Molecular strategies of microbial iron assimilation: from high-affinity complexes to cofactor assembly systems
Marcus
Miethke
*
Department of Chemistry/Biochemistry, Philipps University Marburg, Hans Meerwein Strasse, and Loewe-Center for Synthetic Microbiology, D-35032 Marburg, Germany. E-mail: miethke@staff.uni-marburg.de; Fax: +49 (0)6421 28 22191; Tel: +49 (0)6421 28 25441
First published on 20th November 2012
Abstract
Microorganisms have to cope with restricted iron bioavailability in most environmental habitats as well as during host colonization. The continuous struggle for iron has brought forth a plethora of acquisition and assimilation strategies that share several functional and mechanistic principles. One common theme is the utilization of high-affinity chelators for extracellular iron mobilization, generally known as siderophore-dependent iron acquisition. This basic strategy is related with another central aspect of microbial iron acquisition, which is the release of the mobilized iron from extracellular sources to allow its transfer and incorporation into metabolically active proteins. A variety of mechanisms which are often coupled with high-affinity uptake have evolved to facilitate the removal of iron from siderophore ligands; however, they differ in many key aspects including substrate specificities and release efficiencies. The most sophisticated iron release pathways comprise processes of specific hydrolysis and reduction of ferric siderophores, especially in the case of high-affinity iron complexes with greatly negative redox potentials that often represent crucial factors for virulence development in bacterial and fungal pathogens. During the following steps of iron utilization, the acquired metal is transferred through intracellular trafficking pathways which may include diverse storage compartments in order to be directed to cofactor assembly systems and to final protein targeting. Several of these iron channeling routes have been described recently and provide first insights into the later steps of iron assimilation that characterize an essential part of the cellular iron homeostasis network.
 Marcus Miethke | Marcus Miethke studied Microbiology, Molecular Biology and Biochemistry at the Ernst-Moritz-Arndt University in Greifswald, Germany. After receiving his Diploma in 2004, he started his PhD at the Philipps University of Marburg, where he finished his thesis on iron metabolism and bacterial siderophore pathways in 2007. Then he worked as a postdoctoral fellow at the Technical University of Munich on protein evolution and the biophysics of protein–ligand interactions. In 2009, he became a research associate at Philipps University of Marburg and focuses his studies on molecular mechanisms of metalloenzymes and metal trafficking pathways. Currently, he is affiliated with the Marburg Center for Synthetic Microbiology (SYNMIKRO) and investigates the assembly and flux kinetics of transition metal cofactors in bacterial metalloproteomes. |
1 Introduction
Iron is a mineral nutrient with an outstanding physiological relevance for most forms of life. It is the fourth most abundant element in the earth's crust and exists in a wide range of oxidation states, though +2 and +3 are the most common and form a redox couple that has a standard reduction potential of +0.77 V in water, which is close to that of the O2/H2O redox pair. In this context, iron was most important during the early stages of evolution to drive dissimilatory processes, and it still fulfills this function during microbial ferric iron reduction, representing a key factor of many biogeochemical circuits.1 Beyond its role as an alternative terminal electron acceptor, iron has been established as a versatile redox cofactor in a vast number of primary and secondary metabolic pathways. To drive these iron-related processes efficiently, several mechanisms for extracellular iron mobilization, uptake and intracellular assimilation had to be developed.2 This is in particular the case as the non-limited uptake of soluble ferrous iron is only feasible in a rather small number of anoxic and strongly acidic habitats. In contrast, the vast majority of microorganisms live under conditions that provide soluble iron concentrations below 10−9 M, which is generally the case in aerobic and non-acidic environments as well as in association with many eukaryotic host organisms.3,4 Under such conditions, the utilization of all potential organic and inorganic iron sources is a key determinant of microbial fitness. Iron mobilization includes the secretion of iron cofactor scavenging proteins like hemophores,5 or small molecule chelators called siderophores that are able to sequester ferric iron with enormous affinities from numerous primary sources.6,7 Siderophores are secreted by a vast number of microbes in broad structural variations including scaffold backbones and ligand donor groups and display iron formation constants (Kf) in a range of ∼1020 to ∼1050 M−1.8,9 They are hence compellingly designed to compete very effectively for low-soluble iron pools within a wide range of environmental conditions as well as in tightly iron-regulated host organisms.Ferric iron that is captured by a high-affinity siderophore scaffold is shielded from external ligands and is exchanged very slowly with the environment at physiological pH.10 The extracellularly formed iron–siderophore complexes are delivered to the microbial cell where the iron has to be released to become metabolically available. The release of iron from its external sources is a key process that allows its passage into the so-called labile cellular iron pool,11 from where intracellularly or extracellularly directed trafficking can take place. The main cellular trafficking routes of iron include its long-term storage in proteins or vacuolar compartments, assembly of redox cofactors like iron–sulfur (Fe/S) clusters or heme variants, and its utilization for various informational pathways. However, the limiting external or internal iron release rates from siderophores with moderate (Kf ∼ 1030 M−1) to extremely high (Kf ∼ 1050 M−1) affinities mainly define the flow of iron through these assimilatory pathways.
The release from siderophores can occur either at the cell surface in association with free iron uptake, or after cellular uptake of the ferric siderophore complex by ligand-specific transport systems. In each case, the following basic strategies are utilized to facilitate the removal of iron from its extracellular chelator: (i) the competition of iron coordination with ligand protonation (pH-dependent release), (ii) the hydrolysis of the siderophore backbone (hydrolytic release), and (iii) the reduction of the ferric ion center (reductive release). Both ligand protonation and hydrolytic release are only feasible in a minority of cases due to limitations in changing the intra- and extracellular pH milieus and because of structural requirements such as the introduction of rather unstable ester bonds into the siderophore scaffold. Thus, the majority of iron release processes are reductive, which bears the further advantage of re-utilization of the siderophore scaffold in contrast to a single utilization cycle during a destructive release. However, since the redox potentials of ferric siderophores correlate reciprocally to their formation constants in general (Fig. 1), a combination of release mechanisms is often of vital importance to allow an efficient reduction of high-affinity ferric siderophores. For these cases, highly adapted kinetic mechanisms are required that allow a short-distance electron transfer to the scaffolded ferric iron on the one hand, and they may be coupled on the other hand with an increase of the iron redox potential within the protein–siderophore complex or by additional means of iron complex destabilization. A number of siderophores which are associated with such advanced release mechanisms are produced by important human, animal and plant pathogens as key factors for their virulence development. Hence, a deeper understanding of the molecular mechanisms of iron release and assimilation mechanisms is not only essential for the general characterization of iron acquisition pathways, but also for the development of novel potent therapeutics for iron-dependent pathogen control.
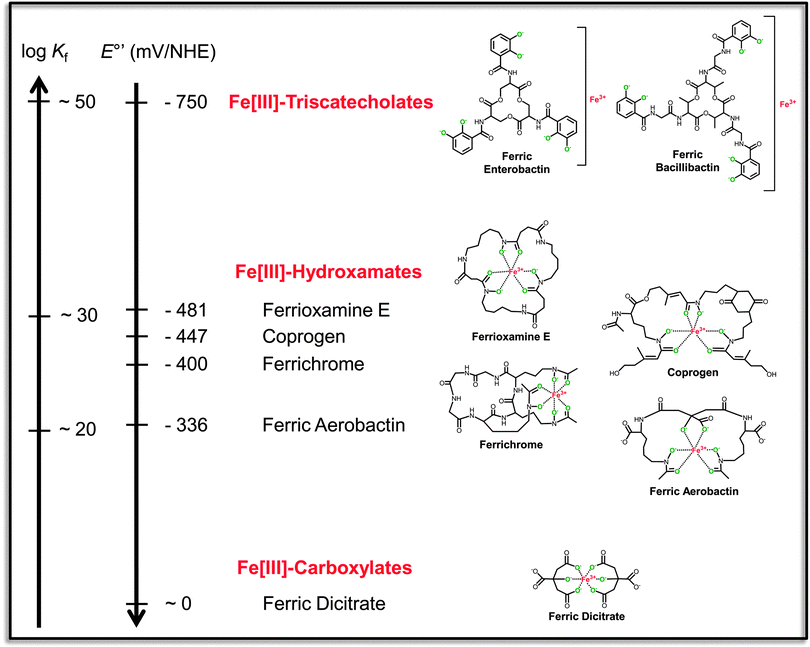 | ||
| Fig. 1 Reciprocally correlated scales of formation constants (Kf) and standard redox potentials at pH 7.0 (E°′) for representative ferric siderophores of different structural classes (indicated in red). The ferric complexes are shown in their hexa-liganded coordination modes with fully deprotonated donor atoms in green. The triscatecholate scaffolds contain representative trilactone tri-L-serine and tri-L-threonine backbones which are accessible to enzymatic hydrolysis. Redox potentials of the ferric complexes are given versus the normal hydrogen electrode (NHE). Affinity constants and redox potentials are taken from the indicated references.13,14,167–171 | ||
2 Extracellular reduction of mobilized iron
The assimilatory reduction of ferric iron complexes in the extracellular environment is common to the majority of microorganisms and is often coupled with high-affinity uptake of the released ferrous iron species. In general, microbes have established two main strategies for extracellular ferric siderophore reduction: (i) reduction mediated by cell surface associated metalloreductases in fungi, and (ii) reduction mediated by secreted redox compounds mainly in bacteria, but also in fungi.Iron reduction at the fungal plasma membrane has been best studied in S. cerevisiae that possesses a series of membrane-integrated ferric reductases (Fre) of the flavocytochrome superfamily. The four reductases Fre1p–Fre4p catalyze ferric siderophore reduction with broadly different substrate specificities and catalytic efficiencies, while the function of two additional homologs, Fre5p and Fre6p, has not been elucidated yet.12 The main reductase activities are provided by Fre1p and Fre2p that reduce Fe(III) bound to scaffolding ligands like citrate, desferrioxamine B, desferrichrome, desferri-triacetyl fusarinine C (TAFC) and rhodotorulic acid. The Fre3p reductase activity for these substrates was found to be ∼40-fold lower, and the Fre4p reductase only facilitated the utilization of iron bound to rhodotorulic acid.12–13 Furthermore, Fre1p and Fre2p were shown to reduce ferric enterobactin, which is remarkable due to the extremely low redox potential of −0.75 V that is associated with one of the highest biological Kf of 1049 M−1 of the free complex at pH 7.0.14,15 However, the reduction is supposed to be coupled with an extracellular acidification that is mediated by the Fre reductases at the same time.16 This in turn leads to a partial destabilization of the ferric triscatecholate complex due to an increased ligand protonation as well as an induced intrinsic switch from the catecholate to the salicylate coordination mode in the lower pH regime.17,18 Thus, the combination of reduction with cell surface acidification likely enhances the electron transfer efficiency to the iron complex. On the other hand, altered iron binding modes including pH-dependent coordination shifts can be of general physiological relevance with respect to complex recognition during further transport and release processes. The fungal cell surface reduction of ferric siderophores is tightly coupled with a high-affinity uptake of ferrous iron by the Fet3p–Ftr1p system. The high-affinity uptake efficiently removes the liberated Fe(II) species from the extracellular equilibrium and hence helps to increase the actual redox potentials of the reductively converted ferric complexes. For the purpose of uptake, the yeast transporter employs the oxygen-dependent multicopper ferroxidase Fet3p, which converts Fe(II) to Fe(III) prior to membrane translocation by the iron permease Ftr1p.19 Functionally related iron uptake systems in bacteria are EfeUOB and FetMP, which either oxidize or reduce free iron prior to its translocation; however, it is not known if they are also closely associated with extracellular ferric siderophore reduction.20–22
Bacteria, in contrast, have developed primarily indirect enzymatic strategies for extracellular ferric siderophore reduction by utilizing mobile electron carriers as redox compounds (Fig. 2). Especially the roles of reduced phenazines and flavins have been addressed in this context.23,24 In soil-dwelling bacteria like Pseudomonas chlororaphis or Shewanella oneidensis, phenazines were shown to mediate electron shuttling to alternative terminal acceptors such as mineralized ferric iron or organic ferric iron chelates, enabling iron mobilization in the reduced form.25,26 Thus, dissimilatory ferric iron reduction can be directly coupled with reductive iron assimilation by increasing the local bioavailability of ferrous iron. Further studies showed that this mechanism is also of importance for opportunistic pathogens such as P. aeruginosa, which produces phenazine derivatives such as phenazine-1-carboxylic acid (PCA) and 5-N-methyl-1-hydroxyphenazine (pyocyanin, PYO). PYO was shown to acquire iron from the human iron chelator transferrin, especially under low oxygen conditions.27 PCA was found to be important for virulence-associated biofilm formation during later stages of infection by neutralizing the activity of Fe(III)-binding host proteins such as conalbumin.28 Remarkably, the redox activity of PCA was found to be tightly coupled with FeoB-mediated high-affinity uptake of Fe(II). A recent study also showed metabolic transformations of P. aeruginosa phenazines by A. fumigatus, resulting in derivatives with alternative properties such as enhanced toxicities and the capability to induce fungal siderophore production.29
![Summary of basic iron transport and release pathways in Gram-negative bacteria. Fundamental transport systems for the main ferric siderophore classes as well as for transferrin- and heme-delivered iron are shown, with emphasis on the E. coli reference model regarding protein nomenclature. The iron transport routes consist of TonB/ExbBD-energized outer membrane receptors that deliver either the intact iron complexes or, as in the case of the transferrin-interacting system, extracellularly abstracted ferric iron into the periplasm, where specific binding proteins are in close association with the receptor complexes for rapid iron–ligand scavenging. The iron sources are delivered by the binding proteins to their specific cytosolic uptake systems, which mainly belong to the class of ABC-type transporters comprising integral membrane permease units that are associated with intracellular nucleotide binding domains that catalyze ATP hydrolysis. Alternatively, some complexes may undergo a periplasmic processing, especially in the case of ferric siderophores with lower iron-binding affinities such as hydroxamates and carboxylates, which can be substrates of periplasmic reductases (indicated by “?”). Ferrous iron that has been liberated in the periplasm as well as during extracellular reduction by mobile electron carriers such as flavins or phenazines can be taken up by high-affinity cytosolic transporters like FeoB that putatively employs GTPase activity as an energy source. On the other hand, ferric siderophore complexes which have been imported into the cytosol are subjected to diverse iron release pathways according to their intrinsic redox potentials. Ferric triscatecholates can either be directly reduced by triscatecholate-specific flavoenzymes such as YqjH or ViuB, belonging to the SIP (siderophore-interacting protein) family of ferric reductases, or can be hydrolyzed prior to reduction by specific esterases such as Fes if they contain cleavable trilactone backbones such as ferric enterobactin. Several ferric hydroxamates can be reduced according to their redox potentials by the loosely membrane-associated FhuF reductase that binds a [2Fe–2S] redox cofactor. Rather unspecific reductases that accept a broader spectrum of substrates including high-potential ferric chelates are represented by flavoenzymes such as Fpr. Exogenous flavin reductases like Fre primarily mediate the reduction of free diffusible flavins, which in turn can react inside or outside the cell with a high number of different substrates including ferric complexes with feasible redox potentials. In contrast, intact heme is cytosolically degraded by heme oxygenases, leading to biliverdin formation and ferrous iron release. The released iron species from all different sources may be further redox converted by yet unknown processes, and are supposed to interact with the labile intracellular iron pool comprising low affinity ligand interactions before they continue to pass through directed cellular trafficking routes for further assimilation.](/image/article/2013/MT/c2mt20193c/c2mt20193c-f2.gif) | ||
Fig. 2 Summary of basic iron transport and release pathways in Gram-negative bacteria. Fundamental transport systems for the main ferric siderophore classes as well as for transferrin- and heme-delivered iron are shown, with emphasis on the E. coli reference model regarding protein nomenclature. The iron transport routes consist of TonB/ExbBD-energized outer membrane receptors that deliver either the intact iron complexes or, as in the case of the transferrin-interacting system, extracellularly abstracted ferric iron into the periplasm, where specific binding proteins are in close association with the receptor complexes for rapid iron–ligand scavenging. The iron sources are delivered by the binding proteins to their specific cytosolic uptake systems, which mainly belong to the class of ABC-type transporters comprising integral membrane permease units that are associated with intracellular nucleotide binding domains that catalyze ATP hydrolysis. Alternatively, some complexes may undergo a periplasmic processing, especially in the case of ferric siderophores with lower iron-binding affinities such as hydroxamates and carboxylates, which can be substrates of periplasmic reductases (indicated by “ ?”). Ferrous iron that has been liberated in the periplasm as well as during extracellular reduction by mobile electron carriers such as flavins or phenazines can be taken up by high-affinity cytosolic transporters like FeoB that putatively employs GTPase activity as an energy source. On the other hand, ferric siderophore complexes which have been imported into the cytosol are subjected to diverse iron release pathways according to their intrinsic redox potentials. Ferric triscatecholates can either be directly reduced by triscatecholate-specific flavoenzymes such as YqjH or ViuB, belonging to the SIP (siderophore-interacting protein) family of ferric reductases, or can be hydrolyzed prior to reduction by specific esterases such as Fes if they contain cleavable trilactone backbones such as ferric enterobactin. Several ferric hydroxamates can be reduced according to their redox potentials by the loosely membrane-associated FhuF reductase that binds a [2Fe–2S] redox cofactor. Rather unspecific reductases that accept a broader spectrum of substrates including high-potential ferric chelates are represented by flavoenzymes such as Fpr. Exogenous flavin reductases like Fre primarily mediate the reduction of free diffusible flavins, which in turn can react inside or outside the cell with a high number of different substrates including ferric complexes with feasible redox potentials. In contrast, intact heme is cytosolically degraded by heme oxygenases, leading to biliverdin formation and ferrous iron release. The released iron species from all different sources may be further redox converted by yet unknown processes, and are supposed to interact with the labile intracellular iron pool comprising low affinity ligand interactions before they continue to pass through directed cellular trafficking routes for further assimilation. ?”). Ferrous iron that has been liberated in the periplasm as well as during extracellular reduction by mobile electron carriers such as flavins or phenazines can be taken up by high-affinity cytosolic transporters like FeoB that putatively employs GTPase activity as an energy source. On the other hand, ferric siderophore complexes which have been imported into the cytosol are subjected to diverse iron release pathways according to their intrinsic redox potentials. Ferric triscatecholates can either be directly reduced by triscatecholate-specific flavoenzymes such as YqjH or ViuB, belonging to the SIP (siderophore-interacting protein) family of ferric reductases, or can be hydrolyzed prior to reduction by specific esterases such as Fes if they contain cleavable trilactone backbones such as ferric enterobactin. Several ferric hydroxamates can be reduced according to their redox potentials by the loosely membrane-associated FhuF reductase that binds a [2Fe–2S] redox cofactor. Rather unspecific reductases that accept a broader spectrum of substrates including high-potential ferric chelates are represented by flavoenzymes such as Fpr. Exogenous flavin reductases like Fre primarily mediate the reduction of free diffusible flavins, which in turn can react inside or outside the cell with a high number of different substrates including ferric complexes with feasible redox potentials. In contrast, intact heme is cytosolically degraded by heme oxygenases, leading to biliverdin formation and ferrous iron release. The released iron species from all different sources may be further redox converted by yet unknown processes, and are supposed to interact with the labile intracellular iron pool comprising low affinity ligand interactions before they continue to pass through directed cellular trafficking routes for further assimilation. | ||
Flavins represent another important group of electron-shuttling compounds in microbes and also higher plants and can also fulfill a functional double role in dissimilatory and assimilatory iron reduction. They form unusually stable complexes with iron in remarkable contrast to further transition metals,30,31 which could support the possibility of a metal-selective inner-sphere electron transfer by these redox-active ligands. Flavin-dependent iron acquisition was shown in Helicobacter pylori that secretes riboflavin to reduce Fe(III) stored in ferritins.32 Similar processes of extracellular iron reduction and the possibility of enhanced iron mobilization by flavins were observed in Shewanella,23Pichia,33 as well as sugar beet and sunflower roots.34 Notably, a great number of so-called “extracellular ferric reductases” in bacteria, which are either secreted into the extracellular milieu or are membrane-bound, belong to the class of NAD(P)H:flavin oxidoreductases (or commonly “flavin reductases”) that can generate either FADH2 or FMNH2 as agents for single electron transfer.13,35 The reduced flavins dissociate in most of the reported cases from the enzymes and hence fulfill an indirect role in unspecific ferric iron reduction. With standard midpoint potentials around −0.2 V, the free flavins can reduce most iron oxides, soluble forms of Fe(III) as well as organic ferric iron chelates including ferric siderophores whose redox potentials are within this range (see Fig. 1).
Another remarkable and wide-spread mechanism of extracellular ferric siderophore reduction is associated with photoreactive siderophores that contain α-hydroxy carboxylates, which are produced by a number of marine bacteria.36,37 These chemical groups permit a sunlight-driven photoreduction of the ferric iron center through the ligand-to-metal charge transfer in ferric siderophores.
3 Periplasmic iron trafficking
Dependent on the nature of the utilized iron source, several specific pathways in Gram-negative bacteria allow trafficking or processing of iron in the periplasm prior to its transport into the cytosol (Fig. 2). Iron acquisition from non-heme iron sources like lactoferrin or transferrin by several Gram-negative pathogens such as Neisseria or Haemophilus is essentially dependent on the periplasmic ferric ion binding protein FbpA, a so-called “bacterial transferrin” due to remarkable structural and functional similarities to mammalian transferrin.38,39 The TonB-dependent surface transferrin receptor complex TbpAB binds transferrin, removes the iron and transports it across the outer membrane to the periplasmic side, where it is bound by the FbpA protein.40–42 The TbpA receptor preferentially interacts with apo-FbpA at the inner side of the outer membrane and releases holo-FbpA,43 which shuttles the iron to the inner membrane transporter FbpBC for cytosolic uptake.44 The iron binding affinity of FbpA is comparable to that of transferrin with a log Kf of about 20 at physiological pH.45 The FbpABC transport system is also involved in TonB-independent iron acquisition from xenosiderophores in Neisseria.46 The initial anchoring of ferric iron in the FbpA binding site depends on a twin-tyrosine motif, whose double mutation is deleterious to the iron sequestration activity.47 Binding of a “synergistic” phosphate anion that completes the iron co-ordination shell of FbpA was further shown to play a crucial role in iron sequestration and release in vitro;48,49 however, mutants of the anion binding site were still capable of mediating iron acquisition in vivo.50Iron-charged siderophores which are transported across the outer membrane through TonB-energized receptors are usually readily captured by their cognate periplasmic binding proteins with dissociation constants (KD's) in the micro- to nanomolar range. Binding proteins like FhuD or BtuF were shown to interact with the TonB part of the outer membrane receptor/transporter complex, their binding sites supposedly facing the periplasmic side of the receptor lumen.51,52 These observations led to the hypothesis of an unidirectional ligand transport through the periplasm to the respective inner membrane ATP-binding-cassette (ABC) transporter that interacts via salt bridges with the holo-form of the binding protein for ligand transfer.53,54 Such a quasi-chaperoned trafficking of most ferric siderophores through the periplasm may reflect that rather few processing mechanisms of siderophores seem to have developed in this compartment. One example of such a periplasmic processing activity is the trilactone esterase IroE, which hydrolyzes triscatecholate siderophores like salmochelin and enterobactin.55,56 Similar to cytosolic trilactone hydrolases, IroE belongs to the α/β-hydrolase superfamily, but it lacks an N-terminal lid region and possesses an atypical catalytic diad comprising only the reactive serine and a conserved histidine.57 In contrast to its cytosolic counterparts, its activity has been mainly related with the single hydrolysis (linearization) of apo-siderophores prior to their secretion, rather than with the complete hydrolysis of ferric siderophores.58–60 However, some groups of proteobacteria like the epsilon subdivision possess only IroE-like hydrolases, whose precise role in the corresponding iron acquisition pathway(s) has not been addressed yet. In addition to hydrolysis, only a marginal number of reports exist about periplasmic ferric siderophore reduction. Such activities have been detected for substrates like ferric citrate in Legionella pneumophila and Vibrio vulnificus,61,62 and for ferripyochelin in Pseudomonas aeruginosa.63 The involved enzymes prefer NAD(P)H or glutathione as reductants.
Further, no evidence about iron release from heme iron sources in the periplasm exists. In contrast to a report about putative activities of periplasmic and cytoplasmic heme dechelation,64 it was shown that the addressed enzymes EfeB and YfeX are rather heme-dependent peroxidases involved in iron transport and protoporphyrinogen oxidation, respectively.65 Heme from extracellular sources is usually not processed in the periplasm, but transported into the cytosol via specific systems that are functionally similar to those of the ferric siderophore transport routes described above.66 If heme is abstracted from host hemoproteins such as hemopexin or methemoglobin, specific uptake pathways are required involving hemophore-mediated transport in many Gram-negative pathogens,5 or cell surface-anchored components such as in the Isd systems of Gram-positive pathogens, that contain high-affinity NEAT domains for peripheral protein binding, heme capturing and directed transfer to the cytosolic importer.67–69
4 Cytosolic iron release
Iron-charged siderophores which have passed the cytoplasmic membrane undergo enzymatic conversions that are essential for iron removal, especially from high-affinity complexes. These reactions are essential in order to allow a subsequent intracellular iron trafficking. The internalized complexes are in part subjected to scaffold backbone hydrolysis or are reduced by different types of oxidoreductases, either directly or after the hydrolytic processing has taken place (Fig. 2).4.1 Ferric siderophore hydrolases
Enzymes that cleave the trilactone rings of ferric siderophores generally belong to the superfamily of α/β-hydrolases. They generate ferric ligand species of higher molecular stoichiometry, which decreases entropy and hence stability of these iron complexes. The release of iron may then occur by a direct competition with ligands of ferric iron binding sites, and can be further enhanced by subsequent complex reductions as discussed below.
E. coli and its close relatives possess the best studied cytosolic siderophore esterases, Fes and IroD, which preferentially hydrolyze the cyclotrimeric scaffolds of ferric enterobactin and its glucosylated derivatives called salmochelins, respectively.55,56 IroD is specifically associated with the iroA gene cluster that contributes to virulence development.70 A further triscatecholate–trilactone hydrolase is the ferric bacillibactin esterase BesA that is conserved in Bacillus spp. and in paenibactin-producing Paenibacillus.71 BesA was found to hydrolyze both tri-L-threonine and tri-L-serine backbones which are present in bacillibactin and enterobactin, respectively, while the Fes esterase cleaves only the tri-L-serine backbone.72,73 The trilactone hydrolases usually possess a conserved Gx![[S with combining low line]](https://www.rsc.org/images/entities/b_char_0053_0332.gif) xG motif that is characteristic for serine esterases and part of a catalytic triad consisting of serine-histidine-glutamate/aspartate. The crystal structures of Shigella flexneri and Salmonella typhimurium Fes (PDB entries 3C87 and 3MGA), and Bacillus cereus BesA (PDB entry 2QM0) further show prominent N-terminal lid domains. At the interfaces of the lid domains and the catalytic domains that bear the reactive serine, several conserved aromatic residues seem to form binding sites that are arranged as a mirror image to the circular plane of the ligand aryl subunits and may hence contribute to hydrophobic interactions. However, the exact molecular mechanisms for substrate recognition as well as for the substrate-specific formations of partial or complete hydrolysis products remain unclear so far. Due to protein sequence analyses, IroD was proposed to be structurally very similar to Fes including the presence of an N-terminal lid domain as well as a catalytic triad.57
xG motif that is characteristic for serine esterases and part of a catalytic triad consisting of serine-histidine-glutamate/aspartate. The crystal structures of Shigella flexneri and Salmonella typhimurium Fes (PDB entries 3C87 and 3MGA), and Bacillus cereus BesA (PDB entry 2QM0) further show prominent N-terminal lid domains. At the interfaces of the lid domains and the catalytic domains that bear the reactive serine, several conserved aromatic residues seem to form binding sites that are arranged as a mirror image to the circular plane of the ligand aryl subunits and may hence contribute to hydrophobic interactions. However, the exact molecular mechanisms for substrate recognition as well as for the substrate-specific formations of partial or complete hydrolysis products remain unclear so far. Due to protein sequence analyses, IroD was proposed to be structurally very similar to Fes including the presence of an N-terminal lid domain as well as a catalytic triad.57
Further biosynthesis gene clusters for putative triscatecholate–trilactone siderophores such as griseobactin from Streptomyces74 or trichrysobactin from Dickeya75 have been identified recently, which are in close association with genes encoding α/β-hydrolases like GriB or CbsH. These hydrolases are closely related to Fes, but they have not been shown yet to cleave trilactones and may possess further hydrolytic activities as in the case of CbsH.74,76 Trivanchrobactin from Vibrio has been isolated as a linear trimer, which could potentially originate from a cyclic trilactone scaffold.77 Also in this case, Fes-homologs such as VabH have been identified and found to be involved in vanchrobactin-mediated iron assimilation.78
Trilactone siderophores with non-catecholic ligands are represented by several trishydroxamates such as fusarinine C (fusigen), TAFC, or neurosporin which are produced and utilized as iron sources by many fungi.79 The IroE-like trilactone esterase EstB was recently identified and found to hydrolyze ferric fusarinine complexes in Aspergillus.80 After EstB-cleavage of fusigen or TAFC, a part of the released iron is usually transferred into the cell vacuole or sequestered by intracellular siderophore compounds for further storage.81–82 Similar hydrolytic release mechanisms can be expected for ferric neurosporin and its bacterial counterpart ferric vicibactin that is produced by Rhizobia.83
4.2 Ferric siderophore reductases
In contrast to siderophore hydrolases, ferric siderophore reductases generate low-affinity ferrous iron complexes without changing the complex stoichiometry. The release of ferrous iron is facilitated by an increased kinetic exchange with the aqueous environment and with iron binding sites of equal or higher affinities. While siderophore hydrolases are rather substrate specific, there are broad variations of substrate specificities among ferric siderophore reductases. Generally, a higher specificity can be associated with an increasing efficiency of iron reduction in low-potential (high-affinity) ferric siderophore complexes.Other types of ferric reductases are independent of exogenous flavin substrates, as they transfer electrons via intrinsically bound flavin cofactors such as E. coli flavohemoglobin Hmp,92 nitroreductase NfnB and ferredoxin–NADP+ reductase Fpr,93Paracoccus FerB,88 or the archaeal ferric reductase FeR from Archaeoglobus fulgidus.94 Generally, these flavoenzymes can act on a broad set of substrates and are often not specifically associated with assimilatory iron reduction. The reaction mechanism for these ferric reductases is assumed to be rather of the nonsequential (“Ping-Pong”) type,13 which has also been suggested for the iron cofactor-directed flow of electrons in ferredoxin–NADP+ reductases.95
A second example is the homologous FchR reductase that was found in the Gram-positive extremophile Bacillus halodurans where it is associated with a ferric schizokinen uptake system, but accepts several further ferric hydroxamates as well as ferric dicitrate as its substrates.98 The midpoint potential of the FchR cluster is ∼ −0.35 V, and its close relationship with FhuF suggests a similar mechanistic mode of action. Electron transfer onto FchR in vitro is possible via a regenerative ferredoxin transfer system. Determination of Km and KD values for several ferric complexes revealed the highest substrate binding affinities for low-potential ferric hydroxamates, while ferric dicitrate was bound with the lowest affinity to the enzyme. The low-potential substrates showed saturated reaction kinetics already at very low concentrations, likely due to rate limitations of electron transfer. Thus, the effective range of electron transfer through the intrinsic Fe/S cofactor basically determines the substrate-dependent turnover rate, hence leading to redox-controlled substrate spectrum. However, the tight binding of several low-potential substrates suggested a mechanism which allows a significant increase of the electron transfer efficiency even onto substrates whose free redox potentials are well below the effective transfer range of the enzyme cofactor. Thus, the actual potentials of both the cofactor and the ferric substrates in the enzyme–substrate complex are of critical importance in these cases.
Regarding the reductive mechanism, an inner-sphere electron transfer from the FchR cofactor to the ferric iron center of the substrate ligand was found to be unlikely since no symmetry changes of the reduced cofactor occurred in the presence of preferred substrates. The general question if a direct inner-sphere electron transfer between cofactor and metal or an outer-sphere transfer via redox-active ligand(s) is favored during enzymatic ferric siderophore reductions has to be addressed in more detail. An outer-sphere transfer might generally be more likely due to the slow ligand exchange rates at the metal center,10 which would cause strong catalytic rate limitations in addition to those limitations that are caused by very low substrate redox potentials. An enzymatic outer-sphere electron transfer may possibly exploit the siderophore-specific ligand-to-metal charge-transfer that is mediated via the inner shell of ligand donor atoms.
A further question that was addressed while studying the FchR kinetic was the influence of product scavengers such as ferrous iron binding apo-proteins. According to the Nernst equation, the actual standard redox potentials of the ferric siderophore substrates would increase in the presence of efficient Fe(II) scavengers which remove the reduced metal ion from the equilibrium by sequestration.99 An increased catalytic efficiency of FchR was indeed observed in the presence of a transition metal binding Fe/S scaffold protein.98 Thus, redox potential shifts due to the presence of efficient iron sinks can be seen as a further indirect mechanism to overcome rate limitations and hence to increase the redox capacities of ferric siderophore reductases.
With respect to the development of counteracting strategies against microbial ferric iron assimilation, siderophores charged with redox-inert metals such as Ga(III)-desferrioxamine were found to be specific inhibitors of FchR and reduced its catalytic activity significantly with inhibition constants in the lower micromolar range.98 The inhibition effect was also observed in bacterial culture and was dependent on the presence of the reductase.
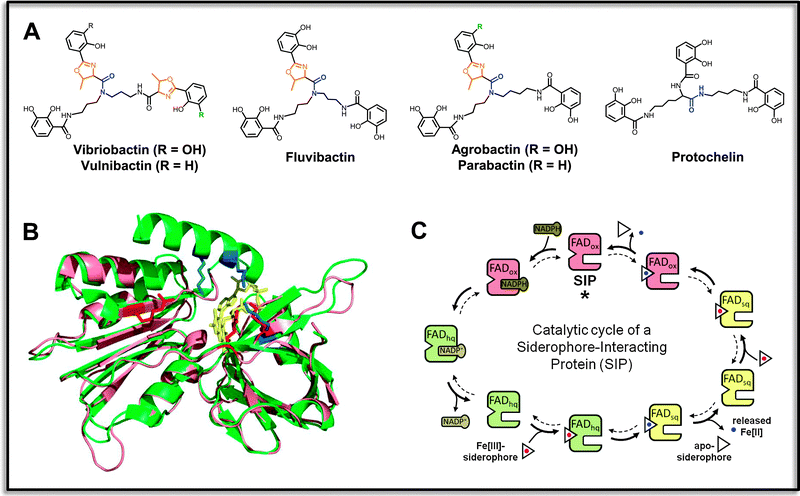 | ||
| Fig. 3 Specific reductases for ferric triscatecholate siderophores. (A) Examples of aryl-capped siderophores with non-hydrolyzable backbones that contain stable amide-linkages (indicated in blue), and frequent oxazoline units adjacent to the aryl groups (shown in orange). Vibriobactin, vulnibactin and fluvibactin are produced by human-pathogenic Vibrio species V. cholerae, V. vulnificus and V. fluvialis, respectively, agrobactin is produced by the plant pathogen Agrobacterium tumefaciens, parabactin by the ecologically important soil denitrifier Paracoccus denitrificans, and protochelin by nitrogen-fixing Azotobacter vinelandii. (B) Superposition of a structural model of ferric triscatecholate reductase YqjH from E. coli (salmon) and the ViuB structure (PDB entry 2GPJ) from S. putrefaciens (green). Conserved basic residues forming part of the ligand binding sites are depicted in red for YqjH and in blue for ViuB. The FAD redox cofactor is shown in yellow. (C) Kinetic mechanism of the YqjH reductase. The enzyme undergoes a double-displacement-type (“Ping-Pong”) reaction, during which the oxidized FAD cofactor (FADox) is at first reduced by NADPH, leading to an enzyme-bound flavohydroquinone (FADhq). The electron-charged enzymes then reduce successively two ferric siderophore species, resulting in the formation of ferrous iron products and the enzyme-bound flavosemiquinone (FADsq) and oxidized FAD species of the resting state, which concludes the cycle. Illustrations are partly adopted from the indicated reference.102 | ||
Additional possibilities for a redox potential increase of ferric triscatecholates may include symmetry changes in their iron coordination modes as well as the involvement of softer ligand donor atoms, especially in the cases of non-hydrolyzable triscatecholate scaffolds. Interestingly, several of these siderophores contain one or two oxazoline rings adjacent to their peripheral aryl-caps (Fig. 3A). In the case of ferric vibriobactin in complex with the binding protein ViuP, an iron coordination was observed in which the iron was liganded by five of the arylic hydroxo groups and by one nitrogen of the second oxazoline ring.109 It is not clear if this binding mode is primarily adopted in particular protein binding sites or if it is also preferred in free solution;110,111 however, such oxazoline-dependent coordination modes in aryl-capped siderophores might play an interesting role during reductive iron release, especially from those siderophores that are equipped with oxazoline rings adjacent to salicylate caps (Fig. 3A). Further, pH-dependent switches between the catecholate and salicylate coordination modes of the iron-binding arylamide groups could contribute to a facilitated reduction, especially in the case of triscatecholates like protochelin possessing neither a hydrolyzable trilactone backbone nor any oxazoline rings. Thus, the mechanistic details of reductive iron release within this group of ferric siderophore reductases have to be studied further, not least since several of their siderophore substrates are important factors for virulence development in a variety of pathogens. A deeper understanding of their iron release mechanisms could lead to the definition of novel target-specific inhibition strategies.
5 Intracellular pathways for iron trafficking and assimilation
The relevance of an efficient cellular iron homeostasis is best reflected by the number of enzymes and processes that require iron as an essential cofactor. Several hundreds of different proteins in a regular cell may require iron binding in various forms, including single iron sites, diiron-oxo centers, heme and siroheme groups, Fe/S clusters or mixed metal sites such as in [NiFe]-hydrogenases or purple acid phosphatases, in which iron can adopt a broad range of oxidation states or may operate as redox factor or activating Lewis acid.112–114 The main intracellular iron channeling routes include trafficking to storage and cofactor assembly systems as well as the directed transfer of the iron cofactors to their target apo-proteins. It is important to note that ferrous iron is relevant for most of these processes, in particular for transfer into ferritins, for de novo heme and iron–sulfur cluster biogenesis,115–118 as well as for intracellular iron sensing and associated regulatory processes.7,119,120As described above, most of the iron is released from siderophore complexes by reduction and is hence available in the ferrous state. Further important iron sources like heme scaffolds can be degraded in the cytosol by heme oxygenases which is accompanied by ferrous iron release,121,122 or could be used directly as cofactors for incorporation into target proteins.123 Iron that is released or imported in the ferric state may be subjected to a number of yet unknown reductive events before entering the main cytoplasmic trafficking routes. It can be assumed that cytosolic reduction of ferric iron also occurs in fungi, either after its cellular import via the Fet3p–Ftr1p system or after its transfer from vacuolar storage sites into the cytosol by homologous transporters like Fet5p–Fth1p in Saccharomyces.124 Another process of intracellular iron reduction could be associated with the release from cytosolic storage compounds such as hyphal ferricrocin or conidial hydroxyferricrocin in Aspergillus.81,82,125
Prior to a directed cytosolic trafficking, imported or released iron may become part of the “labile iron pool” that putatively comprises low-affinity interactions with diverse protein ligands, metabolic intermediates or even phosphorylated sugar compounds.11,126 A direct route from this transient pool can lead to the long-term storage of intracellular iron in the form of a rather inert mineral core in ferritins. Active Fe(II) uptake by bacterial ferritins is generally assumed, while the mechanism of mineral core formation depends on the ferritin subfamily type and can further be coupled with intracellular ROS detoxification.127,128 While a ligand-mediated Fe(II) delivery to bacterial ferritins is not known, the delivery of Fe(II) to mammalian ferritins depends on iron chaperones like PCBP1 and PCBP2, which are also involved in a direct metallation of metabolic target enzymes such as Fe(II)-/2-oxoglutarate-dependent dioxygenases.129,130 The release process of iron from the ferritin mineral core is generally reductive, and may require external reductases. In heme-containing bacterioferritins, the electron transfer processes for both iron core formation and for core reduction appear to be strictly dependent on the intrinsically bound heme cofactors.131,132
It is not yet clear whether the trafficking of iron to cofactor assembly systems principally requires a mediating ligand transfer; however, several mobile scaffolding compounds are known that bind iron with moderate affinity and contribute to its directed trafficking. In eukaryotes, such a ligand-based transport takes place through the bridging [2Fe–2S] centers of monothiol glutaredoxins like Grx3/4, which form independently of an Fe/S biogenesis machinery and may instead deliver iron to components of the cytosolic Fe/S protein assembly (CIA) system.133 In contrast, a targeted iron delivery to mitochondrial Fe/S and heme biosynthesis systems is still speculative. The role of the mitochondrial protein frataxin, which might act as a general iron chaperone as well as a possible iron donor for directed cofactor assembly, is under discussion.115 Frataxin-like proteins are conserved from human to bacteria and possess iron-binding acidic surface regions with KD's in the lower micromolar range.134 While frataxin-mediated iron delivery to assembly components such as the Fe/S scaffold IscU or porphyrin-metallating ferrochelatase has been demonstrated in vitro, its in vivo function for iron trafficking has to be further elucidated.135,136 The role of frataxins to act as allosteric effectors of cysteine desulfurase activities in eukaryotes and in several bacteria including E. coli seems to be established especially in association with ISC-type Fe/S cluster assembly systems.137–139 However, further structural frataxin homologs such as B. subtilis Fra, which can bind both Fe(II) and Fe(III) in similar stoichiometry, seem to be more generally involved in cellular iron homeostasis.140,141 Indeed, B. subtilis Fra appears to be the missing link for iron delivery in a multi-step channeling pathway that includes components of the SUF-type Fe/S assembly machinery (Fig. 4). Nevertheless, several molecular details of the directed iron transfer processes regarding ligand- and component-specific interactions as well as binding site occupations due to possible affinity gradients between different ligand environments remain to be shown.
![A proposed iron channeling pathway in Bacillus subtilis leading to aconitase maturation. The pathway starts with extracellular iron mobilization by the high-affinity ferric iron chelator bacillibactin (Fe-BB; log Kf = 47.6),172 and proceeds via high-affinity uptake of the ferric bacillibactin complex by the FeuABC-YusV transporter (KD of the FeuA/Fe-BB complex which is 27 nM;173 Fe-BB and FeuA/Fe-BB structures are from PDB entry 2WHY105). Cytosolic hydrolysis of the ferric siderophore complex is catalyzed by the BesA esterase (Km(obs) for Fe-BB is ∼0.5 μM;72 the B. cereus BesA structure from PDB entry 2QM0 [green] is shown in a model with the Fe-BB ligand [yellow] attached to the three conserved tryptophan residues [blue] in the lid domain adjacent to the catalytically active serine [red]). Enzymatic hydrolysis leads to the formation of a ferric (2,3-dihydroxybenzoate–glycine–threonine)3 complex, from which iron release is facilitated by ligand exchange with the “labile iron pool” or, alternatively, by interaction with the iron binding sites of apo-frataxin containing high-affinity and lower affinity sites with KD's of 0.1 μM and 2.4 μM for ferrous iron, respectively (B. subtilis frataxin [Fra; formerly YdhG] from PDB entry 2OC6 is shown [salmon] with highlighted conserved acidic surface regions possibly involved in metal binding [blue]).141 A possible step of ferric iron reduction (“?”) might occur prior to iron-charging of frataxin or during the next stage, in which holo-frataxin delivers the bound metal ions to the SUF system for Fe/S biogenesis that includes the cysteine desulfurase SufS and the scaffold protein SufU (SufU from PDB entry 2AZH is depicted [blue] with its three conserved cysteines [green; –SH in yellow] that coordinate a Zn2+ ion in vitro [grey] and form the putative Fe/S binding site in vivo; a flexible loop region that contains the critical Cys41 is possibly involved in interaction with the catalytic center of SufS during sulfide transfer [red]).151,153 During Fe/S cluster formation on SufU, SufS activity is enhanced both by SufU and frataxin (the SufU-dependent Km(app) of SufS is ∼2.6 μM; the specific activity of SufS is ∼20-fold enhanced in the presence of 20-fold molar excess of SufU,152 and still several fold further in the additional presence of Fra [personal communication, A. Albrecht]). During the transfer of the assembled cluster to the apo-aconitase target enzyme (shown is a structure of a homologous holo-aconitase from PDB entry 1FGH [grey] with bound 4Fe–4S cluster [red-yellow]), the in vitro targeting efficiency in the presence of a tripartite holo-Fra/SufSU reconstitution system is about 80% higher after 20 min of transfer than in the presence of the same amounts of free iron and sulfide.140](/image/article/2013/MT/c2mt20193c/c2mt20193c-f4.gif) | ||
Fig. 4 A proposed iron channeling pathway in Bacillus subtilis leading to aconitase maturation. The pathway starts with extracellular iron mobilization by the high-affinity ferric iron chelator bacillibactin (Fe-BB; log Kf = 47.6),172 and proceeds via high-affinity uptake of the ferric bacillibactin complex by the FeuABC-YusV transporter (KD of the FeuA/Fe-BB complex which is 27 nM;173 Fe-BB and FeuA/Fe-BB structures are from PDB entry 2WHY105). Cytosolic hydrolysis of the ferric siderophore complex is catalyzed by the BesA esterase (Km(obs) for Fe-BB is ∼0.5 μM;72 the B. cereus BesA structure from PDB entry 2QM0 [green] is shown in a model with the Fe-BB ligand [yellow] attached to the three conserved tryptophan residues [blue] in the lid domain adjacent to the catalytically active serine [red]). Enzymatic hydrolysis leads to the formation of a ferric (2,3-dihydroxybenzoate–glycine–threonine)3 complex, from which iron release is facilitated by ligand exchange with the “labile iron pool” or, alternatively, by interaction with the iron binding sites of apo-frataxin containing high-affinity and lower affinity sites with KD's of 0.1 μM and 2.4 μM for ferrous iron, respectively (B. subtilis frataxin [Fra; formerly YdhG] from PDB entry 2OC6 is shown [salmon] with highlighted conserved acidic surface regions possibly involved in metal binding [blue]).141 A possible step of ferric iron reduction (“ ?”) might occur prior to iron-charging of frataxin or during the next stage, in which holo-frataxin delivers the bound metal ions to the SUF system for Fe/S biogenesis that includes the cysteine desulfurase SufS and the scaffold protein SufU (SufU from PDB entry 2AZH is depicted [blue] with its three conserved cysteines [green; –SH in yellow] that coordinate a Zn2+ ion in vitro [grey] and form the putative Fe/S binding site in vivo; a flexible loop region that contains the critical Cys41 is possibly involved in interaction with the catalytic center of SufS during sulfide transfer [red]).151,153 During Fe/S cluster formation on SufU, SufS activity is enhanced both by SufU and frataxin (the SufU-dependent Km(app) of SufS is ∼2.6 μM; the specific activity of SufS is ∼20-fold enhanced in the presence of 20-fold molar excess of SufU,152 and still several fold further in the additional presence of Fra [personal communication, A. Albrecht]). During the transfer of the assembled cluster to the apo-aconitase target enzyme (shown is a structure of a homologous holo-aconitase from PDB entry 1FGH [grey] with bound 4Fe–4S cluster [red-yellow]), the in vitro targeting efficiency in the presence of a tripartite holo-Fra/SufSU reconstitution system is about 80% higher after 20 min of transfer than in the presence of the same amounts of free iron and sulfide.140 ?”) might occur prior to iron-charging of frataxin or during the next stage, in which holo-frataxin delivers the bound metal ions to the SUF system for Fe/S biogenesis that includes the cysteine desulfurase SufS and the scaffold protein SufU (SufU from PDB entry 2AZH is depicted [blue] with its three conserved cysteines [green; –SH in yellow] that coordinate a Zn2+ ion in vitro [grey] and form the putative Fe/S binding site in vivo; a flexible loop region that contains the critical Cys41 is possibly involved in interaction with the catalytic center of SufS during sulfide transfer [red]).151,153 During Fe/S cluster formation on SufU, SufS activity is enhanced both by SufU and frataxin (the SufU-dependent Km(app) of SufS is ∼2.6 μM; the specific activity of SufS is ∼20-fold enhanced in the presence of 20-fold molar excess of SufU,152 and still several fold further in the additional presence of Fra [personal communication, A. Albrecht]). During the transfer of the assembled cluster to the apo-aconitase target enzyme (shown is a structure of a homologous holo-aconitase from PDB entry 1FGH [grey] with bound 4Fe–4S cluster [red-yellow]), the in vitro targeting efficiency in the presence of a tripartite holo-Fra/SufSU reconstitution system is about 80% higher after 20 min of transfer than in the presence of the same amounts of free iron and sulfide.140 | ||
The Fe/S assembly on ISC-scaffolds in bacteria and mitochondria is further dependent on ferredoxin, which might be involved in several reductive events such as sulfide generation from cysteine desulfurase-delivered sulfur, reduction of ferric iron sources such as Fe(III)-charged frataxin, and may also permit a reductive [2Fe–2S]2+ to [4Fe–4S]2+ coupling during the later stages of cluster maturation.142,143 The cluster transfer to acceptor proteins in mitochondria is mediated by the monothiol glutaredoxin Grx5 that takes the [2Fe–2S] cluster possibly in complex with glutathione from the Isu assembly scaffold, a process which is enhanced by an ATP-hydrolyzing chaperone system.144,145 The generation of [4Fe–4S] clusters takes place in association with late-acting ISC targeting factors, which also allow the insertion of the cluster into specific target proteins.115 Similarly, monothiol glutaredoxins like GrxS14/S16 in chloroplasts take Fe/S clusters from Nfu-type or Suf-type assembly scaffolds and transfer them to target proteins in this cellular compartment.146
In contrast, bacterial Fe/S cluster transfer is thought to occur via a transient complex formation between a scaffold protein carrying the surface exposed cluster and a target apo-protein by cysteine–thiol ligand exchanges. The transfer reaction and possibly also the subsequent complex dissociation can be enhanced by the ATP-dependent chaperone/co-chaperone system HscA/HscB in ISC-type systems.116,147 In the case of bacterial SUF biogenesis systems with A-type scaffolds,148 primary cluster building is thought to take place on the SufBCD complex which appears to use FADH2 instead of ferredoxin for the reductive assembly.149 The generated clusters are then transferred either directly from SufBCD or through interacting carrier proteins like SufA to their targets.150 SUF systems with U-type scaffolds are mainly present in Gram-positive clades such as the Firmicutes.151 Here, cluster assembly and transfer can be mediated by the SufU scaffold protein in vitro, but it is not clear which function(s) SufU fulfills in vivo, since it has functional relations to both the sulfur carrier SufE and the Fe/S carrier SufA of the distinct Gram-negative SUF systems.152,153
During the biogenesis of heme, a controlled cofactor trafficking is of similar importance for subsequent side-chain modifications and protein targeting. The Fe(II) redox state of ferrochelatase-derived heme b seems to be strictly required for farnesylation reactions that lead to heme o or heme a, as well as for thioether bond formation during cytochrome c maturation.154,155 For the latter process, heme b has to leave the bacterial cytosol or the mitochondrial matrix in eukaryotes. It possibly maintains its ferrous state after crossing the inner mitochondrial membrane, since it requires only the cytochrome c heme lyase (CCHL) for apocytochrome attachment in the intermembrane space.156,157 In contrast, the control of the redox state is more complicated in bacteria. One prominent bacterial heme trafficking pathway employs the CcsAB heme channel, which translocates heme across the cytoplasmic membrane and keeps the ferrous state at the extracellular site by attachment to conserved histidines ligand. Another heme trafficking pathway that is mainly found in α- and γ-Proteobacteria and Archaea utilizes the multilayered Ccm system, in which the membrane-associated CcmABCD complex translocates ferrous heme and delivers it to the periplasmic heme chaperone CcmE.158,159 The membrane-attached chaperone forms a covalent heme adduct which results in cofactor oxidation, and transfers the ferric heme to the cytochrome c synthetase complex CcmF/H, which re-reduces the heme prior to its ligation to the target protein.158,160 In addition to periplasmic heme chaperoning, an ankyrin-containing protein in Campylobacter is proposed to act as a heme chaperone for cytosolic heme trafficking and targeting during intracellular catalase maturation.161
Relatively little is known about directed insertion mechanisms of iron into mononuclear sites such as in non-heme iron dioxygenases or into binuclear sites like diiron-oxo centers associated with ferroxidase activity. In humans, the PCBP chaperones fulfill a role in the maturation of proteins containing those kinds of binding sites.129 In contrast, the situation in microbes is still unclear, and it will be exciting to explore if specific components or rather unspecific cytosolic ligands are involved, and further if gradients of increasing binding affinities are necessary to acquire iron from ligands or chaperones with lower affinities, a mechanism that has been described as a main driving force of directed copper trafficking.162 On the other hand, iron sites containing unusual ligands such as CN− and CO in hydrogenases require highly complex batteries of auxiliary proteins for ligand synthesis, delivery and metal coordination such as the Hyp machinery for [NiFe]-hydrogenase maturation.163–166 Interestingly, the E. coli YqjH ferric siderophore reductase is regulated in response to iron and nickel, which indicates that reductive iron assimilation interacts already at an early stage with nickel homeostasis with yet unknown implications for mixed metal site maturation.106 Altogether, various iron trafficking routes and cofactor assembly systems have been defined today, but the knowledge of cellular iron channeling processes in general is still rather fragmentary. Components which may act as intermediate ligands for trafficking between different protein units and cellular compartments are yet to be identified or further characterized, as well as factors and mechanisms that determine the sequences and directions of the complex trafficking pathways. Further investigations may also comprise the mechanisms of communication between intracellular iron pools as well as different iron cofactors and the various sensor proteins that bind either ionic iron, Fe/S clusters or heme in order to regulate the expression of genes associated with cellular iron homeostasis.
6 Conclusions
Understanding the molecular mechanisms of microbial iron assimilation remains a primary task in light of the vast complexity of iron-driven redox and sensing processes in cellular systems. Bacterial and fungal model organisms, and among them a number of important pathogens, have been employed to establish the basic principles of diverse iron homeostasis strategies. The next layer of studies in this field should entail the various interconnections between the involved system elements, including a steady advancement of the still elementary knowledge of cellular iron trafficking processes. Further in-depth investigations of already known and yet unknown iron acquisition and assimilation components and their functions at both the molecular and the global cellular level are expected to bring about a positive impact on the exploitation of novel iron-dependent targets and the development of novel therapeutic strategies for the purpose of a pathway-specific pathogen defense.Acknowledgements
The Center for Synthetic Microbiology (SYNMIKRO) in Marburg is acknowledged for the current project funding.References
- J. R. Lloyd, FEMS Microbiol. Rev., 2003, 27, 411 CrossRef CAS.
- S. C. Andrews, A. K. Robinson and F. Rodriguez-Quinones, FEMS Microbiol. Rev., 2003, 27, 215 CrossRef CAS.
- U. E. Schaible and S. H. Kaufmann, Nat. Rev. Microbiol., 2004, 2, 946 CrossRef CAS.
- K. N. Raymond, E. A. Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584 CrossRef CAS.
- S. Cescau, H. Cwerman, S. Letoffe, P. Delepelaire, C. Wandersman and F. Biville, Biometals, 2007, 20, 603 Search PubMed.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637 RSC.
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413 CrossRef CAS.
- K. N. Raymond and E. A. Dertz, in Biochemical and physical properties of siderophores, ed. J. H. Crosa, A. R. Mey and S. M. Payne, Washington, DC, 2004 Search PubMed.
- J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66, 223 CrossRef CAS.
- T. P. Tufano and K. N. Raymond, J. Am. Chem. Soc., 1981, 103, 6617 CrossRef CAS.
- W. Breuer, M. Shvartsman and Z. I. Cabantchik, Int. J. Biochem. Cell Biol., 2008, 40, 350 CrossRef CAS.
- C. W. Yun, M. Bauler, R. E. Moore, P. E. Klebba and C. C. Philpott, J. Biol. Chem., 2001, 276, 10218 Search PubMed.
- I. Schroder, E. Johnson and S. de Vries, FEMS Microbiol. Rev., 2003, 27, 427 CrossRef CAS.
- W. R. Harris, C. J. Carrano, S. R. Cooper, S. R. Sofen, A. E. Avdeef, J. V. Mcardle and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 6097 CrossRef CAS.
- L. D. Loomis and K. N. Raymond, Inorg. Chem., 1991, 30, 906 CrossRef CAS.
- E. Lesuisse, M. Casteras-Simon and P. Labbe, Anal. Biochem., 1995, 226, 375 CrossRef CAS.
- R. J. Abergel, J. A. Warner, D. K. Shuh and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 8920 CrossRef CAS.
- S. M. Cohen and K. N. Raymond, Inorg. Chem., 2000, 39, 3624 CrossRef CAS.
- R. Stearman, D. S. Yuan, Y. Yamaguchi-Iwai, R. D. Klausner and A. Dancis, Science, 1996, 271, 1552 CAS.
- J. Cao, M. R. Woodhall, J. Alvarez, M. L. Cartron and S. C. Andrews, Mol. Microbiol., 2007, 65, 857 Search PubMed.
- C. Grosse, J. Scherer, D. Koch, M. Otto, N. Taudte and G. Grass, Mol. Microbiol., 2006, 62, 120 Search PubMed.
- D. Koch, A. C. Chan, M. E. Murphy, H. Lilie, G. Grass and D. H. Nies, J. Biol. Chem., 2011, 286, 25317 Search PubMed.
- E. Marsili, D. B. Baron, I. D. Shikhare, D. Coursolle, J. A. Gralnick and D. R. Bond, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3968 CrossRef CAS.
- L. S. Pierson 3rd. and E. A. Pierson, Appl. Microbiol. Biotechnol., 2010, 86, 1659 CrossRef CAS.
- M. E. Hernandez, A. Kappler and D. K. Newman, Appl. Environ. Microbiol., 2004, 70, 921 CrossRef CAS.
- M. E. Hernandez and D. K. Newman, Cell Mol. Life Sci., 2001, 58, 1562 CrossRef CAS.
- C. D. Cox, Infect. Immun., 1986, 52, 263 Search PubMed.
- Y. Wang, J. C. Wilks, T. Danhorn, I. Ramos, L. Croal and D. K. Newman, J. Bacteriol., 2011, 193, 3606 Search PubMed.
- W. J. Moree, V. V. Phelan, C. H. Wu, N. Bandeira, D. S. Cornett, B. M. Duggan and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13811 Search PubMed.
- A. Albert, Biochem. J., 1950, 47, xxvii Search PubMed.
- A. Albert, Biochem. J., 1953, 54, 646 CAS.
- D. J. Worst, M. M. Gerrits, C. M. Vandenbroucke-Grauls and J. G. Kusters, J. Bacteriol., 1998, 180, 1473 Search PubMed.
- Y. R. Boretsky, O. V. Protchenko, T. M. Prokopiv, I. O. Mukalov, D. V. Fedorovych and A. A. Sibirny, J. Basic Microbiol., 2007, 47, 371 Search PubMed.
- A. Vorwieger, C. Gryczka, A. Czihal, D. Douchkov, J. Tiedemann, H. P. Mock, M. Jakoby, B. Weisshaar, I. Saalbach and H. Baumlein, Planta, 2007, 226, 147 Search PubMed.
- R. E. Cowart, Arch. Biochem. Biophys., 2002, 400, 273 Search PubMed.
- K. Barbeau, Photochem. Photobiol., 2006, 82, 1505 CAS.
- K. Barbeau, E. L. Rue, K. W. Bruland and A. Butler, Nature, 2001, 413, 409 CrossRef CAS.
- H. H. Khun, S. D. Kirby and B. C. Lee, Infect. Immun., 1998, 66, 2330 Search PubMed.
- S. Dhungana, C. H. Taboy, D. S. Anderson, K. G. Vaughan, P. Aisen, T. A. Mietzner and A. L. Crumbliss, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3659 Search PubMed.
- T. A. Mietzner, S. B. Tencza, P. Adhikari, K. G. Vaughan and A. J. Nowalk, Curr. Top. Microbiol. Immunol., 1998, 225, 113 Search PubMed.
- S. D. Gray-Owen and A. B. Schryvers, Microb. Pathog., 1993, 14, 389 Search PubMed.
- S. Banerjee, C. J. Siburt, S. Mistry, J. M. Noto, P. DeArmond, M. C. Fitzgerald, L. A. Lambert, C. N. Cornelissen and A. L. Crumbliss, Metallomics, 2012, 4, 361 RSC.
- C. J. Siburt, P. L. Roulhac, K. D. Weaver, J. M. Noto, T. A. Mietzner, C. N. Cornelissen, M. C. Fitzgerald and A. L. Crumbliss, Metallomics, 2009, 1, 249 RSC.
- D. S. Anderson, P. Adhikari, A. J. Nowalk, C. Y. Chen and T. A. Mietzner, J. Bacteriol., 2004, 186, 6220 Search PubMed.
- A. G. Khan, S. R. Shouldice, S. D. Kirby, R. H. Yu, L. W. Tari and A. B. Schryvers, Biochem. J., 2007, 404, 217 CrossRef CAS.
- H. R. Strange, T. A. Zola and C. N. Cornelissen, Infect. Immun., 2011, 79, 267 Search PubMed.
- H. K. Khambati, T. F. Moraes, J. Singh, S. R. Shouldice, R. H. Yu and A. B. Schryvers, Biochem. J., 2010, 432, 57 Search PubMed.
- H. Boukhalfa, D. S. Anderson, T. A. Mietzner and A. L. Crumbliss, JBIC, J. Biol. Inorg. Chem., 2003, 8, 881 CrossRef CAS.
- M. Gabricevic and A. L. Crumbliss, Inorg. Chem., 2003, 42, 4098 CrossRef CAS.
- A. G. Khan, S. R. Shouldice, L. W. Tari and A. B. Schryvers, Biochem. J., 2007, 403, 43 Search PubMed.
- D. M. Carter, I. R. Miousse, J. N. Gagnon, E. Martinez, A. Clements, J. Lee, M. A. Hancock, H. Gagnon, P. D. Pawelek and J. W. Coulton, J. Biol. Chem., 2006, 281, 35413 CrossRef CAS.
- K. J. James, M. A. Hancock, J. N. Gagnon and J. W. Coulton, Biochemistry, 2009, 48, 9212 CrossRef CAS.
- V. Braun and C. Herrmann, J. Bacteriol., 2007, 189, 6913 CrossRef CAS.
- F. Peuckert, A. L. Ramos-Vega, M. Miethke, C. J. Schworer, A. G. Albrecht, M. Oberthur and M. A. Marahiel, Chem. Biol., 2011, 18, 907 Search PubMed.
- M. Zhu, M. Valdebenito, G. Winkelmann and K. Hantke, Microbiology, 2005, 151, 2363 CrossRef CAS.
- H. Lin, M. A. Fischbach, D. R. Liu and C. T. Walsh, J. Am. Chem. Soc., 2005, 127, 11075 CrossRef CAS.
- N. A. Larsen, H. Lin, R. Wei, M. A. Fischbach and C. T. Walsh, Biochemistry, 2006, 45, 10184 CrossRef CAS.
- M. Luo, H. Lin, M. A. Fischbach, D. R. Liu, C. T. Walsh and J. T. Groves, ACS Chem. Biol., 2006, 1, 29 Search PubMed.
- M. L. Crouch, M. Castor, J. E. Karlinsey, T. Kalhorn and F. C. Fang, Mol. Microbiol., 2008, 67, 971 CrossRef CAS.
- M. Caza, F. Lepine, S. Milot and C. M. Dozois, Infect. Immun., 2008, 76, 3539 Search PubMed.
- M. T. Poch and W. Johnson, Biometals, 1993, 6, 107 Search PubMed.
- R. Mazoy, E. M. Lopez, B. Fouz, C. Amaro and M. L. Lemos, FEMS Microbiol. Lett., 1999, 172, 205 Search PubMed.
- C. D. Cox, J. Bacteriol., 1980, 141, 199 Search PubMed.
- S. Letoffe, G. Heuck, P. Delepelaire, N. Lange and C. Wandersman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11719 CrossRef CAS.
- H. A. Dailey, A. N. Septer, L. Daugherty, D. Thames, S. Gerdes, E. V. Stabb, A. K. Dunn, T. A. Dailey and J. D. Phillips, MBio, 2011, 2, e00248 Search PubMed.
- I. Stojiljkovic and D. Perkins-Balding, DNA Cell Biol., 2002, 21, 281 CrossRef CAS.
- J. C. Grigg, G. Ukpabi, C. F. Gaudin and M. E. Murphy, J. Inorg. Biochem., 2010, 104, 341 CrossRef CAS.
- R. M. Pilpa, E. A. Fadeev, V. A. Villareal, M. L. Wong, M. Phillips and R. T. Clubb, J. Mol. Biol., 2006, 360, 435 CrossRef CAS.
- R. M. Pilpa, S. A. Robson, V. A. Villareal, M. L. Wong, M. Phillips and R. T. Clubb, J. Biol. Chem., 2009, 284, 1166 CAS.
- M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Nat. Chem. Biol., 2006, 2, 132 CrossRef CAS.
- Y. Wen, X. Wu, Y. Teng, C. Qian, Z. Zhan, Y. Zhao and O. Li, Environ. Microbiol., 2011, 13, 2726 Search PubMed.
- M. Miethke, O. Klotz, U. Linne, J. J. May, C. L. Beckering and M. A. Marahiel, Mol. Microbiol., 2006, 61, 1413 Search PubMed.
- R. J. Abergel, A. M. Zawadzka, T. M. Hoette and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 12682 Search PubMed.
- S. I. Patzer and V. Braun, J. Bacteriol., 2010, 192, 426 CrossRef CAS.
- M. Sandy and A. Butler, J. Nat. Prod., 2011, 74, 1207 Search PubMed.
- L. Rauscher, D. Expert, B. F. Matzanke and A. X. Trautwein, J. Biol. Chem., 2002, 277, 2385 CrossRef.
- M. Sandy, A. Han, J. Blunt, M. Munro, M. Haygood and A. Butler, J. Nat. Prod., 2010, 73, 1038 CrossRef CAS.
- M. Balado, C. R. Osorio and M. L. Lemos, Microbiology, 2006, 152, 3517 CrossRef CAS.
- G. Winkelmann and H. Drechsel, in Microbial Siderophores, ed. H. Kleinkauf and H. von Döhren, Weinheim, Germany, 1997 Search PubMed.
- C. Kragl, M. Schrettl, B. Abt, B. Sarg, H. H. Lindner and H. Haas, Eukaryotic Cell, 2007, 6, 1278 Search PubMed.
- H. Haas, M. Eisendle and B. G. Turgeon, Annu. Rev. Phytopathol., 2008, 46, 149 Search PubMed.
- M. Schrettl and H. Haas, Curr. Opin. Microbiol., 2011, 14, 400 Search PubMed.
- J. R. Heemstra Jr., C. T. Walsh and E. S. Sattely, J. Am. Chem. Soc., 2009, 131, 15317 CrossRef.
- M. Ingelman, S. Ramaswamy, V. Niviere, M. Fontecave and H. Eklund, Biochemistry, 1999, 38, 7040 CrossRef CAS.
- J. Coves, M. Eschenbrenner and M. Fontecave, Biochem. Biophys. Res. Commun., 1993, 192, 1403 Search PubMed.
- F. Halle and J. M. Meyer, Eur. J. Biochem., 1992, 209, 621 Search PubMed.
- F. Halle and J. M. Meyer, Eur. J. Biochem., 1992, 209, 613 Search PubMed.
- J. Mazoch, R. Tesarik, V. Sedlacek, I. Kucera and J. Turanek, Eur. J. Biochem., 2004, 271, 553 Search PubMed.
- M. Xia, J. Wei, Y. Lei and L. Ying, Curr. Microbiol., 2007, 55, 71 Search PubMed.
- F. Fieschi, V. Niviere, C. Frier, J. L. Decout and M. Fontecave, J. Biol. Chem., 1995, 270, 30392 CrossRef CAS.
- K. A. Mies, J. I. Wirgau and A. L. Crumbliss, Biometals, 2006, 19, 115 Search PubMed.
- R. K. Poole, N. J. Rogers, A. D'MelloR, M. N. Hughes and Y. Orii, Microbiology, 1997, 143(Pt 5), 1557 Search PubMed.
- K. Takeda, J. Sato, K. Goto, T. Fujita, T. Watanabe, M. Abo, E. Yoshimura, J. Nakagawa, A. Abe, S. Kawasaki and Y. Niimura, Biometals, 2010, 23, 727 Search PubMed.
- H. J. Chiu, E. Johnson, I. Schroder and D. C. Rees, Structure, 2001, 9, 311 Search PubMed.
- N. Carrillo and E. A. Ceccarelli, Eur. J. Biochem., 2003, 270, 1900 Search PubMed.
- K. Muller, B. F. Matzanke, V. Schunemann, A. X. Trautwein and K. Hantke, Eur. J. Biochem., 1998, 258, 1001 Search PubMed.
- B. F. Matzanke, S. Anemuller, V. Schunemann, A. X. Trautwein and K. Hantke, Biochemistry, 2004, 43, 1386 CrossRef CAS.
- M. Miethke, A. J. Pierik, F. Peuckert, A. Seubert and M. A. Marahiel, J. Biol. Chem., 2011, 286, 2245 CrossRef CAS.
- J. L. Pierre, M. Fontecave and R. R. Crichton, Biometals, 2002, 15, 341 Search PubMed.
- C. W. Lee, D. J. Ecker and K. N. Raymond, J. Am. Chem. Soc., 1985, 107, 6920 Search PubMed.
- J. S. Lodge, C. G. Gaines, J. E. Arceneaux and B. R. Byers, Biochem. Biophys. Res. Commun., 1980, 97, 1291 Search PubMed.
- M. Miethke, J. Hou and M. A. Marahiel, Biochemistry, 2011, 50, 10951 Search PubMed.
- J. R. Butterton and S. B. Calderwood, J. Bacteriol., 1994, 176, 5631 Search PubMed.
- V. A. Bamford, M. Armour, S. A. Mitchell, M. Cartron, S. C. Andrews and K. A. Watson, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2008, 64, 792 Search PubMed.
- F. Peuckert, M. Miethke, A. G. Albrecht, L. O. Essen and M. A. Marahiel, Angew. Chem., Int. Ed., 2009, 48, 7924 Search PubMed.
- S. Wang, Y. Wu and F. W. Outten, J. Bacteriol., 2011, 193, 563 CrossRef CAS.
- M. Huyer and W. J. Page, J. Bacteriol., 1989, 171, 4031 Search PubMed.
- S. R. Cooper, J. V. McArdle and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3551 CAS.
- N. Li, C. Zhang, B. Li, X. Liu, Y. Huang, S. Xu and L. Gu, J. Biol. Chem., 2012, 287, 8912 Search PubMed.
- G. L. Griffiths, S. P. Sigel, S. M. Payne and J. B. Neilands, J. Biol. Chem., 1984, 259, 383 CAS.
- J. P. Robinson, E. F. Wawrousek, J. V. Mcardle, G. Coyle and I. Adler, Inorg. Chim. Acta–Bioinorg. Chem., 1984, 92, L19 Search PubMed.
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, J. Biol. Inorg. Chem., 2008, 13, 1205 CrossRef CAS.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823 CrossRef CAS.
- D. E. Wilcox, Chem. Rev., 1996, 96, 2435 CrossRef CAS.
- R. Lill, B. Hoffmann, S. Molik, A. J. Pierik, N. Rietzschel, O. Stehling, M. A. Uzarska, H. Webert, C. Wilbrecht and U. Muhlenhoff, Biochim. Biophys. Acta, 2012, 1823, 1491 Search PubMed.
- M. Fontecave and S. Ollagnier-de-Choudens, Arch. Biochem. Biophys., 2008, 474, 226 CrossRef CAS.
- H. A. Dailey, T. A. Dailey, C. K. Wu, A. E. Medlock, K. F. Wang, J. P. Rose and B. C. Wang, Cell Mol. Life Sci., 2000, 57, 1909 CrossRef CAS.
- S. C. Andrews, Biochim. Biophys. Acta, 2010, 1800, 691 CrossRef CAS.
- E. Pohl, J. C. Haller, A. Mijovilovich, W. Meyer-Klaucke, E. Garman and M. L. Vasil, Mol. Microbiol., 2003, 47, 903 CrossRef CAS.
- J. C. Rutherford, S. Jaron and D. R. Winge, J. Biol. Chem., 2003, 278, 27636 Search PubMed.
- N. Frankenberg-Dinkel, Antioxid. Redox. Signaling, 2004, 6, 825 Search PubMed.
- T. Matsui, M. Unno and M. Ikeda-Saito, Acc. Chem. Res., 2010, 43, 240 CrossRef CAS.
- F. Tiburzi, F. Imperi and P. Visca, IUBMB Life, 2009, 61, 80 Search PubMed.
- J. L. Urbanowski and R. C. Piper, J. Biol. Chem., 1999, 274, 38061 Search PubMed.
- N. G. De Luca and P. M. Wood, Adv. Microb. Physiol., 2000, 43, 39 Search PubMed.
- R. Bohnke and B. F. Matzanke, Biometals, 1995, 8, 223 Search PubMed.
- E. Chiancone and P. Ceci, Biochim. Biophy. Acta, Gen. Subj., 2010, 1800, 798 Search PubMed.
- N. E. Le Brun, A. Crow, M. E. P. Murphy, A. G. Mauk and G. R. Moore, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 732 CrossRef CAS.
- A. Nandal, J. C. Ruiz, P. Subramanian, S. Ghimire-Rijal, R. A. Sinnamon, T. L. Stemmler, R. K. Bruick and C. C. Philpott, Cell Metab., 2011, 14, 647 CrossRef CAS.
- H. Shi, K. Z. Bencze, T. L. Stemmler and C. C. Philpott, Science, 2008, 320, 1207 CrossRef CAS.
- S. G. Wong, R. Abdulqadir, N. E. Le Brun, G. R. Moore and A. G. Mauk, Biochem. J., 2012, 444, 553 Search PubMed.
- S. Yasmin, S. C. Andrews, G. R. Moore and N. E. Le Brun, J. Biol. Chem., 2011, 286, 3473 Search PubMed.
- U. Muhlenhoff, S. Molik, J. R. Godoy, M. A. Uzarska, N. Richter, A. Seubert, Y. Zhang, J. Stubbe, F. Pierrel, E. Herrero, C. H. Lillig and R. Lill, Cell Metab., 2010, 12, 373 CrossRef.
- T. L. Stemmler, E. Lesuisse, D. Pain and A. Dancis, J. Biol. Chem., 2010, 285, 26737 CrossRef CAS.
- P. Subramanian, A. V. Rodrigues, S. Ghimire-Rijal and T. L. Stemmler, Curr. Opin. Chem. Biol., 2011, 15, 312 Search PubMed.
- W. Qi and J. A. Cowan, Coord. Chem. Rev., 2011, 255, 688 Search PubMed.
- C. L. Tsai and D. P. Barondeau, Biochemistry, 2010, 49, 9132 Search PubMed.
- F. Prischi, P. V. Konarev, C. Iannuzzi, C. Pastore, S. Adinolfi, S. R. Martin, D. I. Svergun and A. Pastore, Nat. Commun., 2010, 1, 95 Search PubMed.
- J. Bridwell-Rabb, C. Iannuzzi, A. Pastore and D. P. Barondeau, Biochemistry, 2012, 51, 2506 Search PubMed.
- A. G. Albrecht, H. Landmann, D. Nette, O. Burghaus, F. Peuckert, A. Seubert, M. Miethke and M. A. Marahiel, ChemBioChem, 2011, 12, 2052 Search PubMed.
- W. Qi and J. A. Cowan, Chem. Commun., 2010, 46, 719 RSC.
- K. Chandramouli, M. C. Unciuleac, S. Naik, D. R. Dean, B. H. Huynh and M. K. Johnson, Biochemistry, 2007, 46, 6804 Search PubMed.
- D. J. Netz, M. Stumpfig, C. Dore, U. Muhlenhoff, A. J. Pierik and R. Lill, Nat. Chem. Biol., 2010, 6, 758 Search PubMed.
- U. Muhlenhoff, J. Gerber, N. Richhardt and R. Lill, EMBO J., 2003, 22, 4815 CrossRef.
- M. T. Rodriguez-Manzaneque, J. Tamarit, G. Belli, J. Ros and E. Herrero, Mol. Biol. Cell, 2002, 13, 1109 CrossRef CAS.
- S. Bandyopadhyay, F. Gama, M. M. Molina-Navarro, J. M. Gualberto, R. Claxton, S. G. Naik, B. H. Huynh, E. Herrero, J. P. Jacquot, M. K. Johnson and N. Rouhier, EMBO J., 2008, 27, 1122 CrossRef CAS.
- F. Bonomi, S. Iametti, A. Morleo, D. Ta and L. E. Vickery, Biochemistry, 2008, 47, 12795 Search PubMed.
- M. Fontecave, S. O. Choudens, B. Py and F. Barras, J. Biol. Inorg. Chem., 2005, 10, 713 CrossRef CAS.
- S. Wollers, G. Layer, R. Garcia-Serres, L. Signor, M. Clemancey, J. M. Latour, M. Fontecave and S. Ollagnier de Choudens, J. Biol. Chem., 2010, 285, 23331 Search PubMed.
- V. Gupta, M. Sendra, S. G. Naik, H. K. Chahal, B. H. Huynh, F. W. Outten, M. Fontecave and S. Ollagnier de Choudens, J. Am. Chem. Soc., 2009, 131, 6149 Search PubMed.
- G. P. Riboldi, H. Verli and J. Frazzon, BMC Biochem., 2009, 10, 3 Search PubMed.
- A. G. Albrecht, F. Peuckert, H. Landmann, M. Miethke, A. Seubert and M. A. Marahiel, FEBS Lett., 2011, 585, 465 Search PubMed.
- A. G. Albrecht, D. J. Netz, M. Miethke, A. J. Pierik, O. Burghaus, F. Peuckert, R. Lill and M. A. Marahiel, J. Bacteriol., 2010, 192, 1643 Search PubMed.
- R. G. Kranz, C. Richard-Fogal, J. S. Taylor and E. R. Frawley, Microbiol. Mol. Biol. Rev., 2009, 73, 510 CrossRef CAS.
- K. Saiki, T. Mogi, K. Ogura and Y. Anraku, J. Biol. Chem., 1993, 268, 26041 Search PubMed.
- M. E. Dumont, J. F. Ernst, D. M. Hampsey and F. Sherman, EMBO J., 1987, 6, 235 CAS.
- P. Hamel, V. Corvest, P. Giege and G. Bonnard, Biochim. Biophys. Acta, Mol. Cell Res., 2009, 1793, 125 Search PubMed.
- C. Sanders, S. Turkarslan, D. W. Lee and F. Daldal, Trends Microbiol., 2010, 18, 266 CrossRef CAS.
- C. Richard-Fogal and R. G. Kranz, J. Mol. Biol., 2010, 401, 350 Search PubMed.
- E. M. Harvat, O. Daltrop, F. Sobott, M. Moreau, P. D. Barker, J. M. Stevens and S. J. Ferguson, Metallomics, 2011, 3, 363 RSC.
- A. Flint, Y. Q. Sun and A. Stintzi, J. Bacteriol., 2012, 194, 334 Search PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Nature, 2010, 465, 645 CrossRef CAS.
- M. Ludwig, T. Schubert, I. Zebger, N. Wisitruangsakul, M. Saggu, A. Strack, O. Lenz, P. Hildebrandt and B. Friedrich, J. Biol. Chem., 2009, 284, 2159 Search PubMed.
- C. Pinske and R. G. Sawers, PLoS One, 2012, 7 Search PubMed.
- A. Bock, P. W. King, M. Blokesch and M. C. Posewitz, Adv. Microb. Physiol., 2006, 51, 1 CrossRef.
- L. Forzi and R. G. Sawers, Biometals, 2007, 20, 565 Search PubMed.
- K. N. Raymond and C. J. Carrano, Acc. Chem. Res., 1979, 12, 183 CrossRef CAS.
- W. R. Harris, C. J. Carrano and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 2722 CrossRef CAS.
- E. F. Wawrousek and J. V. Mcardle, J. Inorg. Biochem., 1982, 17, 169 CrossRef CAS.
- I. Spasojevic, S. K. Armstrong, T. J. Brickman and A. L. Crumbliss, Inorg. Chem., 1999, 38, 449 CrossRef CAS.
- G. B. Wong, M. J. Kappel, K. N. Raymond, B. Matzanke and G. Winkelmann, J. Am. Chem. Soc., 1983, 105, 810 CrossRef CAS.
- E. A. Dertz, J. Xu, A. Stintzi and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 22 CrossRef CAS.
- M. Miethke and A. Skerra, Antimicrob. Agents Chemother., 2010, 54, 1580 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |