that the anion of the thiouronium salt had a significant influence on the recognition process: anions with poor hydrogen-bond acceptor abilities led to the best discrimination. The presence of host–guest hydrogen bonding was confirmed in the X-ray crystal structure of S-butyl-N,N′-bis(dehydroabietyl)thiouronium bromide and by computational studies (density functional theory).
:1 recognition mode between the chiral agent (host) and the substrate (guest); binding constants were determined by 1H NMR titrations in solutions of DMSO-d6 in CDCl3. It was also found that the anion of the thiouronium salt had a significant influence on the recognition process: anions with poor hydrogen-bond acceptor abilities led to the best discrimination. The presence of host–guest hydrogen bonding was confirmed in the X-ray crystal structure of S-butyl-N,N′-bis(dehydroabietyl)thiouronium bromide and by computational studies (density functional theory).
Introduction
The chemistry of abiotic receptor compounds for a particular species usually mimics natural systems. In the case of natural anion receptors, the archetypal example of an anion binding host is the guanidinium ion, which naturally occurs as a part of arginine residues.1 Since then, many synthetic guanidinium-based anion hosts have been designed and synthesised,2 including chiral guanidinium derivatives, which feature strong interactions with chiral oxoanions (Fig. 1a).3 The essential feature of the guanidinium cation for attracting the anionic species is the two N–H groups, carrying a delocalised positive charge, which are not only a source of a strong hydrogen bonding, but are also directed in space to form a familiar hydrogen-bond donor motif. The same features may be also recognised in the thiouronium system, which has not been widely explored for anion binding.6![Examples of chiral carboxylate hosts used as NMR enantio-discriminating agents: (a) guanidinium derivative, [A]Cl,3a (b) thiourea derivative, B,4 and (c) imidazolium derivative, [C][NTf2], which is a chiral ionic liquid (with bis(trifluoromethyl)sulfonylamide anion, [NTf2]).5](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f1.gif) | ||
| Fig. 1 Examples of chiral carboxylate hosts used as NMR enantio-discriminating agents: (a) guanidinium derivative, [A]Cl,3a (b) thiourea derivative, B,4 and (c) imidazolium derivative, [C][NTf2], which is a chiral ionic liquid (with bis(trifluoromethyl)sulfonylamide anion, [NTf2]).5 | ||
Thiouronium (formerly known as ‘thiuronium’, ‘pseudothiouronium’ or ‘isothiouronium’) salts are materials used in a range of applications, many of which have been patented.7 They are utilised as stabilisers for vulcanised rubber,7a as chelating agents for noble metals,7b as fire-resistant agents,7c and for protein extraction.7d However, thiouronium salts are easily hydrolysed by bases,8 and thus these conditions must be avoided. Moreover, thiouronium salts have an important role as reaction intermediates in the synthesis of thiols8 and guanidinium salts,9 and they are also used as peptide coupling agents.10 Recently, Abai et al. described a series of S-alkylthiouronium ionic liquids, which were investigated for the desulfurisation of diesel.11 However, in all of these reports, only the achiral thiouronium salts were studied.
While not much interest has been shown in chiral thiouronium salts, their precursors – chiral thioureas – are well known, especially as effective chiral catalysts used in organic transformations such as asymmetric Michael addition.12 Their roles as fluorescent anion sensors13 and amino acid receptors14 have also been reported. Theoretically, it is possible to alter known properties of thioureas through the alkylation of the sulfur atom. The resultant steric hindrance, as well as the Coulombic repulsive forces between the thiouronium cations, could drastically reduce the self-association of the thiourea unit via hydrogen bond interactions.1 Moreover, the hydrogen-bonding relationship of the thiouronium receptor centre with appropriate substrates could then be enhanced when compared to thioureas.15 The easiest way to examine the binding properties of chiral anion receptors, such as novel chiral thiouronium salts, is via NMR analysis of the chiral discrimination. At present, only one report describes structurally simple chiral thioureas as chiral discriminating agents for α-hydroxy and α-amino carboxylates, Fig. 1b.4
There is current interest in searching for new chiral tools that will provide a quick, cheap and facile way of determining enantiomeric purity, and offer an advantage over chiral HPLC.16 Chiral thiouronium salts may be a useful alternative to existing chiral anion binding systems. Additionally, when designed in the appropriate manner, these compounds could be an interesting and novel contribution to the family of chiral ionic liquids that have been applied as chiral agents for the NMR discrimination of chiral analytes (Fig. 1c).5,17 The unique properties of ionic liquids, and their wide ‘tuning’ opportunities combined with a designed functionality, have often delivered a competitive advantage compared to conventional, non-ionic systems.18
Here, we present an effective approach to enantio-recognition of chiral anions using new chiral S-alkylthiouronium salts (Fig. 2), which incorporate a combination of (a) a positively-charged thiourea function as a source of Coulombic attraction to the anionic substrate; (b) a receptor group for chiral carboxylates and analogues as a hydrogen-bonding donor in the form of a thiouronium cation; and (c) the use of a relatively low-cost and easily available source of chirality, e.g. the cation is based on rosin-derived (+)-dehydroabietylamine, denoted Ab.19 Attention here is mainly focussed on the enhancement in binding interactions resulting from introduction of a positive charge on the chiral thioureas.
![A model illustrating how novel chiral salts of the generic type [cation]Y can interact with chiral oxoanions (e.g. carboxylates, sulfonates, or phosphonates) via hydrogen bond recognition; the hydrogen bonding has been encoded using Etter's topographical analysis.20,21](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f2.gif) | ||
| Fig. 2 A model illustrating how novel chiral salts of the generic type [cation]Y can interact with chiral oxoanions (e.g. carboxylates, sulfonates, or phosphonates) via hydrogen bond recognition; the hydrogen bonding has been encoded using Etter's topographical analysis.20,21 | ||
Results and discussion
Synthesis and characterisation
The synthesis of chiral thiouronium salts essentially relies on a three-step reaction path (Scheme 1). In step one, a chiral primary amine was treated with carbon disulfide in the presence of a DCC to form isothiocyanate. This was then treated with a second chiral amine, R′*NH2, to form the disubstituted chiral thiourea; the second amine may or may not be identical to that used in the first step. Treatment of these thioureas with haloalkanes resulted in the formation of novel salts. For uniformity, the alkylation was confined to a butyl group, as the length of simple hydrocarbons does not play a significant role in the binding process and chiral recognition.22 In order to tune the physical properties of the products, a fourth step of anion exchange was conducted in some cases, principally for bis(trifluoromethyl)sulfonylamide (bistriflamide), [NTf2]−. Thus, a series of novel chiral thioureas, 1, and their corresponding chiral S-butylthiouronium salts, [2]Y, with chiral appendages and associated anions were synthesised, as described.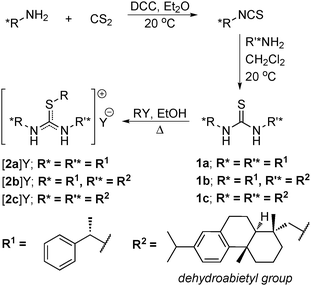 | ||
| Scheme 1 Synthesis of the chiral thioureas, 1, and S-alkylthiouronium salts, 2 (R = butyl, Y = halide, DCC = N,N′-dicyclohexylcarbodiimide). | ||
Collected physical property data (Table 1) for the new compounds show interesting trends. The highest melting points belong to the chiral thioureas 1a and 1c. Alkylation of these compounds to form halide salts causes a drop in the melting point; metathesis to bulkier anions, such as [NTf2]−, lowers the melting point even further. These salts may be classified as belonging to the family of chiral ionic liquids.18a,23 Indeed, while [2a]Br melts at 137.5 °C, [2a][NTf2] is a liquid at room temperature. Also, in the case of the S-butylbis-N,N′-(dehydroabietyl)thiouronium cation, [2c]+, the difference between the melting point of [2c]Br and the other salts, [2c]Y, where Y = [NTf2], [OTf], [CH3CO2], or [NO3], is at least 70 °C. As is typical for ionic liquids, the thermal stability of the thiouronium bistriflamides is much greater than for the halides, as the decomposition mechanism involves nucleophilic attack of the anion on the cation.18a Indeed, [2a][NTf2], [2b][NTf2] and [2c][NTf2] have decomposition temperatures over 200 °C, and they are the most thermally stable salts described here. Measured optical rotations of the products showed significant differences, not only between chiral thioureas and the corresponding thiouronium salts, but also between salts with the same cation but different anions. In this case, molar rotation should be more relevant than specific rotation, as then the molar concentration of the optically active species is taken into account. As Table 1 shows, there is a significant difference in molar rotation between [2a]Br and [2a][NTf2], [2b]Br and [2b][NTf2], and [2c]Br and [2c][NTf2] (69.2, 57.6 and 89.8 units respectively). Surprisingly, the molar rotation of halides with [2c]+ shows an unusual pattern. As the molar mass of the halide increases, the concentration of active species decreases, so we would expect that the values of specific rotation will change monotonically from chloride, through bromide to iodide. In contrast, repeated measurements showed unquestionably that the most laevorotatory is the bromide, then the iodide and lastly the chloride. Whereas for other anions, it is expected that the degree of interaction will change according to their size and shape: for halides they are sterically and electronically similar. However, analysis of the 1H and 13C NMR spectra suggested a likely explanation (vide infra).
| Compound | T m/°C | T dec /°C | [α]20D(c 1.0 (CHCl3)) | [M]20Db |
Form |
|---|---|---|---|---|---|
| a Decomposition temperature determined using thermal gravimetric analysis (TGA). b Molar rotation calculated from [α]20D × M/100, where M is the molar mass. c Product known from the literature4 (Tm 199−200 °C, [α]20D + 114.2 (c 1.05, CHCl3)). d d 20 1.3232 g cm−3, η20 4.704 Pa s with a water content of 186 ppm. e Glass transition temperature (Tg) determined using differential scanning calorimetry (DSC). | |||||
| 1a | 199.6−200.9 | — | +104.4 | +296.9 | White solidc |
| [2a]Br | 137.5 | 151.4 | +125.2 | +527.6 | Creamy solid |
| [2a][NTf2] | –38.1e | 219.1 | +96.0 | +596.8 | Colourless liquidd |
| 1b | 101.6−102.4 | 183.9 | +14.2 | +72.8 | White solid |
| [2b]Br | 79.2 | 158.4 | +34.8 | +203.8 | Yellowish solid |
| [2b][NTf2] | 13.9e | 231.9 | +18.6 | +146.2 | Colourless glass |
| 1c | 139.0−141.0 | 200.5 | +14.3 | +87.6 | White solid |
| [2c]Cl | 58.4e | 160.1 | −11.0 | −77.6 | Yellowish solid |
| [2c]Br | 128.2 | 210.0 | −19.2 | −143.9 | White solid |
| [2c]I | 75.4e | 151.9 | −16.4 | −130.7 | White solid |
| [2c][NTf2] | 44.3 | 227.5 | −5.7 | −54.1 | White solid |
| [2c][NO3] | 49.1 | 169.0 | −19.7 | −144.2 | White solid |
| [2c][OTf] | 54.7 | 189.9 | +8.3 | +68.0 | White solid |
| [2c][CH3CO2] | 49.0 | 158.9 | +16.2 | +118.1 | White solid/glass |
Rotamerism and isomerism in chiral thiouronium salts
Thiourea 1a was formed as a single diastereoisomer, reflecting the high enantiopurity (98%) of the α-(S)-methylbenzylamine (1-phenylethan-1-amine) used in its preparation. Thiourea 1a belongs to the C2 point group, and contains a C2 axis of rotation, hence only one set of peaks was observed for the methyl and methine groups of the amino components in thiourea. On conversion to the thiouronium salt (Scheme 1), the C2 symmetry was reduced to C1, and different signals could now be observed for the methyl, methine, and N–H groups of the two amino components. Because the bond order in the two central C–N and C–S bonds is greater than one, there is also the possibility of slow hindered rotation about these bonds,24 leading to four possible isomeric/rotameric forms namely: syn–syn, syn–anti, anti–syn, and anti–anti of salt [2a]Br (Scheme 2).25 Since each of these forms will produce two sets of amino signals, there is potential for the NMR spectra to be very complex indeed. In reality, the 1H and 13C NMR spectra of compound [2a]Br were remarkably simple in DMSO-d6 as a solvent, giving the impression that only one rotamer was present (Fig. 3). This could mean either that only one of the four possible rotamers was present in solution, or that there was a mixture of rotamers, undergoing rapid interconversion on the NMR time scale, resulting in a time-averaged spectrum. This latter supposition is incorrect, as two sets of signals for the chiral methine *CH and the methyl CH3 groups were observed, ruling out at least some fast bond rotations. It is known that rotation about a partial sulfur–carbon double bond is faster than rotation around a partial nitrogen–carbon double bond, as the NC bond is about 25 kJ mol−1 stronger,24a,b reflecting the poor overlap between a 3p orbital on the sulfur and a 2p orbital on the carbon. It is therefore probable that when the rotation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond is slow on the NMR timescale, rotation around the CS bond will be in the intermediate range.
N bond is slow on the NMR timescale, rotation around the CS bond will be in the intermediate range.
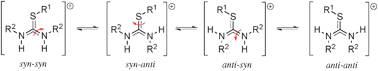 | ||
| Scheme 2 The four possible isomeric/rotameric forms of the thiouronium cation, generated due to hindered rotation about the CN bond and the CS bond. | ||
![The 1H–13C HSQC spectrum (400 and 101 MHz) of [2a]Br (in DMSO-d6 at 20 °C). Red signals indicate CH and CH3 groups; blue signals indicate CH2 groups.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f3.gif) | ||
| Fig. 3 The 1H–13C HSQC spectrum (400 and 101 MHz) of [2a]Br (in DMSO-d6 at 20 °C). Red signals indicate CH and CH3 groups; blue signals indicate CH2 groups. | ||
Furthermore, there was fluxional behaviour observed, demonstrating slow rotation on the NMR timescale around the CS bond leading to chemical exchange between the CHN signals in the proton and carbon NMR spectra. There were four indicators of this. Firstly, the signals, particularly *CH, were very broad, indicating a slow chemical exchange process on the NMR timescale (Fig. 3).26 Secondly, in the HSQC spectrum, each individual methine hydrogen showed correlation to the carbon atom to which it was attached, in both the different environments (Fig. 3). This can only be explained by rotation around the CS bond occurring during the evolution time of the HSQC experiment.
The third indicator was derived from a NOESY experiment (Fig. 4). In general NOESY spectra show cross peaks for nuclei which share a common relaxation pathway. Normally, this involves through-space dipole–dipole relaxation between the nuclei, which is distance dependent and leads to the well known nuclear Overhauser effect. However, chemical exchange is also a relaxation mechanism and can lead to cross peaks in NOESY spectra. These two distinct contributions to NOESY spectra can be distinguished from each other, in small molecules, as dipole–dipole coupling leads to an increase in signal intensity (the nuclear Overhauser effect) whilst chemical exchange leads to a decrease in signal intensity (saturation transfer).27 The NOESY spectrum of compound [2a]Br does indeed show a strong cross peak between the two methine protons in the two different environments (Fig. 4a), proving that these two nuclei are linked by a relaxation pathway. Selective saturation of the methine signal at δ 5.54 ppm led to a decrease in intensity of the other methine signal (at δ 5.16 ppm) to zero, proving that saturation transfer by chemical exchange was the dominant process, confirming slow rotation about the CS bond.
![Chosen fragments of the NOESY spectrum for (a) [2a]Br and its *CH region; and (b) [2c]Br and its NH region. The experiments were performed in (a) DMSO-d6 and in (b) CDCl3 using 1H–1H NMR correlation (400 MHz) at 20 °C.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f4.gif) | ||
| Fig. 4 Chosen fragments of the NOESY spectrum for (a) [2a]Br and its *CH region; and (b) [2c]Br and its NH region. The experiments were performed in (a) DMSO-d6 and in (b) CDCl3 using 1H–1H NMR correlation (400 MHz) at 20 °C. | ||
Finally, it was recognised that the hydrogen atoms in the methylene group adjacent to sulfur are diastereotopic, and give rise to complicated signals at δ 3.25 and 3.18 ppm (Fig. 3). However these signals are sharp, as is the carbon signal with which they are correlated; in other words, neither is displaying any fluxional activity. This can only happen in the syn–syn and anti–anti isomers (Scheme 2), as rotation around the CS bond generates the same molecule and hence is not fluxional with respect to the S–CH2 signal. Intuitively, one would expect a large difference in chemical shift positions for the two *CH peaks in the syn–syn isomer, due to the position of the butyl group. In fact, a large difference (of 0.5 ppm) was observed (Fig. 3). If the compound was the anti–anti isomer, then a large chemical shift difference in the NH peaks would be expected for the same reason, which was not observed.
In further justification of this conclusion, it should be noted that DMSO can readily participate in hydrogen-bonding. It is more than likely that a bifurcated hydrogen-bond interaction between the thiouronium cation and the solvent (Scheme 3) is stabilising the syn–syn rotamer over all other possible forms of this compound (Scheme 2). In addition, the 1H-NMR spectrum of [2a]Br in CDCl3 (not illustrated) was found to be very complex, due to the absence of strong hydrogen bonds, and hence no stabilisation of one specific rotamer.
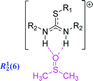 | ||
| Scheme 3 An illustration of hydrogen-bond interaction between the thiouronium cation and dimethylsulfoxide (dmso) stabilising the syn–syn rotamer; the hydrogen bonding has been encoded using Etter's topographical analysis.20,21 | ||
The same detailed analysis of the 1H NMR spectra was performed for the rest of the chiral thiouronium salts. Table 2 shows how the number of isomers can be influenced by both the nature of the N-substituents and by the solvent. In the case of [2a]Y and [2b]Y, the change in the counterion from bromide to bistriflamide does not influence the number of isomers detected, nor the ratio of the isomers (for [2b]Y). These results are in agreement with the findings of Kim and coworkers,24c who demonstrated that the counterion in anthracene derivatives of thiouronium salts had no influence on the ratio of the isomers. For compound [2b]Br, in which the two amino groups are different, rotation about the CS bond leads to a doubling of the peaks. The two resulting isomers are present in the ratio 3:2, as shown in Scheme 4.
| Compound | Solvent | No. of isomers | Ratioa |
|---|---|---|---|
| a Determined by integration of multiple NMR peaks; ratio between syn–syn and syn–anti isomers. b 1H NMR spectrum recorded at 300 MHz. c Ratio between syn–syn and syn–anti isomer with respect to the dehydroabietyl group. d Salt insoluble in DMSO-d6. | |||
| [2a]Brb | DMSO-d6 | 1 | — |
| [2a][NTf2]b | DMSO-d6 | 1 | — |
| [2b]Br | DMSO-d6 | 2 | 3:2c |
| [2b][NTf2] | DMSO-d6 | 2 | 3:2c |
| [2c]Cl | DMSO-d6 | 2 | 1:8 |
| [2c]Cl | CDCl3 | 2 | 1.8:2 |
| [2c]Brd | CDCl3 | 2 | 0.9:2 |
| [2c]Brd | C6D6 | 1 | — |
| [2c]Brd | CDCl2CDCl2 | 2 | 3:1 |
| [2c]I | CDCl3 | 2 | 1:2.6 |
| [2c]I | DMSO-d6 | 1 | — |
| [2c][NTf2] | DMSO-d6 | 1 | — |
| [2c][OTf] | DMSO-d6 | 1 | — |
| [2c][NO3] | DMSO-d6 | 1 | — |
| [2c][MeCO2] | CDCl3 | 1 | — |
![An illustration of the isomers (syn–syn : syn–anti 3 : 2) present in a solution of [2b]Br in DMSO-d6 (pink) which are distinguishable by 1H NMR spectroscopy. Blue and green indicate different environments for the N–H groups. The stereochemistry of the isomers was assigned with respect to the dehydroabietyl group.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-s4.gif) | ||
| Scheme 4 An illustration of the isomers (syn–syn:syn–anti 3:2) present in a solution of [2b]Br in DMSO-d6 (pink) which are distinguishable by 1H NMR spectroscopy. Blue and green indicate different environments for the N–H groups. The stereochemistry of the isomers was assigned with respect to the dehydroabietyl group. | ||
Interestingly, in the case of [2c]Y salts, the number of isomers depends on solvent as well as on counteranion type (Table 2). Even for different halides of [2c]+, changes in ratios between isomers are observable. In general, when using DMSO-d6 as an NMR solvent, the syn–syn isomer is detectable (Schemes 2 and 3). In non-polar solvents, on the other hand, the N–H signal region is more complex, with broader peaks, suggesting the presence of more than one isomer.
Focussing on the bromide salt [2c]Br in CDCl3 as an example, one of the isomers has the N–H groups in two different environments, corresponding to the two equally intense signals at δ 9.06 and 8.62 ppm (Fig. 4b). This feature is found only with the syn–anti isomer (Scheme 2). The second isomer of [2c]Br has only one signal for the N–H groups, at δ 10.35 ppm, weaker in intensity than the other two signals, and represents either a syn–syn or an anti–anti isomer. The NOESY spectrum of [2c]Br shows a strong cross peak between all N–H protons (Fig. 4b). This proves that these nuclei are linked by a relaxation pathway, as was discussed in the case of the methine proton in [2a]Br. Selective saturation of any one of the N–H signals leads to a decrease in intensity of the others to zero, as anticipated. Hence, again, there is fluxional behaviour, demonstrating slow rotation about the CS bond as well as the CN bond, with the CN bond dominating the isomeric distribution.
Examining the single crystal structure of [2c]Br, further evidence for the restricted rotation about the CN bond is found (Fig. 5), where only the anti–syn conformer is observed. Formation of crystals with anti–syn isomerism suggests the high stability of that conformation in the solid state. This contrasts with the situation in solution, where the syn–syn isomer is also observed. Indeed, computational analysis of the differences in energies between the syn–syn, anti–syn and anti–anti conformations of the thiouronium cation in the gas phase showed that the anti–syn conformer possessed the lowest energy (Fig. 6).
![A partial crystal structure of [2c]Br, showing a view of a single pair of ions.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f5.gif) | ||
| Fig. 5 A partial crystal structure of [2c]Br, showing a view of a single pair of ions. | ||
![Energy profile showing energy differences between isomers syn–syn, anti–syn and anti–anti in the gas phase of [2a]+, as an example of a thiouronium cation. The geometries were optimised at the RHF/3-21G* level using Gaussian® 98 (Gaussian Inc.).28](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f6.gif) | ||
| Fig. 6 Energy profile showing energy differences between isomers syn–syn, anti–syn and anti–anti in the gas phase of [2a]+, as an example of a thiouronium cation. The geometries were optimised at the RHF/3-21G* level using Gaussian® 98 (Gaussian Inc.).28 | ||
Noteworthy, when this cation is dissolved in a strong hydrogen-bond acceptor solvent (e.g. DMSO), the formation of a stable hydrogen-bonded motif (R12(6), Scheme 3) inverts the relative stability of these conformers with the syn–syn now becoming the most stable.
To summarise, integration of the N–H signals indicates an excess of syn–anti isomer over syn–syn isomer of [2c]Y in non-polar solvents, such as trichloromethane. Returning to the molar optical rotation results shown in Table 1, the mixture of stable isomers in different ratios (Table 2) for each [2c]Y (Y = Cl, Br or I) observed in trichloromethane at 20 °C is the reason for the unusual order of these values, and probably is connected with the differing hydrogen-bond acceptor abilities of the three halide ions.
Further confirmation of the importance of the anion in the relative stability of the conformers can be found in the case of [2c][MeCO2] in CDCl3, where only one set of sharp signals was observable in the 1H NMR spectrum, and no signal was visible in the region of N–H protons. The most probable explanation here is that the ethanoate ion forms a very strong hydrogen-bond motif (R22(8), Fig. 2) with the two N–H bonds of the cation, even stronger than the R12(6) motif formed with DMSO (Scheme 3).20,21 Thus, there is preferential stabilisation of the syn–syn conformer, resulting in the disappearance (total broadening) of the N–H signal, due to a dynamic process indicating exchange of the proton between oxygen atoms in the ethanoate anion with the nitrogen atoms in the thiouronium cation. So, despite the potential complexity in this system, the syn–syn rotamer can still be locked in as the dominant form.
Crystallography
Crystallographic data (see Experimental section) for [2c]Br reveals many other interesting features than the confirmation of the structure and contributes to the discussion above about isomerism of the thiouronium cation. Strong hydrogen bonding between the anion and the cation is observed (Fig. 7a). The bromide ion is hydrogen bonded to two different cations via a N–H⋯Br⋯H–N pattern (H1⋯Br, 2.54 Å; Br⋯H2, 2.58 Å), as shown in Fig. 7a. Additionally, some weaker C–H⋯Br hydrogen bonding interactions were found (Fig. 5),29 ranging from 2.78 to 2.97 Å (Table 3).![The crystal structure of [2c]Br: (a) hydrogen-bonding interactions of the bromide anion with two adjacent [2c]+ cations (part of one of the [2c]+ cations was omitted for clarity); (b) a perspective view of the anion channel; (c) a perspective view of the cation channel. The prime (') character in atom labels indicates that these atoms are at equivalent positions (−x, −1/2 + y, −z).](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f7.gif) | ||
| Fig. 7 The crystal structure of [2c]Br: (a) hydrogen-bonding interactions of the bromide anion with two adjacent [2c]+ cations (part of one of the [2c]+ cations was omitted for clarity); (b) a perspective view of the anion channel; (c) a perspective view of the cation channel. The prime (') character in atom labels indicates that these atoms are at equivalent positions (−x, −1/2 + y, −z). | ||
The packing of the crystal structure of [2c]Br is dominated by neighbouring cationic and anionic channels along the b axis, Fig. 7b and c. The powder diffraction pattern of [2c]Br is in good agreement with the simulated powder diffraction pattern of the single crystal structure determination (Fig. 8), demonstrating that the crystal examined was typical for the bulk synthetic material. Furthermore, examination of the powder diffraction patterns of [2c]Cl and [2c]Br (Fig. 8) shows that these salts are isostructural.
![Comparison of the powder diffraction patterns of [2c]Cl and [2c]Br (exact, red line, and simulated, black line, from single crystal data).](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f8.gif) | ||
| Fig. 8 Comparison of the powder diffraction patterns of [2c]Cl and [2c]Br (exact, red line, and simulated, black line, from single crystal data). | ||
Chiral NMR discrimination of oxoanions
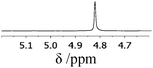
The rotamerism/isomerism of the chiral thiouronium salts revealed an interesting feature connected with the ethanoate anion. In the case of [2c][CH3CO2] the isomerism was not detected at room temperature on the NMR time scale (Table 2). Moreover the 1H NMR spectrum of this compound is sharp and clear, in contrast to the spectra with other anions, which are broad and confusing. This suggested that, probably, the binding of the ethanoate by the thiouronium moiety caused the rigid and stable conformation of [2c]+. As discussed in the Introduction, Fig. 2 is the best representation of the interaction of the thiouronium cation with the oxoanion. Then, as this system is chiral, it may ‘recognise’ enantiomers of the oxoanions differently, and show non-equivalent signals in the 1H NMR spectrum. The first choice of possible substrates was the chiral carboxylates. It is known that when a hydrogen-bond acceptor group is negatively charged, the hydrogen-bond strengths are enhanced.15a,30 Also, in the case of the thiouronium system, it may be expected that a strong interaction with the carboxylates, of the type illustrated in Fig. 2, featuring two parallel hydrogen bonds forming a R22(8) network, would lead to a thermodynamically stable host–guest interaction, locking the structure of the cation into a single isomeric form. Hence, a simple chiral carboxylate was selected as a model substrate, a racemic tetrabutylammonium mandelate, [N4444][3a]: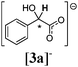
The tetrabutylammonium counterion to the chiral carboxylate was not chosen at random. The first 1H NMR experiments were conducted with a tetramethylammonium salt of mandelate, [N1111][3a], as a substrate, [2a]Br as a chiral agent, and CDCl3 as a solvent. In this case, [N1111]Br precipitated from the solution. In order to prevent this precipitation, a bulkier (more hydrophobic) cation was selected, [N4444]+. When this was used, no precipitation occurred.
The optimum design of the experiment is as follows: the model substrate (0.03 mmol) was dissolved in CDCl3 (0.7 cm3) in the presence of 1a–c or [2a–c]Y (0.03 mmol) and the 1H NMR spectrum recorded. The methine proton from the chiral centre of mandelate is visible in the 1H NMR spectrum as a singlet at around 4.8 ppm. The 1H NMR traces embedded in Table 4 show an evolution of the discriminating properties changing with the structural diversity of the chiral agent. Upon the addition of a chiral agent, the singlet of the methine proton in the mandelate appears now as two resonances, one associated with (R)-mandelate, and the other associated with (S)-mandelate. As each occurs at a different chemical shift, they must each be bound strongly within a stable structural entity. Alkylation of the sulfur atom (to form the novel salts [2a]Br or [2a][NTf2]) causes an increase in the Δδ value (difference of shift values between the peaks of the non-equivalent enantiomers) compared with thiourea 1a (as described in the literature4). Moreover, the chemical shift difference between the two enantiomers is found to be a function of both the cation and the anion of the chiral agent. Thus, if the anion is kept constant and the cation changed i.e. [2a]Br, [2b]Br and [2c]Br, the difference in chemical shift ranges from 0.017 to 0.035 to 0.097 ppm, respectively. Similarly, if the cation is kept the same i.e. [2c]Br and [2c][NTf2], a smaller but still significant change in the chemical shift is observed from 0.097 to 0.110 ppm. A noticeable feature is that a methine proton from the (S)-methylbenzyl group, which is a part of salts [2a]Y and [2b]Y, moved upfield compared to thioureas 1a and 1b respectively.
| Agent | CH peak | Δδb/ppm |
|---|---|---|
| a All samples were prepared by mixing the substrate (0.03 mmol) with a chiral agent (0.03 mmol) in CDCl3 (0.7 cm3). b Difference of CH shift values between peaks of opposite enantiomers of mandelate. c A methine proton signal from the methylbenzyl group present in the chiral agent is visible. | ||
| None |

|
0 |
| 1a |

|
0.013 |
| [2a]Brc |
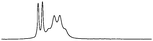
|
0.017 |
| [2a][NTf2]c |
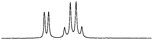
|
0.017 |
| 1b |

|
0 |
| [2b]Brc |

|
0.035 |
| [2b][NTf2]c |

|
0.043 |
| 1c |

|
0.009 |
| [2c]Br |

|
0.097 |
| [2c][NTf2] |

|
0.110 |
| [2c][NTf2] |

|
(R)-Enantiomer |
| [2c][NTf2] |
|
(S)-Enantiomer |
Hence, in some cases, the methine quartet is visible very close to, or even superposing on, the substrate chiral proton peak. This was not the case for [2c]Y salts. Furthermore, the Δδ value increases about ten times when comparing [2c]Br with the thiourea, 1c. The effect intensifies after the anion exchange from bromide to [NTf2]−, as the latter is more weakly coordinating with the thiouronium cation than the bromide anion.31 In conclusion, the best enantio-discriminating results, even on relatively low frequency 1H NMR spectrometers, were obtained with [2c][NTf2] and the assignment of the individual enantiomers was confirmed by using optically pure (R)-mandelate and (S)-mandelate as substrates, which resulted in single resonances being observed (Table 4). Therefore, further research was focussed on this compound as a model chiral agent.
The NMR studies were continued with other oxoanions as substrates and [2c][NTf2] as the chiral agent. As Table 5 shows, a group of fourteen racemic mixtures of chiral carboxylic acids, as well as sulfonic and phosphonic acids, were analysed. All analytes were in the form of tetrabutylammonium salts, [N4444][3a–n], and in each case the complexation process between [2c][NTf2] and the substrate [N4444][3] could be observed by the proton peak non-equivalence of the methine proton connected with the chiral centre of the substrate (with the exception of [N4444][3c] and [N4444][3l]). In some examples, a full resolution of the chiral peaks was observed with no interference, while in other cases partial overlap occurred. The problem of overlapping was particularly significant when the methine group occurred as e.g. overlapping quartets after complexation of [N4444][3b]. However, the situation could be resolved by a partial NMR decoupling experiment, which collapses the quartets into singlets, followed by integration of these singlets to yield the enantiomeric purity of the sample. Nevertheless, the selection of substrates for NMR discrimination relies on avoiding a superposition of important substrate signals with receptor signals in the spectrum. In the case of [2c][NTf2], it provides a useful 1H NMR spectral window for chiral discrimination research in the range from 3.5 ppm to 6.5 ppm, and then above 7.5 ppm, which for α-substituted carboxylates is usually sufficient.
| Code of salt | Racemic acid | Δδb/ppm |
|---|---|---|
| a All samples were prepared by mixing carboxylate salt (0.03 mmol) with [2c][NTf2] (0.03 mmol) in CDCl3 (0.7 cm3). b Difference of shift values between peaks of opposite enantiomers. c Proton peaks from the methoxy group are superimposed on proton peaks from [2c][NTf2] but still visible. d Results of 19F NMR (282 MHz) experiment in CDCl3 at 20 °C. e Results of 1H NMR (400 MHz) experiment in CDCl3 at 20 °C. f Results of 19F NMR (376 MHz) experiment in CDCl3 at 20 °C. | ||
| [N4444][3a] |
|
0.110 |
| [N4444][3b] |
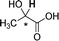
|
0.070 |
| [N4444][3c] |
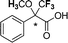
|
0.091c |
| 0.045d | ||
| [N4444][3d] |
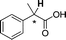
|
0.030 |
| [N4444][3e] |
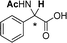
|
0.058 (CH) |
| 0.031 (Ac) | ||
| [N4444][3f] |
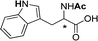
|
0.022 (Ac) |
| 0.107 (Ar-NH) | ||
| [N4444][3g] |
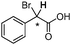
|
0.030 |
| [N4444][3h] |
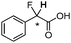
|
0.102e (2JHF = 50.2 Hz) |
| 0.374f (2JHF = 50.3 Hz) | ||
| [N4444][3i] |
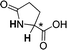
|
0.058 (CH) |
| 0.129 (NH) | ||
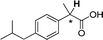
| [N4444][3j] |
|
0.047 |
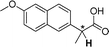
| [N4444][3k] |
|
0.038 |
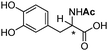
| [N4444][3l] |
|
0.018 |
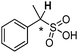
| [N4444][3m] |
|
0.006 |
| [N4444][3n] |
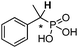
|
0.013 |
The possibility of determining enantiomeric purities was proved by a 400 MHz 1H NMR experiment using the model substrate [N4444][3a] and agent [2c][NTf2] (Fig. 9). Indeed, a linear correlation (R2 = 0.993) was confirmed between the theoretical and observed enantiomeric excess (% ee), when using just one equivalent of agent with respect to the substrate in a CDCl3 solution.
![(a) Determination of the enantiomeric purities of [N4444][(R)-3a] in the presence of [2c][NTf2] using 1H NMR spectroscopy (400 MHz) in CDCl3 at 20 °C. Theoretical % ee values calculated from the mass of enantiomers used for preparing the analytes are given for comparison with practical % ee values, based on the integration of methine peaks (indicated in parentheses). (b) The correlation between the theoretical and practical % ee values.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f9.gif) | ||
| Fig. 9 (a) Determination of the enantiomeric purities of [N4444][(R)-3a] in the presence of [2c][NTf2] using 1H NMR spectroscopy (400 MHz) in CDCl3 at 20 °C. Theoretical % ee values calculated from the mass of enantiomers used for preparing the analytes are given for comparison with practical % ee values, based on the integration of methine peaks (indicated in parentheses). (b) The correlation between the theoretical and practical % ee values. | ||
Molecular recognition mode
NMR study
The molecular recognition behaviour of the new chiral agents was further investigated. There is no doubt that the thiouronium moiety, containing N–H groups as sources of hydrogen bonds, is the receptor centre for the oxoanions, in an analogous way to that already established for guanidinium salts. For the thiouronium complexes with oxoanions, a binding pattern featuring two parallel hydrogen bonds may be described by the R22(8) descriptor (Fig. 2).20,21 The same descriptor is found in the guanidinium system.The fact is that the initial NMR experiments on enantio-discrimination have shown (vide supra) that the complexation results in a locked conformation of the single isomeric form of the cation. This can be see clearly in Fig. 10, by comparing the well resolved and simple spectrum of a 1:1 complex of [2c][NTf2] and [N4444][3a] with the conformational freedom exhibited by the uncomplexed [2c][NTf2]. As the thiouronium moiety recognises the oxoanions, it may be suspected that the 1:1 complexation ratio is an optimum for this system. Indeed, 1H NMR experiments carried out with increasing molar equivalents of [2c][NTf2] added to the model substrate [N4444][3a] confirmed that the optimum stoichiometry of the adduct is indeed 1:1 (Fig. 11). These new reagents are truly competitive with those known from the literature, especially the extant chiral ionic liquids, where an excess of 3 to even 24 equivalents is required for satisfying results in the chiral NMR discrimination.17 Moreover, they may be considered as simple synthetic analogues to the naturally occurring receptor centres in enzymes, since each receptor centre recognises and saturates with a single substrate. They are hence ‘abiotic’ or ‘biomimetic’ receptor systems, analogous to Lehn's archetypal guanidinium system (Fig. 1a).3a Another promising feature is that, for some substrates, less than one equivalent would be sufficient for an effective determination of the ee of the chiral analyte, hence giving increased sensitivity.
![Comparison between the 1H NMR spectrum of uncomplexed [2c][NTf2] (red trace) and complexed with [N4444][3a] (black trace) using 1H NMR spectroscopy (300 MHz) in CDCl3 at 20 °C.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f10.gif) | ||
| Fig. 10 Comparison between the 1H NMR spectrum of uncomplexed [2c][NTf2] (red trace) and complexed with [N4444][3a] (black trace) using 1H NMR spectroscopy (300 MHz) in CDCl3 at 20 °C. | ||
![Determination of the optimum ratio of chiral agent [2c][NTf2] to model substrate [N4444][3a] required for NMR discrimination of using 1H NMR spectroscopy (400 MHz) in CDCl3 at 20 °C.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f11.gif) | ||
| Fig. 11 Determination of the optimum ratio of chiral agent [2c][NTf2] to model substrate [N4444][3a] required for NMR discrimination of using 1H NMR spectroscopy (400 MHz) in CDCl3 at 20 °C. | ||
Thermodynamic binding study
Determination of possible self-association interactions between the chiral agent (host) and the chiral substrate (guest) is important for a full description of the binding process. Hence, the self-association equilibrium for the model agent, here [2a][NTf2], and its molecular analogue, thiourea 1a, was determined (Fig. 12a). Examining Fig. 12a, there is an impression of a general trend of chemical shift increasing with increasing concentration. However, closer inspection reveals that the difference in chemical shift between the lowest and highest concentrations is less than three standard deviations (3σ), and hence there is no significant difference between these values. In other words, within the errors of the NMR experiments, there is no evidence for self-association of either the cation [2a]+ or the neutral thiourea 1a in the range of concentration from 1.7 to 50 mM. Further, when the guest [N4444][3a] was compared with its precursor – mandelic acid (Fig. 12b), the results revealed that, as in the case of host, the self-interactions of [3a]− are also undetectable in these experiments. However, free mandelic acid shows the expected self-association, which is normally found for carboxylic acids.32 In conclusion, for the determination of host–guest interactions by 1H NMR spectroscopy, it is correct to ignore both host–host and guest–guest interactions as these are too weak to be detected.![Dependence of 1H NMR (400 MHz, 20 °C) chemical shifts on concentration for (a) [2a][NTf2] and 1a in DMSO-d6 and (b) [N4444][3a] and mandelic acid in MeOD.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f12.gif) | ||
| Fig. 12 Dependence of 1H NMR (400 MHz, 20 °C) chemical shifts on concentration for (a) [2a][NTf2] and 1a in DMSO-d6 and (b) [N4444][3a] and mandelic acid in MeOD. | ||
Next, the stoichiometry of the host–guest interaction was examined. The Job's plot, Fig. 13a, confirmed the 1:1 stoichiometry of the interaction between [2c]+ and [3a]−, as the maximum appears at ca. 0.5 mol fraction.33 The 500 MHz 1H NMR titration experiments in CDCl3, Fig. 13b, showed that the determination of the binding constants for each enantiomer of the guest [N4444][3a] and optically pure [2c][NTf2] is impossible in this solvent. Sharp NMR isotherms which produce two straight lines that intersect at the [host]/[guest] ratio corresponding to the stoichiometry of the complex (Fig. 13b) indicate that the binding constants are too large to be properly determined by the NMR method (K > 105 M−1).34 Thus, there was a need to use an additive, which will decrease the degree of binding between [3a]− and [2c]+, and then allow for generation of classic 1:1 NMR binding isotherms,34 from which a reduced binding constant can be estimated. A series of trials with different additives (MeOD and DMSO-d6) in different concentrations (1–10% v/v) in CDCl3 showed that the optimum NMR titration data were obtained in a solution of 10% v/v DMSO-d6 in CDCl3, and in this system the reduced binding constant K could be determined for the interaction between [2c][NTf2] and [N4444][3a] (Fig. 13c and Table 6). The methodology for determination of equilibrium constants is explained in detail in the Experimental section. Generally, the method is based on a classic equation for the binding isotherm, adapted to NMR spectroscopy,35 and the curve fitting was accomplished by standard non-linear regression analysis.36 NMR titrations were also carried out with hosts [2a][NTf2] and [2b][NTf2] using [N4444][3a] as a guest (Table 6). Results showed that, for hosts functionalised with an (S)-methylbenzyl group, the binding with a model substrate in CDCl3 is also too strong to have K accurately determined (vide supra).34 Thus the only determination of the preference of binding (R)-[3a]+ over (S)-[3a]+ by [2c][NTf2] is provided by NMR titration experiments in a solvent mixture of 10% v/v DMSO-d6 in CDCl3. Indeed, there is a threefold difference in binding of the (R)-enantiomer of mandelate over the (S)-enantiomer by [2c]+, even for reduced values of binding constants with DMSO-d6. A chiral recognition energy ΔΔG calculated from −RTln(K(S)/K(R)) is equal to 2.51 kJ mol−1 for [2c][NTf2] as a host for mandelate as a substrate.
![1H NMR data (500 MHz; CDCl3; 27 °C) for (a) the Job's plot (■ (S)-enantiomer; □ (R)-enantiomer);33 (b) the titration of [N4444][3a] (cconst = 2 mM) with [3c][NTf2] in CDCl3 expressing the well defined NMR binding isotherms indicative of a binding constant, K > 105 M−1 for the interaction between [2c][NTf2] and [N4444][3a];34 and (c) the 1 : 1 NMR binding isotherms used for estimation of a reduced binding constant K for the interaction between [2c][NTf2] and [N4444][3a] in 10% v/v DMSO-d6 in CDCl3.35 Δδ is the chemical shift difference of the methine proton peaks between free and complexed mandelate; X is the mole fraction; δ is the chemical shift of the methine proton peaks in mandelate; c is the concentration (mol l−1); ceq in the concentration in the equilibrium state required for proper binding constant K estimation. For curves (c), the trend lines were fitted using non-linear regression analysis.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f13.gif) | ||
| Fig. 13
1H NMR data (500 MHz; CDCl3; 27 °C) for (a) the Job's plot (■ (S)-enantiomer; □ (R)-enantiomer);33 (b) the titration of [N4444][3a] (cconst = 2 mM) with [3c][NTf2] in CDCl3 expressing the well defined NMR binding isotherms indicative of a binding constant, K > 105 M−1 for the interaction between [2c][NTf2] and [N4444][3a];34 and (c) the 1:1 NMR binding isotherms used for estimation of a reduced binding constant K for the interaction between [2c][NTf2] and [N4444][3a] in 10% v/v DMSO-d6 in CDCl3.35 Δδ is the chemical shift difference of the methine proton peaks between free and complexed mandelate; X is the mole fraction; δ is the chemical shift of the methine proton peaks in mandelate; c is the concentration (mol l−1); ceq in the concentration in the equilibrium state required for proper binding constant K estimation. For curves (c), the trend lines were fitted using non-linear regression analysis. | ||
| Host | Guest | K /M−1 | NMR solvent | ΔGb/kJ mol−1 |
|---|---|---|---|---|
| a Binding constants were calculated from the equation defined in the experimental part. b Gibbs free energy calculated from −RT ln(K). | ||||
| [2a][NTf2] | (S)-[N4444][3a] | K >105 | CDCl3 | <−28.72 |
| [2a][NTf2] | (R)-[N4444][3a] | K >105 | CDCl3 | <−28.72 |
| [2b][NTf2] | (S)-[N4444][3a] | K >105 | CDCl3 | <−28.72 |
| [2b][NTf2] | (R)-[N4444][3a] | K >105 | CDCl3 | <−28.72 |
| [2c][NTf2] | (S)-[N4444][3a] | K >105 | CDCl3 | <−28.72 |
| [2c][NTf2] | (R)-[N4444][3a] | K >105 | CDCl3 | <−28.72 |
| [2c][NTf2] | (S)-[N4444][3a] | 7413 ± 1235 | 10% v/v DMSO-d6 in CDCl3 | −22.23 |
| [2c][NTf2] | (R)-[N4444][3a] | 20283 ± 5231 |
10% v/v DMSO-d6 in CDCl3 | −24.74 |
The reason for such a strong interaction may lay not only in the combination of the hydrogen bonding and Coulombic forces. The structure of the thiouronium salts may also lead to additional interactions such as stacking interactions, especially in the case of dehydroabietyl as the N-substituent. As seen in Fig. 14a, for [2a][NTf2], there is a much lower probability of direct π–π interactions with the guest, but at the same time a substrate has better access to the receptor centre of the host. A more complex situation exists for [2b][NTf2] (Fig. 14b): on one side of this host centre, there is an α-methylbenzyl group, and on the other side there is a dehydroabietyl group. This case presents a unique combination of more open access for the guest, deriving from the (S)-methylbenzyl substituent, with π–π interactions between the guest and the dehydroabietyl aromatic ring. Finally, [2c][NTf2] has an ideal structure to support the binding of a phenyl-functionalised guest via stacking interactions (Fig. 14c), but at the same time structurally developed substituents, like dehydroabietyl groups, can limit access to the receptor centre.
![The schematic visualisation of possible stacking interactions occurring between the model mandelate anion, [3a]−, and (a) [2a]+, (b) [2b]+ and (c) [2c]+.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f14.gif) | ||
| Fig. 14 The schematic visualisation of possible stacking interactions occurring between the model mandelate anion, [3a]−, and (a) [2a]+, (b) [2b]+ and (c) [2c]+. | ||
Anion influence
When involving a cation that acts as a host for anions, obviously it is in competition with the cation from the guest.1 A similar competition should be recognised between the anions of the guest and the host. Indeed, when comparing the bromide and the bis(trifluoromethyl)sulfonylamide accompanying the same cation (Table 4), there is a noticeable effect on methine peak non-equivalences in [N4444][3a], influenced by the anion of the chiral host. Thus, the 1H NMR spectra were recorded for the S-butyl-N,N′-bis(dehydroabietyl)thiouronium cation with different anions (Table 7). The main factor taken into consideration is again the Δδ of the methine peak from the model substrate (guest): Table 7 clearly shows a strong influence from the nature of the host's anion. An additional interesting observation during this investigation was a significant shift of the N–H peaks in the 1H NMR spectrum of the complexes between the model substrate and [2c]Y salts.| Y− | Δδb/ppm | δ N–H /ppm |
|---|---|---|
| a All samples were prepared by mixing the guest (0.03 mmol) with [2c]Y (0.03 mmol) in CDCl3. b Difference of CH shift values between peaks of opposite enantiomers of mandelate. c Chemical shift of the N–H protons from [2c]+ observed as a broadened peak in the 1H NMR spectrum. | ||
| [CH3CO2]− | 0.013 | 8.935 |
| Cl− | 0.072 | 11.707 |
| [NO3]− | 0.083 | 11.908 |
| Br− | 0.097 | 11.835 |
| I− | 0.101 | 11.861 |
| [OTf]− | 0.107 | 11.976 |
| [NTf2]− | 0.110 | 12.006 |
Organisation of the results in order of increasing Δδ reveals the character of competition between the anions for the thiouronium receptor centre:
| [NTf2]− > [OTf]− > I− > Br− > [NO3]− > Cl− > [CH3CO2]− |
Thus, increasing values of Δδ were observed from ethanoate, through halides, to bis(trifluoromethyl)sulfonylamide. This order appears to reflect the hydrogen-bonding acceptor abilities of the anions, but the precise basicity of anions in ionic liquids is not known. The order as suggested by the Kamlet–Taft solvatochromic β parameter is37,38
| Cl− > Br− > I− > [CH3CO2]− > [NO3]− > [OTf]− > [NTf2]− |
Thus, as may have been anticipated, the competition between anions reflects more than simple basicity, and clearly must feature at least a steric factor and a recognition of the topological pattern (Fig. 2). In general, anion binding selectivity in the absence of specific chemical recognition between the anion and the host cation should follow the order of decreasing anion hydrophobicity (as indicated by the Hofmeister series):1
| I− > [NO3]− > Br− > Cl− ≫ [CH3CO2]− |
When comparing this series with the results for anion competition observed here (note that bistriflamides are extremely hydrophobic), they follow a similar trend.
Results showing that the N–H peak from the host is moving downfield with the higher value of Δδ (Table 7) prove that stronger hydrogen bonding interactions are occurring between the receptor centre and the guest anion, ruling out the host counterion.39 In other words, mandelate is more strongly bonded than the host's anion because of a combination of its hydrophobicity, hydrogen-bond pattern recognition, and steric size.
To conclude, the competition between the host and guest anions for binding with [2c]+ has a significant implication for chiral recognition, viz. the greater the competition, the smaller the discrimination. Thus, in order to examine the substrates with lower hydrogen-bonding acceptor abilities, it is necessary to use non-competing anions in the chiral agent, such as [NTf2]−.
Computational study
Computational optimisation analyses for the two enantiomers of [3a]− with [2c]+ were performed to investigate the origin of the enantio-discrimination connected with the chiral thiouronium salts. The results of density functional theory (DFT) calculations (RB3LYP/STO-3G)28 on the optimised structures of the complexes between [2c]+ and [(S)-3a]−, as well as [(R)-3a]−, are illustrated in Fig. 15. Although it is realised that this is a rather low level calculation, and caution should be exercised when examining the energy predictions, it does well illustrate some of the key features of the system. As the anion approaches the recognition site within the host cation, the cation responds by changing its conformation to accommodate the anion. In other words, the more tightly bound the anion, the more rigid the new conformation of the cation becomes. In addition, as [3a]− possesses an –OH group at the chiral centre, which is spatially close to the carboxylate group, a stabilising intramolecular five-membered ring is formed. This intramolecular hydrogen bond reduces the electron density on the carboxylate oxygen and hence weakens the intermolecular hydrogen bond formed by the same oxygen atom to the thiouronium moiety, Fig. 16a. This is confirmed by analysing the features of the computed structure [2c][3d], Fig. 16b. Here, there is no intramolecular hydrogen bond, as the –OH group was replaced by the –CH3 group, and in addition methyl is electron-releasing, whereas hydroxyl is an electron-attracting group. The net effect is that the oxygen atoms of the carboxylate group are more negatively charged in [2c][3d] than they are in [2c][3a], and therefore the hydrogen bonds from the former are on average 0.03 Å shorter than in the latter.![Optimised structures for (a) [2c][(S)-3a] and (b) [2c][(R)-3a]. Each host–guest pair is illustrated by two different views, the left one emphasising the importance of hydrogen bonding, the right one emphasising the resulting distances between the butyl side chain from the host, [2c]+, and the phenyl group from the guest, [3a]−. The geometries were optimised at the RB3LYP/STO-3G level using Gaussian® 98 (Gaussian Inc.).28](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f15.gif) | ||
| Fig. 15 Optimised structures for (a) [2c][(S)-3a] and (b) [2c][(R)-3a]. Each host–guest pair is illustrated by two different views, the left one emphasising the importance of hydrogen bonding, the right one emphasising the resulting distances between the butyl side chain from the host, [2c]+, and the phenyl group from the guest, [3a]−. The geometries were optimised at the RB3LYP/STO-3G level using Gaussian® 98 (Gaussian Inc.).28 | ||
![Hydrogen bond distances and differences in energies between complexes of [2c]+ and (a) enantiomers of [3a]−; (b) enantiomers of [3d]− obtained by optimisation at the RB3LYP/STO-3G level using Gaussian® 98 (Gaussian Inc.).28 Parts of the [2c]+ cations were omitted for clarity.](/image/article/2013/NJ/c2nj40632b/c2nj40632b-f16.gif) | ||
| Fig. 16 Hydrogen bond distances and differences in energies between complexes of [2c]+ and (a) enantiomers of [3a]−; (b) enantiomers of [3d]− obtained by optimisation at the RB3LYP/STO-3G level using Gaussian® 98 (Gaussian Inc.).28 Parts of the [2c]+ cations were omitted for clarity. | ||
However, the differences in energy for both complexes of the opposite enantiomers of [2c][3a] (0.44 kJ mol−1) and [2c][3d] (0.30 kJ mol−1) are too small to be significant, given the level of the calculation.
Conclusions
A series of chiral thiouronium salts, [2]Y, was developed and used for chiral discrimination of oxoanions, e.g. mandelate, using NMR spectroscopy. The performance of these salts was superior to that of the uncharged molecular precursors, the chiral thioureas.It was confirmed that the alkylation reaction of the sulfur atom induced delocalisation of the positive charge through the π-orbitals of the {CN2S} core. Because of this delocalisation, rotation about the CN bonds exists, but with restricted character. Hence, the thiouronium cations should exhibit discrete rotamers, as confirmed by the broadened and complex NMR signals. Indeed, extensive NMR studies confirmed that the interconversion of the rotamers was dependent on temperature, the nature of the N-substituents, the NMR solvent, and the counterion, as well as influencing the optical rotation.
However, in the case of [2c][CH3CO2], the 1H NMR spectrum revealed a rigid conformation of the cation–anion system, due to strong hydrogen bonding interactions, which produced a sharp set of 1H NMR signals. This observation triggered an extensive study of the chiral discrimination between oxoanions, especially carboxylates, by these new chiral salts.
The best results were obtained with [2c][NTf2] as a chiral agent, and it was found to be comparable, or better, in performance than existing systems in the literature. An additional advantage here is the relatively easy synthesis of this agent with a low cost chiral auxiliary. Apart from other chiral carboxylate substrates, chiral sulfonates and phosphonates could also be recognised and discriminated.
By changing the molar concentration of the chiral agent over the chiral substrate, it was confirmed that a 1:1 ratio is the optimum for the formation of the complex between [2c][NTf2] and the model substrate, and this is enough for an effective discrimination process.
As the binding constants are large in CDCl3 (K > 105 M−1), it was necessary to determine reduced binding constants by the addition of DMSO-d6. Even under these conditions, a three-fold difference in binding of (R)-mandelate over (S)-mandelate was observed, which is competitive with other systems used for chiral discrimination.
Using this kind of receptor may also be useful for the physical separation of racemic mixtures.
Experimental section
General information
All reagents and solvents were obtained from commercial suppliers and were used without further purification unless otherwise stated. (+)-Dehydroabietylamine (60% grade) required simple purification, as described in the literature.19 Dichloromethane was stored closed over molecular sieves, and diethyl ether was stored over sodium. All flash chromatography was performed using Aldrich Silica gel, Merck grade 10180 (70–230 mesh). 1H NMR spectra were recorded at 300, 400 or 500 MHz. 13C NMR spectra were recorded at 101 MHz. 19F NMR spectra were recorded at 376 MHz. Two-dimensional correlation NMR spectra (COSY), heteronuclear single quantum correlation NMR spectra (HSQC) and nuclear Overhauser effect NMR spectra (NOESY) were recorded at 400 MHz (1H NMR traces) and 101 MHz (13C NMR traces). Melting points Tm (mp) were determined in capillaries (uncorrected) or by DSC analysis. Glass transition temperatures Tg were obtained using DSC analysis (N2 atmosphere, 5−10 °C min−1 scan rate). Decomposition temperatures Tdec were determined with TGA analysis (N2 atmosphere, 10 °C min−1 scan rate). Optical rotations [α]20D were determined on a digital polarimeter at 20 °C in CHCl3 as a solvent. The exact mass was obtained using high-resolution mass spectrometry (Waters – LCT PREMIER instrument) with electrospray ionisation (EI). Combustion elemental analysis was performed for all new products. Karl-Fisher titration for water content W, density d20 and viscosity η20 determination was performed for room temperature chiral ionic liquid product ([2a][NTf2]).Crystal data
Crystal data for [2c]Br were collected using a Bruker Nonius Kappa CCD diffractometer with a FR591 rotating anode and a molybdenum target at ca. 120 K in a dinitrogen stream.40 Lorentz and polarisation corrections were applied. The structure was solved by direct methods. Hydrogen-atom positions were located from difference Fourier maps and a riding model, with fixed thermal parameters (Uij = 1.2Ueq for the atom to which they are bonded, 1.5 for methyl), was used for subsequent refinements. The function minimised was Σ[ω(|F0|2 − |Fc|2)] with reflection weights ω−1 = [σ2 |F0|2 + (g1P)2 + (g2P)] where P = [max|F0|2 + 2|Fc|2]/3. The SHELXTL package and OLEX2 were used for structure solution and refinement.41 Formula: C45H69N2SBr; MW: 749.99 g mol−1; crystal size: 0.15 × 0.15 × 0.1 mm; crystal system: monoclinic; space group: P21; a: 11.543(2) Å; b: 11.5238(17) Å; c: 15.689(3) Å; α: 90.00°; β 100.072(4)°; γ 90.00°; V: 2054.8(6) Å3; Z: 2; Dcalc.: 1.212 g cm−3; crystal shape: block; crystal colour: colourless; μ: 1.083 mm−1; F(000): 808; measured reflections: 14473; unique reflections: 12111(Rint = 0.0310); parameters refined: 451; Goof on F2: 0.870; final R indexes [I > 2σ (I)]: R1 = 0.0535, wR2 = 0.0630; final R indexes [all data] R1 = 0.0829, wR2 = 0.0759; Flack parameter 0.018(6), CCDC 882873.
X-ray powder diffraction data
XRD data were collected using a PANalytical's X'PERT Pro MPD powder diffractometer equipped with a Cu-K X-ray (λ = 1.542 Å) source. The 2θ range was 4°−90°, with a step size of 0.016°.General procedure for the synthesis of chiral isothiocyanates
Chiral isothiocyanates were prepared by analogy to reported procedures.42 At 0 °C under N2, to a solution of 1 eq. of N,N′-dicyclohexylcarbodiimide and 1 eq. of chiral amine in dry Et2O ether 3 eq. of CS2 was added dropwise. The reaction was allowed to warm slowly to 20 °C, and then stirred overnight. TLC was used to confirm when the reaction was complete. Then, the precipitated by-product (N,N′-dicyclohexylthiourea) was removed by filtration, and the solvent with an excess of CS2 was evaporated under reduced pressure. The residue was purified by column chromatography (SiO2, hexane).CS), 128.85 (CH), 128.15 (CH), 125.36 (CH), 56.98 (CH), 24.93 (CH3). Calcd for C9H9NS: C 66.22, H 5.56, N 8.58, S 19.64%; found: C 66.03, H 5.60, N 8.55, S 19.44%. HRMS-EI (m/z) calcd for C9H9NS [M]+ 163.0456, found 163.0478.
General procedure for the synthesis of chiral thioureas
Chiral thioureas were prepared by analogy to reported procedures.42 At 0 °C, under dinitrogen, 1 eq. of the chiral amine was added dropwise to a solution of 1 eq. of a chiral isothiocyanate in dry CH2Cl2. Then, the cooling bath was removed and the mixture was stirred overnight at 20 °C. In the case of 1a, the product precipitated during the reaction. When the absence of starting materials in the reaction mixture was confirmed by TLC, the product was collected by filtration, washed with CH2Cl2 and diethyl ether, and dried in vacuo. For the other thioureas, which were readily soluble in CH2Cl2, after the completion of the reaction, the solvent was evaporated and the residue was purified by column chromatography (SiO2, hexane:CH3CO2Et 2:1).
:CH3CO2Et 2:1); mp 199.6–200.9 °C (lit.4 mp 199–200 °C); [α]20D = 104.4 (c = 1.0, CHCl3), (lit.4 [α]25D +114.2 (c 1.05, CHCl3)); 1H NMR (300 MHz, DMSO-d6) δ 7.74 (br s, 2H, 2 × NH), 7.46–7.04 (m, 10H, Ar CH), 5.42 (br s, 2H, 2 × CH), 1.39 (d, J = 6.9 Hz, 6H, 2 × CH3); 13C NMR (75 MHz, DMSO-d6) δ 180.80 (SC), 144.26 (C), 128.26 (CH), 126.67 (CH), 126.02 (CH), 52.24 (CH), 22.44 (CH3); calcd for C17H20N2S: C 71.79, H 7.09, N 9.85, S 11.27%; found: C 71.47, H 7.11, N 9.76, S 11.19%. HRMS-ESI (m/z) calcd for C17H21N2S [M + H]+ 285.1425, found 285.1426.
:CH3CO2Et 2:1); mp 101.6–102.4 °C; [α]20D = 14.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 7.79 (d, J = 6.5 Hz, 1H, N–H), 7.31–7.04 (m, 6H, Ar CH, N–H), 7.14 (d, J = 8.2 Hz, 1H, Ar′ CH), 6.95 (dd, J = 8.1, 1.8 Hz, 1H, Ar′ CH), 6.83 (d, J = 1.5 Hz, 1H, Ar′ CH), 5.42 (br s, 1H, CH), 3.51 (br s, 1H, NCH1), 3.31 (br s, 1H, NCH2), 2.78 (sep, J = 7.0 Hz, 1H, CH), 2.79–2.62 (m, 2H, CH2), 2.25 (br d, J = 12.3 Hz, 1H, CHH), 1.91–1.63 (m, 2H, CH2), 1.63–1.48 (m, 2H, CH2), 1.37 (d, J = 6.9 Hz, 3H, CH3), 1.43–1.25 (m, 3H, CH, CH2), 1.25–1.04 (m, 1H, CHH), 1.17 (d, J = 7.0 Hz, 6H, 2 × CH3), 1.12 (s, 3H, CH3), 0.85 (s, 3H, CH3); 13C NMR (101 MHz, DMSO) δ 182.47 (SC), 147.03 (C), 144.85 (C), 144.35 (C), 134.47 (C), 128.12 (CH), 126.50 (CH), 126.38 (CH), 125.90 (CH), 124.01 (CH), 123.49 (CH), 53.81 (CH2), 52.07 (CH), 44.21 (CH), 37.96 (C), 37.40 (CH2), 36.94 (C), 35.74 (CH2), 32.88 (CH), 29.52 (CH2), 25.07 (CH3), 24.01 (CH3), 23.86 (CH3), 22.25 (CH3), 18.80 (CH3), 18.45 (CH2), 18.20 (CH2); calcd for C29H40N2S: C 77.63, H 8.99, N 6.24, S 7.15%; found: C 78.11, H 8.72, N 6.37, S 7.06%. HRMS-ESI (m/z) calcd for C29H41N2S [M + H]+ 449.2990, found 449.2985.
:CH3CO2Et 2:1); mp 139–141°C; [α]20D = 14.3 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.2 Hz, 2H, 2 × Ar CH), 6.98 (dd, J = 8.1, 1.7 Hz, 2H, 2 × Ar CH), 6.88 (s, 2H, 2 × Ar CH), 5.75 (br s, 2H, 2 × NH), 3.36 (br m, 4H, 2 × NCH2), 2.98–2.74 (m, 4H, 2 × CH2), 2.83 (sep, J = 6.9 Hz, 2H, 2 × CH), 2.27 (br d, J = 12.7 Hz, 2H, 2 × CHH), 1.95–1.55 (m, 8H, 2 × CHH, 2 × CH2, 2 × C′HH), 1.51–1.16 (m, 8H, 2 × C′HH, 2 × CH, 2 × CH2), 1.22 (d, J = 6.9 Hz, 12H, 4 × CH3), 1.20 (s, 6H, 2 × CH3), 0.96 (s, 6H, 2 × CH3); 13C NMR (101 MHz, CDCl3) δ 182.92 (CS), 146.85 (C), 145.68 (C), 134.49 (C), 126.84 (CH), 124.03 (CH), 123.84 (CH), 55.28 (CH2), 45.71 (CH), 38.23 (CH2), 37.73 (C), 37.42 (C), 36.74 (CH2), 33.40 (CH), 29.97 (CH2), 25.17 (CH3), 23.95 (2 × CH3), 19.08 (CH2), 18.65 (CH2), 18.46 (CH3); calcd for C41H60N2S: C 80.33, H 9.87, N 4.57, S 5.23%; found: C 80.30, H 9.69, N 4.52, S 5.38%. HRMS-ESI (m/z) calcd for C41H61N2S [M + H]+ 613.4555, found 613.4569.
General procedure for the synthesis of chiral thiouronium salts
Alkylation reactions were prepared by analogy to reported procedures.11 A solution of thiourea (1 eq.) and 1-halobutane (1.05–6 eq.) in EtOH was heated under reflux for 24 h (1-iodobutane required 65 °C, 1-bromobutane required 80 °C, both reactions in a standard reflux apparatus, and 1-chlorobutane required 90 °C in a sealed glass tube). The consumption of thiourea was monitored by TLC and the reaction time was extended if necessary. When TLC indicated the end of the reaction, the solvent and the excess of haloalkane were evaporated under reduced pressure. Products were dried in vacuo.Note: Since NMR spectra of thiouronium salts are influenced by hindered rotamerism about partial double bonds CN and CS, in some cases a mixture of rotamers/isomers is recognised and 1H NMR is then appropriately described by partial integration of protons or additional notations, e.g. for the same CH3 group available in two isomers: δ 0.90–0.75 (m, 0.5 × 3H, C′′H3), 0.49 (s, 0.5 × 3H, C′′H3).
S), 141.89 (C), 140.87 (C), 128.56 (CH), 127.84 (CH), 127.47 (CH), 126.27 (CH), 125.85 (CH), 55.71 (CH), 53.12 (CH), 32.63 (CH2), 30.73 (CH2), 22.29 (CH3), 21.22 (CH3), 20.80 (CH2), 13.22 (CH3); calcd for C21H29N2SBr: C 59.85, H 6.94, N 6.65, S 7.61%; found: C 60.05, H 6.74, N 6.65, S 7.69%. HRMS-ESI (m/z) calcd for C21H29N2S [M − Br]+ 341.2051, found 341.2064, calcd for [Br]− 78.9183, 80.9163, found 78.9182, 80.9162.
:2), 8.80 (br d, J = 171.2 Hz, 1H, Ab-NH, ratio 3:2), 7.38 (s, 2H, Ar CH), 7.35–6.90 (m, 3H, Ar CH), 7.22–7.01 (m, 1H, Ar CH), 7.01–6.90 (m, 1H, Ar CH), 6.86 (s, 1H, Ar CH), 5.28 (br d, J = 72.7 Hz, 1H, CH, ratio 2:3), 3.74–3.05 (m, 4H, N–CH2, S–CH2), 2.95–2.70 (m, 3H, CH2, CH), 2.36–1.98 (m, 1H, C′HH), 1.98–1.44 (m, 5H, C′′HH, CH2, CH2), 1.54 (br s, 3H, CH3), 1.44–1.27 (m, 4H, C′′HH, CH, CH2), 1.26–1.05 (m, 3H, C′HH, CH2), 1.14 (d, J = 7.4 Hz, 6H, 2 × CH3), 1.02 (s, 3H, CH3), 0.92 (s, 0.6 × 3H, C′H3), 0.90–0.75 (m, 0.5 × 3H, C′′H3), 0.70 (s, 0.3 × 3H, C′H3), 0.49 (s, 0.5 × 3H, C′′H3); 13C NMR (101 MHz, DMSO-d6) δ 166.78 (CS), 146.62 (C), 145.00 (C), 141.86 (C), 133.92 (C), 128.57 (CH), 127.67 (CH), 126.30 (CH), 126.18 (CH), 125.82 (CH), 123.96 (CH), 123.58 (CH), 123.38 (CH), 55.76 (CH), 53.21 (CH), 54.70 (CH2), 52.77 (CH2), 44.47 (CH), 43.50 (CH), 37.89 (C), 37.07 (C), 35.37 (CH2), 32.91 (CH), 32.42 (CH2), 31.13 (CH2), 30.51 (CH2), 29.57 (CH2), 29.13 (CH2), 25.07 (CH3), 23.95 (CH3), 22.21 (CH3), 20.99 (CH2), 18.55 (CH2), 18.19 (CH3), 17.96 (CH2), 13.33 (CH3), 12.90 (CH3); calcd for C33H49N2SBr: C 67.67, H 8.43, N 4.78, S 5.47%; found: C 68.21, H 8.96, N 5.22, S 5.28%. HRMS-ESI (m/z) calcd for C33H48N2SBr [M − H]+ 583.2722, found 583.2732, calcd for [M − Br]+ 505.3616, found 505.3516.
S), 146.74 (C), 145.58 (C), 134.45 (C), 126.76 (CH), 123.89 (CH), 123.67 (CH), 54.53 (CH2), 45.76 (CH′), 44.46 (CH′), 39.64 (C), 38.13 (CH2), 37.42 (CH2), 33.67 (C), 33.40 (CH), 30.74 (CH2), 29.82 (CH2), 25.17 (CH3), 23.94 (CH3), 21.66 (CH2), 19.24 (CH2), 18.34 (CH3), 13.38 (CH3); calcd for C45H69N2SBr: C 72.06, H 9.27, N 3.74, S 4.28%; found: C 72.01, H 9.30, N 3.67, S 4.25%. HRMS-ESI (m/z) calcd for C45H69N2S [M – Br]+ 669.5181, found 669.5143.
S), 146.69 (C), 144.91 (C), 134.14 (C), 126.27 (CH), 123.86 (CH), 123.44 (CH), 54.59 (CH2), 53.41 (CH2), 44.92 (CH), 43.99 (CH), 37.74 (CH2), 36.93 (C), 36.23 (CH2), 35.47 (C), 32.87 (CH), 32.16 (CH2), 30.98 (CH2), 29.37 (CH2), 25.06 (CH3), 23.91 (CH3), 21.12 (CH2), 18.18 (CH2), 18.03 (CH3), 17.86 (CH2), 13.09 (CH3); calcd for C45H69N2SCl: C 76.60, H 9.86, N 3.97, S 4.54%; found: C 76.18, H 10.15, N 3.94, S 4.49%. HRMS-ESI (m/z) calcd for C45H69N2S [M − Cl]+ 669.5181, found 669.5182.
C), 146.69 (C), 145.00 (C), 134.04 (C), 126.27 (CH), 123.88 (CH), 123.53 (CH), 54.96 (CH2), 53.61 (CH2), 45.08 (CH), 44.17 (CH), 38.61 (C), 37.71 (CH2), 36.93 (C), 36.09 (CH2), 32.86 (CH), 32.19 (CH2), 30.84 (CH2), 29.12 (CH2), 24.96 (CH3), 23.91 (CH3), 21.10 (CH2), 18.57 (CH2), 18.17 (CH2), 17.91 (CH3), 17.66 (CH2), 13.09 (CH3); calcd for C45H69N2SI: C 67.81, H 8.73, N 3.51, S 4.02%; found: C 67.34, H 8.39, N 3.53, S 4.04%. HRMS-ESI (m/z) calcd for C45H69N2S [M − I]+ 669.5181, found 669.5093.
General procedure for metathesis reaction
Metathesis reactions were performed by mixing a CH2Cl2 solution of thiouronium bromide (1 eq.) with an aqueous solution (or suspension) of a salt containing the desired anion (1 eq.). The sources of anions were Li[NTf2], Ag[NO3], Ag[OTf] or Ag[CH3CO2]. Then the by-products were collected by filtration, the organic phase was separated, dried with MgSO4, and the solvent removed by evaporation. The product was then dried in vacuo.S), 141.77 (C), 140.72 (C), 128.60 (CH), 127.93 (CH), 126.18 (CH), 125.81 (CH), 124.30 (CH), 119.5 (q, J = 323.2 Hz, CF3), 55.59 (CH), 53.01 (CH), 32.47 (CH2), 30.68 (CH2), 22.15 (CH3), 21.07 (CH3), 20.82 (CH2), 13.17 (CH3); calcd for C23H29F6N3O4S3: C 44.44, H 4.70, N 6.76, S 15.47%; found: C 44.32, H 4.91, N 6.74, S 15.45%. HRMS-ESI (m/z) calcd for C21H29N2S [M − NTf2]+ 341.2051, found 341.2044, calcd for C2NO4S2F6 [NTf2]− 279.9173, found 279.9166.
:2), 8.72 (br d, J = 194.5 Hz, 1H, Ab-NH, ratio 3:2), 7.51–6.91 (m, 5H, Ar 5 × CH), 7.13 (br s, 1H, Ar CH), 6.97 (br s, 1H, Ar CH), 6.88 (s, 1H, Ar CH), 5.21 (quin, J = 6.7 Hz, 1H, CH), 3.65–3.25 (m, 2H, N–CH2), 3.45–3.05 (m, 2H, S–CH2), 3.00 – 2.65 (m, 3H, CH, CH2), 2.36–2.01 (m, 1H, C′HH), 1.90–1.65 (m, 2H, CH2), 1.64–1.45 (m, 5H, CH, CH2, CH2), 1.44–1.28 (m, 4H, C′′HH, CH2, CH), 1.27–1.08 (m, 2H, C′HH, 0.3 × C′H3), 1.16 (br d, J = 6.7 Hz, 6H, 2 × CH3), 1.07–1.00 (m, 1H, C′′HH), 1.04 (s, 2H, 0.6 × C′H3), 0.94 (s, 0.6 × 3H, CH3), 0.89 (br s, 0.5 × 3H, CH3), 0.71 (s, 0.3 × 3H, CH3), 0.52 (s, 0.5 × 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 166.82 (SC), 146.60 (C), 145.02 (C), 141.79 (C), 133.87 (C), 128.58 (CH), 127.70 (CH), 126.28 (CH), 126.07 (CH), 125.72 (CH), 123.95 (CH), 123.58 (CH), 119.45 (q, J = 323.2 Hz, CF3), 55.62 (CH), 54.87 (CH2), 53.14 (CH), 52.79 (CH2), 44.50 (CH), 43.61 (CH), 37.85 (C), 37.10 (C), 35.33 (CH2), 32.89 (CH), 32.32 (CH2), 31.09 (CH2), 30.42 (CH2), 29.50 (CH2), 29.13 (CH2), 25.00 (CH3), 23.91 (CH3), 22.14 (CH3), 20.97 (CH2), 18.53 (CH2), 18.16 (CH3), 17.94 (CH2), 13.26 (CH3), 12.86 (CH3); calcd for C35H49F6N3O4S3: C 53.49, H 6.28, N 5.35, S 12.24%; found: C 53.54, H 5.71, N 5.46, S 11.93%. HRMS-ESI (m/z) calcd for C33H49N2S [M − NTf2]+ 505.3616, found 505.3590, calcd for C2NO4S2F6 [NTf2]− 279.9173, found 279.9173.
S), 146.64 (C), 145.00 (C), 134.04 (C), 126.26 (CH), 123.87 (CH), 123.52 (CH), 119.45 (q, J = 323.3 Hz, CF3), 55.01 (CH2), 53.61 (CH2), 45.04 (CH), 44.18 (CH), 38.56 (C), 37.72 (CH2), 36.93 (C), 36.07 (CH2), 32.87 (CH), 32.13 (CH2), 30.83 (CH2), 29.30 (CH2), 24.98 (CH3), 23.86 (CH3), 21.10 (CH2), 18.55 (CH2), 18.12 (CH2), 17.88 (CH3), 17.68 (CH2), 13.07 (CH3); calcd for C47H69F6N3O4S3: C 59.41, H 7.32, N 4.42, S 10.12%; found: C 59.68, H 7.39, N 4.43, S 10.52%. HRMS-ESI (m/z) calcd for C45H69N2S [M − NTf2]+ 669.5181, found 669.5080, calcd for C2NO4S2F6 [NTf2]− 279.9173, found 279.9162
S), 146.72 (C), 145.02 (C), 134.02 (C), 126.28 (CH), 123.89 (CH), 123.54 (CH), 121.61 (q, J = 132.31 Hz, CF3), 55.01 (CH2), 53.57 (CH2), 45.05 (CH), 44.18 (CH), 38.59 (C), 37.74 (CH2), 36.95 (C), 36.10 (CH2), 32.88 (CH), 32.14 (CH2), 30.85 (CH2), 29.14 (CH2), 25.01 (CH3), 23.88 (CH3), 21.12 (CH2), 18.57 (CH2), 18.20 (CH2), 17.91 (CH3), 17.67 (CH2), 13.10 (CH3); calcd for C46H69F3N2O3S2: C 67.44, H 8.49, N 3.42, S 7.83%; found: C 67.91, H 8.96, N 3.29, S 7.12%. HRMS-ESI (m/z) calcd for C45H69N2S [M − OTf]+ 669.5181, found 669.5120, calcd for CO3F3S [OTf]− 148.9520, found 148.9505.
S), 146.71 (C), 145.01 (C), 134.07 (C), 126.27 (CH), 123.91 (CH), 123.53 (CH), 54.80 (CH), 53.54 (CH), 45.07 (CH2), 44.16 (CH2), 38.71 (C), 37.73 (CH2), 36.95 (C), 36.08 (CH2), 32.87 (CH), 32.15 (CH2), 30.85 (CH2), 29.28 (CH2), 23.91 (CH3), 23.88 (CH3), 21.11 (CH2), 18.58 (CH2), 18.17 (CH2), 17.91 (CH3), 17.69 (CH2), 13.10 (CH3); calcd for C45H69N3SO3: C 73.82, H 9.50, N 5.74, S 4.38%; found: C 73.87, H 9.26, N 5.58, S 4.41%. HRMS-ESI (m/z) calcd for C45H69N2S [M − NO3]+ 669.5181, found 669.5225.
S), 147.37 (C), 145.37 (C), 134.73 (C), 126.75 (CH), 124.16 (CH), 123.69 (CH), 57.57 (CH2), 45.78 (CH), 38.42 (CH2), 38.18 (C), 37.46 (C), 36.43 (CH2), 33.43 (CH), 32.61 (CH2), 32.30 (CH2), 30.19 (CH2), 25.32 (CH3), 23.98 (CH3), 23.28 (CH3CO2), 21.91 (CH2), 19.03 (CH2), 18.68 (CH2), 18.66 (CH3), 13.49 (CH3); calcd for C47H72N2SO2: C 77.42, H 9.95, N 3.84, S 4.40%; found: C 77.84, H 9.92, N 4.08, S 3.86%. HRMS-ESI (m/z) calcd for C45H69N2S [M − CH3CO2]+ 669.5181, found 669.5212.
Preparation of chiral guests
N-Acetylation reactions on phenylalanine, tryptophan,43 and 3,4-dihydroxyphenylalanine44 were performed according to a literature method. Racemisation of (S)-naproxen was performed according to the literature.45 Preparation of 1-phenylethylsulfonic acid,46 and 1-phenylethylphosphonic acid,47 were conducted according to the literature. One equivalent of tetrabutylammonium hydroxide, [N4444][OH], in the form of a 1 M solution in methanol, was added dropwise to one equivalent of a chiral acid dissolved or suspended in methanol. After 1−12 h, the solvent was evaporated and the residue was dried in vacuo.Titration experiments and binding constant determination
The solution of the guest (cconst = 2 mM) was used for preparation of the host solution (c = 11.7–46.6 mM) to maintain a constant concentration of the guest. In an NMR tube, the guest solution (0.5 cm3) was titrated by 5 μl doses of a host solution using a microsyringe; sample zero and each one after addition of a dose was analysed using 1H NMR spectroscopy (500 MHz; 300.0 K). Data from these experiments were plotted to create a 1:1 NMR binding isotherm, which represents the equilibrium:| guest + host ⇌ [(guest)(host)]. |
Using NMR spectroscopy, the latter may be mathematically described by eqn (1) and (2):35,36
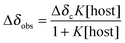 | (1) |
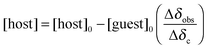 | (2) |
Computational studies
The starting structures for geometry optimisations were generated using HyperChemTM Release 8.0.749 with the MM+ level followed by the semi-empirical PM3 level. Then the final calculation was done using Gaussian® 98 (Gaussian Inc.) with the RHF/3-21G* level for optimisation of thiouronium isomers and the RB3LYP/STO-3G level for optimisations of host–guest complexes.28Acknowledgements
We are very grateful to QUILL and its Industrial Advisory Board for funding, and also wish to thank Prof. A. Prasanna de Silva, Prof. Christopher R. Strauss, Dr Marijana Blesic, Dr Johan Jacquemin, Miłosz Grabski and Tiina Laaksonen for advice, and Richard Murphy and Dr Mark Russell for technical support. P.N. thanks the EPSRC for a RCUK fellowship. The EPSRC UK National Crystallography Service (NCS) and the CBER QUB are acknowledged for single crystal and powder XRD data, respectively.Notes and references
- J. W. Steed, Supramolecular Chemistry, John Wiley & Sons, Ltd, Chichester, 2009 Search PubMed.
- (a) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS; (b) R. J. Fitzmaurice, G. M. Kyne, D. Douheret and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2002, 841–864 RSC; (c) M. D. Best, S. L. Tobey and E. V. Anslyn, Coord. Chem. Rev., 2003, 240, 3–15 CrossRef CAS.
- (a) A. Echavarren, A. Galan, J. M. Lehn and J. De Mendoza, J. Am. Chem. Soc., 1989, 111, 4994–4995 CrossRef CAS; (b) A. Galan, D. Andreu, A. M. Echavarren, P. Prados and J. De Mendoza, J. Am. Chem. Soc., 1992, 114, 1511–1512 CrossRef CAS.
- M. Hernández-Rodríguez and E. Juaristi, Tetrahedron, 2007, 63, 7673–7678 CrossRef.
- B. Altava, D. S. Barbosa, M. Isabel Burguete, J. Escorihuela and S. V. Luis, Tetrahedron: Asymmetry, 2009, 20, 999–1003 CrossRef CAS.
- (a) W.-S. Yeo and J.-I. Hong, Tetrahedron Lett., 1998, 39, 8137–8140 CrossRef CAS; (b) Y. Kubo, M. Tsukahara, S. Ishihara and S. Tokita, Chem. Commun., 2000, 653–654 RSC; (c) H. R. Seong, D. S. Kim, S. G. Kim, H. J. Choi and K. H. Ahn, Tetrahedron Lett., 2004, 45, 723–727 CrossRef CAS; (d) M. Orlowska, M. Mroczkiewicz, K. Guzow, R. Ostaszewski and A. M. Klonkowski, Tetrahedron, 2010, 66, 2486–2491 CrossRef CAS; (e) W. S. Yeo and J. I. Hong, Tetrahedron Lett., 1998, 39, 3769–3772 CrossRef CAS.
- (a) C. Blanchard and S. Pagano, Eur. Pat.,, EP1517799 (A1), 2005 Search PubMed; (b) M. D. Scaia, D. R. Eaton, R. J. Hoekstra and E. A. Haupfear, US Pat., US20060106248A1, 2006 Search PubMed; (c) T. Kazuhiro and I. Terukazu, Ger. Pat., DE2062559A, 1972 Search PubMed; (d) J. V. Hagen, U. Michelsen and M. Wehsling, World Pat., WO2008/58604 A1, 2008 Search PubMed.
- F. A. Carey and R. J. Sundberg, Advanced organic chemistry: Reactions and synthesis, Springer, New York, NY, 2007 Search PubMed.
- D. M. Kneeland, K. Ariga, V. M. Lynch, C. Y. Huang and E. V. Anslyn, J. Chem. Soc., Perkin Trans. 2, 1993, 115, 10042–10055 CAS.
- F. Albericio, M. A. Bailen, R. Chinchilla, D. J. Dodsworth and C. Najera, Tetrahedron, 2001, 57, 9607–9613 CrossRef CAS.
- M. Abai, J. D. Holbrey, R. D. Rogers and G. Srinivasan, New J. Chem., 2010, 34, 1981–1993 RSC.
- Y. Takemoto, Chem. Pharm. Bull., 2010, 58, 593–601 CrossRef CAS.
- T. Gunnlaugsson, A. P. Davis, G. M. Hussey, J. Tierney and M. Glynn, Org. Biomol. Chem., 2004, 2, 1856–1863 CAS.
- G. M. Kyne, M. E. Light, M. B. Hursthouse, J. de Mendoza and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2001, 1258–1263 RSC.
- (a) M. Meot-Ner, Chem. Rev., 2005, 105, 213–284 CrossRef CAS; (b) A. M. Davis and S. J. Teague, Angew. Chem., Int. Ed., 1999, 38, 736–749 CAS.
- T. J. Wenzel, Discrimination of Chiral Compounds Using NMR Spectroscopy, John Wiley & Sons, Inc., Hoboken, New Jersey, 2007 Search PubMed.
- K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235–3250 CrossRef CAS.
- (a) Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed; (b) J. H. Davis Jr., Chem. Lett., 2004, 1072–1077 CrossRef.
- W. J. Gottstein and L. C. Cheney, J. Org. Chem., 1965, 30, 2072–2073 CrossRef CAS.
- (a) M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS; (b) C. B. Aakeroy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- According to the topological method for encoding hydrogen-bond patterns of organic compounds (ref. 20), each type of hydrogen-bond pattern can be easily described. For the thiouronium complexes with oxoanions, a binding pattern featuring two parallel hydrogen bonds may be described by the descriptor (Fig. 2), which shows an intermolecular ring (descriptor R) of 8 atoms, which contains 2 hydrogen bond acceptors (upper descriptor) and 2 hydrogen bond donors (lower descriptor). The same descriptor is found in the guanidinium system.
- This was verified in the laboratory by preparing the S-methyl, S-butyl, S-decyl and and S-hexadecylthiouronium derivatives. As the S-methylthiouronium salt showed slighty lower nonequivalences in the methine peak of racemic mandelate than S-butyl, and S-decyl and S-hexadecylthiourinium showed the same nonequivalences as S-butyl, it was decided to focus on the S-butyl derivative.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
- (a) H. Kessler and H. O. Kalinowski, Angew. Chem., Int. Ed., 1970, 9, 641–643 CrossRef CAS; (b) P. Hanson and D. A. Williams, J. Chem. Soc., Perkin Trans. 2, 1973, 2162–2165 RSC; (c) Q. P. B. Nguyen, J. N. Kim and T. H. Kim, Bull. Korean Chem. Soc., 2009, 30, 2093–2097 CrossRef CAS.
- In the latest report in this area, Kim and coworkers (ref. 24c) suggest that the charge delocalisation in the thiouronium ion contributes to its resonance structures and causes isomerism. This is incorrect.
- M. Ōki, Applications of dynamic NMR spectroscopy to organic chemistry, VCH Publishers, Deerfield Beach, FL, 1985 Search PubMed.
- J. K. M. Sanders and B. K. Hunter, Modern NMR spectroscopy: a guide for chemists, Oxford University Press, 1993 Search PubMed.
- M. J. Frisch, et al., Gaussian 98, Revision A.9, Gaussian, Inc., Pittsburgh, PA, 1998 Search PubMed.
- J. A. van den Berg and K. R. Seddon, Cryst. Growth Des., 2003, 3, 643–661 CAS.
- G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal Engineering: A Textbook, World Scientific Pub Co Inc, Singapore, 2011 Search PubMed.
- J. J. Golding, D. R. Macfarlane, L. Spiccia, M. Forsyth, B. W. Skelton and A. H. White, Chem. Commun., 1998, 1593–1594 RSC.
- J. M. Lehn, Supramolecular chemistry: concepts and perspectives, VCH, Weinheim, 1st edn 1995 Search PubMed.
- (a) P. Job, Ann. Chim., 1928, 9, 113–203 CAS; (b) P. Maccarthy, Anal. Chem., 1978, 50, 2165–2165 CrossRef CAS; (c) V. M. S. Gil and N. C. Oliveira, J. Chem. Educ., 1990, 67, 473–478 CrossRef CAS.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- K. A. Connors, Binding constants: the measurement of molecular complex stability, John Wiley & Sons, Inc., New York, 1987 Search PubMed.
- T. Ramstad, C. Hadden, G. Martin, S. Speaker, D. Teagarden and T. Thamann, Int. J. Pharm., 2005, 296, 55–63 CrossRef CAS.
- (a) H. Zhao, J. Chem. Technol. Biotechnol., 2010, 85, 891–907 CrossRef CAS; (b) T. Welton, in Ionic Liquids in Synthesis, eds. P. Wasserscheid and T. Welton, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, pp. 129–141 Search PubMed.
- (a) M. J. Muldoon, C. M. Gordon and I. R. Dunkin, J. Chem. Soc., Perkin Trans. 2, 2001, 433–435 RSC; (b) W. Linert, R. F. Jameson and A. Taha, J. Chem. Soc., Dalton Trans., 1993, 3181–3186 RSC; (c) A. Bernson and J. Lindgren, Polymer, 1994, 35, 4848–4851 CrossRef CAS.
- P. Schuster, G. Zundel and C. Sandorfy, The Hydrogen bond: recent developments in theory and experiments, North-Holland Pub. Co., Amsterdam, 1976 Search PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef.
- X. Jiang, Y. Zhang, X. Liu, G. Zhang, L. Lai, L. Wu, J. Zhang and R. Wang, J. Org. Chem., 2009, 74, 5562–5567 CrossRef CAS.
- R. Salas-Coronado, A. Vásquez-Badillo, M. Medina-García, J. G. García-Colón, H. Nöth, R. Contreras and A. Flores-Parra, THEOCHEM, 2001, 543, 259–275 CrossRef CAS.
- R. Shchepin, M. N. Moeller, H.-y. H. Kim, D. M. Hatch, S. Bartesaghi, B. Kalyanaraman, R. Radi and N. A. Porter, J. Am. Chem. Soc., 2010, 132, 17490–17500 CrossRef CAS.
- E. J. Ebbers, G. J. A. Ariaans, A. Bruggink and B. Zwanenburg, Tetrahedron: Asymmetry, 1999, 10, 3701–3718 CrossRef CAS.
- R. Yoshioka, O. Ohtsuki, M. Senuma and T. Tosa, Chem. Pharm. Bull., 1989, 37, 883–886 CrossRef CAS.
- N. S. Goulioukina, T. M. Dolgina, I. P. Beletskaya, J. C. Henry, D. Lavergne, V. Ratovelomanana-Vidal and J. P. Genet, Tetrahedron: Asymm., 2001, 12, 319–327 CrossRef CAS.
- SigmaPlot version 11.0, from Systat Software, Inc., San Jose, California, USA, www.sigmaplot.com.
- HyperChem(TM) Professional 7.51, Hypercube, Inc., 1115 NW 4th Street, Gainesville, Florida 32601, USA.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for (S)-α-methylbenzyl isothiocyanate, 1a, [2a][NTf2]; 1H, 13C, COSY and 1H–13C HSQC NMR spectra of dehydroabietyl isothiocyanate, 1b, 1c, [2c]Cl, [2a][NTf2], [2c][NTf2], [2c][CH3CO2], [2c][NO3] and [2c][OTf]; 1H, 13C, COSY, 1H–13C HSQC and 1H–15N HSQC NMR spectra of [2a]Br, [2b]Br and [2c]Br; a table with NMR traces used for determination of peak non-equivalences in tetrabutylammonium salts of chosen racemic acids; Cartesian coordinates of isomeric structures of [2a]+ optimised at the RHF/3-21G* level; Cartesian coordinates of [2c][3a] and [2c][3d] complexes optimised at the RB3LYP/STO-G* level, visualisation of [2c][3d] complexes, and complete ref. 28. CCDC 882873. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2nj40632b |
| ‡ This article is dedicated to Professor A. P. De Silva and Dr Tobias J. Lotz. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |