Anion receptor coordination tripods capped by [9]ane-S3†‡§
Adam M. Todd, Adam N. Swinburne, Andrés E. Goeta and Jonathan W. Steed*
Department of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE. E-mail: jon.steed@durham.ac.uk; Fax: +44 (0)191 384 4737; Tel: +44 (0)191 334 2085
First published on 9th July 2012
Abstract
A series of ruthenium(II) complexes with face-capping [9]ane-S3 ligands are described. The compounds function as supramolecular receptors for anions via three tripodally arranged 3-aminopyridine ligands. The [9]ane-S3 ligand stabilises the tripodal complexes which are more readily prepared and studied than their π-arene ruthenium(II) analogues. Pyrenyl derivative 4 displays some activity as a photophysical anion sensor but the anion response is complicated by the complexes concentration dependent emission behaviour. The receptors bind common anions in relatively polar media forming both 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 host–anion complexes with the CH⋯anion interactions involving the thioether ring being implicated in anion binding as well as the convention NH donors.
:1 and 1:2 host–anion complexes with the CH⋯anion interactions involving the thioether ring being implicated in anion binding as well as the convention NH donors.
Introduction
The use of an octahedral metal centre as the core of a tripodal anion receptor compound has already received some attention owing to the well-defined geometries possible with the inclusion of metal ions, and the potentially rich redox and luminescence properties of coordination complexes.1–4 Generation of a symmetric, metal–organic receptor can be achieved in one of two ways. One is to coordinate three ditopic ligands in a manner related to the well-documented ruthenium or iridium tris-2,2′-bipyridyl (bipy) complexes.5–7 Substitution onto the bipy8–11 or replacement of one bipy group with a similar species, for example biimidazole,12,13 can allow for pendant functional groups which can contain anion binding moieties in a suitable arrangement to engender anion selectivity. The second method relies on blocking three coordination sites of the octahedral metal centre in a facial (fac) manner such that the opposite three sites are positioned in a mutually orthogonal fashion to generate a tripodal anion binding pocket. The best method for achieving such blocking is through the use of a tridentate face-capping ligand, often an arene,14–16 heterocycle,17,18 or claw-like tridentate ligand.19,20 Such a capped metal leads to the range of complexes known as ‘half-sandwich’ or ‘piano-stool’ complexes, named for their distinctive three-legged appearance.The metal ion itself must be relatively inert and exhibit a well-defined preference for an octahedral coordination geometry. In addition a diamagnetic centre is desirable from the point of view of NMR spectroscopic characterisation. These requirements tend to favour low-spin d6 centres such as molybdenum(0), rhenium(I), ruthenium(II), rhodium(III) and iridium(III).14,15,21–25 Square planar palladium(II) and platinum(II) species have been used to form dipodal and tetrapodal molecular tweezers,14,26–28 and other more labile ion such as silver(I) have also been investigated.29–31
In previous work we have produced a range of ruthenium(II) dipodal receptors in which an η6-arene ligand caps three fac coordination site of the octahedral metal centre. Anion binding occurs via two amino- or ureidopyridine ligands while the remaining coordination site is occupied by a chloride ligand.32,33 Synthesis of analogous tripodal complexes is hampered by steric interactions between the three pyridyl ligands, rendering the complexes relatively labile and hence anion complexation studies focussed on the dipodal analogues.34 In the present work we report the synthesis of tripodal tris(pyridyl) ruthenium(II) dicationic receptors in which the hexahapto p-cymene capping group is replaced by the 1,4,7-trithiacyclononane ([9]ane-S3) heterocycle first reported by Schröder and co-workers35 and later by Roche et al.34,36 who achieved coordination of three pyridyl ligands to a [9]ane-S3 capped ruthenium(II) centre. We anticipate therefore that use of the ([9]ane-S3)Ru(II)2+ moiety should allow for the synthesis of tripodal receptors of well-defined geometry. Anion binding by CH⋯anion interactions within the coordinated [9]ane-S3 crown has recently been reported by Bedford and co-workers and offers an interesting additional potential anion binding site.37
Results and discussion
Synthesis and structure
Synthesis of the [9]ane-S3-capped precursor [Ru([9]ane-S3)Cl2(DMSO)] is readily achieved by reaction of [RuCl2(DMSO)4]38 with the thioether. The thioether displaces three of the coordinated DMSO molecules and remains coordinated to the ruthenium centre with a high degree of stability, although coordinated pyridines in the resulting [9]ane-S3 complexes can be displaced in the presence of acetonitrile.36 Metathesis of the counter anions from chloride to hexafluorophosphate was achieved through addition of AgPF6 and subsequent removal of the precipitated AgCl to give an activated solvate complex which was used in situ. Addition of a range of 3-substituted pyridine ligands gave complexes 1–6 (Scheme 1), containing either a secondary amine or a urea as the anion binding functionality. In most cases, the complexes appear green in colour, with the exception of the pyrene-containing 4, which is yellow-ochre. Some problems with purification arose during synthesis of the urea-based receptors 5 and 6 due to the similar solubilities of receptor complex and unbound ligand. These difficulties arose due to the need to have an excess of the ligand present in solution during the reaction to favour formation of the tripod – a threefold excess was found to be insufficient, producing a mixture of three-armed receptor, two-armed receptor, and free ligand. Addition of between 3.5 and 4 equivalents of ligand led to a greater yield of the desired three-armed receptor complexes 5 and 6, but required removal of the excess ligand. For complex 5 this was achieved by dissolution of the crude product in acetone and addition of cold diethyl ether until the complex precipitated. Complex 6 required more careful solvent balance, eventually involving a mixture of acetone, diethyl ether and ethyl acetate to remove the majority of the excess ligand by recrystallisation, although some ligand was still found to be present by 1H-NMR spectroscopy. As a result studies on 6 were not pursued.![[([9]ane-S3)Ru]2+ receptor complexes synthesised in this study. Counter-ions are PF6− in each case.](/image/article/2013/NJ/c2nj40401j/c2nj40401j-s1.gif) | ||
| Scheme 1 [([9]ane-S3)Ru]2+ receptor complexes synthesised in this study. Counter-ions are PF6− in each case. | ||
Receptors 2 and 3 are isomers of one another, the difference being in the placement of the N–H group. Complex 3 potentially has a larger binding pocket within the receptor due to the extra distance from the core and the flexibility provided by the methylene bridge allowing a greater degree of encapsulation of an anion. Receptor 4, with its pyrenyl functionalities, was designed as a potentially fluorescent anion sensor by analogy with related ‘pinwheel’ tripodal receptors.39
While analogous p-cymene-capped complexes were found to be stable as the dipodal species of type [Ru(p-cymene)L2Cl]+,32 formation of the tripodal receptors [Ru(p-cymene)L3]2+ proved difficult, as a result of steric crowding about the metal centre. By contrast, compounds 1–6 based on [9]ane-S3 are significantly more stable than their η6-arene counterparts with respect to solvolysis. Monitoring the 1H-NMR spectra of the complexes showed that after a period of one week in solution, only a small proportion (<10%) of the tripodal complex had degraded to form dipodal complex and free ligand. Even after four weeks, this ratio remains fairly constant, suggesting that the equilibrium tripod and dipod/free ligand is very slow on the NMR timescale and favours the tripodal species.
Complex 1 was obtained as single crystals from the ethanol:water reaction mixture and was characterised by single crystal X-ray crystallography, Fig. 1. The structure adopts space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with an inversion centre linking two complexes into an interdigitated dimer. These two symmetry–equivalent complexes are held together by the hydrogen bond interactions between the aminopyridine groups and two symmetry-related PF6− counter ions, resulting in a tripodal binding pocket for each anion. These anions also interact with the thioether ring of another symmetry equivalent receptor, in a similar manner to that described by Bedford and co-workers,37 such that the anion is involved in six hydrogen bonds, three from N–H groups and three from C–H groups. The second symmetry-independent PF6− anion only engages in one N–H⋯F hydrogen bond (to N(6)H), but is surrounded by three thioether rings, which interacts with the anion via CH⋯F interactions. The NH⋯F distances 3.14–3.41 Å are relatively long compared to typical contacts found in the CSD for NH hydrogen bond donors hydrogen bonding to PF6− which are typically around 3.0 Å, and this may reflect the bifurcation of both the donors and acceptor. The CH⋯anion distances are in the range of 3.1–3.5 Å and are consistent with related systems in the CSD.
, with an inversion centre linking two complexes into an interdigitated dimer. These two symmetry–equivalent complexes are held together by the hydrogen bond interactions between the aminopyridine groups and two symmetry-related PF6− counter ions, resulting in a tripodal binding pocket for each anion. These anions also interact with the thioether ring of another symmetry equivalent receptor, in a similar manner to that described by Bedford and co-workers,37 such that the anion is involved in six hydrogen bonds, three from N–H groups and three from C–H groups. The second symmetry-independent PF6− anion only engages in one N–H⋯F hydrogen bond (to N(6)H), but is surrounded by three thioether rings, which interacts with the anion via CH⋯F interactions. The NH⋯F distances 3.14–3.41 Å are relatively long compared to typical contacts found in the CSD for NH hydrogen bond donors hydrogen bonding to PF6− which are typically around 3.0 Å, and this may reflect the bifurcation of both the donors and acceptor. The CH⋯anion distances are in the range of 3.1–3.5 Å and are consistent with related systems in the CSD.
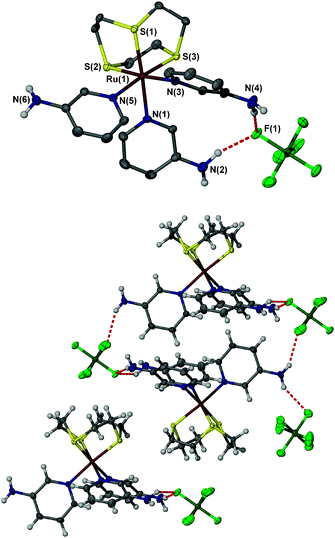 | ||
| Fig. 1 (a) X-ray molecular structure of 1 with hydrogen atoms attached to carbon removed for clarity. Selected H-bond distances (Å): N(4)H–F(1) 3.359(3); N(2)H–F(1) 3.138(3); N(6)H–F(7) 3.408(3). (b) Crystal packing in 1 (50% ellipsoids, CH hydrogen atom omitted for clarity). | ||
The structure shows that the receptor complex, at least in the solid state, exists in the opposite conformation to that expected, with the aminopyridine N–H groups pointing away from a common cavity beneath the metal centre, though this may be an effect of the solid state packing in combination with the relatively large size of the PF6− anion. It seems plausible that rotation about the Ru–N axes could lead to a situation in which three amine N–H groups point to a central position.
Anion binding studies
Binding studies were performed on the receptor complexes using 1H-NMR spectroscopic titration in 30% DMSO-d6–CDCl3 mixture. The inclusion of DMSO was essential to retain solubility of receptor 1 during the titration, as addition of anions to the solution in CDCl3 or acetone-d6 caused precipitation of the complex. For consistency the same solvent system was used throughout the experiments.Complex 1 showed marked broadening of the amine NH resonances upon addition of anions caused either by rotation of the amine group about the C–N bond on a similar timescale to the NMR spectroscopic experiment or, more probably, by amine proton exchange. Similar difficulties were found during experiments with 3, which also experiences increasingly significant broadening as the titration progresses. It is likely that hydrogen bonding to the anion increases the amine acidity. In these cases, the neighbouring pyridyl C–H signal (Hc, Fig. 2) alone was followed during the titration, while for the other amine receptors (2 and 4) the N–H resonance remained sharp in the NMR spectrum and thus could be monitored. In titrations with 4, only the NH resonance was monitored, as the pyridyl CH signal is obscured by the pyrene CH resonances. The NMR spectrum of 4 proved concentration dependent and a dilution study gave a dimerization constant of 2.63(8) which was included as a constant in anion complexation titratons. Titrations with urea-based receptor 5 followed the urea NH resonances exclusively. Job plot analyses suggested a mixture of 1:1 and 1:2 binding in many cases (Fig. 4) and as a result a model including both 1:1 and 1:2 host:anion species proved to be a better fit to the data in most cases. Titration results are summarised in Table 1.
 | ||
| Fig. 2 Chemical shift changes of pyridyl CH resonance of 1 with a variety of anions. | ||
:1 and 1:2 binding modes, refined using HypNMR 200640 in 30% DMSO-d6–CDCl3 mixture. Titrations of receptor 1 with fluoride showed poor binding, so this anion was omitted from further studies. K2 is calculated by subtracting β1 from β12 (where applicable) and represents the second binding step. Data for 4 include the dimerization equilibrium, log K20 = 2.63(8)
| Guest | Host | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| OAc− | β1 | 2.02(1) | 2.05(1) | 2.09(7) | 2.36(1) | 2.5(1) |
| β12 | 3.83(1) | 3.90(2) | — | 4.05(1) | 5.84(3) | |
| (K2) | 1.81 | 1.85 | — | 1.69 | 3.34 | |
| Br− | β1 | 2.04(8) | 2.29(8) | 1.8(1) | 2.68(3) | 3.2(1) |
| β12 | — | — | — | 5.56(5) | 6.29(9) | |
| (K2) | — | — | — | 2.88 | 3.09 | |
| Cl− | β | 1.76(1) | 1.73(1) | 2.2(1) | 2.48(3) | 3.2(1) |
| β12 | 3.52(1) | 3.46(9) | — | 4.6(1) | 5.9(2) | |
| (K2) | 1.76 | 1.73 | — | 2.12 | 2.7 | |
| F− | β1 | <1 | — | — | — | — |
| NO3− | β1 | 1.86(2) | 2.33(6) | 3.2(1) | 2.65(3) | 3.4(1) |
| β12 | 5.3(3) | |||||
From the titration data, it is possible to compare the anion affinities of the receptor complexes. There is only a small difference between the binding affinities of 1 and 2, suggesting that the addition of a benzyl group to the receptor amine has only a marginal effect on the anion binding of the receptor. Interestingly, binding of both of these hosts shows little variation in strength for the addition of the first and second equivalent of anion, especially for chloride, suggesting that there is little cooperativity between ligands.
Changing the position of the N–H group has a much more pronounced effect on the receptor properties. Anions typically bind in a 1:1 ratio with 3 compared to the mixture of 1:1 and 1:2 complexes formed by 2. Compound 3 seems to show a slightly higher affinity for nitrate over the other anions (albeit with large error) perhaps reflecting the larger cavity. The inclusion of the sterically more demanding pyrenyl functionality seems to favour binding of the receptor to chloride (and to a lesser extent acetate), with chloride binding to receptor 4 approximately five to ten fold stronger compared to the smaller receptors 1–3. Receptor 5 with its strongly anion binding urea groups is generally the most effective host. Unusually, receptor 5 appears to show strongest binding with bromide over the other anions studied, despite this anion showing only a small chemical shift change throughout the titration. The higher affinity for bromide compared to acetate may be due to the size and shape complementarity of the receptor to the anion – the spherical bromide is able to interact better with the three binding groups, but has only a small effect on the electronic shielding of the binding protons. The acetate titration data exhibit a marked sigmoidality (Fig. 3) suggesting some degree of positive cooperativity in binding the second anion and indeed K2 is markedly higher than β1. The origins of this effect are not clear but must reflect the more demanding shape of the acetate anion compared to spherical anions such as halides. The nitrate data also showed marked sigmoidality, albeit with a dramatically smaller chemical shift change. This data proved impossible to fit to a 1:1 and 1:2 binding model and a contribution from a 2:1 host:guest species was also included, β21 = 6.5(3) to model the data however the fit remained unsatisfactory.
 | ||
| Fig. 3 Chemical shift change of urea N–H resonance of 5 with a variety of anions. | ||
 | ||
| Fig. 4 Job plot for receptor 1 with TBA chloride. | ||
Overall, the complexes show significantly higher affinity than related metal complexes with only a single pyridyl ligand,23 which are found to bind only very weakly. This is likely to be a reflection of the multiple binding groups and positive charge.
The presence of a 1:2 host:guest species in many cases may indicate the possibility of anions either binding separately to different pyridyl ligands or nestling within the CH binding pocket of the [9]ane-S3 in a similar way to the X-ray crystal structure of 1 and the related [9]ane-S3 capped palladium complex.37 The presence of two different binding modes is consistent with the broadening of the NH resonances observed during some of the NMR titration experiments. Further evidence for the involvement of the [9]ane-S3 CH protons comes from the observation that the resonances assigned to these protons undergo noticeable chemical shift change of up to 0.5 ppm during the titration.
Photophysical studies
Receptor 4 is expected to be fluorescent due to the presence of the photoactive pyrenyl groups.39,41–44 The complex shows an absorbance spectrum similar in structure to that of the free pyrenyl pyridine ligand from which it is derived, albeit red-shifted slightly (Fig. 5), suggesting a lowering in the excitation energy caused by coordination to the metal. As expected, the molar extinction coefficient of 4 is approximately three times greater than that of the free ligand, due to the complex containing three coordinated ligands. | ||
| Fig. 5 UV/Visible absorption profiles of 4 (2 × 10−5 mol dm−3, red) and unbound 3-(1-pyrenemethylamino)pyridine (6 × 10−5 mol dm−3, green) in 30% DMSO/CHCl3 solvent. | ||
Addition of anions to the solution had a negligible impact on the absorption spectrum of the complex. This lack of detectable spectral response to the addition of most anions indicates that the hydrogen bonding functionalities are not electronically coupled to the pyrene chromophore.
Excitation of 4 at the primary absorbance wavelength at 347 nm results in predominantly emission assignable to a monomeric pyrene derivative which shares similarities to the free ligand (Fig. 5). Both free ligand and complex 4 also exhibit broad bands to longer wavelength assignable to pyrene excimer emission.45 This band occurs at 511 nm for the free ligand, while the excimer band for 4 appears to involve two overlapping excimeric species, possibly intra- and intermolecular excimers. The intensity of the excimer band is significantly higher in the case of 4 than the free ligand (at the same concentration). The most feasible explanation is that the band is enhanced by the pyrene groups being forced into close proximity to one another through their coordination to the metal, thus encouraging excimer formation. Addition of acetate, nitrate, chloride and bromide anions to the solution of 4, when followed by fluorimetry, at first glance appears to have a switch-on effect on the emission of the complex, with the emission intensity at 397 nm increasing to almost double the initial value, accompanied by a similar increase in the 500 nm excimer band. Further investigation into this enhancement, however, showed that this is not purely anion driven – a similar emission profile to those found with anions is found from the complex alone at the reduced concentration experienced at the end of the titration experiment (Fig. 6). The increase in emission at lower concentration suggests that some degree of quenching is taking place for receptor 4, since the same effect is not observed for the free ligand.
 | ||
| Fig. 6 Emission profiles for 4 (4 × 10−6 mol dm−3, red) and unbound 3-(1-pyrenemethylamino)pyridine (1.2 × 10−5 mol dm−3, green) in 30% DMSO/CHCl3 solvent, excitation wavelength of 347 nm. | ||
In order to determine the degree of this quenching, the emission intensity of the complex was recorded at a range of concentrations, and in a varying ratio of DMSO:CHCl3 solvent. Dilution of the complex, keeping the solvent ratio the same, follows a non-linear progression, with the highest intensity of emission being observed at extremely low complex concentration (Fig. 7). Below this critical concentration the emission intensity falls off rapidly. Collecting absorbance data over the same range of concentrations shows a linear change in the absorbance with concentration suggestion that the quenching effect is not a result of dimerization or aggregation.
 | ||
| Fig. 7 Normalised emission profiles for 4 (red) and with chloride (green) and acetate (blue) at 4.3 × 10−5 mol dm−3 in 30% DMSO/CHCl3. All three spectra have been normalised relative to the standard before dilution (4 at 5 × 10−5 mol dm−3, black) because of the concentration dependence of the complex's emission intensity. | ||
It was also found that the emission quenching was more significant at lower DMSO content, and less evident in solvent mixtures with a higher proportion of DMSO, suggesting that ion pairing is a possible factor in the quenching process – ion pairing is expected to be less significant in the more polar solvent.
While the emission spectrum of the complex is thus highly dependent on the concentration of the complex, the relative intensity of the excimer emission band is affected by anions, Fig. 8, with acetate giving the most marked effect on the ratio between monomer and excimer bands. An increase in the intensity of this band is observed depending on the identity of the added anion, indicating that the pyrene–pyrene interactions are somewhat influenced enhanced by anion binding. The same effect is not observed by titration of anions into a solution of free 3-(1-pyrenemethylamino)pyridine, and hence it must arise from the close proximity of the pyrenyl groups in the complex.
 | ||
| Fig. 8 Absorbance (red) at 347 nm and emission (normalised, green) and 397 nm of 4 as a function of concentration in 30% DMSO/CHCl3. | ||
Conclusions
In summary, a range of 1,4,7-trithiacyclononane-capped ruthenium(II)-based anion receptor complexes have been synthesised. These receptors, in contrast to similar π-arene capped ruthenium complexes, show a high resistance to degradation from the tripodal ML3 species to a dipodal ML2X species by solvent or anion. The receptors bind common anions in relatively polar media forming both 1:1 and 1:2 host–anion complexes with the CH⋯anion interactions involving the thioether ring being implicated in anion binding as well as the conventional NH donors. Attempts to produce a fluorescent anion sensor based on appended pyrenyl groups were only partially successful because of complications arising from the concentration dependence of the host emission.Experimental
Synthesis
Pyridine-containing ligands were synthesised according to previously published methods.14,31,32,39[Ru([9]ane-S3)Cl2(DMSO)]
RuCl3·3H2O (1.00 g, 3.80 mmol) was dissolved in the minimum amount of DMSO (3 ml) and the solution heated to boiling to remove any water. The DMSO solution was cooled and acetone (25 ml) was added, leading to precipitation of a dark orange material. Filtration of this material left an orange solution which gave the desired RuCl2(DMSO)4 as yellow crystals upon drying. Repeated dissolution of the dark orange precipitate with more DMSO–acetone with heating produced more of the yellow product. Yield 0.80 g, 1.6 mmol, 43%. Anal. calc. for C8H24S4O4RuCl2: C, 19.83; H, 4.99. Found: C, 19.84; H, 4.90. The resulting RuCl2(DMSO)4 (0.48 g, 1.0 mmol) was dissolved in CHCl3 (25 ml) in a round-bottomed flask fitted with a reflux condenser. 1,4,7-Trithiacyclononane (0.20 g, 1.1 mmol) was added and the solution heated to reflux for 90 min with stirring, leading to the precipitation of [Ru([9]ane-S3)Cl2(DMSO)] as a yellow-orange solid. Yield 0.36 g, 0.84 mmol, 84%. Anal. calc. for C8H18S4ORuCl2: C, 22.32; H, 4.21. Found: C, 22.05; H, 4.08.:1, 30 ml) with slight warming of the solution. Silver hexafluorophosphate (0.21 g, 0.85 mmol) was added and the solution stirred at 60 °C for 1 h, over which time the formation of silver chloride could be observed as a grey precipitate. Filtration of this salt left a yellow solution, to which was added 3-aminopyridine (0.11 g, 1.2 mmol). The solution was then heated to reflux for 16 h, during which the colour turned from yellow to blue-green. Cooling and slow evaporation of the solvent led to the formation of the product as deep green needle-shaped crystals. Yield 0.13 g, 0.15 mmol, 37%. 1H NMR (DMSO-d6, J/Hz, δ/ppm): 2.53–2.90 (m, 12 H, CH2), 5.79 (s, 6 H, NH2), 6.98–7.14 (m, 6 H, pyH), 7.41 (d, J = 4.5, 3H, pyH), 7.73 (d, J = 2.2, 3 H, pyH). 13C{1H} NMR (DMSO-d6, δ/ppm): 33.68, 122.46, 126.40, 139.96, 142.31, 146.96. ESI+-MS: m/z 708.8 {([9]ane-S3)Ru(3-aminopyridine)3·PF6]+, 281.7 [([9]ane-S3)Ru(3-aminopyridine)3]2+. Anal. calc. for C21H30N6S3Ru(PF6)2: C, 29.54; H, 3.54; N, 9.85. Found: C, 29.68; H, 3.60; N, 9.52. IR (cm−1): 3391 (ν(N-H)).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)).O)).
O)).O)).General procedure for 1H NMR spectroscopic titration
A solution of the host species of known concentration typically 0.5–1.5 mM, was made up in an NMR tube using the appropriate deuterated solvent (0.5 ml CDCl3–DMSO-d6 (v/v 70/30)). Solutions of the anions, as TBA salts (1 ml) were made ten times the concentration of the host solution. The guest solution was typically added in 10 μl aliquots, representing 0.1 equivalents of the guest with respect to the host. Larger aliquots were used in some cases where no inflection of the trace was evident. Spectra were recorded after each addition and the trace was followed simultaneously. Results were analysed using the curve-fitting program HypNMR 2006.40 For Job plot experiments stock solutions of host and guest concentration 1 mM were mixed in varying proportions to give a constant total volume and the NMR spectra recorded.48Photophysical measurements
UV-vis spectroscopic measurements were carried out using a Perkin-Elmer Lambda 35 spectrometer. Emission and excitation spectra were obtained using a Jobin-Yvon Horiba Fluorolog 3-22 Tau-3 spectrofluorimeter with a right angle illumination method and were corrected for the spectral response of the instrument. Fluorescence spectroscopic titrations used a stock solution of concentration 1.0 × 10−4 mol dm−3 of host in a volumetric flask. A 3 ml sample of host solution was prepared by the desired dilution of the stock solution and titrated with the appropriate guest. Guest solutions were prepared such that 100 μl of guest solution corresponds to 100 equivalents of host.Crystal data
Crystals were grown from 5.1 in ethanol–water (1:1 ratio, 2 ml) by slow evaporation of the solvent. Crystal data for 1: C21H30F12N6P2RuS3, M = 853.70, green needles, 0.02 × 0.02 × 0.2 mm3, triclinic, space group P (No. 2), a = 11.7238(11), b = 11.7563(11), c = 13.1508(12) Å, α = 109.9560(10), β = 94.499(2), γ = 110.922(2)°, V = 1549.4(2) Å3, Z = 2, Dc = 1.830 g cm−3, F000 = 856, Mo-Kα radiation, λ = 0.71073 Å, T = 120(2) K, 2θmax = 61.0°, 27777 reflections collected, 8829 unique (Rint = 0.0280). Final GooF = 0.920, R1 = 0.0306, wR2 = 0.1072, R indices based on 7873 reflections with I > 2σ(I) (refinement on F2), 430 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.910 mm−1.Notes and references
- S. G. Telfer, G. Bernardinelli and A. F. Williams, Chem. Commun., 2001, 1498–1499 RSC.
- S. G. Telfer, G. Bernardinelli and A. F. Williams, Dalton Trans., 2003, 435–440 RSC.
- B. Wu, X. J. Yang, C. Janiak and P. G. Lassahn, Chem. Commun., 2003, 902–903 RSC.
- S. Nieto, J. Pérez, V. Riera, D. Miguel and C. Alvarez, Chem. Commun., 2005, 546–548 RSC.
- N. C. A. Baker, N. McGaughey, N. C. Fletcher, A. V. Chernikov, P. N. Hortonb and M. B. Hursthouse, Dalton Trans., 2009, 965–972 RSC.
- B. Geisser, A. Ponce and R. Alsfasser, Inorg. Chem., 1999, 38, 2030–2037 CrossRef CAS.
- P. L. Arnold and A. C. Scarisbrick, Organometallics, 2004, 23, 2519–2521 CrossRef CAS.
- L. H. Uppadine, F. R. Keene and P. D. Beer, J. Chem. Soc., Dalton Trans., 2001, 2188–2198 RSC.
- X. D. Yu, H. Lin and H. K. Lin, Transition Met. Chem., 2008, 33, 829–834 CrossRef CAS.
- A. Ghosh, S. Verma, B. Ganguly, H. N. Ghosh and A. Das, Eur. J. Inorg. Chem., 2009, 2496–2507 CrossRef CAS.
- X.-F. Shang, J. Li, H. Lin, P. Jiang, Z.-S. Cai and H.-K. Lin, Dalton Trans., 2009, 2096–2102 RSC.
- S. Derossi, H. Adams and M. D. Ward, Dalton Trans., 2007, 33–36 RSC.
- Y. Cui, Y. L. Niu, M. L. Cao, K. Wang, H. J. Mo, Y. R. Zhong and B. H. Ye, Inorg. Chem., 2008, 47, 5616–5624 CrossRef CAS.
- K. J. Wallace, R. Daari, W. J. Belcher, L. O. Abouderbala, M. G. Boutelle and J. W. Steed, J. Organomet. Chem., 2003, 666, 63–74 CrossRef CAS.
- L. Ion, D. Morales, J. Pérez, L. Riera, V. Riera, R. A. Kowenicki and M. McPartlin, Chem. Commun., 2006, 91–93 RSC.
- M. Auzias, B. Therrien, G. Suss-Fink, P. Stepnicka, W. H. Ang and P. J. Dyson, Inorg. Chem., 2008, 47, 578–583 CrossRef CAS.
- A. L. Hector and A. F. Hill, Inorg. Chem., 1995, 34, 3797–3800 CrossRef CAS.
- M. Newell, J. D. Ingram, T. L. Easun, S. J. Vickers, H. Adams, M. D. Ward and J. A. Thomas, Inorg. Chem., 2006, 45, 821–827 CrossRef CAS.
- S. Bolano, J. Bravo, J. Castro, M. M. Rodriguez-Rocha, M. da Silva, A. J. L. Pombeiro, L. GonsalVi and M. Peruzzini, Eur. J. Inorg. Chem., 2007, 5523–5532 CrossRef CAS.
- I. Kuzu, D. Nied and F. Breher, Eur. J. Inorg. Chem., 2009, 872–879 CrossRef CAS.
- J. Pérez and L. Riera, Chem. Soc. Rev., 2008, 37, 2658–2667 RSC.
- J. Pérez and L. Riera, Chem. Commun., 2008, 533–543 RSC.
- L. Ion, D. Morales, S. Nieto, J. Pérez, L. Riera, V. Riera, D. Miguel, R. A. Kowenicki and M. McPartlin, Inorg. Chem., 2007, 46, 2846–2853 CrossRef CAS.
- J. Pérez, D. Morales, S. Nieto, L. Riera, V. Riera and D. Miguel, Dalton Trans., 2005, 884–888 RSC.
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- C. R. Bondy, P. A. Gale and S. J. Loeb, J. Supramol. Chem., 2002, 2, 93–96 CrossRef CAS.
- C. R. Bondy, P. A. Gale and S. J. Loeb, J. Am. Chem. Soc., 2004, 126, 5030–5031 CrossRef CAS.
- M. G. Fisher, P. A. Gale, M. E. Light and S. J. Loeb, Chem. Commun., 2008, 5695–5697 RSC.
- N. Qureshi, D. S. Yufit, J. A. K. Howard and J. W. Steed, Dalton Trans., 2009, 5708–5714 RSC.
- D. R. Turner, B. Smith, E. C. Spencer, A. E. Goeta, I. R. Evans, D. A. Tocher, J. A. K. Howard and J. W. Steed, New J. Chem., 2005, 29, 90–98 RSC.
- D. R. Turner, E. C. Spencer, J. A. K. Howard, D. A. Tocher and J. W. Steed, Chem. Commun., 2004, 1352–1353 RSC.
- S. J. Dickson, S. C. G. Biagini and J. W. Steed, Chem. Commun., 2007, 4955–4957 RSC.
- S. J. Dickson, M. J. Paterson, C. E. Willans, K. M. Anderson and J. W. Steed, Chem.–Eur. J., 2008, 14, 7296–7305 CrossRef CAS.
- S. Roche, C. Haslam, H. Adams, S. L. Heath and J. A. Thomas, Chem. Commun., 1998, 1681–1682 RSC.
- M. N. Bell, A. J. Blake, M. Schröder, H. J. Kuppers and K. Wieghardt, Angew. Chem., Int. Ed. Engl., 1987, 26, 250–251 CrossRef.
- S. Roche, H. Adams, S. E. Spey and J. A. Thomas, Inorg. Chem., 2000, 39, 2385–2390 CrossRef CAS.
- R. B. Bedford, M. Betham, C. P. Butts, S. J. Coles, M. B. Hursthouse, P. N. Scully, J. H. R. Tucker, J. Wilkie and Y. Willener, Chem. Commun., 2008, 2429–2431 RSC.
- I. P. Evans, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 204–209 RSC.
- M. H. Filby, S. J. Dickson, N. Zaccheroni, L. Prodi, S. Bonacchi, M. Montalti, C. Chiorboli, M. J. Paterson, T. D. Humphries and J. W. Steed, J. Am. Chem. Soc., 2008, 130, 4105–4113 CrossRef CAS.
- P. Gans, HypNMR 2006, University of Leeds, Leeds, 2006 Search PubMed.
- I. Suzuki, M. Ui and A. Yamauchi, J. Am. Chem. Soc., 2006, 128, 4498–4499 CrossRef CAS.
- B. Schazmann, N. Alhashimy and D. Diamond, J. Am. Chem. Soc., 2006, 128, 8607–8614 CrossRef CAS.
- S. K. Kim, J. H. Bok, R. A. Bartsch, J. Y. Lee and J. S. Kim, Org. Lett., 2005, 7, 4839–4842 CrossRef CAS.
- S. Nishizawa, Y. Kato and N. Teramae, J. Am. Chem. Soc., 1999, 121, 9463–9464 CrossRef CAS.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- K. J. Wallace, W. J. Belcher, D. R. Turner, K. F. Syed and J. W. Steed, J. Am. Chem. Soc., 2003, 125, 9699–9715 CrossRef CAS.
- D. R. Turner, M. J. Paterson and J. W. Steed, J. Org. Chem., 2006, 71, 1598–1608 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chichester, 2nd edn, 2009 Search PubMed.
Footnotes |
| † This article is included in the All Aboard 2013 themed issue. |
| ‡ Dedicated to Prof. Peter C. Junk on occasion of his 50th birthday. |
| § Electronic supplementary information (ESI) available: Additional titration isotherms and UV-vis absorption spectra. CCDC 882871. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2nj40401j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2013 |