Stabilization of a vanadium(V)–catechol complex by compartmentalization and reduced solvation inside reverse micelles†‡
Brant G.
Lemons
a,
David T.
Richens
a,
Ashley
Anderson
b,
Myles
Sedgwick
b,
Debbie C.
Crans
*b and
Michael D.
Johnson
*a
aDepartment of Chemistry and Biochemistry, New Mexico State University, Las Cruces, NM 88001-3001, USA. E-mail: johnson@NMSU.edu; Fax: +1-575-646-2394; Tel: +1-575-646-3627
bDepartment of Chemistry, Colorado State University, Fort Collins, CO 89523, USA. E-mail: debbie.crans@colostate.edu; Fax: +1-970-491-7635; Tel: +1-970-491-7635
First published on 29th October 2012
Abstract
The kinetics of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex formation and hydrolysis between catechol and 3-substituted catechols with aqueous vanadium(V) at pH ∼ 1 have been investigated in NaAOT (sodium bis(2-ethylhexyl)sulfosuccinate) derived aqueous reverse micelle microemulsions in isooctane. Compared with the reaction in bulk water, the forward rate constant of catechol complexation was modestly accelerated (2 fold) in the reverse micelle microemulsions for even the smallest nanosized water pools (wo = 2). In contrast, the first order reverse (aquation) reaction was significantly suppressed in the water pools below wo = 10. The modest rate acceleration of complex formation within the microemulsions is attributed to changes in water solvation, reaction properties and compartmentalization of the reactants. The dramatic fall off in the rate of catechol dissociation from [VO2(cat)(OH2)2]− is attributed to the reduction in water content as the size of the nanopools is decreased below 200 water molecules. The result is a 10 fold increase in the kinetic formation constant for [VO2(cat)(OH2)2]− under confinement in the RM microemulsion environment. This increase predicts that vanadium catechol complexes will be more stable in vivo and may suggest a general principle for fine-tuning efficacy of metal-based drugs.
:1 complex formation and hydrolysis between catechol and 3-substituted catechols with aqueous vanadium(V) at pH ∼ 1 have been investigated in NaAOT (sodium bis(2-ethylhexyl)sulfosuccinate) derived aqueous reverse micelle microemulsions in isooctane. Compared with the reaction in bulk water, the forward rate constant of catechol complexation was modestly accelerated (2 fold) in the reverse micelle microemulsions for even the smallest nanosized water pools (wo = 2). In contrast, the first order reverse (aquation) reaction was significantly suppressed in the water pools below wo = 10. The modest rate acceleration of complex formation within the microemulsions is attributed to changes in water solvation, reaction properties and compartmentalization of the reactants. The dramatic fall off in the rate of catechol dissociation from [VO2(cat)(OH2)2]− is attributed to the reduction in water content as the size of the nanopools is decreased below 200 water molecules. The result is a 10 fold increase in the kinetic formation constant for [VO2(cat)(OH2)2]− under confinement in the RM microemulsion environment. This increase predicts that vanadium catechol complexes will be more stable in vivo and may suggest a general principle for fine-tuning efficacy of metal-based drugs.
Introduction
The study of the kinetics and mechanisms of ligand replacement reactions at transition metal centers in aqueous solution is now an established field with knowledge based on the accumulated data from 50–60 years of experimental work.1–4 Substitution reactions have been classified as associative, dissociative or interchange with regard to the replacement of the coordinated water ligands at the metal center.5 These designations depend on whether bond making or breaking is important in achieving the transition state or whether a discrete intermediate is formed. Substitution studies of metal complexes having biological or medicinal relevance, such as vanadium,6–9 have added interest in regard to tracking their reactivity pathways in vivo. The insulin-enhancing properties of vanadium are well-known10,11 and representative compounds have been successfully tested in both animals and humans. Bis(maltolato)oxovanadium(IV) (BMOV)12 is one such clinically tested compound that is known to release vanadium ions through ligand dissociation both in vitro and in vivo. Understanding the extent of complex stability under conditions of restricted water confinement is of crucial importance to a better understanding of the therapeutic action of such compounds.In vivo chemical reactivity is generally presumed to be similar to that in aqueous solution and is extrapolated from detailed studies in bulk aqueous media. However, it has become increasingly apparent that within the confined space found in the proximity of an interface, the environment typically found in the interior of a cell, the chemical reactivity of many solutes appears to be significantly different from that established in bulk aqueous media.10–13 Since detailed kinetic studies of the reactivity of components inside cells are difficult to carry out, model systems must be invoked and a variety of these exist ranging from the simplest self-assembling system to complex membranes containing proteins and much more. A growing body of information is emerging from studies carried out with the very simple model systems contained in the nanosized water pools of reverse micelles.13,14 Despite their simplicity, reverse micelles (RMs) can be used to investigate how drugs associate with the cell's interior aqueous environment. As a result, several studies have been carried out demonstrating that highly dynamic nanosized water pools encapsulated by a polar interface still allow polar solutes to penetrate the barrier and travel up into the hydrophobic parts of the interface.15–18
Reverse micelles are formed when a microliter volume of an aqueous analyte is added to a mL volume of a solution of a surfactant (e.g. sodium bis(2-ethylhexyl)sulfosuccinate (AOT), Fig. 1) dissolved in a non-polar solvent such as isooctane or cyclohexane. These mixtures rapidly form transparent and homogeneous solutions containing structures consistent with a spherical reverse micelle shown in Fig. 2(a).12,18–21 The average size, number of AOT molecules per RM and the number of water molecules can vary dramatically, but are controlled by the concentration ratio of water to surfactant, termed wo, as shown in Table 1 below.21 Since the range of sizes and number of water molecules extend over a large range, these systems make for excellent models to examine the effects of confinement near a lipid-interface. There are three regions within a RM system where an analyte can reside, Fig. 2(b): (A) the polar aqueous pool center of the RM, (B) the polar head group region, which, in the case of AOT, are sulfonate groups and (C) the non-polar interfacial region which is comprised of the hydrophobic surfactant tails.12,13,18–23 For NaAOT, the exchange rate of analytes between RMs is rapid and has been measured to be around 106–108 M−1 s−1.14,24–26 The nature of the hydrogen bond between the water molecules inside the micelles has been examined using IR spectroscopy and the core water is surrounded by a water shell that are “bound” to the surface and different from the water molecules in the interior core of the RM.15,20,21
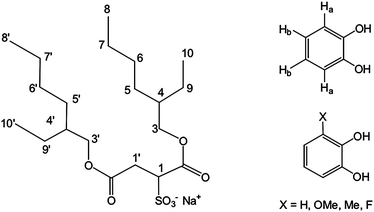 | ||
| Fig. 1 Structure of sodium bis(2-ethylhexyl)sulfosuccinate (AOT) and of the catechol ligands used in this study. | ||
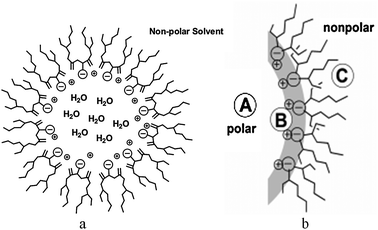 | ||
| Fig. 2 Schematic illustration of AOT-derived reverse micelles. | ||
| w o | n avg | r w (Å) | n w | n M | [RM] |
|---|---|---|---|---|---|
| w o = [H2O]/[AOT], navg = average number of surfactant molecules per RM, nw = average number of water molecules per RM, nM = number of probes per RM if [probe] = 1 mM, [RM]= [AOT]/navg. | |||||
| 4 | 35 | 10 | 140 | 0.0013 | 0.0057 |
| 6 | 50 | 14 | 280 | 0.013 | 0.0040 |
| 8 | 72 | 16 | 578 | 0.026 | 0.0028 |
| 12 | 129 | 22 | 1553 | 0.070 | 0.0015 |
| 20 | 302 | 35 | 6061 | 0.27 | 0.00066 |
Recent studies suggest that lipid-interfaces significantly impact proton transfer reactions.27,28 Rate-enhancement of enzyme reactions anchored to membranes is increased by multiple orders of magnitude. These dramatic rate-enhancements are in part attributed to the changes in solvation as experienced when the solute moves from a largely aqueous environment to the interfacial water–lipid environment. Because of their structures, stabilities and favorable reaction dynamics, RMs are convenient to work with and provide consistent data that is well understood.12,13,15–17 For example, they have been used to study the structure of alpha-helical peptides at a membrane surface, the form of the Jagged-1 interactions with Notch ligand trans-membrane proteins; as well as conformational preferences of Leu-enkephalin.28 They have also been used in dynamic studies to explore proton transport29 and the binding of tryptophan analogs to membrane surfaces.30 However, there have been few studies to date which have focused on the kinetic effects on metal ion complex formation and hydrolysis in the vicinity of a lipid or lipid-like interface and the resulting restrictions in aqueous solvation on fundamental substitution processes.12,31
In this study, we are exploring the complexation reaction between a ligand (catechol) and a metal ion (aqueous vanadium(V); [VO2(OH2)4]+) along with the reverse reactions; aquation of the corresponding vanadium(V)–catechol complex. All of these components are located near, or accessible to, the interface of the RM, location B in Fig. 2b. This complexation reaction and the subsequent much slower oxidation of the bound catechol ligand were studied 40 years ago by Kustin and co-workers32 and provides a well characterized system with which to conduct a detailed kinetic investigation of the effects of confinement within a reverse micelle environment. The kinetic studies carried out with catechol derivatives possessing electron-donating and electron-withdrawing substituents at the 3-position also allowed us to probe whether the fundamental effects expressed in terms of Hammett electronic effects33 are retained on going from bulk aqueous to confined media. These fundamental studies document the changes in reactivity of both free aqueous vanadium(V) and of the vanadium(V)–catechol complex in the vicinity of the interface. As we will show, the vanadium(V)–catechol complex becomes more stable under confinement in the vicinity of the interface compared to bulk aqueous media contrasting with the findings for BMOV which under similar confined conditions is more susceptible to ligand hydrolysis.12 This work demonstrates the importance of understanding the intimate nature of metal–ligand complexation processes under confinement close to a lipid interface if in vivo reactivity is to be reliably predicted.
Experimental
Materials
Sodium orthovanadate (Na3VO4), catechol and 3-substituted catechols were obtained from Aldrich and used as received unless otherwise specified. All other chemicals were reagent grade. A Barnsted Epure system supplied the doubly-distilled, deionized (<18 MΩ cm−2) water used in all experiments. The surfactant, sodium bis-(2-ethylhexyl)sulfosuccinate (Na-AOT, 99%), used in RM preparations, was obtained from Sigma-Aldrich and purified as described previously.341H NMR spectroscopy was used to confirm Na-AOT purity. Isooctane (99%) was purchased from Aldrich and used as received. D2O and CD2Cl2 were obtained from Cambridge Isotope Laboratories Inc.Preparation of reverse micelles (RMs)
Purified Na-AOT was dissolved in isooctane at ambient temperature to make a stock solution of the surfactant. Acidified aqueous stock solutions of [VO2(OH2)4]+ were prepared via dissolution of sodium orthovanadate in perchloric acid. For the vanadium(V) containing reverse micelles, aliquots of the stock solution [VO2(OH2)4]+ adjusted to pH 1.04 with HClO4 were mixed with specific volumes of the 0.2 M solution of NaAOT in isooctane to prepare RMs with wo values from 6 to 30. The wo value is defined as the [H2O]/[AOT] ratio. In each case the [H+] in the final aliquot was 0.092 M. The ternary component systems was added by sonication for 2–3 min or until separation of phases was no longer observed. For the catechol-containing reverse micelles two preparations were employed, methods a and b (see ESI,† Fig. S3). Method a was the same as that used for the vanadium(V) containing reverse micelles and involves the injection of calculated aliquots of the aqueous catechol stock solution, adjusted to pH 1.04 with HClO4, into specific volumes of the 0.2 M solution of NaAOT in isooctane to prepare RMs with wo values from 6 to 30. In method b the pure catechol is initially dissolved into a solution of 0.2 M NaAOT in isooctane followed by the injection of calculated aliquots of water adjusted to pH 1.04 with HClO4 to yield desired wo. The resulting RM mixture is shaken or sonicated until optically clear. Spectral and kinetic measurements were made immediately after preparation. Solutions were purged with argon gas prior to assembly of the RM samples. It was found that the use of air-free conditions or aging of the solutions up to 24 hours had no effect on the resulting kinetics.Kinetic experiments
Reaction rates for the complexation of aqueous vanadium(V) by catechol and the 3-substituted catechols were measured using an OLIS RSM1000 stopped-flow mixing system. The reactions were monitored at 512 nm, the absorption maximum for vanadium(V)–catechol complex. Pseudo first order conditions were used, with excess ligand (range 0.001–0.03 M) over vanadium(V) (∼1 × 10−4 M), or the reverse (excess vanadium(V) over catechol). The absorbance changes (average of 3–5 runs) were fitted using the OLIS data reduction software to yield the observed rate constants. RM sizes ranged between wo 2 to 30. A solution of 0.092 M HClO4 was used throughout to control acidity. The temperature was controlled to within 0.1 °C using a constant temperature bath.Sample preparation for NMR spectroscopy
For the 1-D NMR experiments catechol containing RMs ranging in sizes from wo = 6 to 20 were assembled from a 0.13 M catechol stock solution using 0.2 M NaAOT in isooctane adjusted to a final catechol concentration of 0.01 M and 0.092 M (HClO4). Both methods a and b (above) were employed for the preparation of the RMs. No buffers were used. For the 2-D 1H–1H NOESY experiments the concentration of AOT surfactant was increased to 1.0 M and the final catechol concentration was 0.05 M. In all case the RMs were prepared immediately before acquisition of NMR spectra.NMR spectroscopy: data acquisition and analysis
1H NMR spectra of the catechol ligand in RMs and aqueous solutions were obtained on samples placed in standard 5 mm Wilmad NMR tubes. The data were recorded at ambient temperature on a 400 MHz Varian spectrometer. Routine parameters were used for the 1-D 1H data acquisition. The 1H chemical shifts were referenced to the peak for isooctane at 0.903 pm from sodium DSS (sodium 4,4-dimethyl-4-silapentane-1-sulfonate) used as the internal reference. 19F NMR spectra of 3-fluorocatechol in D2O and in wo = 20 reverse micelles were referenced to hexafluorobenzene (−161 ppm).1H–1H-NOESY (Proton–Proton 2-D Nuclear Overhauser Enhancement Correlation Spectroscopy) NMR experiments were performed using the supplied Varian pulse sequence as reported previously.17 Aqueous solutions of complexes were run in D2O, to provide a lock signal but for studies in RMs H2O and not D2O, was used in these samples.
Dynamic light scattering experiments
Aqueous solutions of the catechol or substituted catechol (in mM) were prepared in pure deionized-water. A catechol stock solution (∼2 mM) was used to yield the desired concentration in the 0.2 M NaAOT RM with a wo value of 10. The DLS experiments were carried out on a Wyatt DynaPro Titan using a temperature-controlled microsampler for the measurements.35 Effective data was obtained using intensities of 80000 to 10000 counts by varying the instrument's GaAs laser power between 10 and 20 mW and collecting the scattered light at 90° and samples were equilibrated in the DLS instrument for 10 min before the sample was measured. The sample was filtered directly into the cuvette through a 0.2 μm filter. Scans were performed at a rate of 10 acquisitions for 100 s with each measurement consisting of no less than 10 runs.
To interpret the DLS data both the refractive index and the viscosity were assumed to be similar to that of the neat organic solvent, isooctane.35 The DynaPro DYNAMICS software (version 6.7.3) was used to evaluate the light scattering data assuming spherical particles. The program fits the autocorrelation to regulated fits and a cumulant fit, which is an average of the regulated fits. On the basis of the exponential fits to the data, the reverse micelle size was obtained with an instrument error of 10%.
Results and discussion
Mixing catechol (H2cat) with acidic solutions of aqueous vanadium(V) results in the appearance of a deep yellow color indicative of the formation of the 1:1 complex [VO2(cat)(OH2)2]− (eqn (1)).32 This catechol complex has a characteristic absorption maximum at | (1) |
| Rate = kobs[V(V)] | (2) |
| kobs = kf[H2cat] + kr | (3) |
| Rate = kobs[H2cat] | (4) |
| kobs = kf[V(V)] + kr | (5) |
In both cases, pseudo-first order rate constants (kobs, s−1) were obtained from exponential fits of the absorbance changes (formation of the complex) and are listed in the ESI.† Plots of kobsversus excess reactant (shown for catechol in Fig. 4) were linear corresponding to a first-order dependence on both reactants. These plots also possess a significant y-intercept which is typical of an equilibrium process where the slope, kf (M−1 s−1), represents the forward rate constant for H2cat complexation with V(V) and the intercept, kr (s−1), is the rate constant for ligand dissociation from [VO2(cat)(OH2)2]−. Values of kf and kr, obtained by linear least squares fits to the plots of kobsversus either excess [V(V)] or [H2cat], were in good agreement indicating no formation of a 2:1 catechol–vanadium(V) complex. Table 2 shows the values of these constants. The values of kf (1.3 ± 0.2 × 104 M−1 s−1) and kr (60 ± 3 s−1) obtained in aqueous media were in good agreement with those reported previously by Kustin (kf = 1.8 ± 0.1 × 104 M−1 s−1; kr = 41.9 ± 3.3 s−1) under somewhat different conditions; [H+] = 0.2 M, I = 1.0 M (HClO4, NaClO4).32 This shows that the reaction rates are generally insensitive to small changes in acidity and ionic strength. This is likely due to the fact that there is no change in vanadium speciation under these conditions and that catechol has no charge. Unfortunately a full pH profile was not possible due to (1) significant hydrolysis of V(V) to anionic species above pH 236,37 and (2) the inability to form RMs below pH 1.
![UV-visible spectral changes following mixing of excess catechol (0.001–0.01 M) at 25 °C with [VO2(OH2)4]+ (1 × 10−4 M) at pH 1.04, I = 0.092 M (HClO4). Inset: a typical stopped-flow trace of the rise in absorbance due to formation of [VO2(cat)(OH2)2]− at 512 nm.](/image/article/2013/NJ/c2nj40524e/c2nj40524e-f3.gif) | ||
| Fig. 3 UV-visible spectral changes following mixing of excess catechol (0.001–0.01 M) at 25 °C with [VO2(OH2)4]+ (1 × 10−4 M) at pH 1.04, I = 0.092 M (HClO4). Inset: a typical stopped-flow trace of the rise in absorbance due to formation of [VO2(cat)(OH2)2]− at 512 nm. | ||
![Typical pseudo-first order plot for the reaction of excess catechol (H2cat) with [VO2(OH2)4]+ (1 × 10−4 M) at pH 1.04 (25 °C), I = 0.092 M (HClO4).](/image/article/2013/NJ/c2nj40524e/c2nj40524e-f4.gif) | ||
| Fig. 4 Typical pseudo-first order plot for the reaction of excess catechol (H2cat) with [VO2(OH2)4]+ (1 × 10−4 M) at pH 1.04 (25 °C), I = 0.092 M (HClO4). | ||
| Medium | 104kf (M−1 s−1) | k r (s−1) | K (M−1) |
|---|---|---|---|
| a RM preparation method a. b RM preparation method b. c Excess vanadium(V). d 24 hour aged solutions prepared via method a | |||
| Aqueous | 1.0 ± 0.1 | 44 ± 2 | 230 ± 30c |
| 1.3 ± 0.2 | 60 ± 3 | 220 ± 40 | |
| w o 30a | 2.4 ± 0.1 | 52 ± 2 | 460 ± 50c |
| w o 20a | 1.9 ± 0.2 | 51 ± 1 | 370 ± 30c |
| 2.3 ± 0.1 | 40 ± 1 | 570 ± 40 | |
| w o 20b | 1.1 ± 0.1 | 74 ± 1 | 150 ± 20 |
| w o 20d | 1.8 ± 0.1 | 70 ± 10 | 260 ± 30 |
| w o 10a | 1.9 ± 0.1 | 45 ± 3 | 425 ± 10 |
| w o 5a | 1.9 ± 0.1 | 28 ± 1 | 680 ± 20 |
| w o 2a | 1.8 ± 0.1 | 8 ± 1 | 2300 ± 300 |
The effect of encapsulation within the RM water on the catechol–vanadium reaction was investigated. Fig. 5 and 6 are plots of kf and kr as a function of the fraction of bound water – which decreases as the RM size decreases (decreased wo). Included in these plots for comparison are the values of kf and kr obtained in bulk aqueous solution. Compared to bulk aqueous solution, the value of kf (1.3 × 104 M−1 s−1) for complexation to catechol roughly doubles in the presence of the largest reverse micelle (wo = 30). As the size of the reverse micelles get smaller, kf gradually decreases to a constant value of 1.8 × 104 M−1 s−1 for the smallest reverse micelle (wo = 2). In contrast, the rate constant for catechol dissociation, kr, shows markedly different behavior. There is little change in kr from the aqueous value for the two largest reverse micelles (wo = 30 and 20) but further decrease in RM size causes a dramatic decrease by a factor of nearly 10 at wo = 2. When combined with the respective kf values this has the effect of increasing the kinetic formation constant (K = kf/kr) for [VO2(cat)(OH2)2]− from 2.3 × 102 M−1 (bulk aqueous) to 2.3 × 103 M−1 (wo = 2 RM).
![Plot of kf for reaction of [VO2(OH2)4]+ with H2cat versus size of the AOT reverse micelles defined by the fraction of bound water (Pb) at the interface and compared to the value in bulk aqueous media under the same conditions, pH 1.04, I = 0.092 M (HClO4). The number labels are the values of wo.](/image/article/2013/NJ/c2nj40524e/c2nj40524e-f5.gif) | ||
| Fig. 5 Plot of kf for reaction of [VO2(OH2)4]+ with H2cat versus size of the AOT reverse micelles defined by the fraction of bound water (Pb) at the interface and compared to the value in bulk aqueous media under the same conditions, pH 1.04, I = 0.092 M (HClO4). The number labels are the values of wo. | ||
![Plot of kr (reaction of [VO2(OH2)4]+ with H2cat) versus size of the AOT reverse micelles defined by the fraction of bound water (Pb) at the interface and compared to the value in bulk aqueous media under the same conditions. pH 1.04, I = 0.092 M (HClO4). The number labels are the respective values of wo.](/image/article/2013/NJ/c2nj40524e/c2nj40524e-f6.gif) | ||
| Fig. 6 Plot of kr (reaction of [VO2(OH2)4]+ with H2cat) versus size of the AOT reverse micelles defined by the fraction of bound water (Pb) at the interface and compared to the value in bulk aqueous media under the same conditions. pH 1.04, I = 0.092 M (HClO4). The number labels are the respective values of wo. | ||
The kinetic data obtained were also independent of the methods used to form the reverse micelles, either dissolution of V(V) and H2cat in the RM water nanopool prior to mixing (method a), or, for H2cat, mixing with the AOT surfactant and isooctane first prior to addition of the acidified water (method b). There was also no change to the kinetic data obtained if the reverse micelles were allowed to stand for 24 hours prior to mixing. To check if ion-pair association of [VO2(OH2)4]+ with the sulfonate head groups of the AOT surfactant was responsible for the kinetic behavior observed, several runs were also carried out in aqueous media containing 0.1 M sodium p-toluenesulfonate. No rate change was observed which indicates either little ion pairing with the sulfonate head group or a lack of sensitivity if an ion pair is formed.
To confirm that reverse micelles were formed under our experimental conditions and to determine their size, we used dynamic light scattering (DLS).35,38 The objective of these experiments was to examine whether the addition of solutes changed the size of the RMs. We found that the presence of catechol, aqueous vanadium(V) and resulting complex did not change the size of the RMs in the region investigated, Table 3. Furthermore, the measured radii of the RMs at wo 10 showed no difference between the two methods for RM preparation, and thus support the interpretation based on kinetic data that the resulting samples are indistinguishable.
To test whether electron withdrawal or donation affects the complexation process, kinetic data were obtained from the reaction of [VO2(OH2)4]+ with various 3-substituted catechols possessing electron-donating and electron-withdrawing groups. As expected, the kinetic data observed in both aqueous media and inside the reverse micelles showed increased electron donation by the substituent. A Hammett plot of the kinetic data obtained in a wo = 10 reverse micelle is shown in Fig. 7 and is linear. The inset table in Fig. 7 also shows that the calculated ρ values in water and at a variety of reverse micelle sizes decrease significantly (from −3.9 in water to −6.0 at wo = 5) as the size of the reverse micelles decrease. When fewer water molecules are available to solvate the catechol, at smaller wo values, our results are consistent with an increased nucleophilicity of the oxygen donors of the catechol ligands.
![Hammett ρ plot (log kX/log kX–Hversus the σ value of the X substituent) for the reaction of [VO2(OH2)4]+ with various 3-X substituted catechols at wo = 10, pH 1.04, I = 0.092 M (HClO4). Inset: values of ρ as a function of reaction media.](/image/article/2013/NJ/c2nj40524e/c2nj40524e-f7.gif) | ||
| Fig. 7 Hammett ρ plot (log kX/log kX–Hversus the σ value of the X substituent) for the reaction of [VO2(OH2)4]+ with various 3-X substituted catechols at wo = 10, pH 1.04, I = 0.092 M (HClO4). Inset: values of ρ as a function of reaction media. | ||
The vanadium(V) (as the [VO2(OH2)4]+ ion) resides in the aqueous pool presumably adjacent to the negatively charged AOT interface as previously reported for other vanadium species.12 However, the neutral catechol ligand has more options for suitable locations within the RM environment. 1-D and 2-D 1H NMR studies were carried out to complement the kinetic studies and provide information on complex and ligand location. These techniques have been successfully used in a number of previous studies to probe reactant location under aqueous confinement.15–17,39Fig. 8 shows 1-D 1H NMR spectra of the two aromatic protons H3 (ortho to the OH groups) and H4 of catechol in both D2O and inside wo = 20 reverse micelles. The conditions were the same as in the kinetic experiments; 0.2 M NaAOT, [H+] = 0.092 (HClO4), [H2cat] = 0.01 M. Inside the RMs, the 1H resonance of the two protons broadened and shifted distinctly upfield from the value in bulk D2O with the biggest upfield shift being apparent for the H4 protons. This shifting has previously been observed for solutes that penetrated interfaces.15–17
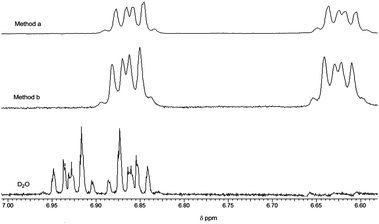 | ||
| Fig. 8 1H-NMR stack plot of catechol in D2O and in wo = 20 reverse micelles. Reverse micelle preparation method a: aqueous (D2O) solution of catechol added to AOT surfactant/isooctane; method b: catechol premixed with AOT surfactant/isooctane prior to addition of D2O. The spectra were referenced to the isooctane peak at 0.903 pm from sodium DSS (sodium 4,4-dimethyl-4-silapentane-1-sulfonate). | ||
As in the DLS experiments, the 1H NMR spectra of catechol were also independent of whether it was added as a solution in D2O to the AOT surfactant–isooctane mixture, method a, or whether catechol was first dissolved in the AOT surfactant/isooctane prior to addition of D2O to form the reverse micelle, method b. The distinct upfield shift in the H4 protons is consistent with penetration of the catechol ligand into the interfacial region.15–17 To confirm the extent of the interfacial penetration a 2-D 1H–1H NOESY experiments were carried out at higher concentrations of AOT surfactant (1.0 M) and catechol (0.05 M). No NOE cross peaks were observed from through space interactions from the H3 and H4 protons of catechol with the 3′–5′ protons of the AOT surfactant in spectra recorded with a range of mixing times.
We also conducted 19F NMR studies with 3-fluorocatechol; these showed little if any shift in the 19F resonance in the reverse micelle compared with D2O. While this result might indicate a residence in water pool for 3-fluorocatechol, it is also consistent with its location at the interface, Fig. 9. When the catechol and vanadium(V) ions are both at the interface, the energy barrier for complexation will be lowered and the reaction rate increased. In addition, a decrease in water solvation can explain the doubling of the forward complexation rate going from bulk aqueous to the largest of the reverse micelles (wo = 30).
 | ||
| Fig. 9 Schematic showing a portion of the interface within the AOT-derived reverse micelles and the likely location of the catechol ligands as indicated by 1H and 19F NMR studies. | ||
Conclusions
This study demonstrates that both the rate and position of the equilibrium governing simple ligand substitution reactions is significantly affected by the confined aqueous environments occurring in AOT-derived reverse micelles. Unlike in bulk aqueous media, metal–ligand substitution reactions taking place within the confined water pools are sensitive to the amount of water present.Because these studies were carried out under conditions with significantly less than one catechol and vanadium(v) per reverse micelle, the formation reaction occurs when RMs collide and their water pool contents mix. Since both components are located at the water–AOT interface, small changes are likely to be observed as the wo size decreases and this is what was observed.
In contrast, the largest effect on the kinetics is the dramatic fall off in the rate constant for the aquation reaction as the size of the reverse micelles are decreased below wo 20. In bulk water, the rate of this process is a constant due to water present in vast excess compared to the complex, i.e., pseudo first order conditions. In the confined water environment inside the reverse micelles, the water concentration is now low and approaches second order conditions. This results in the observed decrease in ligand hydrolysis. This is contrary to the behavior previously found for BMOV, a complex shown in animal studies to undergo ligand dissociation.12
The ratio of these two kinetic processes controls the size of K, the complexation equilibrium constant. Since the forward reaction is insensitive to solvation, the change is mainly due to the decrease in the size of the rate of the reverse reaction (hydrolysis) in RMs. This is attributed to the reduction in water content and pool size as the reverse micelles decrease in size. Indeed, this is an elegant example of the le Chatelier's principle whereby the system compensates by stabilizing the complex upon decreasing the amount of free water.
Acknowledgements
MDJ, and DCC thank NSF for funding this research (CHE 0628260). DTR thanks NMSU for partial funding of this work. Dr Dennis Johnson (NMSU) is thanked for help in recording and interpreting the NMR spectra.References
- T. W. Swaddle, in Physical Inorganic Chemistry; Reactions Processes and Applications, ed. A. Bakac, John Wiley, NY, 2010, 339 Search PubMed.
- R. B. Jordan, Reaction Mechanisms of Inorganic and Organometallic Systems, Oxford University Press, Oxford, 3rd edn, 2007 Search PubMed.
- D. T. Richens, Chem. Rev., 2005, 105, 1961 CrossRef CAS.
- J. Burgess and C. D. Hubbard, Adv. Inorg. Chem., 2002, 54, 71 Search PubMed.
- R. G. Wilkins, The Kinetics and Mechanisms of Reactions of Transition Metal Complexes, Wiley, NY, 2nd edn, 1991 Search PubMed.
- D. C. Crans, Pure Appl. Chem., 2005, 77, 1497 CrossRef CAS.
- D. Rehder, Coord. Chem. Rev., 1999, 182, 297 CrossRef.
- D. Rehder and S. Jantzen, Adv. Environ. Sci. Technol., 1998, 30, 251 Search PubMed.
- D. Mustafi, B. Peng, S. Foxley, M. W. Makinen, G. S. Karczmar, M. Zamor, J. Ejnik and H. Martin, J. Biol. Inorg. Chem., 2009, 14, 1187 CrossRef CAS.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849 CrossRef CAS.
- K. H. Thompson, J. Lichter, C. LeBel and C. Orvig, J. Inorg. Biochem., 2009, 103, 554 CrossRef CAS; K. H. Thompson and C. Orvig, J. Inorg. Biochem., 2006, 100, 1925 CrossRef CAS.
- (a) D. C. Crans, S. Schoeberl, E. Gaidamauskas, B. Baruah and D. A. Roess, J. Biol. Inorg. Chem., 2011, 16, 961 CrossRef CAS; (b) K. H. Thompson, B. D. Liboiron, Y. Sun, K. D. D. Bellman, I. A. Setyawati, B. O. Patrick, V. Karunaratne, G. Rawji, J. Wheeler, K. Sutton, S. Bhanot, C. Cassidy, J. H. McNeill, V. G. Yen and C. Orvig, J. Biol. Inorg. Chem., 2003, 8, 66 CrossRef CAS; (c) S.-Q. Zhang, X.-Y. Zhong, G.-H. Chen, W.-L. Lu and Q. Zhang, J. Pharm. Pharmacol., 2008, 60, 99 CrossRef CAS; (d) D. C. Crans, B. Baruah, A. Ross and N. E. Levinger, Coord. Chem. Rev., 2009, 253, 2178 CrossRef CAS; (e) D. E. Warshawski, A. A. Arnold, M. Beaugrand, A. Gravel, E. Chartrand and I. Marcotte, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1957 CrossRef CAS.
- (a) N. E. Levinger and M. D. Fayer, Annu. Rev. Anal. Chem., 2010, 3, 89–107 Search PubMed; (b) N. E. Levinger, Science, 2002, 298, 1722 CrossRef CAS; (c) R. Bru, A. Sanchez-Ferrer and F. Garcia-Carmona, Biochem. J., 1995, 310, 721 CAS.
- P. L. Luisi, M. Giomini, M. P. Pileni and B. H. Robinson, Biochim. Biophys. Acta, Rev. Biomembr., 1988, 947, 209 Search PubMed.
- D. C. Crans, C. D. Rithner, B. Baruah, B. L. Gourley and N. E. Levinger, J. Am. Chem. Soc., 2006, 128, 4437 CrossRef CAS.
- (a) D. C. Crans B. Baruah, E. Gaidamauskas, B. G. Lemons, B. B. Lorenz and M. D. Johnson, J. Inorg. Biochem., 2008, 102, 1334 CrossRef CAS; (b) M. Vermathen, P. Stiles, S. J. Bachofer and U. Simonis, Langmuir, 2002, 18, 1030 CrossRef; (c) T. Bansagi, v. K. Vanag and I. R. Epstein, Science, 2011, 331, 1309 CrossRef.
- M. D. Johnson, B. B. Lorenz, P. C. Wilkins, B. G. Lemons, B. Baruah, N. Lamborn, M. Stahla, P. B. Chatterjee, D. T. Richens and D. C. Crans, Inorg. Chem., 2012, 51, 2757 Search PubMed.
- S. Dutta Choudhury, M. Kumbhakar, S. Nath, S. K. Sarkar, T. Mukherjee and H. Pal, J. Phys. Chem., 2007, 111, 8842 Search PubMed.
- H. B. Bohidar and M. Behboudnia, Colloids Surf., A, 2001, 178, 313 CrossRef CAS.
- T. K. Jain, M. Varshney and A. Maitra, J. Phys. Chem., 1989, 93, 7409 CrossRef CAS.
- A. Maitra, J. Phys. Chem., 1984, 88, 5122 CrossRef CAS.
- B. Baruah, L. A. Swafford, D. C. Crans and N. E. Levinger, J. Phys. Chem. B, 2008, 112, 10158 CrossRef CAS.
- B. Baruah, J. M. Roden, M. Sedgwick, N. E. Levinger and D. C. Crans, J. Am. Chem. Soc., 2006, 128, 12758 CrossRef CAS.
- P. D. I. Fletcher, A. M. Howe and B. H. Robinson, J. Chem. Soc., Faraday Trans., 1987, 83, 985 Search PubMed.
- T. S. Kale, A. Klaikherd, B. Popere and S. Thayumanavan, Langmuir, 2009, 25, 9660 CrossRef CAS.
- B. H. Robinson, D. C. Steyler and R. D. Tack, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 481 RSC.
- (a) L. Ojemyr, T. Sanden and J. Widengren, Biochemistry, 2009, 48, 2173 Search PubMed; (b) M. Branden, T. Sanden and P. Brezezinski, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19766 CrossRef; (c) D. A. Cherepanov, W. Junge and A. Y. Mulkidjanian, Biophys. J., 2004, 86, 665 CrossRef CAS; (d) G. Nordlund, J. Ng and L. Bergstrom, Nano, 2009, 3, 2639 Search PubMed; (e) S. Serowy, S. M. Saparov, Y. N. Antonenko, W. Kozlovsky, V. Hagen and P. Pohl, Biophys. J., 2003, 84, 1031 Search PubMed.
- S. Rudolph-Bohner, D. Quarzago, M. Czish, J. Ragnarsson and L. Moroder, Biopolymers, 1997, 41, 591 CrossRef CAS.
- S. Y. Park, O. Kwon, T. G. Kim and D. J. Jang, J. Phys. Chem., 2009, 113, 16110 Search PubMed.
- A. Chattopadhyay, A. Arora and D. A. Kelkar, Eur. Biophys. J., 2005, 35, 62 Search PubMed.
- G. Maas, J. Phys. Chem., 1968, 60, 138 Search PubMed.
- (a) K. Kustin, S.-T. Liu, C. Nicolini and D. L. Toppen, J. Am. Chem. Soc., 1974, 96, 7410 CrossRef CAS; (b) K. Kustin, C. Nicolini and D. L. Toppen, J. Am. Chem. Soc., 1974, 96, 7416 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS and references therein.
- M. Stahla, B. Baruah, J. Dustin, M. D. Johnson, N. E. Levinger and D. C. Crans, Langmuir, 2008, 24, 6027 CrossRef CAS.
- M. A. Sedgwick, A. M. Trujillo, N. Hendricks, N. E. Levinger and D. C. Crans, Langmuir, 2011, 27, 948 CrossRef CAS.
- A. Butler and C. J. Carrano, Coord. Chem. Rev., 1991, 109, 61 CrossRef CAS.
- R. Wever and K. Kustin, Adv. Inorg. Chem., 1990, 35, 81 CAS.
- V. R. Vasquez, B. C. Williams and O. A. Graeve, J. Phys. Chem. B, 2011, 115, 2979 Search PubMed.
- D. A. Binks, N. Spencer, J. Wilkie and M. M. Britton, J. Phys. Chem. B, 2010, 114, 12558 Search PubMed.
Footnotes |
| † This article is included in the All Aboard 2013 themed issue. |
| ‡ Electronic supplementary information (ESI) available: Tables of pseudo first order rate constants, schematic of RM preparations. See DOI: 10.1039/c2nj40524e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |