 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal ion release from metallothioneins: proteolysis as an alternative to oxidation†
Estevão A.
Peroza
,
Augusto
dos Santos Cabral
,
Xiaoqiong
Wan‡
and
Eva
Freisinger
*
Institute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: freisinger@aci.uzh.ch; Fax: +41 44 635 6802; Tel: +41 44 635 4621
First published on 9th July 2013
Abstract
Metallothioneins (MTs) are among others involved in the cellular regulation of essential ZnII and CuI ions. However, the high binding affinity of these proteins requires additional factors to promote metal ion release under physiological conditions. The mechanisms and efficiencies of these processes leave many open questions. We report here a comprehensive analysis of the ZnII-release properties of various MTs with special focus on members of the four main subfamilies of plant MTs. ZnII competition experiments with the metal ion chelator 4-(2-pyridylazo)resorcinol (PAR) in the presence of the cellular redox pair glutathione (GSH)/glutathione disulfide (GSSG) show that plant MTs from the subfamilies MT1, MT2, and MT3 are remarkably more affected by oxidative stress than those from the Ec subfamily and the well-characterized human MT2 form. In addition, we evaluated proteolytic digestion with trypsin and proteinase K as an alternative mechanism for selective promotion of metal ion release from MTs. Also here the observed percentage of liberated metal ions depends strongly on the MT form evaluated. Closer evaluation of the data additionally allowed deducing the thermodynamic and kinetic properties of the ZnII release processes. The CuI-form of chickpea MT2 was used to exemplify that both oxidation and proteolysis are also effective ways to increase the transfer of copper ions to other molecules. ZnII release experiments with the individual metal-binding domains of Ec-1 from wheat grain reveal distinct differences from the full-length protein. This triggers the question about the roles of the long cysteine-free peptide stretches typical for plant MTs.
Introduction
Trace metal ions, such as FeII/III, ZnII, and CuI/II, are essential for life. Enzymatic reactions catalyzed by metalloenzymes, signaling, folding and structural stabilization of biomacromolecules are examples of biological processes, where metal ions are involved.1 Therefore, living organisms have developed mechanisms to regulate the homeostasis of essential metal ions, including storage, distribution as well as scavenging of free metal ions. Metallothioneins (MTs) are small ubiquitous proteins that play a major role in the regulation of physiologically important ZnII and CuI ions.2,3 Additionally, they can provide protection against xenobiotic heavy metal ions, e.g. CdII and HgII, and reactive oxygen species (ROS). MTs bind metal ions with high affinity (e.g. rabbit Zn7MT2: K = 3.1 × 1011 M−1)4 mainly via the thiolate groups of their numerous cysteine residues thereby forming metal-thiolate clusters. Histidine residues found in some MTs can also serve as ligands for metal ion coordination.5,6 The field of vertebrate MTs is sustained by 50 years of intensive research; however, despite important recent contributions, the knowledge about plant MTs is comparably limited. Unlike the vertebrate isoforms, which strictly contain 20 Cys and no aromatic residues, the number of Cys residues in the different plant MT forms shows a greater variety (from 10 to 17) and even aromatic amino acids, including histidine, may be present.7 In addition, the Cys-rich regions of plant MTs are generally separated by relatively long Cys-free peptide stretches of ∼15–40 residues length, while the two Cys-rich regions of the vertebrate isoforms are only three amino acids apart. Such remarkable differences between the proteins from these two different kingdoms do most probably account for diverse biological properties and roles, rendering them appealing candidates for a comparative study. But also the members of the four plant MT subfamilies differ significantly from each other, especially with respect to the number and distribution pattern of the Cys residues (Fig. 1). What are the functional impacts of these differences? One aim of our study is to analyze if the properties of a given MT form render it more sensitive to oxidation, which might indicate its potential role in redox processes such as ROS scavenging or oxidation triggered metal ion release as elaborated below.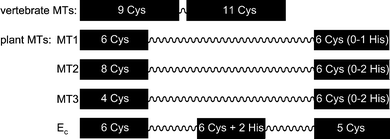 | ||
| Fig. 1 Schematic presentation of the investigated MT species with Cys-rich regions (black boxes, including numbers of Cys and His residues) and Cys-free amino acid linker (wavy lines) indicated. While the vertebrate MT sequences are 61–68 amino acids long, the lengths of the plant MTs vary between roughly 60 and 85 amino acids. | ||
The binding affinities of MTs for metal ions with the electron configuration d10 are very high,4,8 higher than for most other metalloproteins,9 suggesting that, if MTs indeed act as a source of metal ions for other biomolecules as frequently proposed, there must be biochemical mechanisms that regulate the binding and release of metal ions from MTs. As reported previously, naturally occurring oxidizing agents, such as glutathione disulfide (GSSG) or H2O2, promote the release of ZnII from MTs.10,11 Moreover, it has been shown that the combination of GSSG with reduced glutathione (GSH) further increases the transfer of ZnII from MT to the apo-form of sorbitol dehydrogenase,12 yet the precise mode of action of this additional GSH is not known. GSH is the most abundant thiol in mammalian cells and reaches concentrations between 0.5 and 10 mM under normal conditions, while GSSG is present only at micromolar levels.13 Oxidative stress can decrease the GSH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GSSG ratio to values between 10 and 1. In plants, similar values are observed, e.g. 2.7–3.2 mM GSH and approximately 0.1 mM GSSG in the cytosol of Arabidopsis thaliana callus cells grown from stem explants.14 In this report we present a comprehensive study of the ZnII-release properties under various oxidizing conditions of representatives of the four main subfamilies of plant MTs: Cicer arietinum (chickpea) MT1 and MT2, Musa acuminata (banana) MT3, and Triticum aestivum (bread wheat) Ec-1 (cicMT1, cicMT2, musMT3, and Ec-1, respectively, Fig. 1). For comparison, human MT2 (huMT2) was also analyzed under the same conditions. As an alternative to oxidation, we also evaluate the effectiveness of proteolysis to enhance metal ion release from MTs. Situations of elevated cellular proteolytic activity are observed, e.g., during germination,15 apoptosis16 or hypersensitive response in plants.17 To generally probe the hypothesis of protease-facilitated metal ion release, the two well studied proteases proteinase K and trypsin were used in our model systems. Further, we investigated if oxidation and proteolytic digestion have similar effects on the release of copper ions from MTs. For this, cicMT2 was chosen as proteins from this subfamily were shown to be involved in the homeostasis of CuI.3,18 To evaluate the importance of the long Cys-free linker regions for the metal ion binding (in)stability of plant MTs, the ZnII release experiments were repeated for the two separate metal-binding domains of Ec-1, i.e. γ-Ec-1 and βE-Ec-1,5,19–21 and compared to the data obtained with the full-length protein.
:GSSG ratio to values between 10 and 1. In plants, similar values are observed, e.g. 2.7–3.2 mM GSH and approximately 0.1 mM GSSG in the cytosol of Arabidopsis thaliana callus cells grown from stem explants.14 In this report we present a comprehensive study of the ZnII-release properties under various oxidizing conditions of representatives of the four main subfamilies of plant MTs: Cicer arietinum (chickpea) MT1 and MT2, Musa acuminata (banana) MT3, and Triticum aestivum (bread wheat) Ec-1 (cicMT1, cicMT2, musMT3, and Ec-1, respectively, Fig. 1). For comparison, human MT2 (huMT2) was also analyzed under the same conditions. As an alternative to oxidation, we also evaluate the effectiveness of proteolysis to enhance metal ion release from MTs. Situations of elevated cellular proteolytic activity are observed, e.g., during germination,15 apoptosis16 or hypersensitive response in plants.17 To generally probe the hypothesis of protease-facilitated metal ion release, the two well studied proteases proteinase K and trypsin were used in our model systems. Further, we investigated if oxidation and proteolytic digestion have similar effects on the release of copper ions from MTs. For this, cicMT2 was chosen as proteins from this subfamily were shown to be involved in the homeostasis of CuI.3,18 To evaluate the importance of the long Cys-free linker regions for the metal ion binding (in)stability of plant MTs, the ZnII release experiments were repeated for the two separate metal-binding domains of Ec-1, i.e. γ-Ec-1 and βE-Ec-1,5,19–21 and compared to the data obtained with the full-length protein.
Materials and methods
Protein expression and purification
The preparation of pTYB2 vectors encoding cicMT1, cicMT2, musMT3, Ec-1, βE-Ec-1, as well as the expression and purification of the proteins were done as described previously for Ec-1.22 The γ-Ec-1 peptide (MGCDDKCGCA VPCPGGTGCR CTSAR), prepared by solid-phase peptide synthesis, was purchased from Sigma-Genosys (Haverhill, UK) and purified by size-exclusion chromatography. huMT2 was a kind gift from Prof. Milan Vašák (University of Zurich, Switzerland) and obtained as reported.23 After purification, all samples were dialyzed against 1 mM Tris-HCl, pH ∼ 7.5, followed by the determination of the metal-to-protein ratio by flame atomic absorption spectroscopy and thiol quantification.24 Slightly substoichiometric ZnII-loading was adjusted by addition of the required amount of ZnCl2 (Sigma-Aldrich Chemie GmbH, Buchs, Switzerland) producing the fully metalated samples: Zn5cicMT2,25 Zn4musMT3,26 Zn6Ec-1,20 Zn7huMT2,23 Zn4βE-Ec-1, and Zn2γ-Ec-1.20 Although cicMT1 can coordinate up to 5 ZnII ions, the fifth ZnII ion is only weakly coordinated as evident from electrospray ionization mass spectrometry (ESI-MS) analysis,27 and therefore no complement of ZnII was added to the Zn3.9cicMT1 species obtained after dialysis. The metal-to-protein ratios of all samples were finally confirmed by ESI-MS. The CuI-form of cicMT2 was prepared inside a nitrogen-purged glove-box by reconstitution of the apo-form with nine equivalents of [CuI(CH3CN)4](BF4). ESI-MS investigations show that Cu9cicMT2 is one of the major species observed for the CuI-form (ESI†).Metal release experiments
All experiments were performed at 24 °C with two replicates of samples containing 9.0 μM of MT-bound ZnII or 8.0 μM CuI ions, 100 μM 4-(2-pyridylazo)resorcinol (PAR) (Sigma-Aldrich), and 200 mM Tris-HCl, 7.4 (Calbiochem, VWR International AG, Lucerne, Switzerland). PAR was always used in excess to ensure exclusive formation of the 1:2 complex, i.e. Zn(PAR)2. Experiments to evaluate the properties of the combined individual domains of Ec-1 were done with samples containing 6.0 μM of ZnII in the form of Zn4βE-Ec-1 and 3.0 μM of ZnII in the form of Zn2γ-Ec-1. To access the effect of oxidation on metal release, the redox pair GSH and GSSG (Sigma-Aldrich) was added to the samples at concentrations of 1 mM and 0–4.5 mM, respectively. Trypsin (EC 3.4.21.4, porcine; Promega AG, Dübendorf, Switzerland) and Tritirachium album proteinase K (EC 3.4.21.64; Qbiogene, Basel, Switzerland) were used in a ratio of 1:10 MT molecules. Metal ion release from the MTs, or more precisely the metal ion transfer to PAR, was followed by monitoring the changes in PAR absorbance upon metal ion binding at 500 nm for 200 min for experiments with zinc or 100 min for the study of copper release. Preliminary absorptivity measurements with PAR and solutions of [CuI(CH3CN)4](BF4), a relatively oxidation-insensitive CuI complex stabilized with acetonitrile ligands,28 or CuIISO4 showed identical absorptivity values under the experimental conditions employed here. Hence the initial Cu oxidation state has no influence on the measurements. However, molar absorptivity values of the copper–PAR complexes are influenced by the presence of the GSH/GSSG redox couple, probably due to the formation of competing complexes. Accordingly, the respective molar absorptivity values had to be determined for each individual condition: 53200 M−1 cm−1 for the control experiment and the digestion with proteinase K, 26200 M−1 cm−1 for 1 mM GSH/2 mM GSSG, and 12100 M−1 cm−1 for 1 mM GSH/4.5 mM GSSG. For the Zn(PAR)2 complex a molar absorptivity of 65000 M−1 cm−1 was determined irrespective of the conditions used. In analogy to the experiments with the ZnII-MT forms, copper release reactions were performed without exclusion of atmospheric oxygen. All spectra were recorded using a Cary 500 (Varian) scan spectrophotometer.
For the determination of the apparent ZnII binding constants for each MT used (see below) the knowledge of the Zn(PAR)2 concentration at the point of thermodynamic equilibrium is required. Hence in addition fitting of the data was performed where necessary as described in detail in the ESI† resulting in the values for “infinite” time given e.g. in Fig. 3 and 4.
Calculation of apparent ZnII binding constants
Apparent ZnII binding constants for the respective MTs at pH 7.4, Kapp,MT, were calculated according to eqn (1):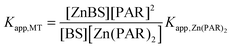 | (1) |
Calculation of first-order rate constants of ZnII release
The integrated first-order law of the ZnII release reaction from the ZnII binding sites in the respective MT can be written as (ref. 34):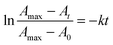 | (2) |
Mass spectrometry
Samples digested with trypsin were subsequently treated with TCEP (10 mM), purified by C4 ZipTip® (Millipore), and analyzed by MALDI-MS. MALDI-MS spectra were recorded on an upgraded Bruker-Daltonics Ultraflex Time-Of-Flight (TOF)/TOF II MALDI mass spectrometer using the control and analysis software Compass v.1.2 (Bruker-Daltonics). Analyses of the peptide fragments found were performed using the FindPept tool of the ExPASy server (http://expasy.org/).35,36Results
Release of ZnII upon MT oxidation
If provided in excess, PAR forms exclusively 2:1 complexes with ZnII and other metal ions and was previously used to follow ZnII transfer reactions from mammalian MTs by UV/Vis spectroscopy.10,29 For all different ZnII-MT forms studied here generally only a small fraction of ZnII ions was released under the control conditions, i.e. in the absence of the GSH/GSSG redox couple, or in the presence of 1 mM GSH. Addition of 0.5–4.5 mM GSSG significantly increased ZnII loss, reaching its maximum at approximately 3 mM GSSG. Representative curves of metal ion release are depicted for musMT3 in Fig. 2. Fig. 3 and 4 show the time-dependent ZnII release for the four plant MTs studied as well as for huMT2 as dependent on the amount of GSH and GSSG added. At the first time point taken, i.e. 0.5 min, approximately the same number of ZnII equivalents (below 1 equiv.) was removed in all samples, irrespective of the MT species and of the amount of GSH/GSSG added (Fig. 3). The picture was similar for the next time point taken, i.e. 2.5 min, with the exception of cicMT2, which showed higher than the average release. The differences in ZnII release, both with respect to the MT species and the amount of GSH/GSSG added, became apparent after 10.5 min and were clearly visible at the end of the data collection, i.e. at 200 min. While some of the samples had reached equilibrium at the end of the measurement, i.e. A500nm remained constant as for example observed for most of the conditions tested with musMT3 (Fig. 2), A500nm for other samples was still increasing, e.g. for Ec-1 (Fig. S1, ESI†). As equilibrium data are required for the determination of apparent ZnII binding constants (see below), additional fitting of the data was performed where necessary as described in detail in the ESI† resulting in the values for “infinite” time given e.g. in Fig. 3 and 4. While differences between the results at 200 min and “infinity” were minor for cicMT2 and musMT3, increased ZnII release of up to 20% was observed for cicMT1 and Ec-1 and even up to 50% for some of the conditions tested with huMT2. In the following we used the data for infinite time unless noted otherwise.
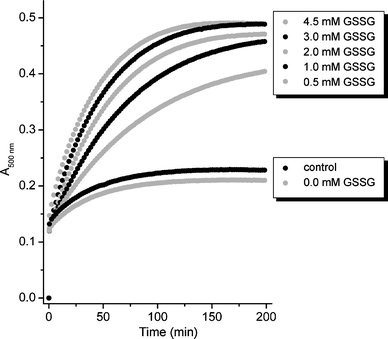 | ||
| Fig. 2 Competition experiment of PAR with Zn4musMT3 followed by UV/Vis spectrophotometry at 500 nm under the different conditions indicated. All solutions, except for the control experiment, contain additionally 1 mM GSH. | ||
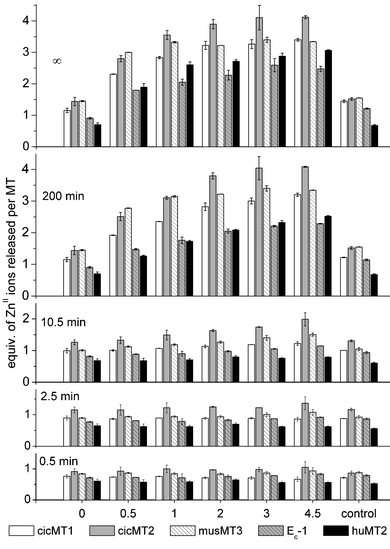 | ||
| Fig. 3 Time-dependence of ZnII release for the different MT species: equivalents of ZnII ions released per MT molecule are plotted against the different conditions indicated, i.e. control conditions and mixtures of 1 mM GSH with 0–4.5 mM GSSG. “Infinity” (∞) data correspond to the values obtained by curve fitting of the data as exemplified in Fig. S1 (ESI†) with equations for exponential association kinetics as described in detail in the ESI.† | ||
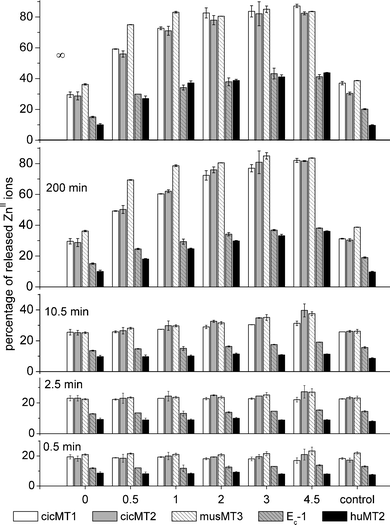 | ||
| Fig. 4 Time-dependence of ZnII release for the different MT species as depicted in Fig. 3, but plotting percentage of ZnII release instead of equivalents. This results in a distinctively different pattern as the total number of metal ions bound per MT molecule differs for the individual species. | ||
The relative effect of GSSG addition and hence of an oxidative environment differs significantly for the individual MT species. While the addition of 4.5 mM GSSG led to an increase of ZnII release by a factor of 2–2.7 in the case of the four plant MTs, a factor of 4.5 was observed for huMT2. Due to the different ZnII binding capacities of the investigated MT species, i.e. 4–7 metal ions per MT molecule (Materials and methods), the plot depicting percentage of ZnII release (Fig. 4) differs from the one presenting released ZnII ions per MT molecule (Fig. 3). While the general trend remains the same with respect to the time dependence and the influence of increasing concentrations of GSH/GSSG, values at 200 min or “infinity” in the percentage plot clearly show that the five different species can be arranged into two groups: group 1 contains the plant MTs cicMT1, cicMT2, and musMT3, which released up to around 80% of their bound ZnII at the highest GSSG concentration tested, while the members of group 2, namely Ec-1 and huMT2, released only around 40%.
Release of ZnII upon proteolysis
Proteases catalyze the hydrolysis of peptide bonds and are involved in a multitude of biochemical processes including degradation and mobilization of storage proteins for seedling development.37 To evaluate the influence of proteolytic cleavage on the ZnII release of MTs, ZnII binding competition experiments were performed for the five different MT species as described above, but this time in the presence of either trypsin or proteinase K. The choice of proteases was based on the fact that all MT species investigated in this work exhibit potential trypsin and proteinase K cleavage sites both within their Cys-rich and Cys-free regions allowing a direct comparison of the results (Fig. 5 and Fig. S2, ESI†). MALDI-MS measurements were performed to analyze the cleavage products, however, due to the much higher number of proteinase K cleavage sites, only the spectra obtained for the trypsin digestions could be analysed in detail. The positions of the potential trypsin cleavage sites and the peptide fragments detected in the mass spectra are depicted in Fig. 5. It has to be noted that the mass limit in these measurements was roughly 1200 Da and that it was accordingly not possible to detect smaller fragments. In Fig. 6 the ZnII release from the five MT species upon treatment with the two different proteases is shown and compared to the amount of ZnII removed in the control experiments and after addition of 4.5 mM GSSG. Treatment with trypsin increased the amount of ZnII released from the four plant MTs compared to the control experiments by 20 (cicMT1 and cicMT2) to 50% (Ec-1), while huMT2 was completely unaffected. In all cases, less ZnII was removed than upon treatment with 4.5 mM GSSG. Digestion with proteinase K increased the ZnII release compared to control conditions by 90 (cicMT1) to 240% (Ec-1) provoking even ZnII release from huMT2 (150% more). Only for Ec-1, the amount of ZnII ions released upon digestion with proteinase K was higher than upon treatment with 4.5 mM GSSG. For all other MT species tested, the amount was lower or equal. Comparing the percentage of ZnII release depicted in Fig. 6(B), a rough separation of the five MT species into two groups can be made with respect to proteolysis-provoked ZnII release: group 1 is formed by the plant MTs releasing around 70–80% ZnII upon incubation with proteinase K and 30–50% upon trypsin treatment, while group 2 consisting of huMT2 as its only member releases only 25 and 10%, respectively. | ||
| Fig. 5 Peptide fragments of the different MT species observed with ESI-MS after trypsin cleavage. For each MT the amino acid sequence of the uncleaved protein is depicted with the Cys-rich regions highlighted in black and the potential trypsin cleavage sites indicated by vertical arrows. Below each sequence, the peptide fragments identified in the ESI-MS spectra are given, gray colored fragments result from unspecific cleavage of the protein. In part (E) of the figure the sequences of the two individually investigated domains of Ec-1 are also given: γ-Ec-1 (left side) separated from βE-Ec-1 by a vertical wavy line. | ||
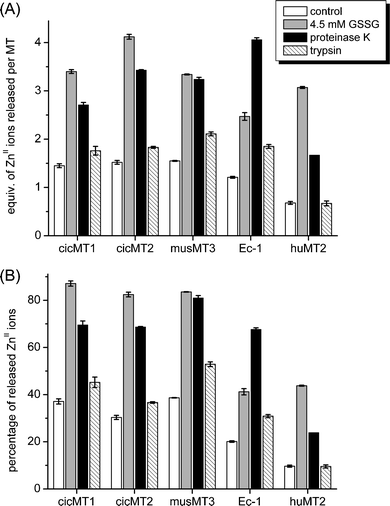 | ||
| Fig. 6 Summary of ZnII release data at equilibrium under control and oxidizing conditions (1 mM GSH/4.5 mM GSSG) as well as upon proteolytic cleavage with proteinase K and trypsin: (A) equiv. of ZnII released per MT, (B) percentage of ZnII release. | ||
Release of ZnII from Ec-1 and its domains
To evaluate the influence of the 11 amino acids long, Cys-free linker region between the smaller N-terminal γ- and the larger C-terminal βE-domain of Ec-1, which are known to fold independently of each other,5,19–21 the separate domains were analysed for their ZnII release properties under different conditions of oxidative stress and proteolytic digestion. Each experiment was performed separately with each individual domain, with the individual domains in a 1:1 mixture, as well as with the full-length protein. For the control experiment, i.e. no addition of GSH or GSSG, equal amounts (within the error range) of ZnII ions were released from the individual domains regardless of the domains being measured in separate solutions or in a 1:1 mixture (Fig. S3, ESI†), while ZnII release from the full-length Ec-1 protein was significantly higher. Clearly the mere concomitant presence of one domain in the 1:1 mixture had no influence on the ZnII release properties of the respective other domain, while the physical connection of both domains by the linker region in the full-length protein affected the metal ion binding properties. Based on this result, the remaining conditions, i.e. addition of GSH and increasing amounts of GSSG, were only evaluated on the separate domains and the full-length protein but not on 1:1 mixtures of the individual domains. The results of the measurements are depicted in Fig. 7. In summary, the amount of ZnII ions released from the full-length Ec-1 protein was consistently higher by 60–100% than the combined amounts released from the individual domains. The equivalents removed from γ-Ec-1 roughly equal the number of ZnII ions removed from βE-Ec-1 under the various conditions. However, as the total number of ZnII ions bound differs, i.e. two ZnII ions in the γ-domain and four in the βE-domain, the percentage of ZnII release per domain differs as well (Fig. S4, ESI†). Direct comparison of these percentage values shows the stronger response of the βE-domain to oxidation compared to γ-Ec-1: Under non-oxidative conditions the γ- and the βE-domain liberate 17 and 7% of their coordinated ZnII ions, respectively. In contrast, in 3–4.5 mM GSSG the γ-domain releases up to 30% of its ZnII (a 2-fold increase with respect to the control condition), while the βE-domain releases about 20% (3-fold increase). When the individual domains were treated with trypsin, the observed combined ZnII release was identical to the one measured for the full-length protein (Fig. S5, ESI†) as trypsin digestion of Ec-1 generated largely the same peptide fragments as digestion of the individual domains (Fig. 5).
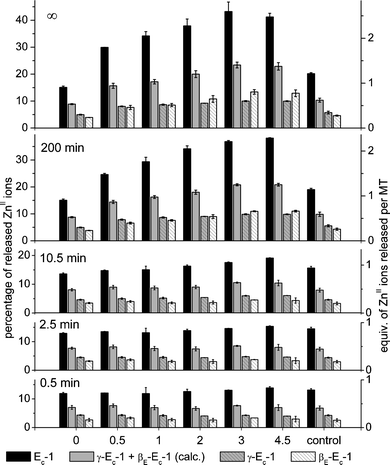 | ||
| Fig. 7 Time-dependence of ZnII release from Ec-1 and its two domains γ-Ec-1 and βE-Ec-1 as affected by the GSSG concentration. The sum of the values obtained for the individual domains is also shown. The percentage values given for the full-length protein as well as for the two separate domains are all relative to the six ZnII ions bound by the full-length protein. See also the legend to Fig. 3. | ||
Release of copper ions from cicMT2
PAR also forms colored complexes with CuII (ref. 38) and, as demonstrated here, can be used to investigate copper ion transfer reactions. The CuI-form of cicMT2, Cu9cicMT2, was prepared under anaerobic conditions. As for the ZnII-form, GSSG also caused release of CuI ions from the protein (Fig. 8). Incubation with 1 mM GSH and 2 mM GSSG more than doubled the transfer of copper ions to PAR compared to the control experiment. Higher GSSG concentration (4.5 mM) tripled the transfer. Incubation with proteinase K had approximately the same effect as intermediate oxidative conditions, i.e. 2 mM GSSG. Overall, regardless of the conditions applied, the percentage of CuI ions released from cicMT2 was approximately tenfold lower compared to the values for ZnII release (Fig. 8).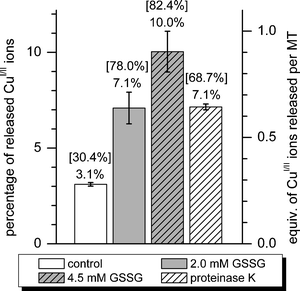 | ||
| Fig. 8 Summary of CuI release data at equilibrium for Cu9cicMT2 under control and oxidizing conditions (1 mM GSH and 2 or 4.5 mM GSSG) as well as upon proteolytic digestion with proteinase K. The exact percentage of CuI release is printed above each column. The numbers in square brackets give the percentage of ZnII release in the corresponding experiments (Fig. 4). | ||
Apparent ZnII binding constants
Extrapolating the absorption data from the competition experiments with the metal ion chelator PAR to equilibrium values allows the calculation of apparent ZnII binding constants. The required apparent ZnII binding constant for the Zn(PAR)2 complex was calculated using literature values. As a number of publications are available each with slightly varying values, the ambiguity of the values chosen here is naturally also transferred to the apparent binding constants calculated from them. Generally, the calculation of apparent metal ion binding constants of MTs poses a number of difficulties, which are often not addressed. MTs feature multiple metal ion binding sites within one protein molecule. Even for the well-studied mammalian forms to date there has been no finite consensus if the ZnII ion affinities of the individual sites are identical within the experimental error or different.39,40 To address this ambiguity we based our calculations on different assumptions:(i) All ZnII binding sites have identical apparent stability constants, Kapp, and do not interact with each other. Accordingly, the starting concentration of ZnII binding sites, [ZnBS]0, equals the concentration of MT-bound ZnII initially added to the solution, i.e. 9 μM. The detailed calculation of the apparent stability constants can be found in the ESI.† As already seen in Fig. 4 and 7, the stability of Zn7huMT2 and Zn4βE-Ec-1 in the control experiments is largest and reflected in logKapp values of 12.50 ± 0.04 and 12.81 ± 0.04, respectively (Table 1, column 2). Zn6Ec-1 and Zn2γ-Ec-1 show lower values of 11.79 ± 0.02 and 11.96 ± 0.09, while the three plant MTs with the long Cys-free linker regions have the lowest stabilities (11.083 ± 0.004–11.36 ± 0.03).
| logKappa |
Eq. ZnII released | logKapp,1 |
logKapp,2d |
logKapp,3e |
|
|---|---|---|---|---|---|
| a Assumption: all ZnII binding sites (ZnBS) have the same Kapp. b Assumption: only 1 ZnBS per protein competes with PAR, the others have higher Kapp and no ZnII is released from them. c Assumption: only 2 ZnBS per protein compete with PAR, the others have higher Kapp and no ZnII is released from them. d Assumption: 1 ZnBS has very low affinity and 1 equiv. (0.9 equiv. for cic-Zn3.9MT1) ZnII is released immediately. Only 1 of the remaining ZnBS per protein competes with PAR, the others have higher Kapp and no ZnII is released from them. e Assumption: 1 ZnBS has very low affinity and 1 equiv. (0.9 equiv. for cic-Zn3.9MT1) ZnII is released immediately, hence competition with PAR is only relevant for the remaining ZnBS, which are all assumed to have the same Kapp. | |||||
| Zn3.9cicMT1 | 11.13 ± 0.03 | 1.45 | 10.49 ± 0.06c | 11.25 ± 0.11 | 11.98 ± 0.08 |
| Zn5cicMT2 | 11.36 ± 0.03 | 1.51 | 10.50 ± 0.07c | 11.44 ± 0.12 | 12.30 ± 0.08 |
| Zn4musMT3 | 11.083 ± 0.004 | 1.55 | 10.348 ± 0.008c | 11.249 ± 0.013 | 11.984 ± 0.009 |
| Zn6Ec-1 | 11.79 ± 0.02 | 1.21 | 11.01 ± 0.03c | 12.53 ± 0.09 | 13.31 ± 0.09 |
| Zn2γ-Ec-1 | 11.96 ± 0.09 | 0.34 | 11.55 ± 0.10b | ||
| Zn4βE-Ec-1 | 12.81 ± 0.04 | 0.28 | 12.10 ± 0.05b | ||
| Zn7huMT2 | 12.50 ± 0.04 | 0.68 | 11.21 ± 0.08b | ||
(ii) (a) The multiple ZnII binding sites within one MT might have different stabilities. This assumption has an influence solely on the starting concentration of ZnII binding sites, [ZnBS]0, used for the calculations. All MTs evaluated release less than two equiv. of ZnII under control conditions. Hence 1–2 equivalents of ZnII ions seem to be bound with lower affinity and compete with PAR, while the other metal ions have much higher affinities to their ZnII binding sites and hence are virtually not affected by PAR. This reduces [ZnBS]0e.g. for Zn6Ec-1 to 3 μM, i.e. 1.2 ZnII ions are released and hence 2 out of totally 6 ZnBS compete with PAR, and for Zn7huMT2 to 1.3 μM, i.e. 0.7 ZnII ions are released and 1 ZnBS competes with PAR. Accordingly, slightly lower apparent stability constants are obtained (Kapp,1, Table 1, column 4).
(ii) (b) For those MTs that release between one and two ZnII ions, i.e. Zn3.9cicMT1, Zn5cicMT2, Zn4musMT3, and Zn6Ec-1, it could also be assumed that the first metal ion is coordinated with very low affinity and hence that only the second ZnII ion competes effectively with PAR reducing [ZnBS]0 even further e.g. to 1.5 μM for Zn6Ec-1, i.e. 1 ZnII ion is released immediately and hence only 1 ZnBS competes with PAR. These calculations yield the slightly higher values (Kapp,2, Table 1, column 5).
First-order rate constants of ZnII release
Rate constants were determined from the semi-log plots described in the experimental part using linear data fits (Fig. 9). Only data from the control experiments, 1 mM GSH and 1 mM GSH/4.5 mM GSSG, were evaluated (Table 2). While differences between most values are significant, it is apparent that they have the same order of magnitude, i.e. 1–9 × 10−4 s−1. As a general trend, rate constants are higher in solutions containing 1 mM GSH, while oxidizing conditions can accelerate (Ec-1, γ-Ec-1), slow down (cicMT2, huMT2) or even do not effect (cicMT1, musMT3, βE-Ec-1) the rate of ZnII release from the different MTs. The initial faster ZnII release step evident from the semi-log plots for rate-constant determination (Fig. 9) could not be quantified due to instrumental limitations. However, for βE-Ec-1 the time frame for this initial step was slightly extended (∼4.5 min) and the respective data (control and 1 mM GSH) indicate an acceleration of release and hence an increase of the corresponding rate-constants by one log unit ((1.8 ± 0.9) × 10−3 and (4 ± 1) × 10−3 s−1).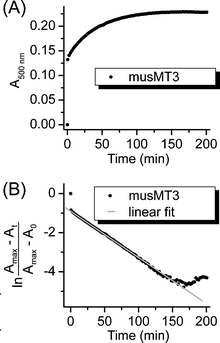 | ||
| Fig. 9 Determination of first-order rate constants exemplified for musMT3 under control conditions. Panel (A) shows the experimental data from the competition experiment with PAR followed by UV/Vis spectroscopy at 500 nm. Panel (B) depicts the semi-log plot of the absorption data against time with Amax denoting the absorption value at equilibrium (A0 equals 0 in this case). The first fast ZnII release step is defined by the time points at 0 and 0.5 min and hence is not sufficiently defined for an analysis. The gray straight line shows the linear fit for the determination of the second slower ZnII release step. | ||
| Control | 1 mM GSH | 1 mM GSH/4.5 mM GSSG | |
|---|---|---|---|
| Zn3.9cicMT1 | (2.09 ± 0.03) × 10−4 | (3.8 ± 0.1) × 10−4 | (2.09 ± 0.01) × 10−4 |
| Zn5cicMT2 | (6.2 ± 0.1) × 10−4 | (8.5 ± 0.2) × 10−4 | (4.42 ± 0.04) × 10−4 |
| Zn4musMT3 | (4.08 ± 0.03) × 10−4 | (4.41 ± 0.02) × 10−4 | (4.11 ± 0.02) × 10−4 |
| Zn6Ec-1 | (1.70 ± 0.02) × 10−4 | (3.3 ± 0.1) × 10−4 | (2.97 ± 0.03) × 10−4 |
| Zn2γ-Ec-1 | (1.79 ± 0.03) × 10−4 | (4.9 ± 0.2) × 10−4 | (2.88 ± 0.02) × 10−4 |
| Zn4βE-Ec-1 | (1.72 ± 0.06) × 10−4 | (4.6 ± 0.5) × 10−4 | (1.87 ± 0.03) × 10−4 |
| Zn7huMT2 | (5.8 ± 0.4) × 10−4 | (9 ± 1) × 10−4 | (1.27 ± 0.01) × 10−4 |
Discussion
Release of metal ions under oxidative conditions and upon proteolysis
Incubation of representatives of the four main plant MT subfamilies as well as human MT2 with 1 mM GSH and increasing GSSG concentrations to mimic the cellular situation of oxidative stress causes increasing release of ZnII ions. Oxidative conditions clearly have a larger effect on the plant MTs from the subfamilies containing long Cys-free linker regions, i.e. cicMT1, cicMT2, and musMT3, than on Ec-1 and huMT2 (Fig. 1 and 4). The data also reveal that the first three MTs respond faster to addition of GSSG. After around 10 min cicMT1, cicMT2, and musMT3 show a significantly higher ZnII release under oxidative conditions compared to the control experiment, while the differences for Ec-1 and huMT2 are much less pronounced. Protective effects of MTs against oxidative stress were shown in several studies, e.g. induction of mouse MT1 in MT-deficient hamster cells decreased the number of oxidative DNA strand breaks.41 In plants, up-regulation of MT mRNA was observed in vivo as a response to ROS, e.g. increased mt1a transcript concentrations in leaves of A. thaliana upon treatment with AgNO342 and higher mt2 transcript levels in cork oak or mt3 levels in cotton seedlings after exposure to H2O2.43,44 The seed-specific plant Ec-1 proteins however have been so far not linked to oxidative stress conditions, but rather associated with ZnII storage for rapid cell division and protein production during seed germination.45,46 In this respect, the pronounced differences observed might indeed allow us to make a correlation to different functions of the plant MT1, MT2, and MT3 proteins versus Ec-1 in vivo. The higher susceptibility of the plant MTs with the long linker regions to oxidation might indicate a role in ROS scavenging. Alternatively or complementarily, oxidation of Cys residues in these proteins could be a specific mechanism to release MT-bound metal ions when required. For instance, Cu/Zn-superoxide dismutase is one of the enzymes overexpressed upon ROS exposure47 and hence, on a speculative basis, MTs could provide the transition metal ions necessary for the functional enzyme. Interestingly, all of the three plant MTs release only roughly 80% of their ZnII ions even at the highest GSSG concentration tested, i.e. in each sub-form approximately one ZnII ion remains bound to the protein. The reason for this can only be speculated on. Maybe this residual metal ion serves the purpose of ensuring a certain level of folding to prevent recognition by cellular control mechanisms that degrade unfolded proteins. In this way, metal ion release from the MT could become a reversible process upon change of the cellular redox state.Evidently, Ec-1 and also huMT2 are less sensitive to oxidation and hence can act as efficient metal ion chelators at different cellular redox states. Nevertheless, if the Ec-1 protein indeed functions as a ZnII reservoir for the onset of germination, the question arises how the metal ions are released in light of the usually lower ZnII affinities of ZnII requiring enzymes. Hence we investigated the possibility of ZnII release promoted by proteolytic digestion as an alternative to oxidative metal ion release. The results are remarkable. While ZnII release from cicMT1, cicMT2, and musMT3 is lower or equal to the values observed under oxidative stress conditions (Fig. 6(B)), digestion of Ec-1 with proteinase K clearly increases the percentage of released ZnII ions up to a value in the range observed for the other three plant MTs under the same conditions. Accordingly, the modulated action of proteases could be an efficient way to liberate ZnII ions from Ec-1, even more as germinating wheat has indeed been shown to have increased proteolytic activity.48,49 Comparing the different degrees of metal ion release upon treatment with trypsin or proteinase K, the correlation with the increased number of cleavage sites is obvious and expected. Being technically polydentate metal ion chelators, cleavage of the peptide backbone can be considered as an inversion of the so-called chelate-effect of coordination chemistry, reducing the binding affinity of a given MT for metal ions. In addition, the exact position of peptide backbone cleavage can also be important and hence the choice of protease can offer a mechanism for the specific regulation of metal ion availability in vivo.
There are strong indications that the plant MTs from the MT1, MT2, and MT3 subfamilies play a role in Cu homeostasis. For example, tissue-specific induction of mt1, mt2, and mt3 gene transcription and MT1, MT2, and MT3 overexpression was observed in A. thaliana upon treatment with CuII.50 In addition, overexpression of cork oak MT2 had a protective effect against Cu toxicity as demonstrated in yeast complementation assays.43 cicMT2 was chosen here as a representative example of a potentially CuI binding plant MT to evaluate the influence of oxidizing conditions and proteolytic cleavage on CuI release. As depicted in Fig. 8, the relative proportions of metal ion release under the different conditions are comparable for Zn5- and Cu9cicMT2. However, the absolute numbers of CuI release amount to only 10% of the values observed for ZnII release. This can be seen as a direct consequence of the higher thiophilicity of CuI ions compared to ZnII and hence of the higher binding affinity to MTs. Hence removal of CuI ions from MTs is clearly more complex for the organism, but on the other hand effective scavenging of redox active CuI ions and prevention of deleterious side reaction are crucial for the cell.
Influence of the Cys-free linker region
Approximately twice the amount of ZnII ions were released from the full-length Ec-1 protein compared to the separate domains βE-Ec-1 and γ-Ec-1 under control conditions and all redox conditions tested (Fig 7). A similar study was previously performed with human MT2 and its two separate domains. But different from Ec-1, the full-length huMT2 protein was found to be less prone to oxidation and ZnII transfer.51 These opposing results illustrate that obviously no generalization is possible with respect to the enhanced or reduced stability of a multi-domain MT compared to its separate domains. On the one hand it is reasonable to assume that the larger the number of potential binding sites within a chelate the greater is the overall affinity for metal ions52 and hence it should be beneficial in terms of metal ion binding to connect multiple domains via linkers to a larger entity. On the other hand it is also conceivable that, especially with shorter linkers, the spatial proximity of one domain interferes with metal ion binding in the respective other domain, e.g. by repulsive charge effects as all domains of Ec-1 and huMT2 have a neutral or slightly negative overall charge. Also steric effects implied by the linker region might play a role, i.e. if a steric strain causes the domain structure to become more solvent accessible and hence more accessible to oxidizing agents and metal chelators. However, this view is contradictory to the experimental results insofar as the shorter linker region in huMT2 should impose the greater steric strain, but huMT2 is more stable against metal ion release and oxidation. Clearly more experimental insights are required, but the general assumption that two domains evolved to stabilize the resulting MT should certainly be viewed critically. It also has to be noted that neither in huMT2 nor in Ec-1 could direct inter-domain contacts be observed, e.g. with NMR spectroscopy. Hence whatever the influence of the respective other domain might be, at most transient contacts can play a role.Ec-1 exhibits not only a linker region between its two metal binding domains, but also an additional Cys-free, 15 amino acids long stretch within the βE-domain (Fig. 1 and 5). Cleavage of this linker during the trypsin digestion experiments clearly has a destabilizing effect of the βE-domain, which is larger than the influence of trypsin cleavage on the γ-domain. Hence, while the percentage of ZnII release from the γ-domain is 2.5 times larger compared to ZnII release from the βE-domain (17 versus 7%, Fig. S5, ESI†) under control conditions, it is nearly equal upon digestion with trypsin (28 versus 32%). The ESI-MS spectra of the digestion products still clearly show signals for the undigested γ-Ec-1 peptide, whereas for βE-Ec-1 only fragments for the two Cys-rich regions were observed, but no signals were observed for the intact domain (Fig. 5).
Apparent stability constants for ZnII binding and rate constants of ZnII release
Numerous apparent stability constants for different vertebrate MT forms have been determined over the years. More recently a binding constant of logKapp 11.2 was reported for each ZnII binding site in rabbit liver MT2 at pH 7.4.40 In another study a series of individual binding constants for the different binding sites within the protein was determined for huMT2 at the same pH.39 Here, one weak binding site (logKapp 7.7), two sites with higher affinity (10.7 and 10.9) as well as 4 ZnII sites with logKapp 11.8 were observed. The different results reported in these two studies of vertebrate MTs were attributed to the differences in protein preparation: rabbit MT2 was isolated from the native source without pH changes, while huMT2 was over-expressed in Escherichia coli and subjected to a reconstitution step at pH 1–2. In the present work, we determined an apparent binding constant for huMT2 of 12.5 (Table 1) calculated under the premise of identical binding sites. Assuming that only one binding site in huMT2 competes with PAR this value is reduced to 11.2. Hence, our investigation could also not reveal the presence of a weaker binding site. The huMT2 form used in our study was also reconstituted at low pH 2. However, this procedure was performed very rapidly as alterations of the physicochemical properties of MTs after prolonged incubation at pH values below 2 have been reported.53 All apparent binding constants calculated for the different plant MT forms are in the same range, i.e. 1.2–9.1 × 1011 M−1, only the value for Zn4βE-Ec-1 is somewhat higher (6.5 × 1012 M−1). These results suggest that the distinctive sequences of the different plant MT subfamilies have not evolved to modulate metal ion binding affinities per se, but rather to fulfill specific functions under the specific cellular conditions they are expressed in. Accordingly, the differences in their metal ion release properties only become manifested under for example oxidative stress or upon the action of proteases.
As a side remark, the order of the observed apparent stability constants, and hence the susceptibility to ZnII release in the control experiments, is consistent with the pH stability of the ZnII–thiolate clusters observed in previous studies: cicMT1 (37% ZnII ions released, apparent pKa values of the Cys residues in the presence of ZnII ions 4.94 ± 0.03), musMT3 (39%, 4.90 ± 0.04) > cicMT2 (30%, 4.78 ± 0.02) > Ec-1 (20%, 4.53 ± 0.01) > huMT2 (10%, 4.36 ± 0.02).7,20,25–27
For the determination of the first-order rate constants of ZnII release, data points between 2.5 and 100 min were used. The preceding faster ZnII release step evident from the semi-log plots (Fig. 9) was not quantified due to instrument limitations. As depicted in Fig. 3 during the first 2.5 min only up to approximately one equiv. of ZnII is released irrespective of the MT form or the condition applied. Hence this first equiv. seems to be kinetically more labile than the other ZnII ions, which display a rate constant that is lower by approximately one order of magnitude. In the literature an experiment performed with huMT2 under conditions closely similar to those of the control experiment reported here resulted in rate constants of 8.4 × 10−4 and 9.9 × 10−5 s−1 for the α-domain, 3.5 × 10−4 and 4.3 × 10−5 s−1 for the β-domain, and “similar” values for full-length huMT2.51 Data points were collected between 0 and 60 min, however, the data range used for the determination of the two different rate constants was not indicated. Nevertheless, the second rate constant is smaller by nearly an order of magnitude than the value reported here. The reason for this is not clear.
Interesting features are also the slightly increased rate constants of ZnII release upon incubation with 1 mM GSH compared to the control experiments. Concurrently, except for Ec-1, the amounts of ZnII released under both conditions are identical within the error limits (3σ) for a given MT. The increased rate constants might be explained by the additional presence of a competing metal ion chelator (GSH), which increases the speed of metal ion release. GSH, however, is not able to compete with PAR for ZnII ions under the conditions applied, i.e. control experiments performed by us showed that addition of GSH to the Zn(PAR)2 complex leads to deviations in the A500nm values of only around 1% and hence validates the applicability of our experimental setup for the quantification of ZnII release. In other words, GSH might aid to remove kinetically labile bound ZnII ions from MTs, but then transfers the ZnII ions immediately to PAR.
In summary, this work shows the distinct differences in oxidative ZnII release between the various plant MTs and huMT2 and highlights proteolytic digestion as an alternative mechanism for controlled metal ion release from MTs. It has to be noted however that the latter mechanism is associated with a higher metabolic cost for the organism requiring synthesis of a protease as well as causing the irreversible degradation of the respective MT.
Abbreviations
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| MT | Metallothionein |
| PAR | 4-(2-Pyridylazo)resorcinol |
| ROS | Reactive oxygen species |
Acknowledgements
We thank Prof. Milan Vašák, University of Zurich, for providing us with the huMT2 sample used in this study and Dr. Serge Chesnov, Functional Genomics Centre Zurich, for MS measurements. This work was supported by the Swiss National Science Foundation (SNSF Professorship to E.F.).References
- Biological Inorganic Chemistry: Structure and Reactivity, ed. I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, University Science Books, Herndon, VA, USA, 1st edn, 2007 Search PubMed.
- N. Romero-Isart and M. Vašák, J. Inorg. Biochem., 2002, 88, 388–396 CrossRef CAS.
- C. Cobbett and P. Goldsbrough, Annu. Rev. Plant Biol., 2002, 53, 159–182 CrossRef CAS.
- D. W. Hasler, L. T. Jensen, O. Zerbe, D. R. Winge and M. Vašák, Biochemistry, 2000, 39, 14567–14575 CrossRef CAS.
- E. A. Peroza, R. Schmucki, P. Güntert, E. Freisinger and O. Zerbe, J. Mol. Biol., 2009, 387, 207–218 CrossRef CAS.
- C. A. Blindauer, M. D. Harrison, J. A. Parkinson, A. K. Robinson, J. S. Cavet, N. J. Robinson and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9593–9598 CrossRef CAS.
- E. Freisinger, Dalton Trans., 2008, 6663–6675 RSC.
- O. I. Leszczyszyn, R. Schmid and C. A. Blindauer, Proteins: Struct., Funct., Bioinform., 2007, 68, 922–935 CrossRef CAS.
- W. Maret and Y. Li, Chem. Rev., 2009, 109, 4682–4707 CrossRef CAS.
- W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3478–3482 CrossRef CAS.
- A. R. Quesada, R. W. Byrnes, S. O. Krezoski and D. H. Petering, Arch. Biochem. Biophys., 1996, 334, 241–250 CrossRef CAS.
- L. J. Jiang, W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3483–3488 CrossRef CAS.
- A. Pastore, G. Federici, E. Bertini and F. Piemonte, Clin. Chim. Acta, 2003, 333, 19–39 CrossRef CAS.
- A. J. Meyer, M. J. May and M. Fricker, Plant J., 2001, 27, 67–78 CrossRef CAS.
- K. Müntz, M. A. Belozersky, Y. E. Dunaevsky, A. Schlereth and J. Tiedemann, J. Exp. Bot., 2001, 52, 1741–1752 CrossRef.
- E. Solary, B. Eymin, N. Droin and M. Haugg, Cell Biol. Toxicol., 1998, 14, 121–132 CrossRef CAS.
- M. C. Heath, Plant Mol. Biol., 2000, 44, 321–334 CrossRef CAS.
- W.-J. Guo, M. Meetam and P. Goldsbrough, Plant Physiol., 2008, 146, 1697–1706 CrossRef CAS.
- E. A. Peroza and E. Freisinger, JBIC, J. Biol. Inorg. Chem., 2007, 12, 377–391 CrossRef CAS.
- E. A. Peroza, A. Al Kaabi, W. Meyer-Klaucke, G. Wellenreuther and E. Freisinger, J. Inorg. Biochem., 2009, 103, 342–353 CrossRef CAS.
- J. Loebus, E. A. Peroza, N. Blüthgen, T. Fox, W. Meyer-Klaucke, O. Zerbe and E. Freisinger, JBIC, J. Biol. Inorg. Chem., 2011, 16, 683–694 CrossRef CAS.
- E. A. Peroza and E. Freisinger, Protein Expression Purif., 2008, 57, 217–225 CrossRef CAS.
- M. Knipp, A. V. Karotki, S. Chesnov, G. Natile, P. J. Sadler, V. Brabec and M. Vašák, J. Med. Chem., 2007, 50, 4075–4086 CrossRef CAS.
- A. O. Pedersen and J. Jacobsen, Eur. J. Biochem., 1980, 106, 291–295 CrossRef CAS.
- X. Wan and E. Freisinger, Metallomics, 2009, 1, 489–500 RSC.
- E. Freisinger, Inorg. Chim. Acta, 2007, 360, 369–380 CrossRef CAS.
- O. Schicht and E. Freisinger, Inorg. Chim. Acta, 2009, 362, 714–724 CrossRef CAS.
- C. Dietrich-Buchecker and J.-P. Sauvage, Tetrahedron, 1990, 46, 503–512 CrossRef CAS.
- C. F. Shaw, III, J. E. Laib, M. M. Savas and D. H. Petering, Inorg. Chem., 1990, 29, 403–408 CrossRef.
- W. J. Geary, G. Nickless and F. H. Pollard, Anal. Chim. Acta, 1962, 27, 71–79 CrossRef CAS.
- J. Ghasemi, S. Ahmadi, M. Kubista and A. Forootan, J. Chem. Eng. Data, 2003, 48, 1178–1182 CrossRef CAS.
- M. Zimmermann, O. Clarke, J. M. Gulbis, D. W. Keizer, R. S. Jarvis, C. S. Cobbett, M. G. Hinds, Z. G. Xiao and A. G. Wedd, Biochemistry, 2009, 48, 11640–11654 CrossRef CAS.
- A. Corsini, Q. Fernando and H. Freiser, Inorg. Chem., 1963, 2, 224–226 CrossRef CAS.
- E. A. Guggenheim, Philos. Mag., 1926, 2, 538–543 CAS.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, in The Proteomics Protocols Handbook, ed. J. M. Walker, Humana Press, 2005, pp. 571–607 Search PubMed.
- A. Gattiker, W. V. Bienvenut, A. Bairoch and E. Gasteiger, Proteomics, 2002, 2, 1435–1444 CrossRef CAS.
- M. Grudkowska and B. Zagdanska, Acta Biochim. Pol., 2004, 51, 609–624 CAS.
- M. Siroki, L. Maric and M. J. Herak, Fresenius' Z. Anal. Chem., 1976, 278, 285–286 CrossRef CAS.
- A. Krezel and W. Maret, J. Am. Chem. Soc., 2007, 129, 10911–10921 CrossRef CAS.
- M. A. Namdarghanbari, J. Meeusen, G. Bachowski, N. Giebel, J. Johnson and D. H. Petering, J. Inorg. Biochem., 2010, 104, 224–231 CrossRef CAS.
- L. S. Chubatsu and R. Meneghini, Biochem. J., 1993, 291, 193–198 CAS.
- S. Navabpour, K. Morris, R. Allen, E. Harrison, S. A-H-Mackerness and V. Buchanan-Wollaston, J. Exp. Bot., 2003, 54, 2285–2292 CrossRef CAS.
- G. Mir, J. Domènech, G. Huguet, W.-J. Guo, P. Goldsbrough, S. Atrian and M. Molinas, J. Exp. Bot., 2004, 55, 2483–2493 CrossRef CAS.
- T. Xue, X. Li, W. Zhu, C. Wu, G. Yang and C. Zheng, J. Exp. Bot., 2009, 60, 339–349 CrossRef CAS.
- I. Kawashima, T. D. Kennedy, M. Chino and B. G. Lane, Eur. J. Biochem., 1992, 209, 971–976 CrossRef CAS.
- L. Ozturk, M. A. Yazici, C. Yucel, A. Torun, C. Cekic, A. Bagci, H. Ozkan, H. J. Braun, Z. Sayers and I. Cakmak, Physiol. Plant., 2006, 128, 144–152 CrossRef CAS.
- E. W. T. Tsang, C. Bowler, D. Herouart, W. Vancamp, R. Villarroel, C. Genetello, M. Vanmontagu and D. Inze, Plant Cell, 1991, 3, 783–792 CAS.
- K. Sutoh, H. Kato and T. Minamikawa, J. Biochem., 1999, 126, 700–707 CrossRef CAS.
- A. Tsuji, M. Tsuji, H. Takami, S. Nakamura and Y. Matsuda, Biochim. Biophys. Acta, Gen. Subj., 2004, 1670, 84–89 CrossRef CAS.
- W. J. Guo, W. Bundithya and P. B. Goldsbrough, New Phytol., 2003, 159, 369–381 CrossRef CAS.
- L. J. Jiang, M. Vašák, B. L. Vallee and W. Maret, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2503–2508 CrossRef CAS.
- T. T. Ngu, A. Easton and M. J. Stillman, J. Am. Chem. Soc., 2008, 130, 17016–17028 CrossRef CAS.
- M. Vašák, Methods Enzymol., 1991, 205, 452–458 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mt00079f |
| ‡ Present address: Key Laboratory of Arable Land Conservation (Middle and Lower Reaches of Yangtse River), Ministry of Agriculture, Huazhong Agricultural University, Wuhan 430070, P. R. China. |
| This journal is © The Royal Society of Chemistry 2013 |