Variation of structural motifs in lanthanoid hydroxo clusters by ligand modification†
Philip C. Andrews
a,
William J. Gee
a,
Peter C. Junk
*b and
Massimiliano Massi
ac
aSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia
bSchool of Pharmacy and Molecular Sciences, James Cook University, Townsville, QLD 4811, Australia. E-mail: peter.junk@jcu.edu.au
cDepartment of Chemistry, Curtin University, Bentley, WA 61002, Australia
First published on 20th September 2012
Abstract
The isolation of multinuclear lanthanoid clusters with different nuclearities and geometries commenced as a serendipitous discovery and is now the focus of intense research, not only from a coordination chemistry point of view, but also in a variety of applied fields. This perspective analyses the structural library of reported lanthanoid hydroxo clusters to find trends linking their structural motifs with factors such as the specific lanthanoid, the bulkiness of the outer shell of ligands, the coordinating nature of the ligands, and the synthetic methodology. While trends are indeed starting to appear, does this signify that the synthetic chemistry of lanthanoid clusters has now progressed from serendipity to a more rational approach?
 Philip C. Andrews | Phil Andrews graduated from the University of Strathclyde in Glasgow in 1992 having completed his PhD studies on alkali metal organometallic complexes under the supervision of Prof. Robert Mulvey. He then took up a Royal Society Postdoctoral Fellowship at Griffith University in Queensland (1993–1994) working with Prof. Colin Raston before moving to Phillips University in Marburg on an Alexander von Humboldt Fellowship (1994–1995), working with Prof. Gernot Boche. Phil then moved back to Australia, this time to Monash University in Melbourne, as a Postdoctoral Research Fellow and was awarded an Australian Research Council QEII Fellowship in 1997. He took up a ongoing academic teaching and research position at Monash in 2004, and is currently an Associate Professor in the School of Chemistry. |
 William J. Gee | Dr William Gee completed his PhD in 2010 working with Prof. Peter Junk and Assoc. Prof. Philip Andrews on the chemistry of lanthanoid oxo/hydroxo clusters at Monash University. He is currently undertaking a postdoctoral fellowship with Prof. Stuart Batten synthesising coordination polymers containing variable length ligands. His research interests include polydentate ligand design and biomimetic inorganic chemistry. |
 Peter C. Junk | Peter Junk graduated from The University of Western Australia in 1984 obtaining a Bachelor of Science with first class Honours in Physical and Inorganic Chemistry, and completed his PhD in Organometallic Chemistry under the supervision of Professor Colin Raston in 1988. He received a D. Sc. from James Cook University in 2007 for his contributions to organometallic chemistry. Peter held Postdoctoral positions at the University of Alabama, UWA and Monash before gaining an academic position at James Cook University in 1997 as a Lecturer in Inorganic Chemistry. He moved to the School of Chemistry at Monash University in 2001 where he was progressively promoted to Professor of Chemistry and since mid-2012 has been the Nevitt Chair in Chemistry at James Cook University. Peter is an Associate Editor for NJC. |
 Massimiliano Massi | Massimiliano Massi obtained his PhD at the University of Bologna in 2005. He then moved to Monash University as a postdoctoral researcher, working on the synthesis of multinuclear lanthanoid clusters in the group of Peter Junk and Phil Andrews. In 2009, he obtained a lectureship at Curtin University, where is now working on the synthesis and photophysical properties of phosphorescent metal complexes. |
Introduction
In the last two decades, there has been steady advancement in the chemistry of multinuclear lanthanoid (Ln) clusters, not only in fundamental areas such as their synthesis and characterisation as novel and fascinating species, but also in their application in a variety of fields by exploiting, for example, the magneto-optical properties associated with the 4f electrons.1,2 Multinuclear metallic clusters are generally viewed as species possessing a metal-rich core, often surrounded by an envelope of various organic ligands, ranging from small molecules such as carbon monoxide to more complex multidentate moieties.3 Despite the fact that this view is generally shared between multinuclear clusters of the transition elements and rare earth elements, their nature is fundamentally different. The most striking difference relates to the metal-rich core, where transition metals are able to engage in metal–metal bonds by covalently sharing their respective nd orbitals. Moreover, the final nuclearity (intended as the number of metal ions present in the cluster core) and shape are often dictated by the preferential coordination number and geometry favoured by the specific metal. By contrast, in the case of lanthanoid elements, the specific coordination number is reliant on the size of the cation (the ionic radii of lanthanoid elements experience a steady decrease upon increase of the nuclear charge, a phenomenon most commonly known as the lanthanoid contraction) and the relative bulkiness of the coordinated ligand.4 These features render lanthanoid coordination complexes more “unpredictable” relative to transition metal elements. The bonding nature between the lanthanoid ions and donor atoms of the ligands is almost purely ionic and rather labile, meaning that in solution these complexes are often characterised by formation and dissociation equilibria.5 Lastly, the inner core nature of the 4f orbitals means that lanthanoid cations have no strict preference in their coordination geometry and favour high coordination numbers, with the most common ranging from 8 to 12.5 This unpredictable nature of the rare earth elements might have somewhat delayed the establishment of the chemistry of multinuclear lanthanoid cluster species. In fact, the majority of the early reports relating to f-block clusters evidence the isolation of these novel assemblies as a serendipitous outcome.6 Since then, many research groups worldwide have focused their attention on the isolation of multinuclear structures. To date, a vast library of examples of f-block clusters has been reported in the literature, containing species with variations in the specific lanthanoid ions and the nature of the surrounding ligands. On examining this library of clusters, trends are starting to become evident. In light of the intense research, the obvious question to ask is whether the chemistry of lanthanoid clusters has now advanced from a merely serendipitous undertaking to a more systematic and rational approach. The breakthrough in lanthanoid cluster chemistry would be the establishment of synthetic methodologies to achieve the isolation of species of defined nuclearities and geometries, starting from specific lanthanoid ions and appropriate ligand design. This possibility would represent a significant advancement, especially in relation to the application of these lanthanoid-rich compounds, as the functional properties of constructed clusters depend on the nuclearity, geometry and specific chemical formulation of the core. However, the rather unpredictable nature of the f-block elements might always represent a limiting point in the establishment of these (more controlled) synthetic methodologies.Lanthanoid hydroxo clusters
While a variety of clusters with different core formulations has been reported to date, this work will exclusively consider the family of the lanthanoid hydroxo clusters. This specific core composition was one of the first isolated and it is still one of the most investigated.7–11 This is not surprising if one considers the highly oxophilic nature of the lanthanoid elements and the synthetic conditions used when these clusters were serendipitously obtained. Lanthanoid hydroxo clusters are generally formed with a protocol termed controlled hydrolysis.4,6 Lanthanoid ions precipitate from aqueous solutions with formation of thermodynamically stable lanthanoid hydroxides, [Ln(OH)3] (these are often poorly defined in terms of stoichiometry and might be described as hydrated oxo/hydroxo compounds of the lanthanoid elements).5 Some of the lanthanoid ions are known to form these hydroxides even when the pH of the solution is just slightly acidic. The early stages of the formation of lanthanoid hydroxo clusters can indeed be regarded as the initial construction of the hydroxo cluster core. The process is then opportunely stopped before the extensive formation of insoluble [Ln(OH)3] takes place. Preventing the pre-formed molecular cluster core from further hydrolysis is the key feature of the controlled hydrolysis approach. This has been achieved effectively with the use of protecting ligands, which are molecular species containing heteroatoms with strong affinity for lanthanoid ions and capable of multidentate binding modes. The role of the protecting ligand is to surround the cluster core once it is formed, thus creating a lipophilic barrier to the OH− anions and effectively protecting the core from further hydroxide attack. The lipophilic nature of the shell also serves the purpose of reducing the solubility of the formed clusters in the aqueous medium, thus promoting the crystallization of species with generic formulation [Lnn(OH)a(H2O)bLc]m+ where L is the protecting ligand. The controlled hydrolysis approach to obtain lanthanoid clusters has also been achieved in the absence of protecting ligands.4,6,12–17 In this case, the pH is slowly raised by the addition of a diluted base (often aqueous NaOH) to the point where the solution becomes persistently turbid. This point is identified as the pH where the equilibrium has reached initial precipitation of lanthanoid hydroxide. The solution is then filtered and left undisturbed to crystallize out defined cluster species containing a lanthanoid hydroxo core. These species are, in general, highly charged, owing to the requirement of anions originating from the starting lanthanoid salt (e.g. Cl−, I−, NO3−, ClO4−) to counteract the positive charge of the core in the outer coordination sphere. The controlled hydrolysis approach was studied in detail by Zheng at pH values close to neutral, where he isolated lanthanoid hydroxo cluster cores using a variety of amino acids as protecting ligands.6These clusters were generally characterised as the cubane motif where four lanthanoid ions assemble within four vertices of a quasi-cubic arrangement, with the remaining four vertices occupied by four OH− anions (Fig. 1). The formulation of the cubane core is [Ln4(μ3-OH)4]8+, a structural motif that is reminiscent of analogous clusters of the transition metal elements such as the cubane Fe–S clusters ubiquitous in biology.18 The Ln cations and the hydroxo anions are alternately arranged so that each lanthanoid cation is bound to three OH− anions. This arrangement is perfectly in agreement with the inability of lanthanoid ions to form metal–metal bonds5 and hence relying on the stabilisation of the structure via ionic bonds. The outer shell of these cubane clusters is composed of amino acids. Not surprisingly, the amino acids are arranged so that their oxygen atoms bind directly to the lanthanoid ions in the vertices of the cube.6 Each carboxylate group bridges between two Ln ions at the opposite corners of each face of the cube. Despite the fact that in amino acids the amino group can also contribute as a donor ligand, albeit nitrogen based, in this series of clusters the amino group is never used for the coordination the lanthanoid cations. This behavior could be explained by the preference for the lanthanoid cations to bind oxygen, coupled with the preference of amino acids to exist in their zwitterionic form at the pH where the cubane cluster was formed (e.g. with the carboxylic acid in the form of anionic carboxylate and the amino group protonated).6 The only exception is constituted by the family of clusters isolated using tyrosine as protecting ligand. For this amino acid, the amino group is directly involved in the stabilisation of the cluster core, along with the two oxygen atoms of the carboxylate group. In this case though, the cluster core isolated was not the cubane-type [Ln4(μ3-OH)4]8+, but an elongated pentadecanuclear cluster with formulation [Eu15(μ3-OH)20(μ5-Cl)(μ3-Tyr)10(OH)2(μ-H2O)5(H2O)18]12+.6 This exemplifies how the change in the binding mode of similar ligands favours the stabilisation of clusters with different nuclearities and geometries. Tyrosine becomes a tridentate ligand (Fig. 2), but due to geometrical restrictions, the amino group cannot be used to bind any of the lanthanoid ions in the former cubane structure. This is especially evident if the two oxygen atoms, belonging to the carboxylate in the same amino acid ligand, are already binding to two lanthanoid ions on the same face of the cubic core. The core of this pentadecanuclear Eu cluster also incorporates water molecules and a chloro ligand, demonstrating that the nature of the protecting ligand has an influence not only on the geometry and nuclearity of the stabilised cluster core, but also on its chemical composition.
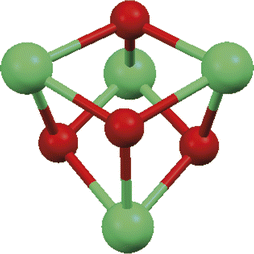 | ||
| Fig. 1 Structure of a cubane cluster core. Green balls identify Ln ions and red balls identify O atoms of the corresponding μ3-OH− ligands. | ||
![Binding mode of tyrosine in [Eu15(μ3-OH)20(μ5-Cl)(μ3-Tyr)10(OH)2(μ-H2O)5(H2O)18]12+.](/image/article/2013/NJ/c2nj40560a/c2nj40560a-f2.gif) | ||
| Fig. 2 Binding mode of tyrosine in [Eu15(μ3-OH)20(μ5-Cl)(μ3-Tyr)10(OH)2(μ-H2O)5(H2O)18]12+. | ||
Although a proper mechanism of formation of the cluster core would be too difficult to depict, it is now commonly accepted that hydroxo clusters are built up by sequential construction of the lanthanoid hydroxo bridged network.19 The water molecules bound to lanthanoid cations are characterised by an enhanced proton dissociation, originating from the strong Lewis acidity of trivalent lanthanoid ions.5 Therefore, upon controlled addition of a base, these water molecules will readily deprotonate and the formed lanthanoid-bound hydroxide can then coordinate to a second lanthanoid ion commencing the formation of hydroxo bridges. This process continues until the cluster is formed, surrounded by the protecting ligands, and eventually isolated from the solution. It is also highly plausible that, due to the lability of Ln coordination compounds, multiple cores of different nuclearities and shapes are formed at equilibrium in solution. The species that eventually precipitates can be exclusively isolated. The presence of these equilibria does indeed play a major role in the unpredictable nature of these species and it is one of the limiting factors in developing the isolation of serendipitous structures into a more rational approach.
The β-diketonate protecting ligands
One of the most studied protecting ligands in lanthanoid cluster chemistry is certainly the β-diketonate moiety, and many clusters with variable nuclearities and geometries have been efficiently stabilised by this ligand. Upon deprotonation of the methylene unit between the two carbonyl groups, the resonance-stabilised anionic β-diketonate ligand is able to form a six membered ring by chelating lanthanoid cations. Moreover, the two oxygen atoms of the ligand are also able to bridge two adjacent lanthanoid cations. This bridging mode seems to be a recurring theme in lanthanoid cluster chemistry, and it has been inferred that this characteristic is somewhat necessary in order to stabilise cluster cores. In fact, the amino acid ligands are found to bind to the cubane motif by bridging between adjacent lanthanoid ions. In these particular structures, the carboxylate anions do not bind with a chelating configuration, which could be explained by the formation of an unfavourable four membered ring arrangement. The early chemistry of lanthanoid clusters stabilised by β-diketonate ligands, was developed by Roesky and others,10,19–22 starting with simple ligands such as dibenzoylmethanide (1−). The reaction of hydrated LnCl3 in absolute alcohol with a weak base in the presence of dibenzoylmethane (1H) yielded neutral hydroxo clusters of formulation [Ln5(OH)5(1)10].19 These pentanuclear clusters are characterised by a square-based pyramidal motif, where each Ln cation is bridged by the hydroxo ligands (Fig. 3). | ||
| Fig. 3 Structure of a pentanuclear square-based pyramidal cluster core. Green balls identify Ln ions and red balls identify O atoms of the corresponding μ4-OH− (base) and four μ3-OH− ligands. | ||
The pathway for the formation of these clusters is assumed to be identical to the controlled hydrolysis in aqueous medium.6 The coordinated water molecules become deprotonated in the presence of the weak base and contribute to the growing of the hydroxo cluster core. As the formed cluster exposes hydrophobic phenyl groups outside the core, these species can be conveniently isolated in non-polar organic solvents such as toluene, where any remaining hydrophilic salt is insoluble. Because of its robustness and the relative stability of the obtained products, this methodology was optimised to access larger scales (up to gram quantities) of hydroxo cluster species.19 Three examples in particular within the work of Hubert-Pfalzgraf and Roesky prompted us to initiate a thorough investigation to understand how a specific cluster geometry and nuclearity is stabilised in relation to the specific lanthanoid cation, the relative bulkiness of the ligand and the incorporation of multiple donor atoms in the ligand structure. The use of allyl acetoacetate (L) as protecting ligand afforded nonanuclear clusters of formulation [Ln9O2(OH)8L10]+;10 the use of a bulkier ligand such as dibenzoylmethanide yielded pentanuclear clusters of formulation [Ln5(OH)5(1)10];19,21 lastly, the replacement of a phenyl ring in the dibenzoylmethanide ligand with a ferrocenyl unit afforded the cubane cluster core.22 It was clear that the cluster nuclearity could be somewhat controlled, or retained, by changing the chemical nature of the ligand at specific locations. Following this idea, we investigated the products obtained by performing controlled hydrolysis with the library of ligands schematised in Fig. 4. With just the exception of 18H, our work mainly focuses on maintaining the diketonate structure to stabilise the various cluster cores. While 1H is commercially available, the ligand series 2H–17H was synthesised via Claisen-type condensation between the corresponding enolate and either an ester or acyl chloride. On the other hand, the ligand 18H was prepared via 1,3-dipolar cycloaddition of NaN3 to 2-cyanopyridine.23
 | ||
| Fig. 4 Library of ligands used for the preparation of polynuclear lanthanoid hydroxo clusters. The diketone structures 1H–17H are represented in their corresponding enol forms. | ||
The dibenzoylmethanide ligand and the lanthanoid contraction effect
The first investigation into a more rational understanding of cluster formation consists of changing the lanthanoid of choice while maintaining unaltered the ligand structure. This investigation was carried out within both Roesky's and our research group using the dibenzoylmethane ligand 1H.20,21,24 The investigation originated from the fact that the structures of the pentanuclear cluster [Ln5(OH)5(1)10] were isolated using trivalent Y, Eu, and Lu. While isolating isostructural motifs for more than one lanthanoid elements is very common, the isolation of the same structure across the entire lanthanoid series is rarer. This is logically explained by the lanthanoid contraction effect, hence as the trivalent lanthanoid ions become smaller, their coordination requirement changes and different core nuclearities and geometries are inevitably preferred. Eu is positioned before the central Gd in the lanthanoid series and trivalent Y has a similar ionic radius to trivalent Ho. The investigation within Roesky's group focused on the synthesis of lanthanoid clusters using Ln of larger ionic radii than Eu. The results show that the bigger lanthanoid cations prefer a tetranuclear planar arrangement (Fig. 5) of formulation [Ln4(OH)2(1)10]. The hydroxo ligands encapsulated in the middle of the cluster core have μ3 bridging modes. The tetranuclear cluster structure was obtained with Pr, Nd, and Sm.20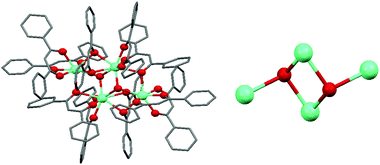 | ||
| Fig. 5 Structure of a tetranuclear cluster (left) with detailed view of the arrangement in the hydroxo core (right). Green balls identify Ln ions and red balls identify O atoms of the corresponding μ3-OH− ligands. | ||
A careful inspection of the tetranuclear and pentanuclear cluster motifs, using 1H as protecting ligand, immediately reveals the difference in the coordination sphere in the two cases. In the tetranuclear core, obtained for the bigger elements, each lanthanoid cation is nonacoordinated, whereas in the pentanuclear core the smaller lanthanoid ions prefer a coordination number of eight. Our studies complement these findings by isolating the pentanuclear [Ho5(OH)5(1)10] cluster.25 Moreover, the same pentanuclear motif was obtained for Gd, Dy, Er and Yb.26,27 Interestingly, we were able to isolate both the pentanuclear and tetranuclear hydroxo cluster cores for Eu.25 The result suggested that the size of the trivalent Eu cation was borderline for the stabilisation of both the tetranuclear core with nonacoordinated Eu ions as well as the pentanuclear core with the octacoordinated Eu ions. The separation of the two structures was possible due to their different solubility in organic solvents.
The exceptions of the dibenzoylmethanide cluster series
While the lanthanoid contraction qualitatively explains the trend on passing from a tetranuclear to a pentanuclear hydroxo cluster core, some exceptions have been reported. The first case was highlighted by our group while we were attempting to complete the lanthanoid dibenzoylmethanide cluster series, and specifically when we focused our attention on isolating the hydroxo cluster core of La.28 According to the trend, the trivalent La cations should prefer a tetranuclear cluster core, since this element is the lightest (hence the biggest) in the lanthanoid series. By performing the same synthetic procedure used to isolate the other clusters, we instead isolated an unprecedented dodecanuclear core (Fig. 6). | ||
| Fig. 6 Structure of the dodecanuclear cluster (top), the cluster core with highlighted the carbonate anions in balls-and-sticks representation (bottom left), and the phenylglyoxylate protecting ligand (bottom right). Colour code: grey C atoms, red O atoms, light blue La atoms. | ||
Interestingly, the formulation of this particular cluster was identified as [La12(OH)12(H2O)4(1)18(Phgly)2(CO3)2], where Phgly = phenylglyoxylate. The noteworthy feature of this exception to the tetra and pentanuclear series was the composition of the cluster core: while the core is predominantly of La hydroxo composition, bearing μ3-chelating OH− ligands, the core motif appears to be templated by two central carbonate anions. The templating effect is understandable by the highly intertwined nature of the CO32− ligands, each displaying a μ6–η1:η1:η1:η1:η1:η2 binding mode. The presence of the carbonate anions was tentatively explained by slow absorption of atmospheric CO2 into the mother liquor. The CO2 can then react with the water/hydroxide molecules coordinated to the La cation. This explanation is supported by other examples where lanthanoid complexes were shown to readily absorb atmospheric carbon dioxide and eventually incorporate carbonate anions into their structures.29,30 The second noteworthy aspect of this dodecanuclear cluster is the presence of two phenylglyoxylate ligands stabilising the inner core along with the rest of the dibenzoylmethanide ligands. Careful examination of the starting dibenzoylmethane reagent by mass spectrometry did not evidence the presence of phenylglyoxylic acid or its corresponding salts. We therefore tentatively explained its presence as an in situ oxidation of the dibenzoylmethanide ligand induced by the presence of atmospheric O2. Whether the presence of the La cations played a role in favouring this oxidation cannot be unambiguously demonstrated, however the group of Roesky had previously reported that pentanuclear clusters of Y are prone to catalyse the oxidation of aldehydes to carboxylic acid in the presence of air.19 They suggested that the mechanism involves the insertion of dioxygen into the aldehydic C–H bond with formation of a peroxyacid. The peroxyacid then rearranges to yield the carboxylic acid. If a similar mechanism is invoked for the dibenzoylmethanide ligand, the final products would result from the cleavage of the C–C bond between the carbonyl C atom and the vinylic C atom (assuming the dibenzoylmethanide is in its enolate form). This explanation is supported by the fact that the presence of benzoic acid was detected by analysing the mother liquor by mass spectrometry (but was not detected in the starting reagent 1H). It remains unclear why the formation of this dodecanuclear hydroxo/carbonato cluster is obtained only in the case of La. It is also not certain whether this particular cluster is obtained by templating a preformed cluster, possibly the tetranuclear hydroxo cluster core, or rather directly grown starting from the simple La precursors and the ligand.
The second exception to the tetranuclear and pentanuclear trend was reported more recently by the group of Holliday.26 Their investigation focused on preparing hydroxo clusters of lanthanoid ions that exhibit luminescence in the IR spectral range, viz. Nd, Er and Yb. While it was not surprising that the pentanuclear cluster core was obtained for Er and Yb, the same core motif was isolated for Nd. The trivalent neodymium cation has a larger ionic radius compared to trivalent europium, so according to the previously described trend, the Nd cations should favour the nonacoordinated environment and hence a tetranuclear arrangement. Interestingly, Holliday's group obtained these pentanuclear clusters following a different methodology. Instead of performing the controlled hydrolysis of the lanthanoid salt precursor, they pre-formed the mononuclear [Ln(1)3(H2O)2] complex to which they added excess lanthanoid ions, in the form of [Ln(OTf)3] where TfO−= trifluoromethanesulphonate. No base was required for the isolation of the final pentanuclear cluster, as according to the authors the triflate anions provide the necessary pH adjustment while the excess of lanthanoid cations favours the growth of the cluster species. The isolation of the pentanuclear clusters by Holliday clearly demonstrates that the experimental methodology employed for preparing the cluster species also has an influence on the obtained product, possibly in shifting the equilibrium of the variety of species in solution to a specific assembly. Therefore, the specific methodology must be properly factored in search of a more rational approach in this area of lanthanoid synthetic chemistry.
Manipulating steric dimensions of the diketonate scaffold
As a general rule, increasing the steric bulk of a ligand will have an adverse effect on the lanthanoid cluster nuclearity. This relationship can be demonstrated with a comparison of clusters stabilised by homoleptic ligand systems from closely related well-reported diketonate families. Such comparisons must be undertaken while retaining the metal identity in order to eliminate the effects of the lanthanoid contraction. The steric bulk of the diketonate scaffold can be increased solely in the plane of the chelation region, or concertedly in multiple planes. The former relates to diketonates where the sum of all functional groups (excluding hydrogen atoms) may align with the chelate plane. The latter relates to ligands unable to minimise their profile along a single plane. Extending the length of the planar diketone ligand acetylacetonate (acac−) shows the effect of extending the bulk of the ligand along a single plane. The successive addition of phenyl rings to acacH serves to crowd the lipophilic barrier around the Ln cores, better shielding the core and limiting aggregation of the pentanuclear subunits (Fig. 7).31–33 | ||
| Fig. 7 Increasing the steric bulk of acac-type ligands along the chelate plane decreases the nuclearity from a tetradecanuclear cluster (acac−) to a nonanuclear subunit (acetylbezoylmethanide) and eventually to a pentanuclear species (1−). Dysprosium metal is used in each example. | ||
Increasing the steric profile of the acacH backbone in multiple planes has a more pronounced impact on the cluster nuclearity, complicating attempts to predetermine the trend. This likely results from greater disruption of closely aligned parallel arrangements often observed around the cluster periphery. An increase in ligand–ligand repulsion culminates in a reduction of the number of ligands that comprise the cluster periphery, typically effecting a contraction in the size of the cluster. Known gadolinium examples highlight this relationship (Fig. 8). Here, acac− stabilises a hexanuclear cluster with a [Gd6(OH)6]12+ core motif.34 Increasing the size of the methyl termini to trifluoromethyl (CF3acac−) yields a cubane [Gd4(OH)4]8+ species,35 while even larger tert-butyl groups (t-Buacac−) stabilise only dimeric species [Gd2]6+ with no hydroxo ligands present.36
![Increasing the steric bulk of the acac− ligand in three dimensions reduces cluster nuclearity as highlighted by the three gadolinium species [Gd6(OH)6(acac)12], [Gd4(OH)4(H2O)4(CF3acac)8] and [Gd2(t-Buacac)6].](/image/article/2013/NJ/c2nj40560a/c2nj40560a-f8.gif) | ||
| Fig. 8 Increasing the steric bulk of the acac− ligand in three dimensions reduces cluster nuclearity as highlighted by the three gadolinium species [Gd6(OH)6(acac)12], [Gd4(OH)4(H2O)4(CF3acac)8] and [Gd2(t-Buacac)6]. | ||
It is evident that reducing the steric bulk of the ligand increases the likelihood of high nuclearity clusters, however, this also imparts upon greater freedom of coordination, potentially giving rise to multiple thermodynamically favoured products. An excellent example has been given above with the acacH ligand, which is known to stabilise multiple species including tetranuclear (Ln = Nd), hexanuclear (Ln = Gd), nonanuclear (Ln = Sm, Eu, Tb, Yb) and tetradecanuclear (Ln = Tb, Eu, Dy) clusters.31,34,37,38 In such instances, the identity of the product is largely influenced by the reaction conditions (i.e. solvent choice, crystallization conditions, etc.) and ionic radius of the lanthanoid cation.
para-Functionalised β-diketonate as protecting ligands
The next step in our investigation focused on the isolation of hydroxo clusters using functionalised dibenzoylmethanide ligands. The goal of this investigation was to improve the understanding on how variations in the phenyl rings influence the stabilisation of a specific cluster core. Moreover, we wanted to isolate lanthanoid hydroxo clusters whose outer sphere contains reactive functionalities, so that the formed cluster could be used as a precursor for further reactions or for the preparation of functional materials. We focused our attention on grafting small substituents into the para positions of the two phenyl rings: the substituents chosen were the methoxy, ethoxy and allyloxy groups thus yielding ligands 2H, 3H and 4H, respectively.25,39The isolation of cluster motifs using 2− as protecting ligand proved unsuccessful, typically yielding very insoluble mixtures. Only in the case of Nd, were we able to obtain crystals of X-ray quality from evaporating DMF solutions after several months. The structure was identified as the mononuclear complex [Nd(2)3(DMF)2],25 as shown in Fig. 9. The isolation of this mononuclear complex could be explained either by the fact that (i) a tetra or pentanuclear insoluble cluster was obtained in the reaction but when the cluster is dissolved in the highly polar DMF, the cluster core fragments and the product rearranges into the mononuclear complex; or (ii) the mononuclear complex is formed immediately and prevents the formation of any other structures, including hydroxo clusters, due to its insolubility. By comparing our results with available data reporting that lanthanoid clusters were rather unstable in aqueous medium but stable in highly polar organic solvents such as DMF and DMSO,40 we concluded that the formation of the mononuclear complex [Nd(2)3(DMF)2] was exclusively driven by its precipitation.
![Structure of the mononuclear complex [Nd(2)3(DMF)2].](/image/article/2013/NJ/c2nj40560a/c2nj40560a-f9.gif) | ||
| Fig. 9 Structure of the mononuclear complex [Nd(2)3(DMF)2]. | ||
When 3− was used as protecting ligand, we were able to isolate the pentanuclear core for Eu and Ho (Fig. 10).25 The core of this hydroxo cluster is isostructural with all the previously reported examples of pentanuclear clusters, demonstrating that the addition of ethoxy functionalities in the para positions of the phenyl rings does not interfere with the stabilisation of the core. On considering the crystal structure of both clusters, it can be easily rationalised why the same cluster motif is obtained using either 1− or 2− as protecting ligand: the ethoxy chains are oriented towards the outside of the outer shell and are not spatially close to the hydroxo core, thus they are not hindering the formation of this specific core motif.
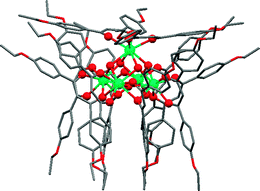 | ||
| Fig. 10 Structure of the pentanuclear cluster with the ethoxy functionalised ligands. The Ho cluster structure is used in this example. | ||
As expected, the same pentanuclear motif was obtained starting from ligand 4−, which contains an extra carbon atom in the functional groups with respect to 3−. Again, the allyloxy moieties are pointing towards the outside of the cluster assembly, thus the formation of the pentanuclear core is not hindered. We were able to obtain pentanuclear clusters of Eu, Tb, and Ho (Fig. 11).39
 | ||
| Fig. 11 Structure of the pentanuclear cluster with the allyloxy functionalised ligands. The Ho cluster structure is used in this example. | ||
From these results it can be concluded that functional groups can be introduced into the protecting ligands at positions that do not impose on the cluster core region. Isostructural motifs in the hydroxo clusters can be obtained with these families of differently functionalised ligands. However, other effects might govern the formation of a specific product, as in the case of the mononuclear [Nd(2)3(DMF)2] complex that was prevented from undergoing controlled hydrolysis due to insolubility. The other difficulty pertaining to the synthesis of hydroxo cluster species from para-functionalised dibenzoylmethanide ligands is the structural identification of the final product. As the complexity and flexibility of the added functional group increases, obtaining high quality single crystals suitable for X-ray diffraction becomes challenging. In fact, from the perspective of these functional methoxy, ethoxy and allyloxy chains, these cluster species begin to resemble spherical dendrimers originating from a lanthanoid hydroxo core. While X-ray data may be more difficult to obtain, preliminary results have shown that Raman spectroscopy is a useful tool in the identification of a specific cluster core, especially when considering the low wavenumber region (200–900 cm−1) where the Ln–O vibrations appear.39,41 It has been recognised that isostructural cluster cores possess a fingerprint series of bands, which remain unaltered upon modification of the outer ligands. Moreover, different cluster geometries and nuclearities possess fingerprint peaks that can be differentiated. Of course, the obvious requirement is to construct a library of Raman fingerprint spectra obtained from lanthanoid hydroxo clusters that have been previously structurally characterised via X-ray diffraction.
 | ||
| Fig. 12 Structure of the trinuclear Ho cluster obtained with the pyridyl functionalised ligand. Light green atoms are the two chloro ligands directly coordinated to the Ho centres. | ||
Pyridyl-functionalised β-diketonate as protecting ligands
After investigating the effect of para-functionalised ligands, we focused on including extra donor atoms in the aromatic rings of the dibenzoylmethanide structure. To maintain the steric profile of the protecting ligand, we decided to exchange pyridyl substituents for the two phenyl rings.42 We first synthesised the ligands 5H and 6H: the former contains the N donor atom adjacent to the carbonyl group, hence having the potential to interact with the cluster core, whereas the N donor atom in the latter is in position 4′. Due to the interaction of the pyridyl N atoms with the lanthanoid centres, no tetranuclear or pentanuclear motifs were isolated.In the case of 5−, a cationic trinuclear Ho cluster of formulation [Ho3(5)3(μ3-OH)2(H2O)4Cl2]Cl2 was structurally characterised (Fig. 12).42 The hydroxo ligands stabilise the planar cluster core by binding both sides in a μ3-coordinating fashion, analogous to that reported for the tetranuclear and pentanuclear hydroxo core. The structure is also stabilised by the three protecting ligands 5−, each of which uses the two O atoms of the carbonyl groups as well as one the two pyridyl N atoms. The stabilisation of this particular triangular core seems to occur due to the chelating and bridging capability of the ligand 5−. Each Ho centre is octacoordinated, binding to either water molecules or chloro anions to complete its coordination sphere. Interestingly, an isostructural cluster core was reported by Powell, using ortho-vanillin (namely 2-hydroxy-3-methoxy-benzaldehyde) as protecting ligand.43 The core of this trinuclear Dy cluster possesses identical μ3-coordinating fashion for the hydroxo ligands. Each Dy cation is again octacoordinated, binding to two ortho-vanillin ligands, two hydroxo ligands and completing the coordination sphere with water molecules and chloro anions. The most interesting feature of these two similar cluster species is the fact that an identical core motif was stabilised by two chemically different ligands that possess the donor atoms in identical positions (Fig. 13).
 | ||
| Fig. 13 Comparison of the binding modes of the 5− and the ortho-vanillin ligand. | ||
The controlled hydrolysis with ligand 6H did not result in the isolation of any polynuclear cluster. Instead, monodimensional polymeric chains were obtained in the case of Gd.42 The structure of this species was characterised by octacoordinated mononuclear Gd complexes. The coordination sphere of each Gd centre is composed of three η2 bidentate ligands 6−, one water molecule, and one N atom of the pyridyl ring belonging to an adjacent Gd complex. This motif is repeated to yield monodimensional chains [Gd(6)3(H2O)]∞ (Fig. 14). The individual chains are held together via hydrogen bonding between the non-coordinating N atom on one of the two pyridyl rings and the coordinated water molecule of a Gd complex in the adjacent chain. The result indeed demonstrates that the addition of donor atoms on the opposite side of the main coordinating pocket, the diketonate in this case, favours the formation of mono or multidimensional networks. The formation of this extended network is now competing with the formation of discrete cluster species. In the case presented, the high degree of hydrogen bonding further stabilises the polymeric chains favouring their exclusive crystallization. An analogous result was previously obtained by our group when we tried to stabilise cluster cores with benzoic acids possessing diol-terminated chains in the para position.44 Like in the case of [Gd(6)3(H2O)]∞, the isolated product was a hydrogen-bonded network of monodimensional chains composed of mononuclear La complexes.
 | ||
| Fig. 14 Structure of the Gd monodimensional polymer obtained with the pyridyl functionalised ligand. | ||
Thiophene-functionalised β-diketonate as protecting ligands
Substituting a phenyl ring of dibenzoylmethane for a thiophene group yields 7H, a model used to determine the effect of a softer donor atom adjacent to the chelation region. The protonated ligand 7H represents an ideal dibenzoylmethane substitute, to such an extent that identical unit-cell dimensions are observed for both in the crystalline state.45 The interchangeable nature of the ligand was confirmed when [Ln4(OH)2(7)10] (Ln = Nd, Eu) and [Er5(OH)5(7)10] motifs were isolated, of which both were structurally similar to the motifs shown in Fig. 3 and 5.46 Only a single crystallographically characterised example of a lanthanoid–thiophene interaction has been isolated to date which required tethering of the thiophene group at a capping position by rigid naphthalenol arms to effect, hence this result was not surprising.47ortho-Functionalised β-diketonate as protecting ligands
Imparting functionality at the ortho-phenyl position of dibenzoylmethane was found to increase the steric bulk of the resultant ligands in more than a single plane, owing to the ensuing restriction of the torsion angle of the functionalised ring.48 The parent ligands, 1H and 8H, were found to be suited to adopting a planar conformation owing to the extended conjugation between both rings and the resonance coupled diketones. The cluster chemistry of 1− has been extensively discussed above and 8− was found to stabilise both the tetranuclear [Nd4(OH)2(8)10] and the nonanuclear hourglass [Er9O2(OH)8(8)10]+ motifs. In each example close packing of diketonate ligands around the cluster periphery was observed.By contrast, the addition of functional groups (nitro, methoxy, fluoro) to ortho ring positions (9H, 10H, 11H and 12H in Fig. 4) was found to sterically block planar conformation owing to electron pair repulsion between the ortho group and a chelating oxygen from the diketonate. This small restriction in the conformational freedom of the ligand impacts on how the ligands align at the cluster periphery, ultimately influencing the cluster motif. Each of the ligands 9H, 10H, 11H and 12H were found to stabilise cubane clusters with a general composition of [Ln4(OH)4(H2O)x(L)8] (x = 0, 2; L=9, 10, 11, 12) (Fig. 15). The ortho-nitro variant of dibenzoylmethane, 9H, was found to yield a dihydrate, [Er4(OH)4(H2O)2(7)8]. The retention of aqua ligands in clusters ligated by diketonate ligands is rarely seen. This is attributed to the propensity of the diketonate ligands to adopt bridging modes and hence displace coordinated water molecules. Intra-cluster hydrogen bonding between both the hydroxo groups of the cubane core and the coordinated aqua ligands was observed, stabilising and directing ligand arrangement around the cluster periphery. The smaller, cyclopentanone-based ligand 10H also yielded a family of cubane clusters of composition [Ln4(μ3-OH)4(10)8] (Ln = Gd, Tb, Dy, Er). Each cluster was found to contain a crystallographically imposed ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) symmetry and each metal was seven-coordinate, derived from three μ-OH interactions (Fig. 15) and chelation to two ligands in a η2(O,O′) fashion.
symmetry and each metal was seven-coordinate, derived from three μ-OH interactions (Fig. 15) and chelation to two ligands in a η2(O,O′) fashion.
 | ||
| Fig. 15 Comparison of the cubane-type clusters obtained with ligands 9H, 10H, 11H and 12H (from top left to bottom right). | ||
The nitro group was next varied to methoxy in the ligand 11H. The resultant cubane [Er4(μ3-OH)4(11)8] was unusual in that examples of both hepta and octacoordinated metal centres were observed within the core, owing to only three of a possible four diketonate ligands participating in a μ–η2 bridging mode. Intra-cluster hydrogen-bonding was again observed as a stabilising force within the cluster, this time between two core μ3-OH groups and two inward facing methoxy groups from adjacent ligands. It is likely that ligands attempting to adopt torsion angles suitable for hydrogen bonding result in interligand crowding thus disrupting the final μ–η2 bridging mode. Longer hydrogen bond distances were observed relative to [Er4(OH)4(H2O)2(7)8], possibly due to variation in sterics and the ionic nature of the nitro group.
The final ortho-fluoro substituted ligand, 12H, a nonsymmetrical pentafluorinated variant of dibenzoylmethane, completes the cubane families. The cluster is a hydrate of composition [Er4(OH)4(H2O)2(12)8]. Similarities between the steric profile of 9− and 12− appear to mediate retention of the aqua ligands, with the size of the dibenzoylmethanide scaffold likely restricting the number of bridging modes possible. Weak intra-cluster hydrogen bonding was again inferred between the fluorinated rings and the coordinated aqua ligands, stabilising the ligand arrangement.
These results suggest that a targeted synthesis of the cubane cluster motif using diketonate ligands is viable by restricting the ligand from adopting a planar conformation, which we achieved by the inclusion of ortho-phenyl ring functionality. The size of the ligand scaffold was also found to determine the efficiency of aqua displacement, likely owing to inhibition of ligand bridging modes by larger scaffolds.
Aldol extension of the β-diketonate chelation region
There are instances where cluster species with increased nuclearities have been achieved by the addition of further coordinating atoms into the ligand scaffold. This is exemplified by a recent example where a phenyl ring of dibenzoylmethane was substituted with a o-phenol group, creating a hexanuclear yttrium cluster by elongating the wings of the tetranuclear butterfly motif shown in Fig. 16.20,49 A one-pot Claisen condensation aldol addition, yielding 13H and 14H after workup, allowed us to undertake a similar investigation.50 The aldol addition imparts a tertiary alcohol framed by a cyclopentane ring, giving a 1,3-diketo-5-hydroxy binding region. This kind of 1,3,5 array is analogous to the diformylphenolato ligand that commonly serves to coordinate two lanthanide ions.51,52 The phenyl species, 13H, was found to stabilise cationic tetranuclear clusters in a tetrahedral arrangement around a μ4-oxo core with the composition [Ln4(μ2-Cl)2(μ4-O)(13)6]Cl2 (Ln = Nd, Ho, Tb, Er) (Fig. 16). Retention of coordinated chloride was unexpected, as previous examples of the [Ln4(μ2-Cl)2(μ4-O)] core were only isolable using air-sensitive conditions or by partial hydrolysis of air-sensitive materials.53,54 In this instance, the bridged chloride products were generated using hydrated lanthanoid salts and wet solvent mixtures. The rationale for the unexpected inclusion of bridging chloride was a combination of the steric properties of the cyclopentane ring capping the alcohols, coupled with the bridging ability of the chloro ligand. Triethylamine was used to deprotonate 13H, which was sterically blocked from accessing the 5-hydroxy arm of the ligand by the shielding effect of the cyclopentanone ring when coordinated to lanthanoid metals. If the 5-hydroxy arm were to detach from the metal, such that it were base-accessible, the loss of electrophilic coordination imparted by the metal would deactivate the alcohol to deprotonation. Consequently, 13− becomes a highly-shielding tridentate ligand amenable to effecting aqua ligands displacement while remaining singly deprotonated. Conversion of an aqua ligand to a hydroxo bridge by triethylamine can be inferred to be reliant upon activation of the former by three Lewis-acidic lanthanoid metals, based on observed cluster motifs and prevalence of μ2 bridging aqua ligands, hence the inclusion of chloride anions in the μ2 bridging positions. Two free chloride anions, hydrogen bonding to the tertiary alcohols, balance the charge of the cluster.![Tetranuclear [Ln4(μ2-Cl)2(μ4-O)(13)6]Cl2 clusters in a tetrahedral arrangement around a μ4-oxo core.](/image/article/2013/NJ/c2nj40560a/c2nj40560a-f16.gif) | ||
| Fig. 16 Tetranuclear [Ln4(μ2-Cl)2(μ4-O)(13)6]Cl2 clusters in a tetrahedral arrangement around a μ4-oxo core. | ||
The inclusion of a para-methoxy group to the ligand, 14H, was found to stabilise a dinuclear motif of composition [Ln2(14)5]Cl (Ln = La, Nd, Tb, Dy, Er) (Fig. 17). A single cationic charge remains on the dimer, balanced by a hydrogen bonded chloride. Contraction of the core serves to minimise the cationic charge due to fewer trivalent lanthanoid ions as well as increasing the ratio of anionic ligands, bringing the dinuclear species closer to neutrality. It is likely that solubility differences imparted by the methoxy functionality are the cause of crystallization of the dimeric species as opposed to the cluster (as in the case of the mononuclear [Nd(2)3(DMF)2] complex),25 however steric or electronic arguments cannot be completely discounted.
![Structure of the dinuclear [Ln2(14)5]Cl cluster.](/image/article/2013/NJ/c2nj40560a/c2nj40560a-f17.gif) | ||
| Fig. 17 Structure of the dinuclear [Ln2(14)5]Cl cluster. | ||
Carbohydrate-functionalised β-diketonate as protecting ligands
The creation of chiral metal clusters represents a logical direction for the development of lanthanoid cluster chemistry, owing to demonstrated catalytic activity and applicability towards molecular probes.55–58 Shrouding the core in chiral carbohydrate-based ligands may allow for catalytic reactions to be rendered enantioselective and aid in camouflaging in vivo probes from the immune system. Work in this area has evolved from chiral mononuclear lanthanoid compounds, once used as NMR shift reagents, to a number of well-characterised amino acid ligated species, some of which have the ability to cleave phosphate diester bonds. This ability has prompted these species to be viewed as artificial enzymes.59,60 Our work focuses on three ligands that incorporate a protected D-galactose functionality (ligands 15H, 16H, 17H in Fig. 4).61,62 Protection of the chiral diols as acetonide groups was enacted so as to extend the chiral profile of the ligands as well as prevent lanthanoid diol interactions that could interfere with the cluster motif, as previously demonstrated by our group.44 Methylated 15H represents a compromise between steric crowding (acetylated galactose) and coordinative freedom (methyl terminus) that, from our ortho-substituted ligand study, was postulated to form a cubane architecture, if steric crowding did not negate cluster formation. In fact, a cubane species [Ho4(μ3-OH)4(η2-15)7(μ-η2-15)(μ-η2-Ac)(H2O)2] was formed, albeit with an unexpectedly exposed route to the core found to be occupied by an acetate co-ligand (Fig. 18). The incorporation of acetate, which was not present in the reaction mixture or used during the synthesis of the precursor material, was likely produced in situ by deesterification of ethyl acetate, mediated by coordination to the cluster core. The ability of lanthanoid species to effect transesterification is well known,63 and parallels can be drawn with the accelerated hydrolysis of phosphodiester bonds by the dinuclear [Ln2(OH)2]4+ motif. Isolation of this chiral species containing a chiral cleft exposing the reactive [Ln4(OH)4)]8+ core represents an important step towards the creation of artificial enzymes. We envisage that fusing the various coordinating ligands into a much larger single polydentate ligand will be essential in overcoming the labile nature of diketonate ligands and promote controlled syntheses of analogous clusters.![Structure of the cubane cluster cubane species [Ho4(μ3-OH)4(η2-15)7(μ-η2-15)(μ-η2-Ac)(H2O)2].](/image/article/2013/NJ/c2nj40560a/c2nj40560a-f18.gif) | ||
| Fig. 18 Structure of the cubane cluster cubane species [Ho4(μ3-OH)4(η2-15)7(μ-η2-15)(μ-η2-Ac)(H2O)2]. | ||
A planar increase in ligand size effects a contraction in the size of the lanthanoid product, further evidencing the general trend detailed in Fig. 7. The phenyl and pyridyl ligands 16H and 17H were found to stabilise identical [Ln2L6] dinuclear motifs and consequently only the phenyl derivative will be discussed here (Fig. 19). This species exhibited lanthanoid coordination to the pyranose ring, with two ligand binding in a tridentate μ2-η2(O,O′)η2(O′,PyO) mode. As with the cubane species, addition of the bulky carbohydrate functionality imparts a chiral cavity into the known [Ln2L6] motif,64 once again allowing extraneous molecules to access the reactive dinuclear core. In the crystal lattice, the chiral pocket is filled by a DMF molecule that was likely introduced as part of the crystallization process. Preliminary studies show that in solution the complex was found to effect an enantioselective thio-Michael reaction in 95% ee, albeit in very modest yield.62
 | ||
| Fig. 19 Dinuclear cluster motifs obtained with ligands 16H and 17H. | ||
Importantly, in all three instances the incorporation of protected carbohydrates resulted in conversion of known motifs with well-defined ligand barriers to motifs with chiral pockets in the ligand periphery. The final challenge, inhibiting ligand lability, would make this strategy viable in generating families of new enantioselective catalysts.
 | ||
| Fig. 20 Dinuclear hydroxo cluster obtained with ligand 18H. | ||
2-Pyridyltetrazolate as protecting ligands
Along with the diketonate-type coordination ligands described, we also investigated tetrazole-based protecting ligands in the formation of lanthanoid clusters via controlled hydrolysis.65 We reasoned that, despite the fact that lanthanoid ions are oxophilic, ligands such as bipyridine and phenanthroline are able to coordinate to rare earth elements. Therefore, the anionic 2′-pyridyltetrazolate ligand 18− should be able to bind to lanthanoid cations. Moreover, like in the diketonate case, the ligand 18− is capable of both chelating and bridging binding modes. Despite these characteristics, the only hydroxo clusters we were able to obtain were dimeric hydroxo-bridged units in the case of Y and Yb (Fig. 20). On the other hand, in the case of La, Gd, and Ho, cationic disubstituted complexes of the type [Ln(18)2(H2O)n]Cl (n = 4, 5) were obtained. In all cases, the lanthanoid cation binds to two bidentate ligands 18−. The remaining coordination sphere is occupied by water molecules, suggesting that the oxophilic nature of the lanthanoid elements stabilises mononuclear hydrated complexes rather that cluster cores. In fact, the dimeric hydroxo species of Y and Yb can be viewed as the conjuction of two [Ln(18)2(H2O)n]Cl after deprotonation of one water molecule on each complex and bridge formation with the two resulting hydroxo ligands.Remarkably, to tentatively favour the formation of a cluster core, an excess of ligand 18H was used, but an analogous disubstituted structure was obtained and identified as [Y(18)2(H2O)n]18·18H·4H2O.66 All these structures evidence an extended tridimensional hydrogen-bonded network formed between the non-coordinating tetrazolate N atoms and either coordinated or lattice water molecules. This hydrogen bonding network seems to be the another dominating factor in the predominant stabilisation and isolation of these disubstituted complexes. In the case of [La(18)2(H2O)n]Cl, the hydrogen bonding network was also able to support reversible hydrogelation.67
Perspectives
This perspective gives an overview of the large library of lanthanoid hydroxo clusters reported in the literature. The synthesis of lanthanoid hydroxo clusters has enormously advanced in the last couple of decades, and it is now possible to establish relationships between ligand design and expected structural cluster motif. However, as demonstrated in this account, the final structure is clearly dependent on a variety of factors that not only relate to the nature of the ligand and the specific lanthanoid elements. It is evident that the isolation of cluster structures is no longer exclusively dictated by serendipity, albeit predictions of precise structures are still rather difficult to obtain. The next obvious step in this field relates to the application of these species. Indeed, the main feature to be considered is the presence of multiple lanthanoid centres within a confined core, so obviously these molecular entities can become functional materials due to the intrinsic properties of the lanthanoid cations. The three main characteristics of trivalent lanthanoid elements are their Lewis acid nature, their magnetic properties associated with the presence of unpaired electrons, and their luminescent properties associated with f–f electronic transitions.The examples of catalytic roles of lanthanoid cluster species relate to the oxidation of aldehydes to carboxylic acids and the hydrolysis of phosphodiester bonds.6,19 The difficulty in advancing the area of homogeneous catalysis with lanthanoid hydroxo clusters is probably due to the fact that in solution these species exist as equilibria between fragmented smaller cluster cores and/or aggregated clusters of higher nuclearities. Additionally, substrate access to the reactive lanthanide core will require a degree of ligand lability that is also not high enough to allow the decomposition of the core. As previously illustrated, the fact that a specific cluster structure is identified in the solid state with X-ray diffraction techniques does not exclude that in solution, due to the usually labile nature of the Ln–X bond (where X is a donor atom), multiple different structures can simultaneously exist at equilibrium.
A large number of fundamental magnetic studies have appeared specifically for lanthanoid hydroxo clusters, for lanthanoid clusters of various formulations, and also for mixed 3d–4f magnetic systems.1 It is indeed clear that these species have promising properties that could be applicable in the fields of molecular-based magnetism and magnetic materials. For example, the pentanuclear Dy hydroxo cluster stabilised by dibenzoylmethanide and the triangular Dy hydroxo cluster stabilised by ortho-vanillin exhibit single-molecule magnet (SMM) behavior, where blocking anisotropy favours relaxation and quantum tunneling of the magnetisation.27,43,68
Luminescence studies on lanthanoid hydroxo clusters have just started to appear. Reported results have shown how the intrinsic luminescent properties of single lanthanoid elements can be transferred into cluster species. Examples of green-emitting Tb and red-emitting Eu clusters of various nuclearities have been reported, with systematic studies linking the fine structures of the emission profile with the specific geometry and nuclearity of the cluster core.41 Moreover, examples of IR-emitting clusters using Er have appeared.26 Aside from the fundamental studies on the photophysical properties of these cluster systems, pentanuclear red-emitting Eu clusters have been used in the preparation of hybrid materials.39 Our studies have demonstrated how the species [Eu5(OH)5(4)10] can be used a crosslinker in the radical polymerisation of methyl methacrylate, yielding reinforced polymethyl methacrylate (PMMA) hybrid materials that emit red light when photoexcited with radiation around 410 nm. The obtained spectral profile from the emission of the functional PMMA suggest that the cluster core remained intact while the outer allyl groups of the ligand participated into the radical polymerisation thus cross-linking the growing chains. On the other hand, a similar result was later obtained by the group of Weiss, where they simply embedded luminescent lanthanoid clusters of the type [Eu5(OH)5(1)10] in various polymeric matrices forming luminescent nanoparticles and thin films.69 The [Eu5(OH)5(1)10] cluster was also recently shown to be stable when suspended in a copper metallogel.70 We have also demonstrated solvent-modulated luminescence from the thiophene containing [Eu4(OH)2(7)10].46
Acknowledgements
The authors wish to thank the Australian Research Council for financial support.References
- R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328 CrossRef CAS.
- S. V. Eliseeva and J.-C. G. Bünzli, New J. Chem., 2011, 35, 1165 RSC.
- T. Fehlner, J.-T. Halet and J.-Y. Saillard, Molecular Clusters, Cambridge University Press, 2007 Search PubMed.
- Z. Zheng, Cluster Compounds of Rare-Earth Elements, Elsevier, Oxford, 2010 Search PubMed.
- S. Cotton, Lanthanide and Actinide Chemistry, Wiley, West Sussex, 2006 Search PubMed.
- Z. Zheng, Chem. Commun., 2001, 2521 RSC.
- M. R. Burgstein, H. Berberich and P. W. Roesky, Chem.–Eur. J., 2001, 7, 3078 CrossRef CAS.
- R. C. Mehrotra, A. Singh and U. M. Tripathy, Chem. Rev., 1991, 91, 1287 CrossRef CAS.
- L. G. Hubert-Pfalzgraf, Coord. Chem. Rev., 1998, 178–180, 967 CrossRef CAS.
- L. G. Hubert-Pfalzgraf, N. Miele-Pajot, R. Papiernik and J. Vaissermann, J. Chem. Soc., Dalton Trans., 1999, 4127 RSC.
- R. C. Mehrotra, R. Bohra and D. P. Gaur, Metal beta-Diketonates and Allied Derivatives, Academic Press, London, 1978 Search PubMed.
- R. Wang, M. D. Carducci and Z. Zheng, Inorg. Chem., 2000, 39, 1836 CrossRef CAS.
- G. Calvez, O. Guillou, C. Daiguebonne, P. E. Car, V. Guillerm, Y. Gisérault, F. Le Dret and N. Mahé, Inorg. Chim. Acta, 2008, 361, 2349 CrossRef CAS.
- O. Guillou, C. Daiguebonne, G. Calvez, F. Le Dret and P. E. Car, J. Alloys Compd., 2008, 451, 329 CrossRef CAS.
- G. Calvez, C. Daiguebonne, O. Guillou and F. Le Dret, Eur. J. Inorg. Chem., 2009, 3172 CrossRef CAS.
- G. Calvez, C. Daiguebonne and O. Guillou, Inorg. Chem., 2011, 50, 2851 CrossRef CAS.
- Z. Zak, P. Unfried and G. Giester, J. Alloys Compd., 1994, 205, 235 CrossRef CAS.
- A. D. Tsaousis, S. Ollagnier de Choudens, E. Gentekaki, S. Long, D. Gaston, A. Stechmann, D. Vinella, B. Py, M. Fontecave, F. Barras, J. Lukes and A. J. Roger, Proc. Natl. Acad. Sci. U. S. A., 2012,(26), 10426 CrossRef CAS.
- P. W. Roesky, G. Canesco-Melchor and A. Zulys, Chem. Commun., 2004, 738 RSC.
- V. Baskar and P. W. Roesky, Z. Anorg. Allg. Chem., 2005, 631, 676 CrossRef.
- S. Datta, V. Baskar, H. Li and P. W. Roesky, Eur. J. Inorg. Chem., 2007, 4216 CrossRef CAS.
- V. Baskar and P. W. Roesky, Dalton Trans., 2006, 676 RSC.
- K. Koguro, T. Oga, S. Mitsui and R. Orita, Synthesis, 1998, 910 CrossRef CAS.
- D. T. Thielemann, I. Fernandez and P. W. Roesky, Dalton Trans., 2010, 39, 6661 RSC.
- P. C. Andrews, T. Beck, B. H. Fraser, P. C. Junk, M. Massi, B. Moubaraki, K. S. Murray and M. Silberstein, Polyhedron, 2009, 28, 2123 CrossRef CAS.
- X.-Y. Chen, X. Yang and B. J. Holliday, Inorg. Chem., 2010, 49, 2583 CrossRef CAS.
- M. T. Gamer, Y. Lan, P. W. Roesky, A. K. Powell and R. Clerac, Inorg. Chem., 2008, 47, 6581 CrossRef CAS.
- P. C. Andrews, T. Beck, C. M. Forsyth, B. H. Fraser, P. C. Junk, M. Massi and P. W. Roesky, Dalton Trans., 2007, 5651 RSC.
- G. B. Deacon, T. Feng, D. C. R. Hockless, P. C. Junk, B. W. Skelton and H. W. Allan, Chem. Commun., 1997, 341 RSC.
- L. Natrajan, J. Pecaut and M. Mazzanti, Dalton Trans., 2006, 1002 RSC.
- R. Wang, D. Song and S. Wang, Chem. Commun., 2002, 368 RSC.
- G. Xu, Z.-M. Wang, Z. He, Z. Lu, C.-S. Liao and C.-H. Yan, Inorg. Chem., 2002, 41, 6802 CrossRef CAS.
- R. G. Xiong, J. L. Zuo, Z. Yu, X. Z. You and W. Chen, Inorg. Chem. Commun., 1999, 2, 490 CrossRef CAS.
- A. A. Pandey, P. Mayer, W. J. Parak and A. B. Samaddar, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 263 CAS.
- J. C. Plakatouras, I. Baxter, M. B. Hursthouse, K. M. A. Malik, J. McAleese and S. R. Drake, Chem. Commun., 1994, 2455 RSC.
- I. I. Baxter, S. R. Drake, M. B. Hursthouse, K. M. A. Malik, J. McAleese, D. J. Otway and J. C. Plakatouras, Inorg. Chem., 1995, 34, 1384 CrossRef CAS.
- M. Addamo, G. Bombieri, E. Foresti, M. D. Grillone and M. Volpe, Inorg. Chem., 2004, 43, 1603 CrossRef CAS.
- O. Poncelet and L. G. Hubert-Pfalzgraf, Polyhedron, 1989, 8, 2183 CrossRef CAS.
- P. C. Andrews, D. H. Brown, B. H. Fraser, N. T. Gorham, P. C. Junk, M. Massi, T. G. St Pierre, B. W. Skelton and R. C. Woodward, Dalton Trans., 2010, 39, 11227 RSC.
- N. Mahé, O. Guillou, C. Daiguebonne, Y. Gérault, A. Caneschi, C. Sangregorio, J. Y. Chane-Ching, P. E. Car and T. Roisnel, Inorg. Chem., 2005, 44, 7743 CrossRef.
- S. Petit, F. Baril-Robert, G. Pilet, C. Reber and D. Luneau, Dalton Trans., 2009, 6809 RSC.
- P. C. Andrews, G. B. Deacon, R. Frank, B. H. Fraser, P. C. Junk, J. G. MacLellan, M. Massi, B. Moubaraki, K. S. Murray and M. Silberstein, Eur. J. Inorg. Chem., 2009, 744 CrossRef CAS.
- J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729 CrossRef CAS.
- P. C. Andrews, C. M. Forsyth, B. H. Fraser, P. C. Junk, M. Massi and M. Silberstein, CrystEngComm, 2007, 9, 282 RSC.
- M. M. Conradie, A. J. Muller and J. Conradie, S. Afr. J. Chem., 2008, 61, 13 CAS.
- P. C. Andrews, G. B. Deacon, W. J. Gee, P. C. Junk and A. Urbatsch, Eur. J. Inorg. Chem., 2012,(20), 3273 CrossRef CAS.
- E. Grunova, E. Kirillov, T. Roisnel and J.-F. Carpentier, Dalton Trans., 2010, 39, 6739 RSC.
- P. C. Andrews, W. J. Gee, P. C. Junk and J. G. MacLellan, Dalton Trans., 2011, 40, 12169 RSC.
- A. K. Jami, P. V. V. Kishore and V. Baskar, Polyhedron, 2009, 28, 2284 CrossRef CAS.
- P. C. Andrews, W. J. Gee, P. C. Junk and J. G. MacLellan, Inorg. Chem., 2010, 49, 5016 CrossRef CAS.
- P.-H. Lin, M. Leclere, J. Long, T. J. Burchell, I. Korobkov, R. Clerac and M. Murugesu, Dalton Trans., 2010, 39, 5698 RSC.
- M. A. Singh-Wilmot, I. A. Kahwa, A. J. P. White, D. J. Williams and A. J. Lough, Polyhedron, 2010, 29, 270 CrossRef CAS.
- W. J. Evans, M. S. Sollberger and T. P. Hanusa, J. Am. Chem. Soc., 1988, 110, 1841 CrossRef CAS.
- C. Pi, L. Wan, Y. Gu, W. Zheng, L. Weng, Z. Chen and L. Wu, Inorg. Chem., 2008, 47, 9739 CrossRef CAS.
- H. C. Aspinall, Chem. Rev., 2002, 102, 1807 CrossRef CAS.
- M. A. Giardello, V. P. Conticello, L. Brard, M. Sabat, A. L. Rheingold, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10212 CrossRef CAS.
- M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187 CrossRef CAS.
- G. Muller, Dalton Trans., 2009, 9692 RSC.
- N. H. Williams, B. Takasaki, M. Wall and J. Chin, Acc. Chem. Res., 1999, 32, 485 CrossRef CAS.
- P. Hurst, B. K. Takasaki and J. Chin, J. Am. Chem. Soc., 1996, 118, 9982 CrossRef CAS.
- P. C. Andrews, W. J. Gee, P. C. Junk and J. G. MacLellan, Polyhedron, 2011, 30, 2837 CrossRef CAS.
- W. J. Gee, J. Hierold, J. G. MacLellan, P. C. Andrews, D. W. Lupton and P. C. Junk, Eur. J. Inorg. Chem., 2011, 3755 CAS.
- T. Okano, K. Miyamoto and K. Kiji, Chem. Lett., 1995, 246 CrossRef CAS.
- S. Bruck, M. Hilder, P. C. Junk and U. H. Kynast, Chem. Commun., 2000, 666 CAS.
- P. C. Andrews, T. Beck, B. H. Fraser, P. C. Junk and M. Massi, Polyhedron, 2007, 26, 5406 CrossRef CAS.
- D. D'Alessio, L. E. Karagiannidis, B. W. Skelton, M. Massi and M. I. Ogden, Aust. J. Chem., 2012, 65, 819 CrossRef CAS.
- P. C. Andrews, P. C. Junk, M. Massi and M. Silberstein, Chem. Commun., 2006, 3317 RSC.
- I. J. Hewitt, J. Tang, N. T. Madhu, C. E. Anson, Y. Lan, J. Luzon, M. Etienne, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2010, 49, 6352 CrossRef CAS.
- C. P. Hauser, D. T. Thielemann, M. Adlung, C. Wickleder, P. W. Roesky, C. K. Weiss and K. Landfester, Macromol. Chem. Phys., 2011, 212, 286 CrossRef CAS.
- W. J. Gee and S. R. Batten, Chem. Commun., 2012, 48, 4830 RSC.
Footnote |
| † This article is included in the All Aboard 2013 themed issue. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |