 Open Access Article
Open Access ArticleThe use of X-ray absorption and synchrotron based micro-X-ray fluorescence spectroscopy to investigate anti-cancer metal compounds in vivo and in vitro
Alfred A.
Hummer
and
Annette
Rompel
*
Institut für Biophysikalische Chemie, Universität Wien, Althanstr. 14, 1090 Wien, Austria. E-mail: annette.rompel@univie.ac.at; Fax: +43-1-4277-9525; Tel: +43-1-4277-52502
First published on 8th March 2013
Abstract
X-ray absorption spectroscopy (XAS) and micro-synchrotron based X-ray fluorescence (micro-SXRF) are element specific spectroscopic techniques and have been proven to be valuable tools for the investigation of changes in the chemical environment of metal centres. XAS allows the determination of the oxidation state, the coordination motif of the probed element, the identity and the number of adjacent atoms and the absorber–ligand distances. It is further applicable to nearly all types of samples independent of their actual physical state (solid, liquid, gaseous) down to μM concentrations. Micro-SXRF can provide information on the distribution and concentration of multiple elements within a sample simultaneously, allowing for the chemical state of several elements within subcellular compartments to be probed. Modern third generation synchrotrons offer the possibility to investigate the majority of the biologically relevant elements. The biological mode of action of metal-based compounds often involves interactions with target and/or transport molecules. The presence of reducing agents may also give rise to changes in the coordination sphere and/or the oxidation state. XAS and micro-SXRF are ideal techniques for investigating these issues. This tutorial review introduces the use of XAS and micro-SXRF techniques into the field of inorganic medicinal chemistry. The results obtained for platinum, ruthenium, gallium, gold and cobalt compounds within the last few years are presented.
 Alfred A. Hummer | Alfred A. Hummer obtained his bachelor's and master's degrees in Biotechnology at the University of Natural Resources and Life Sciences (BOKU), Vienna. He joined the Institute of Biophysical Chemistry at the University of Vienna in 2009, where he is currently completing his PhD in Chemistry. The main focus of his work is on the structure and function elucidation of anti-cancer ruthenium and gallium compounds in biological tissues and in the presence of possible target molecules, applying X-ray absorption and micro-X-ray fluorescence techniques based on synchrotron radiation. |
 Annette Rompel | Annette Rompel has been the head of the Institute of Biophysical Chemistry at the University of Vienna since 2008, when she joined the Faculty of Chemistry. She studied chemistry at the University of Münster where she received her PhD. Besides research at the University of California, Berkeley and the Lawrence Berkeley National Laboratory she was a visiting scientist at RIKEN, The Institute of Physical and Chemical Research, Sendai, Japan and the University of Southern Denmark, Odense. Her main research interests are metal containing enzymes from natural sources and their structure and function elucidation using synchrotron based techniques. |
1. Introduction
Metal compounds have been used in the treatment of a various number of diseases, but it was in 1965 when Rosenberg et al. first encountered the anti-tumoural activity of cisplatin.1 Since then intensive research in the field of tumour inhibiting metal complexes has been undertaken. X-ray absorption spectroscopy (XAS) is an important tool for the examination of structurally undefined heterogeneous systems. There are several review articles on the anti-tumoural activity of metal complexes2–6 as well as the application of XAS7–14 and micro-SXRF15–17 in biological systems or bioinorganic chemistry. This review will provide a glimpse into the possibilities, applications and outcomes of these spectroscopic techniques in metal based cancer research.It focuses on the metal complexes of platinum, gallium, ruthenium, gold and cobalt which were recently investigated using XAS and micro-SXRF, and are promising candidates for use as anti-cancer agents.
The first section of this article will give an overview of XAS and micro-SXRF and their underlying principles. The following sections will discuss the proposed modes of action of compounds comprised of the aforementioned metals and present the latest results of XAS and micro-SXRF investigations and their contributions to the clarification of the in vivo pathways.
1.1. X-ray absorption spectroscopy in a nutshell
XAS is a method which probes the core–shell electrons of an atom (1s, 2s or 2p) by means of X-rays. Corresponding to the energy levels of the core shell electrons of the probed element, the X-rays will be absorbed at distinct energies displayed in a sudden rise in the absorption spectrum. This increase in absorption is called the X-ray absorption edge or edge. According to the energy state of the excited electron the edges are named K (1s), LI (2s) and LII and LIII (2p1/2 and 2p3/2). The sector from 30 electron volt (eV) below and up to 50 eV above the edge is called the X-ray absorption near edge structure (XANES) and the ongoing spectral features are called extended X-ray absorption fine structure (EXAFS). In the XANES regime the electrons are excited into the lowest unoccupied bound state orbitals (LUMO) just above the Fermi energy. In the case of EXAFS the electrons are promoted into higher energy states (the so called continuum), creating a photoelectron and interacting with the surrounding atoms.9 The constructive and destructive interactions of the photoelectron with the backscattering from adjacent atoms give rise to the well known oscillations in EXAFS (see Fig. 1). XANES gives important information about the oxidation state, the electronic structure of the absorber–ligand environment and the coordination motif of the excited atom, whereas EXAFS is suitable to determine the absorber–ligand distances and the number and the type of the nearest neighboring atoms (up to about 4 Å). | ||
| Fig. 1 Center: XAS spectrum of ruthenium compound 19 KP1019 collected in transmission mode (structural formula see Fig. 7). Right: extracted fine structure (EXAFS) and Fourier transform (FT) thereof. Left: the normalized XANES spectrum and the normalized derivative f′(x). | ||
The analysis of the absorption spectra, their features and the underlying principles are discussed elsewhere.18–21 For further reading introductory books of Bunker,18 de Groot and Kotani,22 and Stöhr,23 and review articles19,24,25 covering the underlying principles, applications and analysis of XAS can be recommended. Furthermore, the articles of Rehr and Albers26 and Ankudinov et al.27 discuss the theoretical models applied to XAS in detail. A historical overview of the development of XAS is given in the reviews of Lytle28 and Stumm von Bordwehr.29
1.2. Microprobe synchrotron based X-ray fluorescence
Micro-synchrotron based X-ray fluorescence (micro-SXRF) or micro-synchrotron radiation induced X-ray emission (micro-SRIXE)30 uses X-ray beams focused to spot sizes ranging from hundreds of μm2 down to the nm2 range. The sample stage is moved in a fixed distance horizontally and vertically relative to the incoming beam to raster the desired sample area. Depending on the beam size and collection time per spot the distribution and concentration profiles of larger sample areas or subcellular structures can be monitored. When collecting SXRF data all elements with lower edge energies than the incident one are excited and the corresponding spatial distribution data of these elements are collected at once. Micro-SXRF is applied in anti-cancer research to study the two dimensional distribution and concentration of metals and other biologically important elements, like phosphorus or sulfur, in cell or tissue samples. Thus, the direct assignment of the metal distribution to specific biological functions and targets is possible, e.g. DNA binding or accumulation in membrane regions. To quantify the elements in the tissue or cells the collected spectra and peak intensities must be calibrated against a standard of known composition and concentrations of the elements in question.15,16,311.3. Application of XAS in biological systems
XAS can be applied independently of the actual physical state (solid, liquid, gaseous) and does not require any long range order. It has evolved into a standard measurement technique in the study of amorphous materials and in bioinorganic chemistry.14,24 Especially in the investigation of spectroscopically silent metals like Au(I), Zn(II), Ga(III) and K(I), Ca(II), Mg(II) with a full or an empty d-shell it is one of the few techniques which can obtain valuable structural information.32 XAS is elemental specific and the elements Pt, Ru, Ga, and Au are not naturally abundant chemical species in biological tissues. This makes XAS an ideal choice for the study of anti-tumoural compounds.332. Platinum compounds
In the last few years the research in the field of Pt(II) complexes focused on cisplatin (1),1 carboplatin (2),34 oxaliplatin (3),35 and nedaplatin (4).36–38 These compounds are available commercially and are in clinical use worldwide (1 cisplatin, 2 carboplatin and 3 oxaliplatin, Fig. 2), or at least in one country: 4 nedaplatin (Japan, Fig. 2),36,37 heptaplatin (5, Republic of Korea, Fig. 2)36,37 and lobaplatin (6, China, Fig. 2).39 Today these drugs are mostly used in combined treatment plans.40,41platinum(ii) (3, oxaliplatin), (diammine[hydroxyacetato(2-)-O,O′]platinum(ii) (4, nedaplatin). Center: [propanedioato(2-)-O,O′][2-(1-methylethyl)-1,3-dioxolane-4,5-dimethanamine-N,N′]platinum(ii) (5, heptaplatin), [2-hydroxypropanoato(2-)-O1,O2][1,2-cyclobutanedimethanamine-N,N′]platinum(ii) (6, lobaplatin), and the intercalators [Pt(5,6-dimethyl-1,10-phenanthroline)(trans-1R,2R-diaminocyclohexane)]2+ (7, [56MERR]), and [Pt(5,6-dimethyl-1,10-phenanthroline)(trans-1S,2S-diaminocyclohexane)]2+ (8, [56MESS]). Bottom: the Pt(ii) complexes: [trisamminechloroplatinum(ii)]+ [Pt(ii)Cl(NH3)3]+ (9), Pt[N(p-HC6F4)CH2]2py2 (12, Pt103), cis-ammine(3-bromopyridine)dichloroplatinum(ii) [PtCl2(3-Brpyr)(NH3)] (16, Brpyr = 3-bromopyridine), and a platinum complex with an anthrachinone ligand [PtCl2(2C3)(NH3)] (18, Pt-2C3).](/image/article/2013/MT/c3mt20261e/c3mt20261e-f2.gif) | ||
| Fig. 2 Top: the most prominent Pt(II) anti-cancer drugs today: cis-diamminedichloroplatinum(II) (1, cisplatin), cis-diammine(cyclobutane-1,1-dicarboxylate-O,O′)platinum(II) (2, carboplatin), [(1R,2R)-cyclohexane-1,2-diamine](ethanedioato-O,O′)platinum(II) (3, oxaliplatin), (diammine[hydroxyacetato(2-)-O,O′]platinum(II) (4, nedaplatin). Center: [propanedioato(2-)-O,O′][2-(1-methylethyl)-1,3-dioxolane-4,5-dimethanamine-N,N′]platinum(II) (5, heptaplatin), [2-hydroxypropanoato(2-)-O1,O2][1,2-cyclobutanedimethanamine-N,N′]platinum(II) (6, lobaplatin), and the intercalators [Pt(5,6-dimethyl-1,10-phenanthroline)(trans-1R,2R-diaminocyclohexane)]2+ (7, [56MERR]), and [Pt(5,6-dimethyl-1,10-phenanthroline)(trans-1S,2S-diaminocyclohexane)]2+ (8, [56MESS]). Bottom: the Pt(II) complexes: [trisamminechloroplatinum(II)]+ [Pt(II)Cl(NH3)3]+ (9), Pt[N(p-HC6F4)CH2]2py2 (12, Pt103), cis-ammine(3-bromopyridine)dichloroplatinum(II) [PtCl2(3-Brpyr)(NH3)] (16, Brpyr = 3-bromopyridine), and a platinum complex with an anthrachinone ligand [PtCl2(2C3)(NH3)] (18, Pt-2C3). | ||
The pharmacological mode of action is supposed to be similar for 1 cisplatin, 2 carboplatin, 3 oxaliplatin and 4 nedaplatin.42 The complexes can be seen as pro-drugs activated through hydrolysis, ending in an active diamine-diaquo-platinum complex (diaminocyclohexane-diaquo-platinum for 3 oxaliplatin).42 The difference in their cytotoxicity is found to be due to their differing pharmacokinetic behaviour, mainly governed by the velocity of aquation of the leaving ligands chloride (1 cisplatin), cyclobutane (2 carboplatin), the oxalate group (3 oxaliplatin) and the glycolate moiety (4 nedaplatin).39,42 The tumour inhibiting Pt(II) complexes are supposed to form adducts with amine groups of proteins, RNA and DNA. Mainly the crosslinking of guanine (N7 position) and adenine residues in DNA is thought to be the major cause of cytotoxicity. The crosslinking occurs within the same strand (intrastrand) and between different strands (interstrand). The bending of the DNA strand prohibits proper DNA transcription and the large number of crosslinks overwhelms the DNA repair mechanisms.43 Thus, cells with enhanced DNA repair mechanisms tend to exhibit a resistance to 1 cisplatin.42,43 Although DNA is a major target for platinum drugs it is not necessarily the only cause of cell death and treated cells showing no DNA damage have undergone apoptosis.44
Apart from the aforementioned Pt(II) complexes, there exists much interest in the use of Pt(IV) complexes as pro-drugs.45,46 Their mode of action is less understood and reduction of the Pt(IV) precursor to a Pt(II) pro-drug followed by a hydrolysis step is argued (Fig. 3).47 The higher stability of octahedral Pt(IV) complexes under physiological conditions reduces unwanted interactions with non-DNA biological nucleophiles and Pt related severe side effects.46,48 The main goals in the development of Pt(IV) complexes are: (i) oral application of Pt(IV) compounds, (ii) increase of its target selectivity (compared to Pt(II) complexes) via the modification of the axial ligands, and (iii) decrease in non-specific interactions (particularly with thiols) en route to the target sites, by increasing its kinetic inertness.47,48 One approach to overcome the early reduction through gastric juices is the use of nanobased delivering systems like carbon nanotubes, gold based nanoparticles and polymeric nanoparticles.5 Nonetheless, up to now, no Pt(IV) compound has reached the status of an approved cancer drug (albeit entered clinical trials).5,37,39
![The platinum(iv) complexes. Top: trans,mer-[Pt(iv)Cl(OH)2(NH3)3]+ (10), cis,trans,cis-[PtCl2(OAc)2(NH3)(NBA)] (NBA = n-butylamine) (11), cis-[Pt(iv)Cl4(NH3)2] (13), cis,trans,cis-[Pt(iv)Cl2(OAc)2(NH3)2] (14). Bottom: cis,trans,cis-[Pt(iv)Cl2(OH)2(NH3)2] (15), and cis,trans,cis-[PtCl2(OAcBr)2(NH3)2] (OAcBr = bromacetato) (17), and the Pt(iv) to Pt(ii) reduction scheme.](/image/article/2013/MT/c3mt20261e/c3mt20261e-f3.gif) | ||
| Fig. 3 The platinum(IV) complexes. Top: trans,mer-[Pt(IV)Cl(OH)2(NH3)3]+ (10), cis,trans,cis-[PtCl2(OAc)2(NH3)(NBA)] (NBA = n-butylamine) (11), cis-[Pt(IV)Cl4(NH3)2] (13), cis,trans,cis-[Pt(IV)Cl2(OAc)2(NH3)2] (14). Bottom: cis,trans,cis-[Pt(IV)Cl2(OH)2(NH3)2] (15), and cis,trans,cis-[PtCl2(OAcBr)2(NH3)2] (OAcBr = bromacetato) (17), and the Pt(IV) to Pt(II) reduction scheme. | ||
Metallointercalators are a new class of platinum(II) DNA-damaging compounds active against 1 cisplatin and 3 oxaliplatin resistant cell lines.49–51 Dillon reported the synthesis and the mechanism of action of metallointercalator compounds, among them the platinum complexes [56MERR] (7, Fig. 2) and [56MESS] (8, Fig. 2).527 [56MERR] and 8 [56MESS] are isomers possessing planar aromatic rings which can be inserted between the DNA base pairs. This non-covalent interaction unwinds the DNA double helix and is further stabilized through an overall positive charge of the complex. The resulting high selectivity for DNA make them promising highly toxic anti-cancer agents.52
2.1. XAS and micro-SXRF investigations on platinum
One of the first investigations on Pt anti-cancer compounds by means of XAS dates back to 1978 when Teo and Lippard et al. studied the binding of 1 cisplatin to calf thymus DNA.53 Since then, XAS has proven to be an invaluable technique in probing the reduction of Pt(IV) to Pt(II), as well as the changing coordination environment of the Pt(II) in cellulo and in different media environments. | ||
| Fig. 4 Left: XANES spectra of 1 cisplatin (Pt(II), dashed) and 15 (Pt(IV), solid) complexes. Right: linear fit of the peak height ratio a/b extracted from 15 (Pt(IV))/1 (Pt(II)) standard mixtures. Reprinted (adapted) with permission from M. D. Hall, G. J. Foran, M. Zhang, P. J. Beale, and T. W. Hambley, J. Am. Chem. Soc., 2003, 125, 7524–7525. Copyright 2003 American Chemical Society. | ||
In a recent paper56 the validity of the Pt(IV)/Pt(II) ratio method was further expanded to a number of platinum(IV) and platinum(II) complexes with different coordination spheres, and in different isomeric arrangements.56 The XANES spectra of six Pt(II) and twelve Pt(IV) complexes were collected in phosphate buffered saline (PBS buffer). The standard curves were constructed with solutions of the Pt(IV) complexes and their corresponding Pt(II) analogues in fixed molar ratios from 0% to 100% Pt(IV). The obtained ratios a/b were the same for all Pt(II) complexes. In the case of the Pt(IV) compounds the ratios a/b increased with the number of oxygen ligands in the first coordination sphere. Nevertheless in all cases the linear relationship between the ratio a/b and the Pt(IV) content was preserved. The reduction of Pt(IV) was monitored in cell growth medium (after 2, 14 and 24 h) and in A2780 ovarian cancer cells treated with the cis- and trans-Pt(IV) anti-tumoural compounds (after 2 and 24 h). The overall stability of the Pt(IV) complexes in cell growth medium was higher, presumably as a result of the decreasing reducing power of the medium over time due to the oxidative effects of air. The stability of the compounds in A2780 cells was lower, with most of the trans-Pt(IV) complexes reduced after 2 h, whereas the cis-Pt(IV) complexes were revealed to be more stable.56
Studies on the interaction of 1 cisplatin and 2 carboplatin with reduced glutathione (GSH) were conducted by Obata et al.58 As explained in Section 7, elements next to each other in the periodic table are hard to distinguish in XAS analysis. However, the replacement of light scatterers (e.g. N/O) through heavy scatterers (e.g. S/Cl) is possible and was studied with Pt LIII-edge EXAFS in this article. The experiments were carried out in PBS buffer at room temperature in the presence of 10-fold excess GSH. The N/O peaks in the Fourier transform (FT) of the EXAFS at around 1.6 Å decreased monotonically whereas the Cl/S peak at around 2.0 Å increased in the same manner. Following 180 and 930 min, a vast majority of the N/O ligands were observed to be exchanged for 1 cisplatin and 2 carboplatin. The advantage of EXAFS in monitoring these highly heterogenic systems without the need of labelling with expensive isotopes like 15N or 195Pt was clearly shown.
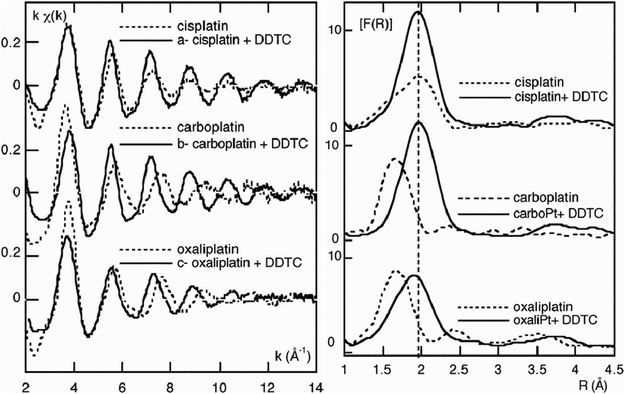 | ||
| Fig. 5 Left: comparison of EXAFS spectra before and after reaction between diethyl-dithiocarbamate (DDTC) and 1 cisplatin, 2 carboplatin, or 3 oxaliplatin. Right: comparison of Fourier transform amplitudes before and after reaction between DDTC and 1 cisplatin, 2 carboplatin, or 3 oxaliplatin. The replacement of N/O through Cl/S introduces a change in the FT peaks. Reprinted (adapted) with permission from: D. Bouvet, A. Michalowicz, S. Crauste-Manciet, D. Brossard, and K. Provost, Inorg. Chem., 2006, 45, 3393–3398. Copyright 2006 American Chemical Society. | ||
The cellular distributions of 1 cisplatin and its Pt(IV) analogues 13, 14, and 15 (Fig. 3) were analyzed using micro-SXRF by Hambley et al.65 These four compounds display a similar cellular distribution with Pt localized in the nucleus. Micro-XANES spectra collected on areas of high Pt concentration in the cells revealed that the complex 15 with dihydroxo axial ligands is the most resistant against reduction to Pt(II).
In a subsequent step A2780 cancer cells treated with the bromine labelled Pt(II) compound 16 (Fig. 2) and the Pt(IV) compound 17 (Fig. 3) were investigated with micro-SXRF.66 The bromine labelled ligands have replaced an amine ligand in the Pt(II) complex 16 and an axial ligand in the Pt(IV) complex 17. The dissociation of the bromine labelled axial ligands from 17 was used to monitor reduction to Pt(II). In the case of compound 16 the Pt and Br distributions were the same confirming the amine ligands as non-leaving groups. In the case of 17 the bromine distribution was more diffuse pointing toward the reduction of a substantial amount of the Pt(IV) complexes to Pt(II). However, it could not be determined if the reduction took place extra- or intracellularly. Furthermore, control A2780 cells treated with the unlabelled 1 cisplatin showed unexpected endogenous levels of bromine, but at substantially lower concentrations.66 Micro-SXRF investigations were undertaken on mouse tumour thin sections (4T1.2 neo 1 mammary tumour) derived from 15 (Fig. 3) treated Balb/c mice and on A2780 ovarian cancer cells treated with the intercalating complex 18 (Pt-2C3, Fig. 2).66 Due to the phosphate backbones of DNA, the phosphorous distribution within the A2780 cells is likely to correspond to the distribution of cellular DNA. The Pt distribution exhibited high co-localization with the phosphorous distribution, consistent with the selective DNA targeting abilities of the intercalating ligand.66
Davis et al.67 applied micro-SXRF to thin sections of A549 lung cancer cells treated with 1 cisplatin and the metallointercalators 7 [56MERR] and 8 [56MESS] (Fig. 2). The Pt in 7 [56MERR] and 8 [56MESS] treated cells was clearly co-localized with phosphorus, which points toward a main interaction with the densely packed DNA in the heterochromatin regions of the nucleus (see Fig. 6c and d).
![Micro-SXRF elemental maps for A549 cells following treatment (4 h) with: (a) cell medium, (b) 1 cisplatin, (c) 7 [56MERR] and (d) 8 [56MESS]. Reprinted (adapted) with permission from K. J. Davis, J. A. Carrall, B. Lai, J. R. Aldrich-Wright, S. F. Ralph, and C. T. Dillon, Dalton Trans., 2012, 41, 9417. Copyright 2012 Royal Society of Chemistry.](/image/article/2013/MT/c3mt20261e/c3mt20261e-f6.gif) | ||
| Fig. 6 Micro-SXRF elemental maps for A549 cells following treatment (4 h) with: (a) cell medium, (b) 1 cisplatin, (c) 7 [56MERR] and (d) 8 [56MESS]. Reprinted (adapted) with permission from K. J. Davis, J. A. Carrall, B. Lai, J. R. Aldrich-Wright, S. F. Ralph, and C. T. Dillon, Dalton Trans., 2012, 41, 9417. Copyright 2012 Royal Society of Chemistry. | ||
Zhang et al.68 used SXRF tomography and 2D X-ray fluorescence microscopy (2D-XFM) to study the bio-distribution of several anti-cancer platinum(II) complexes, among them 1 cisplatin, in a solid tumour model. Cryo-sectioning and cryo-fixing techniques were revealed to be less invasive than formalin fixing and more suitable for distribution studies of metal compounds in biological tissues. SXRF tomography allowed the drug distribution to be studied without the use of chemical fixatives, physical sectioning and lyophilisation. For the first time the good penetration of 1 cisplatin through DLD-1 colorectal cancer cell spheroids could be shown. SXRF tomography has a complicated experimental setup, and 2D-XFM was pointed out as a suitable, less complex alternative showing only minor changes in the observed metal distribution patterns in comparison to SXRF tomography.68
3. Ruthenium compounds
The most promising Ru compounds investigated so far show anti-tumour activity exceeding that of Pt-compounds or other cytostatic agents (e.g. in colorectal carcinomas in vivo and a variety of primary explanted human tumours in vitro).2,69,70 Compared to Pt compounds the Ru complexes cause less side effects and cellular resistance occurs to a lesser extent.2,71 The overall chemical and pharmacokinetic behaviour of Ru is quite different from Pt compounds,72 as reflected in extensive protein binding which questions the relevance of DNA interactions in vivo.3.1. Octahedral ruthenium complexes (KP1019/KP1339 and NAMI-A)
The Ru-complex KP1019 (19, Fig. 7)73 has shown promising anti-cancer activity70 and its sodium analogue KP1339 (20, Fig. 7)74 is in a clinical phase I–IIa study,75 whereas NAMI-A (21, Fig. 7)76,77 has shown activity mainly against metastases.72,78 Fast uptake of 19 KP1019 and 20 KP1339 by the cell was shown and initiation of apoptosis was indicated by caspase activation.75 The uptake mechanism of 19 KP1019 and 20 KP1339 may involve the iron transport protein transferrin (Tf) and the Tf receptor, which is over-expressed in tumour cells to meet their increased demand for iron.79 Another possible transport protein is human serum albumin (HSA).80 Once the ruthenium compound is inside the cell a pro-drug function is hypothesised and the reduction of Ru(III) to Ru(II) within this process is under debate. The electrochemical potential of Ru(III)/Ru(II) is accessible under physiological conditions and GSH and single-electron transfer proteins are able to reduce Ru(III) in the presence of NADH.79![Top: the Ru anti-cancer drugs indazolium-trans-[tetrachlorobis(indazole)ruthenate(iii)] (19, KP1019), sodium-trans-[tetrachlorobis(indazole)ruthenate(iii)] (20, KP1339), and imidazolium trans-[imidazoledimethyl-sulfoxide-tetrachlororuthenate] (21, NAMI-A). Center: the general formula of η6-arene complexes, the η6-arene thiophenolate complex [(η6-arene)Ru(en)(SOnR)]+, and [Ru(η6-p-cymene)Cl2(pta)] (22, RAPTA-C, pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decanephosphine). Bottom: the photoactive bipyridine complex [Ru(bpy)2(4AP)2]2+ (23, bpy = 2,2′-bipyridine, 4AP = 4-aminopyridine), and the Ru(ii) complex cis,fac-[dichlorobis(trisdimethylsulfoxide)imidazole-ruthenate(ii)] (24). en ethylenediamine, pyr pyridine, acac acetylacetonato.](/image/article/2013/MT/c3mt20261e/c3mt20261e-f7.gif) | ||
| Fig. 7 Top: the Ru anti-cancer drugs indazolium-trans-[tetrachlorobis(indazole)ruthenate(III)] (19, KP1019), sodium-trans-[tetrachlorobis(indazole)ruthenate(III)] (20, KP1339), and imidazolium trans-[imidazoledimethyl-sulfoxide-tetrachlororuthenate] (21, NAMI-A). Center: the general formula of η6-arene complexes, the η6-arene thiophenolate complex [(η6-arene)Ru(en)(SOnR)]+, and [Ru(η6-p-cymene)Cl2(pta)] (22, RAPTA-C, pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decanephosphine). Bottom: the photoactive bipyridine complex [Ru(bpy)2(4AP)2]2+ (23, bpy = 2,2′-bipyridine, 4AP = 4-aminopyridine), and the Ru(II) complex cis,fac-[dichlorobis(trisdimethylsulfoxide)imidazole-ruthenate(II)] (24). en ethylenediamine, pyr pyridine, acac acetylacetonato. | ||
The main targets of 21 NAMI-A are located in the extracellular matrix81–83 and several studies on different tumour types and cell lines confirmed this main site of action.84,85 The compound alters the extracellular matrix of the solid tumours, accompanied by accumulation of the tumour cells in the G1/G085 or G286 phase and a decrease of cells present in the S phase of the cell cycle.
3.2. Organometallic ruthenium arene compounds
Organo-metallic photoactive Ru–bipyridine and Ru–η6-arene complexes draw increasing interest due to their selective anti-proliferative properties.87,88 The η6-arene Ru compounds of the form [(η6-arene)Ru(en)Cl]+ (see Fig. 7) for example are activated through hydrolysis in vivo in environments low in Cl−, like the cell nucleus.87 The hydrolyzed species [(η6-arene) Ru(en)(H2O)]2+ is capable of binding to DNA, with high affinity to the N7 position of guanine bases,87 forming only monofunctional DNA adducts.89 In general, the cytotoxicity of Ru–arene complexes can be influenced by the nature of the η6-arene ligand, the bidentate chelating ligand (Y–Z) and the leaving group X (see Fig. 7).87 Complexes with extended η6-arene ligands (e.g. biphenyl) show higher ability to bind to DNA due to an increased intercalating effect.88 Complexes with a readily aquated leaving group X (X = halide) are more active than those with a non-aquating ligand (X = pyridine, pyr).90 An exception is [(η6-arene)Ru(en)(SOnR)]+ (Fig. 7) with a thiophenolate leaving ligand X. Thiophenolate is relatively inert to hydrolysis, but the complex shows anti-proliferative effects of the order of 1 cisplatin and 2 carboplatin.91 The activation may involve the oxidation of the thiolate (SR−), which was studied by XAS.92 Another promising Ru η6-arene compound is RAPTA-C (22, Fig. 7),93 which has two hydrolysable Cl− binding sites and pta (pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decanephosphine), an interesting alkylation site which can be used to modify the lypophilicity of the compound.943.3. Photoactivatable polypyridyl ruthenium complexes
The bipyridine Ru complexes are a class of photoactivatable metal complexes offering new possibilities of selective prodrug activation.95–98 The crucial step is the light induced dissociation of one or more ligands and the subsequent binding to biological target molecules. 23 (Fig. 7) is such a compound which transforms into a disubstituted product after photodissociation of two 4APs (4AP = 4-aminopyridine).97,99 The two new binding sites are possible targets for an adduct formation with DNA and the introduction of lethal DNA crosslinks.993.4. XAS and micro-SXRF investigations on Ru
After 8 h of incubation in CS buffer the first shell environment and/or the oxidation state of the Ru center changed dramatically, which is displayed by a significant decrease in the edge energy position. In the presence of five-fold excess GSH (5 h in CS buffer), the best matching first shell coordinations were Ru(II)Cl3N2S and Ru(III)ClN2S3. In comparison to CS buffer, the spectrum of 19 KP1019 in carbonate buffer (30 min, pH 7.4, 37 °C) was slightly shifted to higher energies. A possible interaction with apo-transferrin (apoTf) was indicated by a further shift to higher energies when apoTf was present under the same incubation conditions (30 min, pH 7.4, 37 °C). Independently of the used dosages and application schemes, 19 KP1019 and 20 KP1339 gave the same spectra in the mouse model, most consistent with a Ru(III)Cl3N2(O/N) and Ru(II)ClN2(O/N)3 first shell.100
McFarlane et al.101 studied a spectrochemical series of Ru(III) and Ru(II) compounds, among them the anti-cancer complexes 19 KP1019 and 21 NAMI-A. Ruthenium LIII/II-edge and chlorine K-edge XAS and density functional theory (DFT) calculations were used to investigate the electronic structure around the Ru metal center. The spectra of the Cl K-edge (2822 eV) and Ru LIII- and LII-edge (LIII = 2838 eV and LII = 2967 eV) were recorded at once. All Cl K-edge spectra for both Ru(III) and Ru(II) complexes exhibited pre-edge features, owing to the presence of vacant Ru d orbitals (see Fig. 8). Because of the strong ligand-field splitting of the Ru valence d-orbitals, the simple ground state configuration provides information about the spectral features. The chlorine pre-edges of the Ru(III) complexes are double-peaked and separated by 1.5 eV from each other (Fig. 8 left), representing the gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the Ru metal center. These findings were also reproduced through the DFT calculations. The changes in the numbers of dimethyl sulfoxide (DMSO) molecules and imidazole ligands within the measured complexes had no significant effect on the overall electronic structure. This was partly ascribed to the large effective nuclear charge of Ru(III) and the counteracting effect of σ-donors and π-accepting roles of trans DMSO sulfur ligands. The variations in the Ru LIII-edge peak positions and intensities of the Ru(III) compounds were greater than in the Cl K-edge spectra, with 19 KP1019 showing the highest peak energy and intensity. This was consistent with the highest effective nuclear charge for 19 KP1019 which lowers the 2p level and raises the LIII-edge energy transition, reflected in a 12% higher LIII pre-edge feature than for its imidazole analogue trans-[tetrachlorobis-(1H-imidazole)ruthenate(III)] (KP418).102 The spectral differences of the Ru(II) complexes were less pronounced and the LIII edge positions showed only slight variations within the experimental error. The high similarities are explained as a limitation of the ligand-to-metal donations when reaching a combination of ligands around two DMSO, two Cl− and two imidazoles coordinated to the Ru(II) center.
 | ||
| Fig. 8 Cl K-edge X-ray absorption near edge structure spectra with corresponding molecular orbital diagrams of the d manifold for (a) Ru(III) compound 21 NAMI-A and (b) Ru(II) compound 24 (see also Fig. 7). Reprinted (adapted) with permission from: T. V. Harris, R. K. Szilagyi, and K. L. McFarlane Holman, J. Biol. Inorg. Chem., 2009, 14, 891–898. With kind permission from Springer Science and Business Media. Copyright 2009. | ||
Lay and coworkers103 postulated that 21 NAMI-A binds to bovine serum albumin (BSA) via surface-donor groups, like histidine and methionine residues, rather than to a specific binding site. Freeze dried samples of 21 NAMI-A/BSA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mM), 21 NAMI-A/BSA (4:1 mM), 21 NAMI-A/bovine serum (0.5:0.6 mM) and 21 NAMI-A in cell culture medium supplemented with 2% fetal calf serum were investigated with Ru K-edge XAS. The resulting spectra were fitted with linear combinations of spectra from Ru(III) complexes with [Ru(NH3)6]Cl3 (Ru(III)N6), [Ru3O(OAc)6(OH2)3](OAc) (Ru(III)O6) and K2[RuCl5(OH2)]·H2O (Ru(III)Cl5O) first shell environments. The resulting fits pointed toward a replacement of the S/Cl ligands of 21 NAMI-A in biological media mainly through N/O ligands. In the linear regression analysis of 21 NAMI-A in cell culture medium a significant proportion of 16% was constituted by Ru(III)Cl5O, explained with incomplete S/Cl replacement or binding to Cl− ions present in the cell culture medium.103
:1 mM), 21 NAMI-A/BSA (4:1 mM), 21 NAMI-A/bovine serum (0.5:0.6 mM) and 21 NAMI-A in cell culture medium supplemented with 2% fetal calf serum were investigated with Ru K-edge XAS. The resulting spectra were fitted with linear combinations of spectra from Ru(III) complexes with [Ru(NH3)6]Cl3 (Ru(III)N6), [Ru3O(OAc)6(OH2)3](OAc) (Ru(III)O6) and K2[RuCl5(OH2)]·H2O (Ru(III)Cl5O) first shell environments. The resulting fits pointed toward a replacement of the S/Cl ligands of 21 NAMI-A in biological media mainly through N/O ligands. In the linear regression analysis of 21 NAMI-A in cell culture medium a significant proportion of 16% was constituted by Ru(III)Cl5O, explained with incomplete S/Cl replacement or binding to Cl− ions present in the cell culture medium.103
Ascone et al.104 used ruthenium K- and LIII-edge, chlorine K-edge and sulfur K-edge XAS to identify the oxidation state and the ligand exchange reactions during the 21 NAMI-A/BSA adduct formation. The Ru LIII-edge spectra resembled two distinct peaks separated by 3 ± 0.3 eV (denoted A and B bands). The A-bands of the model ruthenium(III) acetylacetonate (Ru(acac)3), 21 NAMI-A and the 21 NAMI-A/BSA adduct are at the same position. The B-bands of the 21 NAMI-A/BSA adduct and Ru(acac)3 are shifted to higher energies by +0.8 eV compared to 21 NAMI-A. The Ru K-edge spectra of the 21 NAMI-A/BSA adduct and the Ru(acac)3 spectra are shifted by +4 eV to higher energies with respect to the spectrum of 21 NAMI-A. The shifts in the Ru K- and LIII-edge spectra confirm the unchanged Ru(III) oxidation state. The Cl K-edge spectra of 21 NAMI-A and the 21 NAMI-A/BSA adduct are quite different with a missing pre-edge and a decrease of about −0.5 eV in energy for the latter. This indicates a release of chlorine ligands when BSA binding took place. The sulfur K-edge data were dominated by 40 sulfur atoms of the BSA forming 17 disulfide bonds. The single-atom signal from 21 NAMI-A contributes only to a minor extent to the overall sulfur XAS spectrum.104
Microprobe X-ray fluorescence of tumour (SW480) thin sections from mice treated with 20 KP1339 revealed a strong penetration of the Ru into all regions of the tumour tissue, with the highest concentrations near blood vessels and in the edge regions of the tissue samples.100
Micro-SXRF investigations on 19 KP1019 and 21 NAMI-A in single SH-SY5Y human neuroblastoma cells105 showed the correlation of the Ru and Fe distribution in 19 KP1019 treated cells, whereas for 21 NAMI-A this was not the case. About 50% of 19 KP1019 was concentrated in the nuclear region. After treatment of the cells with 19 KP1019 the Fe distribution was altered and resembled the Ru one, with unchanged total Fe concentrations inside the cells. In contrast, no significant uptake into the cell was seen for 21 NAMI-A, which supports the assumption of an extracellular main site of action. The Cu concentration was significantly increased within the total cell and in the cell nucleus of 21 NAMI-A and 19 KP1019 treated cells.105
![Sulfur K-edge spectra of Ru(ii) [(η6-arene)Ru(en)(SOnR)]+ (A) thiolato 1a and 1b, (B) sulfenato 2a and 2b, and (C) sulfinato 3a and 3b complexes. An asterisk (*) represents features due to thiolate-based impurities in the sulfenato–sulfinato complexes. Reprinted (adapted) with permission from: T. Sriskandakumar, H. Petzold, P. C. A. Bruijnincx, A. Habtemariam, P. J. Sadler, and P. Kennepohl, J. Am. Chem. Soc., 2009, 131, 13355–13361. Copyright 2009 American Chemical Society.](/image/article/2013/MT/c3mt20261e/c3mt20261e-f9.gif) | ||
| Fig. 9 Sulfur K-edge spectra of Ru(II) [(η6-arene)Ru(en)(SOnR)]+ (A) thiolato 1a and 1b, (B) sulfenato 2a and 2b, and (C) sulfinato 3a and 3b complexes. An asterisk (*) represents features due to thiolate-based impurities in the sulfenato–sulfinato complexes. Reprinted (adapted) with permission from: T. Sriskandakumar, H. Petzold, P. C. A. Bruijnincx, A. Habtemariam, P. J. Sadler, and P. Kennepohl, J. Am. Chem. Soc., 2009, 131, 13355–13361. Copyright 2009 American Chemical Society. | ||
4. Gallium compounds
Gallium(III) displays coordination characteristics similar to group 13 metal ions, but also to group 8 metal ions as iron(III).111 Gallium can bind to proteins which require the trivalent form of iron, but not to proteins requiring the divalent one. The uptake of Ga(III) into the cell is thought to be coupled to Tf and to compete with the iron transport process.112 The gallium bound to Tf is taken up by receptor mediated endocytosis.113 Although Fe(III) and Ga(III) have similar uptake mechanisms, they differ from each other in terms of intracellular transport processes. Iron is bound mainly to ferritin and undergoes a reduction and re-oxidation process. Gallium shows no change in the oxidation state under physiological conditions and is mostly present in the labile gallium pool.114 The above described uptake mechanism is generally based on experiments conducted with Ga-nitrate (Ga(NO3)3) or other Ga-salts. Up to now it has not been fully clarified if the Tf coupled uptake process also applies to the Ga tumour inhibiting complexes such as 25 (KP46, Fig. 10) and 26 (gallium maltolate, Fig. 10).2 | ||
| Fig. 10 Anti-cancer gallium compounds tris(8-quinolinolato)gallium(III) (25, KP46), and tris(3-hydroxy-2-methyl-4H-pyran-4-one)gallium(III) (26, gallium maltolate). | ||
4.1. XAS and SXRF investigations on gallium
The interaction of the anti-cancer complex 25 KP46 with possible transport molecules and the distribution of the compound in tumour and liver tissue were investigated with XAS and micro-XAS.115 The coordination environment of the metal center remains intact in the presence of apoTf and HSA (Fig. 11). The gallium distribution pattern in tumour and liver tissue revealed high similarities to the distribution patterns of Zn and Fe, minor similarities to Cu and Ni, and none to Ca. The complex was observed to be very stable under physiological conditions, in cell culture media and in tissue samples.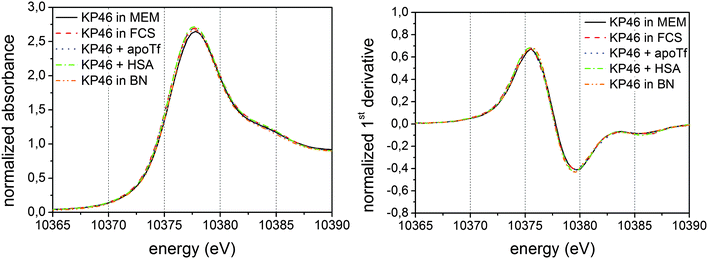 | ||
| Fig. 11 Left: XANES spectrum of 25 KP46 in minimum essential medium (MEM), fetal calf serum (FCS), in the presence of apoTf and HSA and in boron nitride (BN). Right: first derivative thereof. Reprinted (adapted) with permission from A. A. Hummer, C. Bartel, V. B. Arion, M. A. Jakupec, W. Meyer-Klaucke, T. Geraki, P. D. Quinn, A. Mijovilovich, B. K. Keppler, and A. Rompel, J. Med. Chem., 2012, 55, 5601–5613. Copyright 2012 American Chemical Society. | ||
The study of Bockman et al.116 is the first application of micro-X-ray fluorescence to Ga(NO3)3 and its interaction with the Ca pathway in cancer induced hypercalcemia. The incorporation of trace amounts of Ga into the bone material and the elemental distribution have been studied in vitro and in vivo. It was found that Ga is mainly reabsorbed in the metabolic active regions at the interface of the organic (collagen-associated) and the mineral components, where mineralization occurs.116 The coordination of Ga in newly formed bone material could be elucidated as Ga bound in a distorted octahedron with Ga–O distances of around 1.91 Å.117
5. Gold compounds
The sole clinical application of gold compounds presently is in the treatment of rheumatoid arthritis.118–120 Amongst these, the Au(I) complex, auranofin (27, Fig. 12), also exhibits anti-tumoural activity.120–122 Other gold(I) compounds used in chrysotherapy are gold(I) thioglucose (solganol),123 gold(I) sodium thiomalate (myochrysin),123 gold(I) thiopropanol sulphonate (allochrysine)124 and gold(I) bis(thiosulfate) (sanochrysine),124 which are all hydrophilic, except 27 auranofin which is hydrophobic.124![Investigational anti-cancer Au(i) and Au(iii) compounds triethylphosphine(2,3,4,6-tetra-O-acetyl-β-1-d-thiopyranosato-S)gold(i) (27, auranofin), [Au(iii)(2,2′-bipyridine)(OH)2]PF6 (28, aubipy), [Au2(6,6′-dimethyl-2,2′-bipyridine)2(μ–O)2](PF6)2 (29, auoxo6).](/image/article/2013/MT/c3mt20261e/c3mt20261e-f12.gif) | ||
| Fig. 12 Investigational anti-cancer Au(I) and Au(III) compounds triethylphosphine(2,3,4,6-tetra-O-acetyl-β-1-D-thiopyranosato-S)gold(I) (27, auranofin), [Au(III)(2,2′-bipyridine)(OH)2]PF6 (28, aubipy), [Au2(6,6′-dimethyl-2,2′-bipyridine)2(μ–O)2](PF6)2 (29, auoxo6). | ||
The most investigated gold compounds are Au(I) complexes with phosphine and carbene ligands, Au(III) dithiocarbamate compounds, Au(III) porphyrinates and organogold(III) compounds. The important oxidation states for gold in pharmacological applications are gold(I) (d10 linear dicoordinated) and gold(III) (d8 square planar).124 The interchange between these two oxidation states is physiologically accessible and the high similarity to Pt(II) coordination and the electronic configuration (d8) make Au(III) compounds very promising candidates for new anti-cancer drugs.125 In the last 20 years a variety of Au(III) compounds have been synthesized and tested against Pt resistant tumour cell lines.126,127 The main drawbacks of the Au(III) complexes are their instability under physiological conditions and their fast reduction to elemental gold. Au(III) is a strongly oxidizing “borderline” cation which shows preference for nitrogen donors, but also for “soft” donor ligands (like S, Se or cyanide), similar to the “soft” cation Au(I).121,124,127,128
Two major biological targets could be elucidated for Au(I) compounds, the mitochondrial membrane and the selenoenzyme thioredoxin reductase (TrxR).124,129 The Cys and selenocysteine present in the active site of TrxR were revealed as an optimal target for the Au(I) complexes which show soft acid character.124 As TrxR protects the cell against oxidative stress, its inhibition causes the increase of reactive oxygen species and the damage of mitochondrial functions.130,131 Some of the Au(I) compounds like N-heterocyclic carbene complexes combine selective TrxR inhibition and mitochondrial membrane permeabilisation properties.132
The cytotoxic activity of Au(III) compounds is thought to be similar to the cytotoxicity proposed for Au(I), by preferentially targeting thiol, imidazole and selenol groups of proteins.127 The major cause of cytotoxicity is strongly dependent on the Au(III) ligands. For aurobipyridines (see Fig. 12) TrxR was observed as a major target.127 The Au(III) porphyrins instead showed a significantly higher affinity to DNA, causing fragmentation of genomic DNA.126 In the context of other possible targets in the cell the enzymes lipoamide dehydrogenase, glutathione reductase and mercuric ion reductase are mentioned, which are all members of the family of pyridine nucleotide oxidoreductases like TrxR.133 Messori et al.134 synthesized two promising anti-cancer agents bearing the bipyridyl motif:123,135 the Au(III) centered aubipy (28, Fig. 12) and the oxobridged dinuclear auoxo6 (29, Fig. 12).136–138 These gold bipyridine complexes showed considerable cytotoxic activity against 1 cisplatin resistant cell lines.127,131
5.1. XAS investigations on gold
A combination of Au LIII-edge XANES and DFT was used to study the electronic configuration of 28 aubipy.139 A structure optimized with DFT calculations was used as an input for the calculation of theoretical XANES spectra. The HOMO–LUMO gap and the orbital shapes were determined as well.139The three gold(I/III) compounds 27 auranofin, 28 aubipy and 29 auoxo6 were investigated by Messori et al. by means of Au LIII-edge XAS.134 The binding of these three complexes to the potential transport proteins apoTf (of 27 auranofin and 29 auoxo6) and BSA (of 28 aubipy) and their potential changes in the oxidation state after 24 h in solution were studied (50 mM phosphate buffer, pH 7.4, 1:1 10−4 M). The XANES spectra of the Au(III) 29 auoxo6/apoTf and 28 aubipy/BSA adducts clearly showed a reduction to Au(I) through a remarkable decrease in white line intensity (Fig. 13) and a shift of about +1 eV to higher energies, which is special for Au and reverse to an expected shift to lower energies in the case of a reduction.123,140 The EXAFS analysis of 29 auoxo6/apoTf and 28 aubipy/BSA revealed a decrease in coordination number from four to two, for each of the complexes. For 29 auoxo6/apoTf the loss of the Au–Au peak in the FT was interpreted as a binding to apoTf of the now mononuclear 29 auoxo6 via 1.9 ± 0.4 O or N ligands.134 In the presence of apoTf the Au(I) compound 27 auranofin preserved its oxidation state +1 and the coordination number was still two, with slightly increased average S/P distances pointing toward an 27 auranofin/apoTf adduct formation via a protein thiol or thioether bond, most likely replacing the thiosugar ligand.
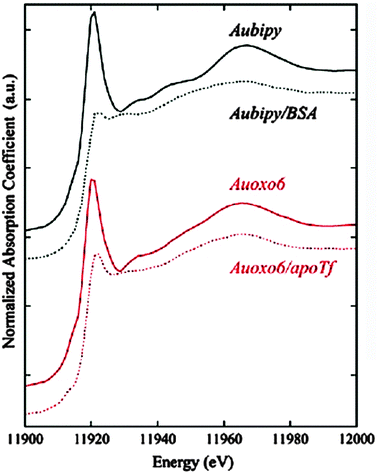 | ||
| Fig. 13 Relevant changes in the oxidation states of 29 auoxo6 and 28 aubipy compared with their respective protein adducts are shown by XANES spectra. Reprinted (adapted) with permission from L. Messori, A. Balerna, I. Ascone, C. Castellano, C. Gabbiani, A. Casini, C. Marchioni, G. Jaouen, and A. C. Castellano, J. Biol. Inorg. Chem., 2010, 16, 491–499. With kind permission from Springer Science and Business Media. Copyright 2010. | ||
Hill et al.141 used XAS to specify the binding partners of seleno-auranofin (Et3PAuSe-tagl, tagl = tetraacetylglucose, S in 27 auranofin replaced through Se) by measuring the Au LIII- and Se K-edge of a solid powder sample. Gold was found to be in the oxidation state +1 and bound to one phosphor ligand, according to the peak intensity at 11928 eV, whose intensity is directly proportional to the number of Au–P bonds. The lack of a strong 2p → 5d transition “white line” characteristic of Au(III) and the absence of the two peaks at 11945 eV and 11967 eV distinct for Au(0) clearly indicated the Au(I) state. The best fit of the FT could be achieved with 0.9 P at 2.28 Å and 1.6 Se at 2.41 Å. At the Se K-edge three distinct peaks in the FT became visible. The peaks were best fitted with 1.2 C at 1.91 Å, 0.9 Au at 2.39 Å and a 1.4 second shell P at a non-bonded distance of 4.61 Å. The difference in the Au–Se distances determined over the Au LIII- and Se K-edge EXAFS is only 0.02 Å. The sum of the Au–P and Au–Se distances from the Au LIII EXAFS data is 4.69 Å, which is in agreement with the direct fitted second shell P distance of 4.61 Å from the Se K-edge XAS data. Furthermore, this high similarity in bond distances points toward a nearly linear P–Au–Se grouping.141
6. Cobalt compounds
Cobalt complexes serve as prodrugs and release cytotoxic ligands upon reduction. Research into this area has so far concentrated on four types of compounds: (i) hexacarbonyldicobalt complexes with alkyne ligands, (ii) [Co(III)(NH3)6]Cl3, (iii) Co(III) complexes with Schiff base ligands, (iv) Co(II) and/or Co(III) complexes with cytotoxic mustamine, mithramycin, and thiouracil ligands.6 In many solid tumours the rapid growth of the tumour tissue leads to insufficient vascularization and hypoxic conditions. Ware et al. studied the activation of Co(III) compounds containing N-mustards under these conditions.142–145 The Co(III) center is reduced to Co(II) and the N-mustard ligand is released.146 The workgroup of Hambley developed the complex 30 (Fig. 14), which works as a carrier system for marimastat (mmst).147,148 Mmst itself is an inhibitor of matrix metalloproteinases, which are involved in tumour metastasis.149 It has been suggested that the selective activation depends on the rate of reoxidation. In normal oxygenated environments the complex is reduced and activated, but is then rapidly reoxidized and deactivated. Under hypoxic conditions the reoxidation rate is low and the activated anti-cancer agent is able to induce its action.6,70,150![Chemical structures of the cobalt–tpa marimastat complex. [Co(iii)(mmst)tpa]+ (mmst = marimastat, tpa = tris(methylpyridyl)amine) (30), [Co(iii)Cl(H2O)tpa]2+ (31), [Co(aha-H)(tpa)]PF6 (32, aha = acetohydroxamate), trans-[CoCl2(en)2]Cl (33), and of the diNOsar complex cobalt-1,8-dinitro-3,6,10,13,16,19-hexaazabicyclo[6.6.6]eicosane (34, diNOsar = dinitrosarcophagine). In the lower part the Co–Cys complexes trans(N,S)-bis(cysteinato)diaquacobalt(iii) (35, Co:2Cys) and one possible diastereoisomer of tris(cysteinato)cobalt(iii) (36, Co:3Cys), and one possible structure of bis(N-(2-mercaptopropionyl)glycinato)triaquacobalt(ii) (37, Co:1MPG).](/image/article/2013/MT/c3mt20261e/c3mt20261e-f14.gif) | ||
| Fig. 14 Chemical structures of the cobalt–tpa marimastat complex. [Co(III)(mmst)tpa]+ (mmst = marimastat, tpa = tris(methylpyridyl)amine) (30), [Co(III)Cl(H2O)tpa]2+ (31), [Co(aha-H)(tpa)]PF6 (32, aha = acetohydroxamate), trans-[CoCl2(en)2]Cl (33), and of the diNOsar complex cobalt-1,8-dinitro-3,6,10,13,16,19-hexaazabicyclo[6.6.6]eicosane (34, diNOsar = dinitrosarcophagine). In the lower part the Co–Cys complexes trans(N,S)-bis(cysteinato)diaquacobalt(III) (35, Co:2Cys) and one possible diastereoisomer of tris(cysteinato)cobalt(III) (36, Co:3Cys), and one possible structure of bis(N-(2-mercaptopropionyl)glycinato)triaquacobalt(II) (37, Co:1MPG). | ||
6.1. XAS investigations on cobalt
:Co(III) oxidation states in a mixture of unknown composition was tested.151 Aqueous solutions of known ratios of Co(II)Cl2:[Co(III)(NH3)6]Cl3 and Co(II)Cl2:31 Co(III) (Fig. 14) were investigated according to their changes in energy positions and peak heights. With increasing Co(III) content in the aqueous solutions the maximum at approx. 7720 eV decreased and the edge energy shifted toward higher energies. The relationship between the edge maximum to post edge minimum ratio a/b in the XANES spectra and the proportion of Co(II):Co(III) appeared to be parabolic. The determination of the Co(II):Co(III) ratios in unknown mixtures through XANES was stated as feasible, as long as ligand exchange reactions in the Co(III) state are slow relative to reduction to Co(II).151
Bresson and coworkers studied the toxicity of cobalt toward HaCaT keratinocyte cells.153–158 Co K-edge XANES and EXAFS in combination with molecular dynamics (MD) calculations gave an insight into the binding behaviour of Co(II)Cl2 to biologically important thiol containing molecules.153–155 Cobalt model complexes were synthesized with the ligands Cys and N-(2-mercaptopropionyl)glycine (MPG), a synthetic analogue of the right hand portion of GSH.153 The complexes Co:2Cys (35, Fig. 14), Co:3Cys (36, Fig. 14) and Co:1MPG (37, Fig. 14) were measured in aqueous solutions at the Co K-edge, resulting in Co(II) and Co(III) XANES spectra showing the same features as described in the paragraph before in the studies of Bonnitcha and Hall.150,15135 Co:2Cys and 36 Co:3Cys were identified to be in the Co(III) oxidation state, 37 Co:1MPG to be Co(II) upon comparison of the pre-edge positions.153 The oxidation states were dependent on the pH of the solutions (pH 11 for 35 Co:2Cys and 36 Co:3Cys, and pH 7 for 37 Co:1MPG).155 The EXAFS analysis of these compounds confirmed the proposed coordination of 2 Cys to 35 Co:2Cys and 3 Cys to 36 Co:3Cys in the Co(III) state (Fig. 14), respectively.153 In the case of 37 Co:1MPG a dinuclear cobalt compound bridged through the two sulfur atoms of the bidentate MPG ligands was the structure that best fitted the EXAFS data (Fig. 14). In a subsequent publication155 the structures of tris(cysteinato)cobalt(II) (38, Co(II):3Cys) and 36 Co(III):3Cys were optimized through MD calculations156 and used as structural models for the EXAFS analysis.155 EXAFS analysis of 36 Co(III):3Cys with MD calculated and X-ray diffraction structural data gave the same results.155In situ XANES measurements were used to follow the oxidation of 38 Co(II):3Cys to 36 Co(III):3Cys (see Fig. 15), resulting in the progressive changes in the edge features toward an edge shape typical for a Co(III) species, visible through a decrease in the edge height and the development of a double-peaked edge shape.155
 | ||
| Fig. 15 XANES in situ monitoring of the oxidation of 38 Co(II):3Cys to 36 Co(III):3Cys at the Co K-edge. An intermediate mixture of Co(II) and Co(III) is represented by the middle dotted spectrum. Reprinted (adapted) with permission from C. Bresson, R. Spezia, S. Esnouf, P. L. Solari, S. Coantic, and C. D. Auwer, New J. Chem., 2007, 31, 1789–1797. Copyright 2007 Royal Society of Chemistry. | ||
7. Limitations of XAS and micro-SXRF
In XANES and EXAFS, there is a need to acquire data for an extensive range of solid samples for modelling purposes. If the concentration of the element of interest is not high enough XAS is limited to the XANES regime and/or the resulting spectra are very noisy. The processing and fitting of the EXAFS data is non-trivial, and it is hard to distinguish between elements of nearly the same atomic number Z, as it is the case with S and Cl, and O and N. Like other spectroscopic techniques XAS probes only the average coordination sphere of the metals in a particular biological system, and much information is lost on the minor species present in the sample. In SXRF, physical fixation of the cell samples can lead to elemental rearrangement and loss of information. Sometimes, the data are limited by biologically relevant concentrations of the compounds in the biological systems, and higher dosages are needed to see the elements in the systems.8. Conclusion
XAS has proven to be a valuable tool for the investigation of the anti-cancer metals Pt, Ru, Ga, Au and Co in biological environments. The near edge structure (XANES) provides information about the oxidation state, the electronic structure of the absorber–ligand environment and the coordination motif, while the extended fine structure (EXAFS) gives information about the amount, the type and the distance of the next neighbouring atoms up to a distance of about 4 Å. In the presence of the reducing agents ascorbate, GSH and/or Cys the reduction of Ru(III) to Ru(II), Pt(IV) to Pt(II) and Co(III) to Co(II) was investigated by means of XANES spectroscopy. For Pt(IV)/Pt(II) and Co(III)/Co(II) a method was presented to determine the ratio of these oxidation states in samples of unknown composition. This method was also applied to follow the reduction of metal based drugs in tissue and cell samples and to test the drug stability under physiological conditions. XAS is one of the few methods that allow the monitoring of exchange reactions of atoms at the absorber, like O/N for Cl/S or vice-versa, and the accompanying possible changes in the oxidation state. This fact was used in interaction studies of the metal based drugs with possible transport proteins in vivo like HSA, BSA and apoTf and to test their stability in solutions. The stability of platinum drugs in infusion solution was tested and the proposed biochemical pathways of Ru, Au and Ga complexes were studied. Theoretical structural models optimized with DFT calculations have shown to be valuable tools for the analysis of XAS spectra and to identify the main chemical constitution of the element in an unknown sample or where detailed X-ray diffraction data are missing. This approach was used to obtain insight into the photo-chemistry of Ru–bipyridine complexes and the activation mechanism of thiophenolate containing Ru–arene complexes. Micro-SXRF studies ranging from several μm2 spotsize to the subcellular level have been undertaken on tissues derived from Pt, Ru and Ga treated mice and on cell lines treated with Pt and Ru anti-cancer agents. The transport into and within cells, the main leaving ligands and the major site of action as well as the penetration into the tissue and the correlation with other biologically important elements can be monitored. Micro-XAS allows the additional elucidation of the oxidation state and coordination at micro-metric spatial resolution in the sample.In conclusion it can be said that the synchrotron radiation spectroscopic techniques presented here will play a vital role in medicinal chemistry in the future. With the approach of the fourth generation of synchrotron radiation facilities and their improved and more sensitive instrumentation, investigations in the sub-μm range at physiological concentrations will be possible at a high level of spectroscopic details. Examination methods like X-ray tomography will reduce the need for detrimental fixation and sectioning of tissue samples. The combination of micro-XAS and micro-SXRF will allow for the simultaneous investigation of the metal distribution and its metabolism. The further progress in the theory of XAS and the theoretical calculation of XAS spectra from artificially constructed structures will minimize the need for extensive collection of model compounds for analysis.
Abbreviations
| 4AP | 4-aminopyridine |
| acac | acetylacetonate |
| aha | acetohydroxamate |
| apoTf | apo-transferrin |
| BN | boron nitride |
| bpy | 2,2′-bipyridine |
| Brpyr | 3-bromopyridine |
| BSA | bovine serum albumin |
| CS buffer | citrate saline buffer |
| Cys | cysteine |
| DDTC | diethyl-dithiocarbamate |
| DFT | density functional theory |
| diNOsar | 1,8-dinitro-3,6,10,13,16,19-hexaazabicyclo-[6.6.6]-eicosane |
| en | ethylenediamine |
| eV | electron volt |
| EXAFS | extended X-ray absorption fine structure |
| FT | Fourier transform |
| GSH | glutathione |
| HOMO | highest occupied molecular orbital |
| HSA | human serum albumin |
| HSQC-NMR | heteronuclear single quantum coherence nuclear magnetic resonance |
| LUMO | lowest unoccupied molecular orbital |
| MD | molecular dynamics |
| micro-SRIXE | micro-synchrotron radiation induced X-ray emission |
| micro-XAS | micro-X-ray absorption spectroscopy |
| micro-SXRF | micro-synchrotron based X-ray fluorescence |
| mmst | marimastat |
| MPG | N-(2-mercaptopropionyl)glycine |
| NBA | n-butylamine |
| OAc | acetate |
| OAcBr | bromacetato |
| PBS | phosphate buffered saline |
| pta | 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decanephosphine |
| py | pyridine |
| RAPTA | ruthenium arene pta |
| Sec | selenocysteine |
| SO2R− | sulfinate |
| SOR− | sulfenate |
| SR− | thiolate |
| tagl | tetraacetylglucose |
| Tf | transferrin |
| tpa | tris(methylpyridyl)amine |
| TrxR | thioredoxin reductase |
| XANES | X-ray absorption near edge structure |
| XAS | X-ray absorption spectroscopy |
Acknowledgements
The research was funded by the Austrian Science Fund (FWF): P23711-N19. This research was also funded by a short-term grant abroad (KWA) of the University of Vienna (call 15.10.2011). The results reported in this work are based on experiments carried out at synchrotron radiation facilities. A.A.H. and A.R. are grateful for the support through Diamond Light Source (SP1640, SP6303), the European Synchrotron Radiation Facility (MD-514, MD-678), the Angströmquelle Karlsruhe (OTH-88), and the Deutsche Elektronensynchrotron.References
- B. Rosenberg, L. Vancamp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- B. Desoize, Anticancer Res., 2004, 24, 1529–1544 CAS.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Antioxid. Redox Signaling, 2011, 15, 1085–1128 CrossRef CAS.
- S. P. Best and M. H. Cheah, Radiat. Phys. Chem., 2010, 79, 185–194 CrossRef CAS.
- R. W. Strange and M. C. Feiters, Curr. Opin. Struct. Biol., 2008, 18, 609–616 CrossRef CAS.
- J. Yano and V. K. Yachandra, Photosynth. Res., 2009, 102, 241–254 CrossRef CAS.
- G. N. George, Curr. Opin. Struct. Biol., 1993, 3, 780–784 CrossRef CAS.
- B. Akabayov, C. J. Doonan, I. J. Pickering, G. N. George and I. Sagi, J. Synchrotron Radiat., 2005, 12, 392–401 CrossRef CAS.
- I. Nicolis, E. Curis, P. Deschamps and S. Bénazeth, J. Synchrotron Radiat., 2003, 10, 96–102 CrossRef CAS.
- M. Groessl and P. J. Dyson, Curr. Top. Med. Chem., 2011, 11, 2632–2646 CrossRef CAS.
- R. Ortega, A. Carmona, I. Llorens and P. L. Solari, J. Anal. At. Spectrom., 2012, 27, 2054–2065 RSC.
- R. Ortega, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 231, 218–223 CrossRef CAS.
- R. Ortega, J. Anal. At. Spectrom., 2010, 26, 23–29 RSC.
- S. Majumdar, J. R. Peralta-Videa, H. Castillo-Michel, J. Hong, C. M. Rico and J. L. Gardea-Torresdey, Anal. Chim. Acta, 2012, 755, 1–16 CrossRef CAS.
- G. Bunker, Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy, Cambridge University Press, Cambridge, 1st edn, 2010 Search PubMed.
- S. D. Kelly, D. Hesterberg and B. Ravel, Methods of Soil Analysis. Part 5. Mineralogical Methods., American Society of Agronomy, Madison, 2008, vol. 5, pp. 387–463 Search PubMed.
- D. C. Koningsberger, B. L. Mojet, G. E. Van Dorssen and D. E. Ramaker, Top. Catal., 2000, 10, 143–155 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- F. de Groot and A. Kotani, Core Level Spectroscopy of Solids, Crc Pr Inc., 2008 Search PubMed.
- J. Stöhr, NEXAFS Spectroscopy, Springer, 1st edn 1992, Corr. 2nd printing 2003, 1992 Search PubMed.
- J. E. Penner-Hahn, Coord. Chem. Rev., 1999, 190, 1101–1123 CrossRef.
- F. de Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- F. W. Lytle, J. Synchrotron Radiat., 1999, 6, 123–134 CrossRef CAS.
- R. S. von Bordwehr, Ann. Phys., 1989, 14, 377–465 CrossRef.
- P. Ilinski, B. Lai, Z. Cai, W. Yun, D. Legnini, T. Talarico, M. Cholewa, L. K. Webster, G. B. Deacon, S. Rainone, D. R. Phillips and A. P. J. Stampfl, Cancer Res., 2003, 63, 1776–1779 CAS.
- R. Ortega, P. Moretto, A. Fajac, J. Benard, Y. Llabador and M. Simonoff, Cell. Mol. Biol. (Noisy-le-grand), 1996, 42, 77–88 CAS.
- J. E. Penner-Hahn, Coord. Chem. Rev., 2005, 249, 161–177 CrossRef CAS.
- S. J. Lippard and J. M. Berg, Principles of bioinorganic chemistry, University Science Books, 1994 Search PubMed.
- B. D. Evans, K. S. Raju, A. H. Calvert, S. J. Harland and E. Wiltshaw, Cancer Treat. Rep., 1983, 67, 997–1000 CAS.
- Y. Kidani and K. Inagaki, U.S. Pat. 4169846, 1979 Search PubMed.
- T. W. Hambley, Dalton Trans., 2007, 4929–4937 RSC.
- E. Reisner, V. B. Arion, B. K. Keppler and A. J. L. Pombeiro, Inorg. Chim. Acta, 2008, 361, 1569–1583 CrossRef CAS.
- O. Shiratori, H. Kanai, Y. Uchida, Y. Takeda, T. Totani and K. Sato, Recent Advances in Chemotherapy, University of Tokyo Press, Tokyo, 1985, pp. 635–636 Search PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- F. Muggia, Gynecol. Oncol., 2009, 112, 275–281 CrossRef CAS.
- L. M. Pasetto, M. R. D'Andrea, A. A. Brandes, E. Rossi and S. Monfardini, Crit. Rev. Oncol. Hematol., 2006, 60, 59–75 CrossRef.
- B. Desoize and C. Madoulet, Crit. Rev. Oncol. Hematol., 2002, 42, 317–325 CrossRef.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS.
- H. Zorbas and B. K. Keppler, ChemBioChem, 2005, 6, 1157–1166 CrossRef CAS.
- M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS.
- P. Gramatica, E. Papa, M. Luini, E. Monti, M. B. Gariboldi, M. Ravera, E. Gabano, L. Gaviglio and D. Osella, JBIC, J. Biol. Inorg. Chem., 2010, 15, 1157–1169 CrossRef CAS.
- S. van Zutphen and J. Reedijk, Coord. Chem. Rev., 2005, 249, 2845–2853 CrossRef CAS.
- J. Moretto, B. Chauffert, F. Ghiringhelli, J. R. Aldrich-Wright and F. Bouyer, Invest. New Drugs, 2011, 29, 1164–1176 CrossRef CAS.
- J. Ruiz, V. Rodríguez, N. Cutillas, A. Espinosa and M. J. Hannon, J. Inorg. Biochem., 2011, 105, 525–531 CrossRef CAS.
- C. Sanchez-Cano and M. J. Hannon, Dalton Trans., 2009, 10702–10711 RSC.
- C. T. Dillon, Aust. J. Chem., 2012, 65, 204–217 CrossRef CAS.
- B.-K. Teo, P. Eisenberger, J. Reed, J. K. Barton and S. J. Lippard, J. Am. Chem. Soc., 1978, 100, 3225–3227 CrossRef CAS.
- M. D. Hall, G. J. Foran, M. Zhang, P. J. Beale and T. W. Hambley, J. Am. Chem. Soc., 2003, 125, 7524–7525 CrossRef CAS.
- H. L. Daly, M. D. Hall, T. W. Failes, M. Zhang, G. J. Foran and T. W. Hambley, Aust. J. Chem., 2011, 64, 273–278 CrossRef CAS.
- M. D. Hall, H. L. Daly, J. Z. Zhang, M. Zhang, R. A. Alderden, D. Pursche, G. J. Foran and T. W. Hambley, Metallomics, 2012, 4, 568–575 RSC.
- A. Nemirovski, I. Vinograd, K. Takrouri, A. Mijovilovich, A. Rompel and D. Gibson, Chem. Commun., 2010, 46, 1842–1844 RSC.
- M. Obata, M. Harada, H. Ohi, S. Hirohara, M. Gottchaldt and S. Yano, Chem. Pharm. Bull., 2009, 57, 1107–1109 CrossRef CAS.
- K. Provost, D. Bouvet-Muller, S. Crauste-Manciet, J. Moscovici, L. Olivi, G. Vlaic and A. Michalowicz, Biochimie, 2009, 91, 1301–1306 CrossRef CAS.
- E. Curis, K. Provost, I. Nicolis, D. Bouvet, S. Bénazeth, S. Crauste-Manciet, F. Brion and D. Brossard, New J. Chem., 2000, 24, 1003–1008 RSC.
- E. Curis, K. Provost, D. Bouvet, I. Nicolis, S. Crauste-Manciet, D. Brossard and S. Bénazeth, J. Synchrotron Radiat., 2001, 8, 716–718 CrossRef CAS.
- E. C. Beret, K. Provost, D. Müller and E. S. Marcos, J. Phys. Chem. B, 2009, 113, 12343–12352 CrossRef CAS.
- D. Bouvet, A. Michalowicz, S. Crauste-Manciet, D. Brossard and K. Provost, Inorg. Chem., 2006, 45, 3393–3398 CrossRef CAS.
- D. Bouvet, A. Michalowicz, S. Crauste-Manciet, E. Curis, I. Nicolis, L. Olivi, G. Vlaic, D. Brossard and K. Provost, J. Synchrotron Radiat., 2006, 13, 477–483 CrossRef CAS.
- M. D. Hall, C. T. Dillon, M. Zhang, P. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, JBIC, J. Biol. Inorg. Chem., 2003, 8, 726–732 CrossRef CAS.
- M. D. Hall, R. A. Alderden, M. Zhang, P. J. Beale, Z. Cai, B. Lai, A. P. J. Stampfl and T. W. Hambley, J. Struct. Biol., 2006, 155, 38–44 CrossRef CAS.
- K. J. Davis, J. A. Carrall, B. Lai, J. R. Aldrich-Wright, S. F. Ralph and C. T. Dillon, Dalton Trans., 2012, 41, 9417–9426 RSC.
- J. Z. Zhang, N. S. Bryce, A. Lanzirotti, C. K. J. Chen, D. Paterson, M. D. de Jonge, D. L. Howard and T. W. Hambley, Metallomics, 2012, 4, 1209–1217 RSC.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- P. Heffeter, M. Pongratz, E. Steiner, P. Chiba, M. A. Jakupec, L. Elbling, B. Marian, W. Körner, F. Sevelda, M. Micksche, B. K. Keppler and W. Berger, J. Pharmacol. Exp. Ther., 2005, 312, 281–289 CrossRef CAS.
- E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525–1535 CrossRef CAS.
- K.-G. Lipponer, E. Vogel and B. K. Keppler, Met.-Based Drugs, 1996, 3, 243–260 CrossRef CAS.
- W. Peti, T. Pieper, M. Sommer, B. K. Keppler and G. Giester, Eur. J. Inorg. Chem., 1999, 1551–1555 CrossRef CAS.
- P. Heffeter, K. Böck, B. Atil, M. Reza Hoda, W. Körner, C. Bartel, U. Jungwirth, B. Keppler, M. Micksche, W. Berger and G. Koellensperger, JBIC, J. Biol. Inorg. Chem., 2010, 15, 737–748 CrossRef CAS.
- G. Mestroni, E. Alessio and G. Sava, International Patent PCT C 07F 15/00, A61K 31/28 WO 98/00431, 1998 Search PubMed.
- A. Bergamo, R. Gagliardi, V. Scarcia, A. Furlani, E. Alessio, G. Mestroni and G. Sava, J. Pharmacol. Exp. Ther., 1999, 289, 559–564 CAS.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine, G. Pezzoni, L. Sartor and S. Garbisa, Clin. Cancer Res., 2003, 9, 1898–1905 CAS.
- M. A. Jakupec, E. Reisner, A. Eichinger, M. Pongratz, V. B. Arion, M. Galanski, C. G. Hartinger and B. K. Keppler, J. Med. Chem., 2005, 48, 2831–2837 CrossRef CAS.
- N. Cetinbas, M. I. Webb, J. A. Dubland and C. J. Walsby, JBIC, J. Biol. Inorg. Chem., 2009, 15, 131–145 CrossRef.
- J. B. Aitken, A. Levina and P. A. Lay, Curr. Top. Med. Chem., 2011, 11, 553–571 CrossRef CAS.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 RSC.
- A. Bergamo and G. Sava, Dalton Trans., 2007, 1267–1272 RSC.
- F. Frausin, V. Scarcia, M. Cocchietto, A. Furlani, B. Serli, E. Alessio and G. Sava, J. Pharmacol. Exp. Ther., 2005, 313, 227–233 CrossRef CAS.
- B. Gava, S. Zorzet, P. Spessotto, M. Cocchietto and G. Sava, J. Pharmacol. Exp. Ther., 2006, 317, 284–291 CrossRef CAS.
- S. Zorzet, A. Bergamo, M. Cocchietto, A. Sorc, B. Gava, E. Alessio, E. Iengo and G. Sava, J. Pharmacol. Exp. Ther., 2000, 295, 927–933 CAS.
- A. F. A. Peacock and P. J. Sadler, Chem.–Asian J., 2008, 3, 1890–1899 CrossRef CAS.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 4003–4018 CrossRef CAS.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernández, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS.
- H. Petzold, J. Xu and P. J. Sadler, Angew. Chem., Int. Ed., 2008, 47, 3008–3011 CrossRef CAS.
- T. Sriskandakumar, H. Petzold, P. C. A. Bruijnincx, A. Habtemariam, P. J. Sadler and P. Kennepohl, J. Am. Chem. Soc., 2009, 131, 13355–13361 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- P. J. Dyson, Chimia, 2007, 61, 698–703 CrossRef CAS.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466–3479 CrossRef CAS.
- S. Betanzos-Lara, O. Novakova, R. J. Deeth, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec, P. J. Sadler and A. Habtemariam, JBIC, J. Biol. Inorg. Chem., 2012, 17, 1033–1051 CrossRef CAS.
- L. Zayat, C. Calero, P. Alborés, L. Baraldo and R. Etchenique, J. Am. Chem. Soc., 2003, 125, 882–883 CrossRef CAS.
- L. Zayat, M. Salierno and R. Etchenique, Inorg. Chem., 2006, 45, 1728–1731 CrossRef CAS.
- L. Salassa, C. Garino, G. Salassa, C. Nervi, R. Gobetto, C. Lamberti, D. Gianolio, R. Bizzarri and P. J. Sadler, Inorg. Chem., 2009, 48, 1469–1481 CrossRef CAS.
- A. A. Hummer, P. Heffeter, W. Berger, M. Filipits, D. Batchelor, G. Büchel, M. A. Jakupec, B. K. Keppler and A. Rompel, J. Med. Chem., 2013, 56, 1182–1196 CrossRef CAS.
- T. V. Harris, R. K. Szilagyi and K. L. M. Holman, JBIC, J. Biol. Inorg. Chem., 2009, 14, 891–898 CrossRef CAS.
- B. K. Keppler, W. Rupp, U. M. Juhl, H. Endres, R. Niebl and W. Balzer, Inorg. Chem., 1987, 26, 4366–4370 CrossRef CAS.
- M. Liu, Z. J. Lim, Y. Y. Gwee, A. Levina and P. A. Lay, Angew. Chem., Int. Ed., 2010, 49, 1661–1664 CrossRef CAS.
- I. Ascone, L. Messori, A. Casini, C. Gabbiani, A. Balerna, F. Dell'Unto and A. C. Castellano, Inorg. Chem., 2008, 47, 8629–8634 CrossRef CAS.
- J. B. Aitken, S. Antony, C. M. Weekley, B. Lai, L. Spiccia and H. H. Harris, Metallomics, 2012, 4, 1051–1056 RSC.
- L. Salassa, D. Gianolio, C. Garino, G. Salassa, E. Borfecchia, T. Ruiu, C. Nervi, R. Gobetto, R. Bizzarri, P. J. Sadler and C. Lamberti, J. Phys.: Conf. Ser., 2009, 190, 012141 CrossRef.
- L. Salassa, E. Borfecchia, T. Ruiu, C. Garino, D. Gianolio, R. Gobetto, P. J. Sadler, M. Cammarata, M. Wulff and C. Lamberti, Inorg. Chem., 2010, 49, 11240–11248 CrossRef CAS.
- L. Salassa, T. Ruiu, C. Garino, A. M. Pizarro, F. Bardelli, D. Gianolio, A. Westendorf, P. J. Bednarski, C. Lamberti, R. Gobetto and P. J. Sadler, Organometallics, 2010, 29, 6703–6710 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- K. Getty, M. U. Delgado-Jaime and P. Kennepohl, Inorg. Chim. Acta, 2008, 361, 1059–1065 CrossRef CAS.
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2534 CrossRef CAS.
- C. R. Chitambar and Z. Zivkovic, Cancer Res., 1987, 47, 3929 CAS.
- D. R. Richardson and P. Ponka, Biochim. Biophys. Acta, 1997, 1331, 1–40 CrossRef CAS.
- N. P. Davies, Y. S. Rahmanto, C. R. Chitambar and D. R. Richardson, J. Pharmacol. Exp. Ther., 2006, 317, 153 CrossRef CAS.
- A. A. Hummer, C. Bartel, V. B. Arion, M. A. Jakupec, W. Meyer-Klaucke, T. Geraki, P. D. Quinn, A. Mijovilovich, B. K. Keppler and A. Rompel, J. Med. Chem., 2012, 55, 5601–5613 CrossRef CAS.
- R. Bockman, M. Repo, R. Warrell, J. Pounds, G. Schidlovsky, B. Gordon and K. Jones, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 4149 CrossRef CAS.
- M. Korbas, E. Rokita, W. Meyer-Klaucke and J. Ryczek, JBIC, J. Biol. Inorg. Chem., 2004, 9, 67–76 CrossRef CAS.
- A. S. Thakor, J. Jokerst, C. Zavaleta, T. F. Massoud and S. S. Gambhir, Nano Lett., 2011, 11, 4029–4036 CrossRef CAS.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS.
- E. R. T. Tiekink, Gold Bull., 2003, 36, 117–124 CrossRef CAS.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 RSC.
- C.-M. Che and R. W.-Y. Sun, Chem. Commun., 2011, 47, 9554–9560 RSC.
- R. C. Elder and M. K. Eidsness, Chem. Rev., 1987, 87, 1027–1046 CrossRef CAS.
- A. Casini and L. Messori, Curr. Top. Med. Chem., 2011, 11, 2647–2660 CrossRef CAS.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550–580 CAS.
- C. Gabbiani, A. Casini and L. Messori, Gold Bull., 2006, 40, 73–81 CrossRef.
- A. Casini, C. Hartinger, C. Gabbiani, E. Mini, P. J. Dyson, B. K. Keppler and L. Messori, J. Inorg. Biochem., 2008, 102, 564–575 CrossRef CAS.
- A. Molter and F. Mohr, Coord. Chem. Rev., 2010, 254, 19–45 CrossRef CAS.
- F. Caruso, R. Villa, M. Rossi, C. Pettinari, F. Paduano, M. Pennati, M. G. Daidone and N. Zaffaroni, Biochem. Pharmacol., 2007, 73, 773–781 CrossRef CAS.
- Y. Omata, M. Folan, M. Shaw, R. L. Messer, P. E. Lockwood, D. Hobbs, S. Bouillaguet, H. Sano, J. B. Lewis and J. C. Wataha, Toxicol. In Vitro, 2006, 20, 882–890 CrossRef CAS.
- M. Coronnello, E. Mini, B. Caciagli, M. A. Cinellu, A. Bindoli, C. Gabbiani and L. Messori, J. Med. Chem., 2005, 48, 6761–6765 CrossRef CAS.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642–1647 CrossRef CAS.
- E. S. J. Arnér and A. Holmgren, Eur. J. Biochem., 2000, 267, 6102–6109 CrossRef.
- L. Messori, A. Balerna, I. Ascone, C. Castellano, C. Gabbiani, A. Casini, C. Marchioni, G. Jaouen and A. C. Castellano, JBIC, J. Biol. Inorg. Chem., 2010, 16, 491–499 CrossRef.
- G. Marcon, L. Messori, P. Orioli, M. A. Cinellu and G. Minghetti, Eur. J. Biochem., 2003, 270, 4655–4661 CrossRef CAS.
- A. Casini, M. A. Cinellu, G. Minghetti, C. Gabbiani, M. Coronnello, E. Mini and L. Messori, J. Med. Chem., 2006, 49, 5524–5531 CrossRef CAS.
- C. Gabbiani, A. Casini, L. Messori, A. Guerri, M. A. Cinellu, G. Minghetti, M. Corsini, C. Rosani, P. Zanello and M. Arca, Inorg. Chem., 2008, 47, 2368–2379 CrossRef CAS.
- M. A. Cinellu, L. Maiore, M. Manassero, A. Casini, M. Arca, H.-H. Fiebig, G. Kelter, E. Michelucci, G. Pieraccini, C. Gabbiani and L. Messori, ACS Med. Chem. Lett., 2010, 1, 336–339 CrossRef CAS.
- M. A. Soldatov, I. Ascone, A. C. Castellano, L. Messori, M. A. Cinellu, A. Balerna, A. V. Soldatov and G. E. Yalovega, J. Phys.: Conf. Ser., 2009, 190, 012210 CrossRef.
- R. C. Elder, M. K. Eidsness, M. J. Heeg, K. G. Tepperman, C. F. Shaw and N. Schaeffer, Platinum, Gold, and Other Metal Chemotherapeutic Agents, American Chemical Society, 1983, vol. 209, pp. 385–400 Search PubMed.
- D. T. Hill, A. A. Isab, D. E. Griswold, M. J. DiMartino, E. D. Matz, A. L. Figueroa, J. E. Wawro, C. DeBrosse, W. M. Reiff, R. C. Elder, B. Jones, J. W. Webb and C. F. Shaw, Inorg. Chem., 2010, 49, 7663–7675 CrossRef CAS.
- D. C. Ware, P. J. Brothers, G. R. Clark, W. A. Denny, B. D. Palmer and W. R. Wilson, J. Chem. Soc., Dalton Trans., 2000, 925–932 RSC.
- D. C. Ware, W. R. Wilson, W. A. Denny and C. E. F. Rickard, J. Chem. Soc., Chem. Commun., 1991, 1171–1173 RSC.
- D. Ware, H. Palmer, F. Pruijn, R. Anderson, P. Brothers, W. Denny and W. Wilson, Anti-Cancer Drug Des., 1998, 13, 81–103 CAS.
- D. C. Ware, B. D. Palmer, W. R. Wilson and W. A. Denny, J. Med. Chem., 1993, 36, 1839–1846 CrossRef CAS.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588–7598 RSC.
- M. Hidalgo and S. G. Eckhardt, J. Natl. Cancer Inst., 2001, 93, 178–193 CrossRef CAS.
- T. W. Failes, C. Cullinane, C. I. Diakos, N. Yamamoto, J. G. Lyons and T. W. Hambley, Chem.–Eur. J., 2007, 13, 2974–2982 CrossRef CAS.
- N. Johansson, M. Ahonen and V.-M. Kähäri, Cell. Mol. Life Sci., 2000, 57, 5–15 CrossRef CAS.
- P. D. Bonnitcha, M. D. Hall, C. K. Underwood, G. J. Foran, M. Zhang, P. J. Beale and T. W. Hambley, J. Inorg. Biochem., 2006, 100, 963–971 CrossRef CAS.
- M. D. Hall, C. K. Underwood, T. W. Failes, G. J. Foran and T. W. Hambley, Aust. J. Chem., 2007, 60, 180–183 CrossRef CAS.
- M. D. Hall, T. W. Failes, N. Yamamoto and T. W. Hambley, Dalton Trans., 2007, 3983–3990 RSC.
- C. Bresson, S. Esnouf, C. Lamouroux, P. L. Solari and C. D. Auwer, New J. Chem., 2006, 30, 416–424 RSC.
- C. Bresson, C. Lamouroux, C. Sandre, M. Tabarant, N. Gault, J. L. Poncy, J. L. Lefaix, C. Den Auwer, R. Spezia, M.-P. Gaigeot, E. Ansoborlo, S. Mounicou, A. Fraysse, G. Deves, T. Bacquart, H. Seznec, T. Pouthier, P. Moretto, R. Ortega, R. Lobinski and C. Moulin, Biochimie, 2006, 88, 1619–1629 CrossRef CAS.
- C. Bresson, R. Spezia, S. Esnouf, P. L. Solari, S. Coantic and C. D. Auwer, New J. Chem., 2007, 31, 1789–1797 RSC.
- R. Spezia, C. Bresson, C. D. Auwer and M.-P. Gaigeot, J. Phys. Chem. B, 2008, 112, 6490–6499 CrossRef CAS.
- R. Ortega, C. Bresson, A. Fraysse, C. Sandre, G. Devès, C. Gombert, M. Tabarant, P. Bleuet, H. Seznec, A. Simionovici, P. Moretto and C. Moulin, Toxicol. Lett., 2009, 188, 26–32 CrossRef CAS.
- N. Gault, C. Sandre, J.-L. Poncy, C. Moulin, J.-L. Lefaix and C. Bresson, Toxicol. In Vitro, 2010, 24, 92–98 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |