 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysosomal metal, redox and proton cycles influencing the CysHis cathepsin reaction
Thomas D.
Lockwood
*
Dept. of Pharmacology, School of Medicine, Wright State University, Dayton, Ohio 45435, USA. E-mail: thomas.lockwood@wright.edu; Tel: +1 937 775 4402
First published on 9th January 2013
Abstract
In the 1930's pioneers discovered that maximal autolysis in tissue homogenates requires metal chelator, sulfhydryl reducing agent and acid pH. However, metals, reducing equivalents and protons (MR&P) have been overlooked as combined catalytic controls. Three categories of lysosomal machinery drive three distinguishable cycles importing and exporting MR&P. Zn2+ preemptively inhibits CysHis catalysis under otherwise optimal protonation and reduction. Protein-bound cell Zn2+ concentration is 200–2000 times the non-sequestered inhibitory concentration. Following autophagy, lysosomal proteolysis liberates much inhibitory Zn2+. The vacuolar proton pump is the driving force for Zn2+ export, as well as protonation of the peptidolytic mechanism. Other machinery of lysosomal cycles includes proton-driven Zn2+ exporters (e.g. SLC11A1), Zn2+ channels (e.g. TRPML-1), lysosomal thiol reductase, etc. The CysHis dyad is a sensor of the vacuolar environment of MR&P, an integrator of these simultaneous variables, and a catalytic responder. Rate-determination can shift between autophagic substrate acquisition (swallowing) and substrate degradation (digesting). Zn2+ recycling from degraded proteins to new proteins is a fourth cycle that might pace lysosomal function under some conditions. Heritable insufficient or excess functions of CysHis cathepsins are associated with dysfunctional inflammation and immunity/auto-immunity, including diabetic pathogenesis.
 Thomas D. Lockwood | Dr Lockwood received a BA in Biology from Gettysburg College, Gettysburg Pennsylvania, and a PhD from the School of Medicine of the University of Rochester, Rochester, New York. Before joining the Medical School of Wright State University in Dayton, Ohio, he was a research associate at the Salk Institute in La Jolla, California, and assistant professor at the Massachusetts Institute of Technology in Cambridge, Massachusetts. His research interests are the enzymology and physiology of protein degradation, and medical intervention against pathogenic dysfunction. |
1. Introduction
1.1. Four lysosomal cycles
Despite differences in substrate preferences and functions, lysosomal CysHis proteases share much in common. In enzyme assay their maximal catalytic rate requires EDTA and DTT under acid-optimal pH. Recent advances reveal a correspondence between this chemical technology and Nature's engineering. Metals, redox and protons (MR&P) are regulated by lysosomal machinery as well as enzymologists. These combined variables can grade CysHis catalysis from completely “off” to maximally “on”1,2 (Fig. 1). Many lysosomal enzymes cooperate in hydrolysis of macromolecular bonds; however, the triple responsiveness of CysHis cathepsins is unique.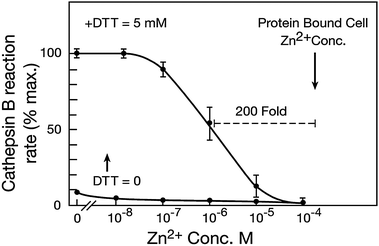 | ||
| Fig. 1 Zn2+ concentration inhibiting cathepsin B under optimal reduction and protonation. Zn2+ was removed from purified bovine cathepsin B by EDTA followed by exhaustive dialysis. The protease was preliminarily oxidized to the presumptive sulfoxide by incubation in air-equilibrated water. Reaction rate was assayed as previously described by standard methods using fluorimetric detection of the hydrolysis of carbobenzyloxy-ArgArg-AMC (previously reported in Lockwood 2010). The inactive enzyme was placed in reaction buffer (optimal pH of 5.5) containing the indicated Zn2+ concentrations; and substrate was added. A low activity of the oxidized enzyme was observed (bottom trace). DTT reducing agent (5 mM) was then added to the same reactions; and the reaction rate was again measured. These measurements reveal that a low concentration of Zn2+ preemptively inhibits cathepsin B in the presence of excess DTT activator and optimal pH. Thus, Zn2+ can be an overriding factor limiting the peptidolytic mechanism. Findings were indistinguishable with several L type CysHis cathepsins from various species. Zn2+ concentration bound to cell proteins. Gross, protein-bound Zn2+ concentration per unit of cell volume was measured in perfused myocardial tissue by standard methods using atomic absorption after complete acid hydrolysis of proteins (Lockwood 2010). Extracellular fluid was preliminarily replaced by Zn2+ free perfusion; and results were corrected for extracellular volume. The protein-bound Zn2+ concentration/unit cell volume (200 μM) is 200–2000 times the range of Zn2+ concentrations that inhibits CysHis proteases (0.1–1.0) μM or less. However, DTT binds added Zn2+ and decreases its inhibitory potency (see text). Without DTT, Zn2+ partially inhibits at the lowest concentration shown (0.01 μM). | ||
Long ago our group wondered whether MR&P factors might influence CysHis catalysis in lysosomes as well as enzyme assays. We developed a reliable bioassay to measure the release of 3H-leucine from degraded proteins of a perfused tissue. Interventions in redox metabolism or metal homeostasis were affirmative3,4 (not fully reviewed here). In the following decades, the genome was sequenced; and proteins regulating lysosomal MR&P have been identified. Dysregulations of lysosomal MR&P and CysHis cathepsins are now known to be a causative factor in major human diseases.
This perspective integrates the catalytic properties of CysHis cathepsins with disparate advances in machinery regulating lysosomal MR&P. Normal functions of CysHis cathepsins are revealed by the consequences of their heritable dysfunction. First, deletion of a single CysHis cathepsin gene can slow lysosomal catalysis.5 CysHis cathepsins can serve as lysosomal “pacemakers” under some conditions. Second, single and multiple CysHis cathepsin gene deletions protect against progressive auto-immune tissue damage,6,7 and inflammatory damage from pro-catabolic cytokines.8 Inflammation and auto-immunity are “double trouble” underlying the pathogenesis of several diseases, including diabetic progression.9 Third, heritable dysregulations of lysosomal MR&P in certain cell types are associated with diseases of insufficient or excessive lysosomal function (see below).
Metallomics and redox proteomics have become routine concepts.10–13 However, “multiomics” has not yet entered the lexicon. Thus far, no unifying redox code or metal code has been deciphered in prokaryotes or the compartmentalized cells of eukaryotes. Primordial “omics” has been complicated by the evolution of higher-order protein structure, and separately regulated compartments. There is one type of catalytic mechanism, in one type of compartment, which retains a primitive code of omic control. A proton-responsive enzyme with cross-membership in the redox proteome and metal-interactive proteome is controlled by several simultaneous variables. Semanticists have not yet characterized pH-dependent proteins as the “protonosome”. However, the lysosomal compartment regulates these three variables; and these variables regulate CysHis catalysis. Cys confers redox-responsiveness, and His adds metal-responsiveness; together they confer proton-responsiveness. It is proposed here that the CysHis dyad is (a) a sensor of the vacuolar environment of metals, redox and protons, (b) an integrator of these simultaneous variables, and a (c) catalytic responder to cell regulation of the three categories of factors.
Understanding the control of CysHis cathepsins requires decoding the combined effects of metals, redox and protons on the peptidolytic mechanism. In the absence of metals the activity is maximal under optimal pH near 5.5, and reductive activation by several mM DTT (Fig. 1). Any of the three members of the governing triumvirate can limit CysHis peptidolysis. However, Zn2+ (0.1–1.0 μM) is a preemptive inhibitor despite maximal stimulation by protonation and reduction. The disulfhydryls of DTT bind Zn2+; therefore the actual inhibitory Zn2+ concentration in Fig. 1 is much less than the added concentration. Prior to addition of DTT some amount of inhibition can be observed at the lowest Zn2+ concentration shown (0.01 μM) (Fig. 1, bottom trace). The Zn2+ sensitivities of several B- and L-type cathepsins were similar. The triple responsiveness of CysHis cathepsins corresponds to the three categories of lysosomal import–export cycles (Fig. 2). These inputs and outputs displace vacuolar MR&P from equilibrium conditions. CysHis cathepsins are “wired” to the currents of metals, reducing equivalents and protons created by these cycles. A fourth lysosomal cycle transfers Zn2+ from degraded protein binding sites to new proteins.
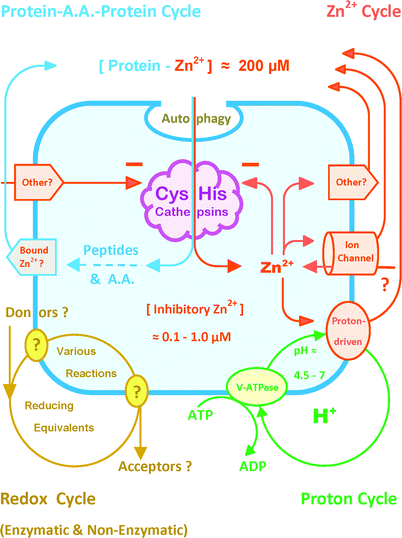 | ||
| Fig. 2 Recognition of four lysosomal cycles for future correlation with CysHis cathepsin activity. The peptidolytic chemistry of Fig. 1 challenges molecular biology to account for lysosomal metal, redox and proton (MR&P) cycles that determine the catalytic environment of CysHis cathepsins. This simplified composite is intended to accommodate future advances in the machinery involved and its integrated control. Following substrate uptake, intra-vacuolar proteolysis liberates a large amount of Zn2+ from macromolecular binding sites; other inhibitory metals might be relevant (see text). Without export, lysosomal Zn2+ would rapidly accumulate to an inhibitory concentration (Fig. 1); and CysHis catalysis would be self-limited. The recycling of released metals from degraded proteins to new proteins might be a rate-determining factor in recycling amino acids into new proteins. The V-ATPase is a major driving force for the export of Zn2+ and other metals via the proton-driven Slc11A1 and other transporters. Conceivable importers and exporters labeled “other” include ATP-binding cassette (ABC) transporters which transport a wide variety of substances that bind Zn2+, e.g. peptides, glutathione, organic cations etc. (and see text). Some TRP channels passively conduct Zn2+ down its electrochemical gradient inward as well as outward. Some TRP channels are phosphorylated or pH dependent; and some rectify conductance. Multiple, enzymatic and non-enzymatic redox reactions might influence CysHis cathepsins. Pathway(s) transferring reducing equivalents into and out of the lysosome are presently unknown. This abbreviated summary excludes such relevant topics as the cystine exporter, cystinosin, GSH/GSSG transport, lysosomal dipeptidase etc. (see text). | ||
1.2. Scope and limitations of the abstract theory
Prior to consideration of the present theory, several misperceptions must be dispelled. It is generally presumed that autophagy is the rate-determining step in lysosomal function. However, autophagy and intra-vacuolar degradation can be compared to swallowing and digesting. If intra-vacuolar hydrolysis were always faster than substrate acquisition, then lysosomes would never become loaded with un-degraded macromolecules. In most images of the cell, some lysosomes appear loaded. Micro- and macro-autophagy have been reviewed many times in recent years, (but not here!). In contrast, intra-vacuolar catalysis has evaded attention thus far. The rate-limitation on lysosomal function can shift between substrate acquisition and substrate degradation under various conditions.In the absence of inhibitory metal concentration CysHis peptidolysis can function outside the lysosome or cell at a submaximal rate until the enzyme becomes oxidized14 (illustrated by the bottom trace in Fig. 1 under low metal concentration). Diverse lysosomal functions have evolved in relation to the diversity of specialized cells that employ them. CysHis cathepsins have extra-cellular functions, e.g. degradation of the extra-cellular matrix. This review is generalized to the essential CysHis reaction mechanism within vacuoles. Several topics are postponed until better understanding of their relevance to lysosomal cycles, e.g. the integrity of the lysosomal membrane,15 activation of the “inflammasome” cascade,16 endogenous protease inhibitors,17 metabolic and post-receptor signals regulating the lysosomal machinery, etc.
The prokaryotic CysHis catalytic mechanism evolved into cathepsins, caspases and calpains. MR&P control of intra-vacuolar catalysis does not conflict with present understanding of extra-lysosomal CysHis proteases (see Section 7).
Lysosomal electrophysiology is presently controversial;18–21 and, understanding of integrated Zn2+ transport is presently at the beginning stages. Acceptance of this incomplete theory of catalytic control does not require knowledge of the lysosomal membrane potential or control of all ions. Indeed, this theory provides a guide to unanswered questions.
When disparate advances from many investigators are assembled, Nature's logic becomes self-evident. The present theory is difficult to distinguish from a fact; however, lysosomal MR&P cycles are initially described here with broad simplicity. Introduction at the preliminary stage is justified by the immediate relevance to pathology and therapeutics.2
2. A lysosomal cycle importing and exporting protons
Names for different structures of the lysosomal system include: dense granules, pre-lysosomes, endosomes, phagosomes, pinocytic vesicles, macro-autophagic and micro-autophagic vacuoles, endoplasmic reticulum-lysosome fusion bodies, lysosomal-mitochondrial fusion bodies (“lysondrion”), expired lysosomes, residual bodies, exosomes and even “regurgisomes”. These various structures have a wide range of pH, redox properties, metal contents and catalytic activities. Here, the criterion of a functional lysosome is a catalytically active vesicle hydrolyzing peptide bonds.2.1. Import of lysosomal protons
Enzymatic and non-enzymatic peptide bond hydrolysis (“splitting by addition of water”) is highly dependent upon proton concentration. However, lysosomal proton transport has acquired additional significance in light of other processes that depend on it (Fig. 2). Inactive lysosomes can exist with neutral pH; and active vesicles can decrease pH below 5. Progress in regulation of the vacuolar proton pump has been slow due to the complexity of this machine. The structure and function of the V-ATPase in relation to other ions has been reviewed in detail.21If the proton pump is completely inactive, the non-sequestered vacuolar Zn2+ concentration at pH of 7 might be similar to the cytoplasm. When the proton pump is active, Zn2+ export lowers the inhibitory metal to the permissive concentration for CysHis cathepsin activity (Fig. 1 and 2). The non-sequestered, vacuolar Zn2+ concentration presumably varies inversely with the proton gradient that drives export. However, Zn2+ and proton concentrations might not exist in direct inverse proportionality. Increasing proton concentration drives Zn2+ export while simultaneously protonating the CysHis reaction mechanism, and releasing large amounts of Zn2+ from degraded binding sites. Various TRP channels can be influenced or controlled by Zn2+, Mg2+, pH, phosphorylation and others.22,23
2.2. Export of lysosomal protons
A significant route of proton export is direct coupling to metal export (see below). The ability of lysosomes to acidify implies that proton export and leak are small relative to import under maximal function of the V-ATPase.3. A lysosomal cycle importing and exporting reducing equivalents, and reducing and oxidizing CysHis cathepsins
The chemistry of protein thiolation–dethiolation corresponds to the pharmacology of Kosower's sulfhydryl oxidizing agent (diamide) vs. Cleland's sulfhydryl reducing agent (dithiothreitol). Diamide accepts reducing equivalents from two sulfhydryls in a two-step oxidation catalyzing the formation of disulfides.24,25 DTT undergoes internal oxidations of its disulfhydryls to disulfides as it transfers to acceptors. Thiolation–dethiolation was demonstrated using one dimensional gel electrophoresis of tissues exposed to diamide.26,27 An increase in glutathione disulfide causes corresponding increase in protein-mixed disulfides. An idealized theory suggests that the GSSG/GSH redox ratio controls the functions of some proteins. Redox buffering remains valid in purified reactions; however, application to the compartmentalized cell under non-equilibrium conditions requires modification.25,28Evidence for lysosomal machinery associated with thiolation–dethiolation can be summarized with a few findings. In 1993 Stephen et al.29 reported that an inactive papain disulfide can be reductively reactivated by NADPH, thioredoxin reductase and thioredoxin in a reconstituted reaction. It was simultaneously reported that pharmacologic thiolation–dethiolation caused by diamide and DTT resulted in large changes in protein degradation in perfused tissue.3,4 A lysosomal thioredoxin was then described,30 and found to activate lysosomal proteases.31 Cathepsin B is responsive to the redox ratio of GSSG/GSH32 as well as a variety of other oxidants. Assembly of these findings suggests transfer of extra-lysosomal reductive energy to oxidized vacuolar CysHis cathepsins. The exact intermediaries and pathways transferring extra-lysosomal reducing energy into the vesicle are unknown. The “redox cycle” shown in Fig. 2 is a generalized composite representing multiple intermediaries and pathways in various cell types and species.
It is now known that thioredoxins and glutaredoxins comprise the major pathways to protein reduction in various compartments. However, thioredoxins and glutaredoxins exhibit cross talk, and some redundant functions.33 Indeed, thioredoxins of some compartments are glutathionylated. Glutaredoxins and thioredoxins can both reduce GSSG and PrSSG as well as other oxidation products. Oxidized and reduced glutathione are found in lysosomes as well as the endoplasmic reticulum (ER). Lysosomal transport of some form(s) of glutathione is suspected. Thus far, functional glutathione reductase and glutaredoxins have not been reported within lysosomes.
3.1. The oxidation of CysHis cathepsins and export of lysosomal reducing equivalents
Only the reduced sulfhydryl of CysHis cathepsins is catalytically active. Submaximal activity results from partition of the population of enzyme molecules among the reduced active state and all oxidized inactive states. The partition of catalytic cysteine is the net result of all opposing enzymatic and non-enzymatic redox reactions. Catalytic sulfur can exist in a shifting partition among reversible states until it is irreversibly oxidized. The catalytic partner can oxidize to 2-oxo-histidine.34,35 Extensive histidine oxidation seems likely under extreme conditions in some species. However, the significance of histidine redox in mammalian CysHis peptidolysis represents an important gap in present knowledge.The best single answer to the question of vacuolar CysHis redox is that there is no single answer. CysHis redox within the lysosome can be compared to a busy intersection with cross-traffic from many transferring pathways. The simplified redox cycle of (Fig. 2) represents a multitude of non-enzymatic and enzymatic redox interactions with proteases. This problem is comparable to other cell compartments.36 The chemical and biochemical contents of the lysosomal system are undefined to state the least. Lysosomal contents can include any exogenous particle or solute that can be taken up by fluid pinocytosis, phagocytosis or receptor-mediated endocytosis. Moreover, the catabolic vacuole can contain the entire content of the mitochondrion or endoplasmic reticulum. Ongoing processes in mitochondria and ER do not cease instantaneously upon fusion with lysosomes.
Much of what is now known about protein sulfur redox was originally learned using a CysHis protease as experimental tool. Papain permits determination of the redox state of a single protein sulfur site using protease activity as an indicator. This botanical protease has long been available in crystallized form, thereby permitting defined reactions without impurities.37 Papain can be “gently” oxidized under conditions that do not cause irreversible inactivation. Far more is known about papain chemistry in purified reactions than papain biology in the cell. Oxidized derivatives of papain sulfur include sulfenic, sulfinic and sulfonic acid (PrSO, PrSO2, PrSO3),38 glutathionylations (PrSSG),39,40 nitrosylations (PrSNO).41–48 Some “deep” oxidation states are not reversible under biological conditions, e.g. PrSO3.
Reactive oxygen species (ROS), reactive nitrogen species (RNS) and the redox ratio of GSSG/GSH are known to be cell signals. Protein sulfoxygenations, nitrosylations and glutathionylations control the functions of some proteins. However, defined redox reactions in purified reconstituted systems can differ greatly from in vivo reactions. An experimental sulfhydryl oxidant can first oxidize and deplete protective GSH, and then oxidize the unprotected protease cysteine directly. In purified enzyme solutions, hydrogen peroxide produces protein sulfenic acids; and nitric oxide donors produce nitrosylations. When competing GSH is present, these oxidants first produce GSSG, and then increasingly oxidize proteins as GSH is depleted.47–49 Any agent that oxidizes GSH to GSSG can secondarily oxidize Protein-SH to Protein-SSG. Therefore, diverse oxidants act initially to increase the GSSG/GSH ratio and secondarily to oxidize protein sulfhydryls.
Pathways transferring reducing equivalents from lysosomal sulfhydryls to oxygen, or other acceptors, are uncharacterized. Work to be done is illustrated by comparison of known features of redox transfer in the endoplasmic reticulum with corresponding unknowns of the lysosome. The ER has an enzymatic relay system transferring reducing equivalents from internal protein sulfhydryls to oxygen in the process of forming internal disulfide bonds of the folded protein.50–53 The ER has multiple pathways of sulfhydryl oxidation in addition to the ER oxidoreductin system (ERO). Dehydroascorbic acid/ascorbic acid redox has been implicated in ER-associated degradation as well as lysosomal degradation.36,53 In contrast to cytoplasm, the ER compartment has a high content of glutathionylated proteins and GSSG. Thus far, there has been no suggestion that the ERO relay system is operative in the lysosome after compartmental fusions. Redox implications of lysosomal fusions with other compartments are virtually unstudied.
Speciated metals could participate in the pathways transferring from reduced sulfhydryls of proteins or glutathione to oxygen and other acceptors. (The oxidation state of Zn2+ does not change under biological conditions; although some binding sites are redox-responsive.) Speciated metals are mutually reactive with protein sulfhydryls and reactive oxygen species or diatomic oxygen. Fe3+/Fe2+ or Cu2+/Cu1+ can act as cycling intermediates, and accelerate the transfer of electrons from CysHis cathepsins to oxygen or other acceptors. Speciated metals accelerate the oxidation of CysHis cathepsins in air-equilibrated water. In addition, Fe3+ and Cu2+ (oxidized) directly inhibit CysHis cathepsins.1 In contrast, Fe2+ and Cu1+ (reduced) are not potent inhibitors of CysHis cathepsins. Under extreme oxidative conditions (or reductive deficiency) in lower species, ferric and cupric states almost certainly attain sufficient concentrations to directly inhibit CysHis cathepsins.
ROS and RNS are suspected of an influence on CysHis cathepsin function in some specialized cell types.38,41,43–48 The superoxide radical is reported to have a role in controlling CysHis cathepsins in macrophages and dendritic cells.54–57 Net superoxide levels result from the relative rates of production by NADPH oxidase vs. elimination by superoxide dismutase as well as non-enzymatic reactions. The complicated reactions derived from the superoxide radical can produce an oxidative signal in low amounts and pathogenic oxidations in higher amounts. ROS or RNS might also interact with some forms of Fe or Cu to oxidize CysHis cathepsins. GSH and the anti-oxidant enzymes, superoxide dismutase, catalase, and peroxidase might all be involved in lysosomal redox of various specialized cell types.58 Mitophagy transfers the entire content of the mitochondrion to the catabolic fusion vacuole with unknown consequences.
3.2. The import of lysosomal reducing equivalents and reduction of CysHis cathepsins
In the absence of glutaredoxins, GSH activates CysHis cathepsins with less potency than equimolar DTT.32,49 Several mM GSH alone can cause an appreciable activation of cathepsins in reaction without metals. Cathepsin B activity is responsive to the ratio of GSSG/GSH as expected of a typical “redox-buffered” protein i.e. increasing GSSG causes increasing inhibition.32 Cathepsin L from various species exhibits less sensitivity to the redox ratio of GSSG/GSH (unpublished observation). It should be cautioned that GSSG peptide might compete with peptide substrates without forming a true disulfide bond.In viable tissue, the responsiveness of proteolysis to diamide vs. DTT is consistent with a contribution of thiolation–dethiolation to the control of proteases.3,4 In perfused myocardial tissue non-toxic diamide concentrations (100 μM or less), reversibly inhibited all of lysosomal and some of extra-lysosomal proteolysis (3H-leucine release from proteins). Following discontinuation of short diamide exposures, cell reductive metabolism could reverse the inhibitory action. After prolonged diamide exposures of 1–2 hours inhibitions were not completely reversible. Following oxidation, GSSG is rapidly exported from this tissue.59 Exogenous GSH is not rapidly taken up. Therefore, perfused tissue was exposed to supra-physiologic extracellular GSH concentration in order to replete cell GSH following diamide exposure. Despite toxicity of 1 mM GSH, this high concentration reversed the anti-proteolytic effect of diamide beginning after a lag period.
Consistent with studies on perfused tissue it was reported that reducing equivalents from NADPH can be transferred to the reduction of oxidized papain disulfide via thioredoxin reductase and thioredoxin.29 A lysosomal thioredoxin was later identified by its mannose-phosphate destination tag.30 The lysosomal reductase co-localizes with vacuolar proteases.31 This reductase reduces internal disulfide bonds and unfolds antigens in specialized cells of the immune system.30,31,60 However, the lysosomal reductase has been identified in other cell types. Thioredoxins exhibit broad substrate specificity. A reductase that can reduce internal disulfide bonds of proteins can also reduce oxidized sulfur at a protease surface. The lysosomal reductase can activate pro-CysHis cathepsin B and D31 (and see Section 6). Genetic polymorphism in the expression of the lysosomal thiol reductase is associated with diabetic pathogenesis.61
The extra-lysosomal origins of vacuolar reducing equivalents, and pathways into the vesicle are unknown (represented as “various reactions” in Fig. 2). The source of reduction of extra-lysosomal thioredoxins is NADPH via thioredoxin reductase. Reduction of the intra-lysosomal thioredoxin-type reductase is puzzling insofar as NADPH dos not cross membranes. Multiple enzymatic and non-enzymatic factors might serve as redox shuttles from extra-lysosomal compartments into the lysosome.32,62 Much about redox-dependent proteolysis might be learned from the unusual metabolism of apicomplexan (malarial) parasites63 and other parasites.
Many active transporters are capable of moving a wide variety of endogenous and xenobiotic molecules with molecular weights of several hundred e.g. the ABC transporters (see below). The lysosome is believed to import and export various peptides with broad specificity64,65 (Fig. 2). Cystinosis is a lysosomal storage disease causing accumulation of vacuolar cystine and lysosomal insufficiency. Cystinosis involves heritable dysfunction of the lysosomal cystine exporter “cystinosin”. Cystinosis reveals the normal function of this transporter (not shown in Fig. 2). Lysosomal export of cystine is apparently necessary because the thiol reductase cannot reduce this disulfide bond or cannot reduce it fast enough. Cystine/cysteine is part of redox signaling.66 However, the possible roles of cystine/cysteine and GSSG/GSH in lysosomal redox have not been characterized. The lysosome might import and export many redox-active or metal-binding substances that are yet to be discovered.
4. Lysosomal cycles importing and exporting Zn2+ and other metals
A slight elevation of extra-cellular Zn2+ can decrease lysosomal proteolysis47. Therefore, lysosomal Zn2+ regulation somehow communicates with extra-lysosomal and extra-cellular Zn2+.67 Lysosomal Zn2+ regulation involves trans-membrane Zn2+ gradients; therefore, it is responsive to Zn2+ in other cell compartments. In addition, cytoplasmic Zn2+ might influence signal networks and autophagy.A vast literature on metal regulation is complicated by several factors. Compartmental Zn2+ regulation results from an interaction among multiple transport mechanisms with opposing vectorial directions. Active lysosomes function under non-equilibrium conditions, with an import/export current of metal ions (Fig. 2). Reported driving forces for lysosomal Zn2+ transport include a proton gradient, a bicarbonate gradient,68 the Zn2+ electrochemical gradient, ATP and perhaps others. Different metals share some of the many transport mechanisms summarized below. For example, Zn2+ interacts competitively with the regulation of Fe2+.69–72 Although Zn2+ is emphasized here, Cu2+, Fe3+ and some other metals can also inhibit CysHis cathepsins.
It is difficult to determine the subcellular location and function of a transporter or channel. The position and transport direction of membrane-associated protein can change under membrane fusions.73 The identical metal transporter might be found in the plasma membrane-early endosome of one cell type, and the lysosome of another. Indeed endosomes can recycle to the cell surface or fuse with lysosomes. The locations and functions of various metal transporters, and the primary metal(s) transported might differ in various specialized cells.
The electrophysiology integrating lysosomal Zn2+ translocation with other lysosomal ion regulation is virtually unstudied. This topic involves the lysosomal membrane potential and regulation of all other ions, e.g. bicarbonate, chloride, magnesium, calcium, etc.74The present perspective defers to references18–23,75,76 for discussion of lysosomal electrophysiology. If Zn2+ binding to macromolecules or peptides is tight, and dissociation is slow, transport does not conform to principles of ionic electro-physiology. In addition, metallothioneins have a redox-dependent Zn2+ release mechanism.77,78
Recent progress in Zn2+ biology has been explosive. Confirmed and likely lysosomal transporters are described by several names due to the history of independent discoveries. The Transporter Classification Database (TCDB) used here is an organization approved by the International Union of Biochemistry and Molecular Biology (IUBMB).79 Many metal translocators might be directly or indirectly relevant to the lysosome. Accordingly, Fig. 2 accommodates anticipated progress by representing lysosomal Zn2+ regulation with known and unknown driving mechanisms.
As a beginning, lysosomal Zn2+ homeostasis must be organized as (a) the regulation of the “mobile” or non-bound metal ion, and (b) the regulation of the bound metal that is co-transported with other substances. Cell Zn2+ regulation includes compartmental sequestration, high affinity protein binding, and low affinity buffering involving many substances e.g. citrate, phosphate, amino acids etc.80–83 Terms used to describe the biologically effective Zn2+ concentration include “free”, “active”, “non-bound”, “un-buffered”, “non-sequestered”, “mobile”, “chelatable” etc. Perfectly free biological Zn2+ is believed to be in the picoMolar range. For ionic Zn2+ the relevant consideration is the effective electrochemical Zn2+ gradient across the lysosomal membrane, and not the total Zn2+ concentration. The cytoplasmic activity of Zn2+ is mimicked by addition of approximately 1–3 μM Zn2+ to a mixture of endogenous Zn2+ buffers at pH 7. In catalytically active lysosomes the non-bound Zn2+ concentration is presumably maintained near the permissive concentration of 0.1 μM (Fig. 1). In inactive vesicles at neutral pH the Zn2+ concentration might be much higher without the driving force of the proton gradient.
Zn2+ has six coordination sites. Binding can decrease effective collisions and “potency” of Zn2+ without eliminating all reactivity of sites remaining exposed. Therefore, the proper consideration for enzyme inhibition is the effective concentration of interactive Zn2+ sites remaining exposed under all endogenous ligands at some particular pH. Indeed, for some enzymes the effective inhibitory potency of some Zn2+ complexes can be greater than the free ion.84 Coordination of Zn2+ with some binding sites need not eliminate fluorophore interactions of ligands remaining exposed. Imaging with cytofluorimetric Zn2+ indicators does not distinguish the sequestered and non-sequestered lysosomal Zn2+ concentration, or electrochemical gradient.85–88
Several types of Zn2+ translocators have been identified; however, their driving forces, controls and functions are not well understood. Transporters of non-bound Zn2+ include several families. The mammalian genome codes a family of 14 Zip transporters (TCDB:Slc39a) and 9 ZnT transporters (TCDB:slc30a).82 Slc39a14 (or Zip 14) has been reported to be a metal/bicarbonate symporter.89 Slc30a (ZnT) transporters function as Zn2+/H+ exchangers. Slc39a transporters are reportedly found in the plasma membrane, whereas Slc30a are have been found in various organelles. The two divalent metal ion transporters (Slc11A1 and Slc11A2) couple the uphill transport of Zn2+ and downhill transport of protons. These proteins also transport Fe2+ in some cell types. ATP binding cassette (ABC) transporters couple metal transport to ATP hydrolysis. ABC transporters are fundamental to metal regulation in bacteria;90,91 however, this mechanism is not well characterized in eukaryotes. Humans have more than 50 ABC transporters. Mammalian ABC transporters are known to transport a wide variety of substances that bind Zn2+(see below). In addition to transporters, mammals have 8 passive Transient Receptor Potential (TRP) channels. These trans-membrane channels conduct non-sequestered metal ions down their electrochemical gradients. Calcium channels have been reported to pass Zn2+.92,93 Omitting the ABC transporters and calcium channels, the combined number of transporters and channels involved in ionic Zn2+ regulation exceeds 30. Many of these might influence lysosomal regulation directly or indirectly. At present, the locations, vectorial orientations, and interactions among these many transporters are not known.
Lysosomal import and export of Zn2+ that is bound to other substances can differ markedly from transport of ionic Zn2+.94 Approximately 10% of cell proteins and many other substances can bind Zn2+ with a wide range of affinities.95 It is suggested that Zn2+ might exchange between high-affinity protein binding sites directly without dissociation into solvent.96 The stable Zn2+ interactions with 2Cys2His of many Zn2+ finger proteins are well studied;97 however, Zn2+ interactions with 1Cys1His sites of proteases have received little attention. Regulatory Zn2+ structures with Cys near His include an ion channel;98 and metallothionein99 as well as the familiar Zn2+ finger motifs.
The ABC transporters include diverse members with a broad range of specificities. Peptides are exported from lysosomes and also imported into lysosomes, probably by ABC transporters.64,65 GSH and GSSG form protective complexes with metals including Zn2+; and these might share Zn2+ transport via ABC transporters.100–102 ABC transporters are involved in defenses against metal toxicity in eukaryotic cells.103,104 Much about lysosomal metal regulation might be learned from defense against overload of biological and non-biological metals. The broadly specific, ATP-dependent, organic cation transporters might transport Zn2+ that is bound to other substances. Finally, bound Zn2+ might accompany the translocation of peptides via endocytosis, exocytosis and organelle fusions e.g. mitophagy or autophagy. Indeed, a fraction of most proteins can be found in lysosomes, presumably with any Zn2+ that is bound.
4.1. Selected topics in the lysosomal import of Zn2+ and other metals
It can be readily estimated that a major route of lysosomal Zn2+ acquisition in active lysosomes is autophagic uptake of protein-bound Zn2+ followed by release upon proteolysis. Approximately 10% of the proteome has Zn2+ binding sites; and many more proteins bind with lower affinity. Metallothioneins exhibit differential protease susceptibility and intra-lysosomal metal release.105 The gross, protein-bound Zn2+ concentration is 200 μM (Fig. 2). Basal protein turnover of a typical cell is approximately 2–3% h−1. Therefore, each hour 2–3% of 200 μM protein-bound Zn2+ is released from binding sites. This release rate would increase non-sequestered cell Zn2+ concentration by 4–6 μM per hour if it were uniformly distributed. Approximately half of total protein degradation occurs within lysosomes in most cell types. The active lysosomal volume is only 10–15% of total cell volume. Therefore, 2–3 μM Zn2+ is liberated from degraded proteins into 10–15% of cell volume each hour. This liberated Zn2+ concentration would be 200–2000 times the concentration that inhibits CysHis cathepsins (0.1–1.0 μM, Fig. 1) if released from binding sites without vacuolar export. Without Zn2+ buffering by 5 mM DTT, some inhibition was observed at 0.01 μM Zn2+ (Fig. 1, bottom trace). The 0.01 μM inhibitory Zn2+ concentration is approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold below the protein-bound cell Zn2+ concentration of 200 μM. The lifetimes of lysosomes in various cells are not known with precision. However, if vesicles persist for only 0.5–1.0 h, then the Zn2+ liberated from degraded proteins would rapidly accumulate to an inhibitory concentration in the absence of export. Possible inaccuracies of these estimates are quite conservative. Thus, active lysosomes function under a cycle of massive Zn2+ acquisition on substrate proteins, liberation by proteolysis, and export by multiple routes (Fig. 2).
000 fold below the protein-bound cell Zn2+ concentration of 200 μM. The lifetimes of lysosomes in various cells are not known with precision. However, if vesicles persist for only 0.5–1.0 h, then the Zn2+ liberated from degraded proteins would rapidly accumulate to an inhibitory concentration in the absence of export. Possible inaccuracies of these estimates are quite conservative. Thus, active lysosomes function under a cycle of massive Zn2+ acquisition on substrate proteins, liberation by proteolysis, and export by multiple routes (Fig. 2).
Non-lysosomal compartments might import ionic Zn2+, and then fuse with lysosomes secondarily. Indeed, yeast cells have a Zn2+ storage vacuole for future growth under Zn2+ deficiency.106 Some mammalian cells sequester Zn2+ in vacuoles.107
4.2. Selected topics in the lysosomal export of Zn2+ and other metals
TRPM1–TRPM8 comprise a group of eight ion channels which can pass metals in either direction down the electrochemical gradient.108 Zn2+ is reported to regulate its own conductance through a TRP channel with allosteric regulatory kinetics.109–115 Zn2+ conductance through some TRP channels is believed to be partially rectified;110,111 although the present relevance is unknown. Some TRP channels are responsive to pH and phosphorylation.22,23,109–114 The TRPML-1 channel is of great interest to lysosomal function.114–117The TRPML-1 is found in the lysosomal membranes of diverse cell types. A telling feature is that heritable deficiency of TRPML-1 channel function results in a lysosomal storage disease known as mucolipidosis-IV.117 Many variants of lysosomal storage diseases affect all cell types; however, the life-limiting injury is to neural tissue. Mucolipidosis results in accumulation of undegraded macromolecules in large lysosomes of the brain. Eichelsdoerfer et al.118 reported: …“the loss of TRPML1 function results in intracellular chelatable zinc dyshomeostasis. We propose that chelatable zinc accumulation in large lysosomes and membranous vacuoles may contribute to the pathogenesis of the disease and progressive cell degeneration in ML-IV patients”… This disease of denatured protein accumulation implies that a normal function of the TRPML-1 is net export of lysosomal Zn2+ and perhaps other cell Zn2+ regulation. Mitochondria are also affected. The drosophila ortholog of TRPML-1 is necessary for normal lysosomal function.119 The TRPML-1 does not account for all routes of lysosomal Zn2+ export. Complete loss of lysosomal Zn2+ export would presumably be embryolethal. Loss of TRPML-1 channel function is also reported to cause disruption of lysosomes and release of cathepsin B into the cytoplasm.81,120 The normal TRPML-1 channel somehow serves in ionic and volume control of the vacuole.121–124 The TRPML-1 channel convincingly reveals that Zn2+ export is related to lysosomal function as well as the CysHis cathepsin reaction mechanism (Fig. 1 and 2).
The macrophage protein named “Natural Resistance Associated Membrane Protein 1 (NRAMP1 or SLC11A1) is also fundamentally related to lysosomal function. This protein was originally discovered by the association of microbial infections with loss of its function, hence the name. This gene family has two members: Slc11A1 and Slc11A2; the former is of most interest here. Slc11A1 is expressed in phagocytes, and exports metals from the phagosome. The driving force for uphill lysosomal metal transport is coupling to downhill proton transport. Curiously, insufficient function of Slc11A1 is somehow associated with inability to defend against microbes; and excessive function is associated with a variety of hyper-immune immune syndromes independent of microbes.
Heritable or regulatory alteration of protein function can be caused by structural gene mutations, variations in gene copy number, altered regulation of gene transcription, alternative transcript splicing, RNA interference etc. Beyond the present scope, these combined mechanisms can lead to a wide range of protein content and function among individuals. Regardless of causative mechanism(s), the present theory is the only way to account for all pathogenic results of both insufficient and excess functions of lysosomal metal export.
Slc11A1 can export multiple divalent metal cations from lysosomes, including Zn2+ and Fe2+. The normal function of Slc11A1 was believed to be starvation of phagocytosed microbes for Fe2+ as a growth requirement in specialized phagocytes. This microbicidal mechanism is currently known as “metal withdrawal defense”.125 The mechanism of immune deficiency that is associated with decreased Slc11A1 function is currently believed to be a failure to starve phagocytosed microbes for Fe2+ and other metals. It has not been appreciated that metal withdrawal defense might interact with an additional microbicidal mechanism of the Slc11A1 exporter. Normal export of inhibitory metals from phagosomes/lysosomes could activate CysHis cathepsins, and increase the degradation of captured microbes while starving them for metal requirements. Conversely, deficiency in the lysosomal export of inhibitory metals would explain protection of endocytosed microbes against destruction by CysHis cathepsins.
Whereas deficient Slc11A1 function is associated with immune deficiency, heritable up-regulation is associated with hyper-immunity. The hyper-immunity that is associated with excessive expression of Slc11A1 is obviously not related to starvation of phagocytosed microbes for Fe. Therefore, the theory of “metal withdrawal defense” cannot account for the pathogenic results of excessive function of the Slc11A1 transporter. It is well known that lysosomal CysHis cathepsins are required for proteolytic antigen processing to immunogenic peptides and presentation to specialized cells of the immune system.126–128 Most investigators believe that excessive cell content of the Slc11A1 somehow increases lysosomal proteolytic processing of auto-antigens to immunogenic peptides, thereby increasing auto-immunity. Several CysHis cathepsins are involved in antigen processing.7 Autoimmunity can be associated with heritable up-regulation of Slc11A1 expression. Many studies demonstrate a relationship between excessive expression of the Slc11A1 transporter and the auto-immunity of diabetic progression.129–142 The result of excessive function of Slc11A1 confirms the relevance of metal export to lysosomal proteolytic function.
Hyper-immunity might be also be associated with excessive lysosomal export of inhibitory Zn2+ and Fe2+via Slc39A8 (Zip-8). Slc 39A8/ZIP-8 is localized to the lysosomes of at least some cell types. In other cells Slc 39A8 is located on the cell surface and in early endosomes.69–72 In a model of T-cell activation and antigen processing, lysosomal Slc 39A8 is increased in association with interferon production.143 A role of Slc39A8 in lysosomal Zn2+ export upon T-cell activation has been suggested. Such an interpretation would be consistent with an associated role of Slc39A8 in cathepsin activation.
In summary, the normal functions of lysosomal metal regulation in catabolic control are revealed by the results of dysregulations. Defective TRP channel structure, or down-regulation of the proton-driven exporter alone can decrease lysosomal Zn2+ export, and cause immune deficiency or a lysosomal storage disease. At the other extreme, excessive function of proton-driven, lysosomal, metal export is associated with excessive lysosomal metal export, and diverse hyper-immune syndromes, e.g. progressive diabetic tissue damage. Optimal lysosomal function exists within some intermediate range between pathogenic excess and insufficiency. All of these observations can be explained by a regulatory contribution of Zn2+, and other metals, to CysHis cathepsin function.
5. The energy dependence of lysosomal cycles
The hydrolytic reaction catalyzed by CysHis cathepsins is driven by energy released from the peptide bond. Therefore, the catalyzed reaction is ultimately exergonic. However, maintenance of the maximally active CysHis catalyst has a continual energy requirement. ATP is required for metal export and protonation of the CysHis reaction; and reducing energy is required to oppose protease oxidations. The high-energy phosphate network and redox network are distinguishable systems. After a century of investigation, relationships between the high-energy phosphate network and the redox network involve unanswered questions.144,145 Nonetheless, interventions with traditional agents of known actions provide limited information for correlation with future advances.Experimental demonstration of an ATP requirement for a hypothetical process can lead to the conclusion that fluctuations in cell ATP content control that process. However, a requirement for ATP does not necessarily demonstrate regulation of the process by the permissible range of cell ATP fluctuations. Many observed phenomena are co-variables of injury and death under extreme experimental ATP depletion. If the ATP content of a tissue can be decreased to the minimal amount required for sustained viability without changing lysosomal function, then the ATP requirement for proton pumping would not seem to control lysosomal cycles. In perfused myocardium, repeated mitochondrial poisoning and ATP depletion did not decrease lysosomal proteolysis until immediately before functional demise and death.4 Thus, the minimal amount of ATP needed to sustain viability of myocardial tissue also suffices to sustain lysosomal cycles. In marked contrast, interventions with non-toxic concentrations of sulfhydryl oxidants and reductants cause large fluctuations in bulk protein degradation with absolutely no change in tissue ATP content or contractile function. Unnecessarily high or prolonged exposures to redox active agents were injurious. Conclusions from mammalian myocardial tissue might not typify the wide range of metabolic conditions observed in all plants, animals and microbes. It is likely that severe, sublethal ATP depletion can limit lysosomal proton pumping and catalysis in some species under some conditions.
6. Beyond the fact that pro-cathepsin B is a member of the Zn2+ – binding “metallome”
Most of the cell content of lysosomal hydrolases consists of inactive pro-enzymes. The simultaneous activation of more than 60 acid hydrolases by cleavage of pro-regions remains unexplained.14 Active lysosomal proteases can cleave the pro-regions of inactive pro-proteases and other pro-hydrolases. Therefore, all lysosomal proteases can be considered to be convertases activating other lysosomal enzymes. For this reason, it has been presumed that the activation of pro-hydrolases is determined by the ongoing rate of vacuolar proteolysis; and this is not inconsistent with the present theory.The control of ongoing CysHis peptidolysis by MR&P cycles could influence normal or abnormal activation of other pro-lysosomal enzymes. Some investigators suspect that lowering pH alone causes increasing activation of pro-hydrolases.21 However, lysosomal pH is only one component of the MR&P triumvirate. It has been suggested that the lysosomal reductase system can contribute to activation of pro-cathepsins B and D.31 However, it has not been suggested that all three lysosomal cycles conspire to determine the activation of all pro-lysosomal enzymes. The present theory agrees that reducing energy and acidity are positive influences on pro-protease activation; however, it has not been considered that Zn2+ might preemptively oppose these activating influences.
Pro-cathepsin B was discovered objectively in a screen of the Zn2+ – binding proteome.10 A speculative possibility is that cathepsin B is a primary “trigger protease” for the activation of other lysosomal enzymes, at least in cell types where it is abundant. Cathepsin B can be promptly activated ex vivo either by non-specific inter-molecular proteolytic cleavage of the pro-region or decreasing pH below 5. Cathepsin B has a unexplained structure called the occluding loop. The pro-region of Cathepsin B is folded over its catalytic dyad. It has been suggested that cathepsin B auto-activates by self-cleavage of its own pro-region under a pH-dependent conformational change in the occluding loop.146 Bound Zn2+ at the catalytic dyad might suppress such self-cleavage of the pro-region by enzymatic activity.
It remains to be determined whether cathepsin B is the primary convertase activating other proteases in response to lysosomal cycles. Metallomic techniques might not detect metal binding to other pro-proteases that are present in lower amounts. Curiously, gene deletion reveals that cathepsin B is not critical to the life of the mouse due to a degree of functional redundancy. Therefore, cathepsin B cannot be the only converting mechanism activating lysosomal enzymes. The question of whether pro-hydrolase activation by MR&P involves cathepsin B or all CysHis cathepsins might depend upon the relative enzyme contents of particular cell types. The liver has a very high content of cathepsin B. Massive hepatic failure causes the lethal, life-limiting tissue damage 12–24 h following overdose of TNFα. As described above, knockout of the cathepsin B gene alone protects against this lethal receptor-mediated liver damage.8
7. The recycling of amino acids from old proteins to new proteins in association with recycling of metals from degraded binding sites to new binding sites
Recycling of Zn2+ from degraded proteins to new proteins has all features expected of a crude coordinator of lysosomal and perhaps some extra-lysosomal pathways of proteolysis. Limited studies indicate that extra-lysosomal proteolysis is indeed responsive to redox and Zn2+. This responsiveness includes caspases,147 calpains,148 and the ubiquitin-proteasome pathway.149–153 Cathepsin B has been found in extra-lysosomal compartments, and is also suspect of extra-lysosomal functions. Oxidative stress is a widely used term that has not been defined. Moderate oxidative stress (or reductive insufficiency) can be a non-injurious part of metabolic control. Moderate oxidative conditions can decrease apoptosis in some cell types.154 However, changes induced by Zn2+ and/or redox can involve higher order enzyme structure as well as the catalytic mechanisms discussed here.149,150The reaction mechanism of an active caspase or calpain might serve its specialized function at a small fraction of the maximal catalytic rate. Indeed, non-injurious operation of CysHis cathepsins in vivo might correspond to a small fraction of the optimized rate shown in Fig. 1. Relative sensitivities of lysosomal and extra-lysosomal proteolytic pathways to Zn2+ and redox are uncharacterized. Zn2+ might contribute to coordination of multiple proteolytic pathways. Interestingly, metalloproteases and metallopeptidases require Zn2+ for activity, and do not require a reductive activator. Lysosomal dipeptidase is such a metal-redox independent enzyme.155 In summary, diverse extra-lysosomal proteolytic systems are sensitive to Zn2+ and redox; however, this responsiveness includes higher-order protein structure.
Pharmacologic interventions against excessive CysHis cathepsin activity can have effective anti-inflammatory actions against diabetic pathogenesis. The most effective drug against progressive diabetic pathogenesis is an anti-lysosomal “zincophore”.2 However, drug development156,157 must be cautious. Normal lysosomal function requires a mid-range between excessive and insufficient function. Many other causes of denatured protein accumulation might interact with excessive anti-lysosomal therapy e.g. Parkinson's disease,158 Alzheimers disease,159 and many lysosomal storage diseases.160
References
- T. D. Lockwood, Cys-His proteases are among the wired proteins of the cell, Arch. Biochem. Biophys., 2004, 432, 12–24 Search PubMed.
- T. D. Lockwood, The lysosome among targets of metformin: new anti-inflammatory uses for an old drug?, Expert Opin. Ther. Targets, 2010, 14, 467–478 Search PubMed.
- D. P. Thorne and T. D. Lockwood, Four proteolytic processes of myocardium, one insensitive to thiol reactive agents and thiol protease inhibitor, Am. J. Physiol., 1993, 265, E10–E19 Search PubMed.
- T. D. Lockwood, Redox-dependent and redox-independent subcomponents of protein degradation in perfused myocardium, Am. J. Physiol., 1999, 276, E945–E954 Search PubMed.
- J. Dennemärker, T. Lohmüller, S. Müller, S. V. Aguilar, D. J. Tobin, C. Peters and T. Reinheckel, Impaired turnover of autophagolysosomes in cathepsin L deficiency, Biol. Chem., 2010, 391, 913–922 Search PubMed.
- R. Maehr, J. D. Mintern, A. E. Herman, A. M. Lennon-Dumenil, D. Mathis, C. Benoist and H. L. Ploegh, Cathepsin L is essential for onset of autoimmune diabetes in NOD mice, J. Clin. Invest., 2005, 115, 2934–2943 Search PubMed.
- L. C. Hsing, E. A. Kirk, T. S. McMillen, S. H. Hsiao, M. Caldwell, B. Houston, A. Y. Rudensky and R. C. LeBoeuf, Roles for cathepsins S, L, and B in insulitis and diabetes in the NOD mouse, Autoimmunity, 2010, 34, 96–104 Search PubMed.
- M. E. Guicciardi, H. Miyoshi, S. F. Bronk and G. J. Gores, Cathepsin B knockout mice are resistant to tumor necrosis factor-alpha mediated hepatocyte apoptosis and liver injury: implications for therapeutic applications, Am. J. Pathol., 2001, 159, 2045–2054 Search PubMed.
- J. I. Odegaard and A. Chawla, Connecting Type 1 and Type 2 Diabetes through Innate Immunity, Cold Spring Harbor Perspect. Med., 2012, 2(3), a007724 Search PubMed.
- J. S. Becker, S. Mounicou, M. V. Zoriy, J. S. Becker and R. Lobinski, Analysis of metal-binding proteins separated by non-denaturating gel electrophoresis using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) and laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS), Talanta, 2008, 76, 1183–1188 CrossRef CAS.
- S. M. Marino, Y. Li, D. E. Fomenko, N. Agisheva, R. L. Cerny and V. N. Gladyshev, Characterization of surface-exposed reactive cysteine residues in Saccharomyces cerevisiae, Biochemistry, 2010, 49, 7709–7721 Search PubMed.
- M. Thamsen and U. Jakob, The redoxome: Proteomic analysis of cellular redox networks, Curr. Opin. Chem. Biol., 2011, 15, 113–119 Search PubMed.
- D. P. Jones and Y. M. Go, Mapping the cysteine proteome: analysis of redox-sensing thiols, Curr. Opin. Chem. Biol., 2011, 15, 103–112 Search PubMed.
- J. Reiser, B. Adair and T. Reinheckel, Specialized roles for cysteine cathepsins in health and disease, J. Clin. Invest., 2010, 120, 3421–3431 Search PubMed.
- M. J. Sousa, F. Azevedo, A. Pedras, C. Marques, O. P. Coutinho, A. Preto, H. Gerós, S. R. Chaves and M. Côrte-Real, Vacuole-mitochondrial cross-talk during apoptosis in yeast: a model for understanding lysosome-mitochondria-mediated apoptosis in mammals, Biochem. Soc. Trans., 2011, 39, 1533–1537 Search PubMed.
- K. Niemi, L. Teirilä, J. Lappalainen, K. Rajamäki, M. H. Baumann, K. Öörni, H. Wolff, P. T. Kovanen, S. Matikainen and K. K. Eklund, Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway, J. Immunol., 2011, 186, 6119–6128 Search PubMed.
- M. Volpicella, C. Leoni, A. Costanza, F. De Leo, R. Gallerani and L. R. Ceci, Cystatins, serpins and other families of protease inhibitors in plants, Curr. Protein Pept. Sci., 2011, 12, 386–398 Search PubMed.
- B. E. Steinberg, K. K. Huynh, A. Brodovitch, S. Jabs, T. Stauber, T. J. Jentsch and S. Grinstein, A cation counterflux supports lysosomal acidification, J. Cell Biol., 2010, 189, 1171–1186 Search PubMed.
- L. Wartosch and T. Stauber, A role for chloride transport in lysosomal protein degradation, Autophagy, 2010, 6, 158–159 Search PubMed.
- J. E. DiCiccio and B. E. Steinberg, Lysosomal pH and analysis of the counter ion pathways that support acidification, J. Gen. Physiol., 2011, 137, 385–390 Search PubMed.
- J. A. Mindell, Lysosomal acidification mechanisms, Annu. Rev. Physiol., 2012, 74, 69–86 Search PubMed.
- J. A. Kozak, M. Matsushita, A. C. Nairn and M. D. Cahalan, Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels, J. Gen. Physiol., 2005, 126, 499–514 Search PubMed.
- R. H. Chokshi, M. Matsushita and J. A. Kozak, Detailed examination of Mg2+ and pH sensitivity of human TRPM7 channels, Am. J. Physiol.: Cell Physiol., 2012, 302, C1004–C1011 Search PubMed.
- N. S. Kosower and E. M. Kosower, Diamide: an oxidant probe for thiols, Methods Enzymol., 1995, 251, 123–133 CAS.
- R. E. Hansen, D. Roth and J. R. Winther, Quantifying the global cellular thiol-disulfide status, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 422–427 Search PubMed.
- L. M. Grimm, M. W. Collison, R. A. Fisher and J. A. Thomas, Protein mixed-disulfides in cardiac cells, S-thiolation of soluble proteins in response to diamide, Biochim. Biophys. Acta, 1985, 844, 50–54 Search PubMed.
- M. W. Collison, D. Beidler, L. M. Grimm and J. A. Thomas, A comparison of protein S-thiolation (protein mixed-disulfide formation) in heart cells treated with t-butyl hydroperoxide or diamide, Biochim. Biophys. Acta, 1986, 885, 58–67 Search PubMed.
- A. J. Cooper, J. T. Pinto and P. S. Callery, Reversible and irreversible protein glutathionylation: biological and clinical aspects, Expert Opin. Drug Metab. Toxicol., 2011, 7, 891–910 Search PubMed.
- A. G. Stephen, R. Powls and R. J. Beynon, Activation of oxidized cysteine proteinases by thioredoxin-mediated reduction in vitro, Biochem. J., 1993, 291(Pt 2), 345–347 Search PubMed.
- B. Arunachalam, U. T. Phan, H. J. Geuze and P. Cresswell, Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT), Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 745–750 CrossRef CAS.
- O. G. Goldstein, L. M. Hajiaghamohseni, S. Amria, K. Sundaram, S. V. Reddy and A. Haque, Gamma-IFN-inducible-lysosomal thiol reductase modulates acidic proteases and HLA class II antigen processing in melanoma, Cancer Immunol. Immunother., 2008, 57, 1461–1470 Search PubMed.
- T. D. Lockwood, Cathepsin B responsiveness to glutathione and lipoic acid redox, Antioxid. Redox Signaling, 2002, 4, 681–691 Search PubMed.
- S. Prast-Nielsen, H. H. Huang and D. L. Williams, Thioredoxin glutathione reductase: its role in redox biology and potential as a target for drugs against neglected diseases, Biochim. Biophys. Acta, 2011, 1810, 1262–1271 Search PubMed.
- C. Schöneich, Mechanisms of metal-catalyzed oxidation of histidine to 2-oxo-histidine in peptides and proteins, J. Pharm. Biomed. Anal., 2000, 21, 1093–1097 CrossRef CAS.
- D. A. Traoré, A. El Ghazouani, L. Jacquamet, F. Borel, J. L. Ferrer, D. Lascoux, J. L. Ravanat, M. Jaquinod, G. Blondin, C. Caux-Thang, V. Duarte and J. M. Latour, Structural and functional characterization of 2-oxo-histidine in oxidized PerR protein, Nat. Chem. Biol., 2009, 5, 53–59 CrossRef CAS.
- N. J. Bulleid and L. Ellgaard, Multiple ways to make disulfides, Trends Biochem. Sci., 2011, 36, 485–492 Search PubMed.
- B. Turk, V. Turk and D. Turk, Structural and functional aspects of papain-like cysteine proteinases and their protein inhibitors, Biol. Chem., 1997, 378, 141–150 Search PubMed.
- A. T. Saurin, H. Neubert, J. P. Brennan and P. Eaton, Widespread sulfenic acid formation in tissues in response to hydrogen peroxide, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17982–17987 CrossRef CAS.
- M. Fratelli, E. Gianazza and P. Ghezzi, Redox proteomics: identification and functional role of glutathionylated proteins, Expert Rev. Proteomics, 2004, 1, 365–376 Search PubMed , Review.
- J. M. Hansen, H. Zhang and D. P. Jones, Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions, Free Radicals Biol. Med., 2005, 40, 138–145 Search PubMed.
- G. Venturini, E. Fioravanti, M. Colasanti, T. Persichini and P. Ascenzi, Cys25-nitrosylation inactivates papain, Biochem. Mol. Biol. Int., 1998, 46, 425–428 Search PubMed.
- M. Xian, X. Chen, Z. Liu, K. Wang and P. G. Wang, Inhibition of papain by S-nitrosothiols, Formation of mixed disulfides, J. Biol. Chem., 2000, 275, 20467–20473 CrossRef CAS.
- R. Sengupta and A. Holmgren, The role of thioredoxin in the regulation of cellular processes by S-nitrosylation, Biochim. Biophys. Acta, 2012, 1820, 689–700 Search PubMed.
- M. W. Foster, M. T. Forrester and J. S. Stamler, A protein microarray-based analysis of S-nitrosylation, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18948–18953 CrossRef CAS.
- G. Venturini, E. Fioravanti, M. Colasanti, T. Persichini and P. Ascenzi, Cys25-nitrosylation inactivates papain, Biochem. Mol. Biol. Int., 1998, 46, 425–428 Search PubMed.
- N. J. Kettenhofen and M. J. Wood, Formation, reactivity, and detection of protein sulfenic acids, Chem. Res. Toxicol., 2010, 23, 1633–1646 Search PubMed.
- M. D. Percival, M. Ouellet, C. Campagnolo, D. Claveau and C. Li, Inhibition of cathepsin K by nitric oxide donors: evidence for the formation of mixed disulfides and a sulfenic acid, Biochemistry, 1999, 38, 13574–13583 Search PubMed.
- W. S. Lin and D. A. Armstrong, Glutathione mediation of papain inactivation by hydrogen peroxide and hydroxyl radicals, Radiat. Res., 1997, 69, 434–441 Search PubMed.
- G. Lemaire, O. Guittet, M. F. Vesin, M. Lepoivre and M. H. Cottet, Glutathione depletion reveals impairment of antigen processing and inhibition of cathepsin activity by nitric oxide in antigen-presenting cells, Mol. Immunol., 2008, 46, 1100–1108 Search PubMed.
- T. J. Tavender and N. J. Bulleid, Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum, Antioxid. Redox Signaling, 2010, 13, 1177–1187 Search PubMed.
- S. Kim, D. P. Sideris, C. S. Sevier and C. A. Kaiser, Balanced Ero1 activation and inactivation establishes ER redox homeostasis, J. Cell Biol., 2012, 196, 713–725 Search PubMed.
- G. Bánhegyi, E. Margittai, A. Szarka, J. Mandl and M. Csala, Crosstalk and barriers between the electron carriers of the endoplasmic reticulum, Antioxid. Redox Signaling, 2012, 16, 772–780 Search PubMed.
- T. D. Lockwood, Inactivation of intracellular proteolysis and cathepsin B enzyme activity by dehydroascorbic acid and reactivation by dithiothreitol in perfused rat heart, Biochem. Pharmacol., 1997, 54, 669–675 Search PubMed.
- E. Godat, V. Hervé-Grvépinet, F. Veillard, F. Lecaille, M. Belghazi, D. Brömme and G. Lalmanach, Regulation of cathepsin K activity by hydrogen peroxide, Biol. Chem., 2008, 389, 1123–1126 Search PubMed.
- F. Meissner, K. Molawi and A. Zychlinsky, Superoxide dismutase 1 regulates caspase-1 and endotoxic shock, Nat. Immunol., 2008, 9, 866–872 Search PubMed.
- J. M. Rybicka, D. R. Balce, M. F. Khan, R. M. Krohn and R. M. Yates, NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes, Proc. Natl. Acad. Sci. U. S. A., 2011, 107, 10496–10501 Search PubMed.
- J. M. Rybicka, D. R. Balce, S. Chaudhuri, E. R. Allan and R. M. Yates, Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner, EMBO J., 2011, 31, 932–944 Search PubMed.
- M. P. Murphy, A. Holmgren, N. G. Larsson, B. Halliwell, C. J. Chang, B. Kalyanaraman, S. G. Rhee, P. J. Thornalley, L. Partridge, D. Gems, T. Nyström, V. Belousov, P. T. Schumacker and C. C. Winterbourn, Unraveling the biological roles of reactive oxygen species, Cell Metab., 2011, 13, 361–366 Search PubMed.
- T. Ishikawa, H. Esterbauer and H. Sies, Role of cardiac glutathione transferase and of the glutathione S-conjugate export system in biotransformation of 4-hydroxynonenal in the heart, J. Biol. Chem., 1986, 261, 1576–1581 CAS.
- R. Singh and P. Cresswell, Defective cross-presentation of viral antigens in GILT-free mice, Science, 2010, 328, 1394–1398 Search PubMed.
- V. Turcot, L. Bouchard, G. Faucher, A. Tchernof, Y. Deshaies, L. Pérusse, P. Marceau, F. S. Hould, S. Lebel and M. C. Vohl, A polymorphism of the interferon-gamma-inducible protein 30 gene is associated with hyperglycemia in severely obese individuals, Hum. Genet., 2011, 131, 57–66 Search PubMed.
- M. A. Feeney, K. Veeravalli, D. Boyd, S. Gon, M. J. Faulkner, G. Georgiou and J. Beckwith, Repurposing lipoic acid changes electron flow in two important metabolic pathways of Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7991–7996 Search PubMed.
- S. C. Maughan, M. Pasternak, N. Cairns, G. Kiddle, T. Brach, R. Jarvis, F. Haas, J. Nieuwland, B. Lim, C. Müller, E. Salcedo-Sora, C. Kruse, M. Orsel, R. Hell, A. J. Miller, P. Bray, C. H. Foyer, J. A. Murray, A. J. Meyer and C. S. Cobbett, Plant homologs of the Plasmodium falciparum chloroquine-resistance transporter, PfCRT, are required for glutathione homeostasis and stress responses, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2331–2336 Search PubMed.
- M. Thamotharan, Y. B. Lombardo, S. Z. Bawani and S. A. Adibi, An active mechanism for completion of the final stage of protein degradation in the liver, lysosomal transport of dipeptides, J. Biol. Chem., 1997, 272, 11786–11790 Search PubMed.
- I. Bangert, F. Tumulka and R. Abele, The lysosomal polypeptide transporter TAPL: more than a housekeeping factor?, Biol. Chem., 2011, 392, 61–66 Search PubMed.
- Y. M. Go and D. P. Jones, Cysteine/cystine redox signaling in cardiovascular disease, Free Radicals Biol. Med., 2011, 50, 495–509 Search PubMed.
- J. Gibon, P. Tu, S. Bohic, P. Richaud, J. Arnaud, M. Zhu, G. Boulay and A. Bouron, The over-expression of TRPC6 channels in HEK-293 cells favours the intracellular accumulation of zinc, Biochim. Biophys. Acta, 2011, 1808, 2807–2818 Search PubMed.
- D. W. Nebert, M. Gálvez-Peralta, E. B. Hay, H. Li, E. Johansson, C. Yin, B. Wang, L. He and M. Soleimani, ZIP14 and ZIP8 zinc/bicarbonate symporters in Xenopus oocytes: characterization of metal uptake and inhibition, Metallomics, 2012, 4, 1218–1225 RSC.
- J. J. Pinilla-Tenas, B. K. Sparkman, A. Shawki, A. C. Illing, C. J. Mitchell, N. Zhao, J. P. Liuzzi, R. J. Cousins, M. D. Knutson and B. Mackenzie, Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron, Am. J. Physiol.: Cell Physiol., 2011, 301, C862–C871 CrossRef CAS.
- A. C. Illing, A. Shawki, C. L. Cunningham and B. Mackenzie, Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1, J. Biol. Chem., 2012, 287, 30485–30496 Search PubMed.
- S. Jenkitkasemwong, C. Y. Wang, B. Mackenzie and M. D. Knutson, Physiologic implications of metal-ion transport by ZIP14 and ZIP8, Biometals, 2012, 25, 643–655 Search PubMed.
- C. Y. Wang, S. Jenkitkasemwong, S. Duarte, B. K. Sparkman, A. Shawki, B. Mackenzie and M. D. Knutson, ZIP8 Is an Iron and Zinc Transporter Whose Cell-surface Expression Is Up-regulated by Cellular Iron Loading, J. Biol. Chem., 2012, 287, 34032–34043 Search PubMed.
- A. C. Cerny and A. Huber, Regulation of TRP signalling by ion channel translocation between cell compartments, Adv. Exp. Med. Biol., 2011, 704, 545–572 Search PubMed.
- A. Shawki and B. Mackenzie, Interaction of calcium with the human divalent metal-ion transporter-1, Biochem. Biophys. Res. Commun., 2010, 393, 471–475 Search PubMed.
- T. Stauber and T. J. Jentsch, Chloride in Vesicular Trafficking and Function, Annu. Rev. Physiol., 2012, Oct, 17 Search PubMed.
- M. Toei, R. Saum and M. Forgac, Regulation and isoform function of the V-ATPases, Biochemistry, 2010, 49, 4715–4723 Search PubMed.
- W. Maret, Redox biochemistry of mammalian metallothioneins, J. Biol. Inorg. Chem., 2011, 16, 1079–1086 CrossRef CAS.
- W. Maret, New perspectives of zinc coordination environments in proteins, J. Inorg. Biochem., 2012, 111, 110–116 Search PubMed.
- M. H. Saier Jr, M. R. Yen, K. Noto, D. G. Tamang and C. Elkan, The Transporter Classification Database: recent advances, Nucleic Acids Res., 2009, 37, D274–D278 Search PubMed.
- K. A. Jackson, R. A. Valentine, L. J. Coneyworth, J. C. Mathers and D. Ford, Mechanisms of mammalian zinc-regulated gene expression, Biochem. Soc. Trans., 2008, 36, 1262–1266 CrossRef CAS.
- K. Kiselyov, G. A. Colletti, A. Terwilliger, K. Ketchum, C. W. Lyons, J. Quinn and S. Muallem, TRPML: Transporters of metals in lysosomes: essential for cell survival?, Cell Calcium, 2011, 50, 288–294 Search PubMed.
- T. Fukada and T. Kambe, Molecular and genetic features of zinc transporters in physiology and pathogenesis, Metallomics, 2011, 3, 662–674 RSC.
- D. E. Sutherland and M. J. Stillman, The “magic numbers” of metallothionein, Metallomics, 2011, 3, 444–463 RSC.
- D. Sweeney, M. L. Raymer and T. D. Lockwood, Antidiabetic and antimalarial biguanide drugs are metal-interactive antiproteolytic agents, Biochem. Pharmacol., 2003, 66, 663–677 CrossRef CAS.
- E. M. Nolan and S. J. Lippard, Small-molecule fluorescent sensors for investigating zinc metalloneurochemistry, Acc. Chem. Res., 2009, 42, 193–203 CrossRef CAS.
- E. Tomat and S. J. Lippard, Imaging mobile zinc in biology, Curr. Opin. Chem. Biol., 2010, 14, 225–230 CrossRef CAS.
- M. D. Pluth, E. Tomat and S. J. Lippard, Biochemistry of mobile zinc and nitric oxide revealed by fluorescent sensors, Annu. Rev. Biochem., 2011, 80, 333–355 CrossRef CAS.
- J. E. Kwon, S. Lee, Y. You, K. H. Baek, K. Ohkubo, J. Cho, S. Fukuzumi, I. Shin, S. Y. Park and W. Nam, Fluorescent Zinc Sensor with Minimized Proton-Induced Interferences: Photophysical Mechanism for Fluorescence Turn-On Response and Detection of Endogenous Free Zinc Ions, Inorg. Chem., 2012, 51, 8760–8774 Search PubMed.
- K. Girijashanker, L. He, M. Soleimani, J. M. Reed, H. Li, Z. Liu, B. Wang, T. P. Dalton and D. W. Nebert, Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter, Mol. Pharmacol., 2008, 73, 1413–1423 CrossRef CAS.
- J. S. Klein and O. Lewinson, Bacterial ATP-driven transporters of transition metals: physiological roles, mechanisms of action, and roles in bacterial virulence, Metallomics, 2011, 3, 1098–1108 RSC.
- C. M. Vahling-Armstrong, H. Zhou, L. Benyon, J. K. Morgan and Y. Duan, Two plant bacteria, S. meliloti and Ca. Liberibacter asiaticus, share functional znuABC homologues that encode for a high affinity zinc uptake system, PLoS One, 2012, 7, e37340 Search PubMed.
- S. Hou, L. E. Vigeland, G. Zhang, R. Xu, M. Li, S. H. Heinemann and T. Hoshi, Zn2+ activates large conductance Ca2+ -activated K+ channel via an intracellular domain, J. Biol. Chem., 2010, 285, 6434–6442 Search PubMed.
- G. Kovacs, T. Danko, M. J. Bergeron, B. Balazs, Y. Suzuki, A. Zsembery and M. A. Hediger, Heavy metal cations permeate the TRPV6 epithelial cation channel, Cell Calcium, 2011, 49, 43–55 Search PubMed.
- S. A. Sinclair and U. Krämer, The zinc homeostasis network of land plants, Biochim. Biophys. Acta, 2012, 1823, 1553–1567 Search PubMed.
- L. Decaria, I. Bertini and R. J. Williams, Zinc proteomes, phylogenetics and evolution, Metallomics, 2010, 2, 706–709 RSC.
- L. C. Costello, C. C. Fenselau and R. B. Franklin, Evidence for operation of the direct zinc ligand exchange mechanism for trafficking, transport, and reactivity of zinc in mammalian cells, J. Inorg. Biochem., 2011, 105, 589–599 Search PubMed.
- T. Simonson and N. Calimet, Cys(x)His(y)-Zn2+ interactions: thiol vs. thiolate coordination, Proteins, 2002, 49, 37–48 CrossRef CAS.
- H. Hu, M. Bandell, M. J. Petrus, M. X. Zhu and A. Patapoutian, Zinc activates damage-sensing TRPA1 ion channels, Nat. Chem. Biol., 2009, 5, 183–190 Search PubMed.
- O. I. Leszczyszyn, C. R. White and C. A. Blindauer, The isolated Cys2His2 site in EC metallothionein mediates metal-specific protein folding, Mol. Biosyst., 2010, 6, 1592–1603 RSC.
- M. W. Carew, H. Naranmandura, C. B. Shukalek, X. C. Le and E. M. Leslie, Monomethylarsenic diglutathione transport by the human multidrug resistance protein 1 (MRP1/ABCC1), Drug Metab. Dispos., 2011, 39, 2298–2304 CrossRef CAS.
- A. Kreżel, J. Wójcik, M. Maciejczyk and W. Bal, Zn(II) complexes of glutathione disulfide: structural basis of elevated stabilities, Inorg. Chem., 2011, 50, 72–85 Search PubMed.
- E. M. Leslie, Arsenic-glutathione conjugate transport by the human multidrug resistance proteins (MRPs/ABCCs), J. Inorg. Biochem., 2012, 108, 141–149 Search PubMed.
- Y. Long, Q. Li, Y. Wang and Z. Cui, MRP proteins as potential mediators of heavy metal resistance in zebrafish cells, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2011, 153, 310–317 Search PubMed.
- S. H. Hahn, O. J. Yoo and W. A. Gahl, Effect of metal ions on the stability of metallothionein in the degradation by cellular fractions in vitro, Exp. Mol. Med., 2001, 33, 32–36 Search PubMed.
- J. M. McKim Jr, S. Choudhuri and C. D. Klaassen, In vitro degradation of apo-, zinc-, and cadmium-metallothionein by cathepsins B, C, and D, Toxicol. Appl. Pharmacol., 1992, 116, 117–124 Search PubMed.
- C. Simm, B. Lahner, D. Salt, A. LeFurgey, P. Ingram, B. Yandell and D. J. Eide, Saccharomyces cerevisiae vacuole in zinc storage and intracellular zinc distribution, Eukaryotic Cell, 2007, 6, 1166–1177 Search PubMed.
- H. C. Roh, S. Collier, J. Guthrie, J. D. Robertson and K. Kornfeld, Lysosome-related organelles in intestinal cells are a zinc storage site in C. elegans, Cell Metab., 2012, 15, 88–99 Search PubMed.
- B. Nilius and G. Owsianik, The transient receptor potential family of ion channels, Genome Biol., 2011, 12, 218 Search PubMed.
- S. Lambert, A. Drews, O. Rizun, T. F. Wagner, A. Lis, S. Mannebach, S. Plant, M. Portz, M. Meissner, S. E. Philipp and J. Oberwinkler, Transient receptor potential melastatin 1 (TRPM1) is an ion-conducting plasma membrane channel inhibited by zinc ions, J. Biol. Chem., 2011, 286, 12221–12233 Search PubMed.
- W. Yang, P. T. Manna, J. Zou, J. Luo, D. J. Beech, A. Sivaprasadarao and L. H. Jiang, Zinc inactivates melastatin transient receptor potential 2 channels via the outer pore, J. Biol. Chem., 2011, 286, 23789–23798 Search PubMed.
- W. Yang, J. Zou, R. Xia, M. L. Vaal, V. A. Seymour, J. Luo, D. J. Beech and L. H. Jiang, State-dependent inhibition of TRPM2 channel by acidic pH, J. Biol. Chem., 2010, 285, 30411–30418 Search PubMed.
- X. Cheng, D. Shen, M. Samie and H. Xu, Mucolipins: Intracellular TRPML1-3 channels, FEBS Lett., 2010, 584, 2013–2021 Search PubMed.
- S. Vergarajauregui, R. Oberdick, K. Kiselyov and R. Puertollano, Mucolipin 1 channel activity is regulated by protein kinase A-mediated phosphorylation, Biochem. J., 2008, 410, 417–425 Search PubMed.
- S. Vergarajauregui, J. A. Martina and R. Puertollano, LAPTMs regulate lysosomal function and interact with mucolipin 1: new clues for understanding mucolipidosis type IV, J. Cell Sci., 2011, 124, 459–468 Search PubMed.
- A. A. Soyombo, S. Tjon-Kon-Sang, Y. Rbaibi, E. Bashllari, J. Bisceglia, S. Muallem and K. Kiselyov, TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity, J. Biol. Chem., 2006, 281, 7294–7301 Search PubMed.
- D. A. Zeevi, S. Lev, A. Frumkin, B. Minke and G. Bach, Heteromultimeric TRPML channel assemblies play a crucial role in the regulation of cell viability models and starvation-induced autophagy, J. Cell Sci., 2010, 123, 3112–3124 Search PubMed.
- C. Curcio-Morelli, F. A. Charles, M. C. Micsenyi, Y. Cao, B. Venugopal, M. F. Browning, K. Dobrenis, S. L. Cotman, S. U. Walkley and S. A. Slaugenhaupt, Macroautophagy is defective in mucolipin-1-deficient mouse neurons, Neurobiol. Dis., 2010, 40, 370–37 Search PubMed.
- J. L. Eichelsdoerfer, J. A. Evans, S. A. Slaugenhaupt and M. P. Cuajungco, Zinc dyshomeostasis is linked with the loss of mucolipidosis IV-associated TRPML1 ion channel, J. Biol. Chem., 2010, 285, 34304–34308 Search PubMed.
- T. Sun, X. Wang, Q. Lu, H. Ren and H. Zhang, CUP-5, the C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes, Autophagy, 2011, 7, 1308–1315 Search PubMed.
- G. A. Colletti, M. T. Miedel, J. Quinn, N. Andharia, O. A. Weisz and K. Kiselyov, Loss of lysosomal ion channel transient receptor potential channel mucolipin-1 (TRPML1) leads to cathepsin B-dependent apoptosis, J. Biol. Chem., 2012, 287, 8082–8091 Search PubMed.
- K. Abe and R. Puertollano, Role of TRP channels in the regulation of the endosomal-lysosmal pathway, Physiology, 2006, 26, 14–2 Search PubMed.
- Z. Zhu, Z. Luo, S. Ma and D. Liu, TRP channels and their implications in metabolic diseases, Pfluegers Arch., 2011, 461, 211–223 Search PubMed.
- C. Grimm, S. Hassan, C. Wahl-Schott and M. Biel, Role of TRPML and TPC channels in endolysosomal cation homeostasis, J. Pharmacol. Exp. Ther., 2012, 342, 236–244 Search PubMed.
- B. Lelouvier and R. Puertollano, Mucolipin-3 regulates luminal calcium, acidification, and membrane fusion in the endosomal pathway, J. Biol. Chem., 2011, 286, 9826–9832 Search PubMed.
- M. F. Cellier, P. Courville and C. Campion, Nramp1 phagocyte intracellular metal withdrawal defense, Microbes Infect., 2007, 9, 1662–1670 CrossRef CAS.
- J. M. Vyas, A. G. Van der Veen and H. L. Ploegh, The known unknowns of antigen processing and presentation, Nat. Rev. Immunol., 2008, 8, 607–618 CrossRef CAS.
- C. Watts, The endosome-lysosome pathway and information generation in the immune system, Biochim. Biophys. Acta, 2012, 1824, 14–21 Search PubMed.
- C. Stoeckle, P. Quecke, T. Rückrich, T. Burster, M. Reich, E. Weber, H. Kalbacher, C. Driessen, A. Melms and E. Tolosa, Cathepsin S dominates autoantigen processing in human thymic dendritic cells, J. Autoimmun., 2012, 38, 332–343 Search PubMed.
- N. J. Hill, P. A. Lyons, N. Armitage, J. A. Todd, L. S. Wicker and L. B. Peterson, NOD Idd5 locus controls insulitis and diabetes and overlaps the orthologous CTLA4/IDDM12 and NRAMP1 loci in humans, Diabetes, 2000, 49, 1744–1747 Search PubMed.
- L. S. Wicker, G. Chamberlain, K. Hunter, D. Rainbow, S. Howlett, P. Tiffen, J. Clark, A. Gonzalez-Munoz, A. M. Cumiskey, R. L. Rosa, J. M. Howson, L. J. Smink, A. Kingsnorth, P. A. Lyons, S. Gregory, J. Rogers, J. A. Todd and L. B. Peterson, Fine mapping, gene content, comparative sequencing, and expression analyses support Ctla4 and Nramp1 as candidates for Idd5.1 and Idd5.2 in the nonobese diabetic mouse, J. Immunol., 2004, 173, 164–173 Search PubMed.
- K. Takahashi, J. Satoh, Y. Kojima, K. Negoro, M. Hirai, Y. Hinokio, Y. Kinouchi, S. Suzuki, N. Matsuura, T. Shimosegawa and Y. Oka, Promoter polymorphism of SLC11A1 (formerly NRAMP1) confers susceptibility to autoimmune type 1 diabetes mellitus in Japanese, Tissue Antigens, 2004, 63, 231–236 Search PubMed.
- M. Nishino, H. Ikegami, T. Fujisawa, Y. Kawaguchi, Y. Kawabata, M. Shintani, M. Ono and T. Ogihara, Functional polymorphism in Z-DNA-forming motif of promoter of SLC11A1 gene and type 1 diabetes in Japanese subjects: association study and meta-analysis, Metabolism, 2005, 54, 628–633 Search PubMed.
- P. Courville, R. Chaloupka and M. F. Cellier, Recent progress in structure-function analyses of Nramp proton-dependent metal-ion transporters, Biochem. Cell Biol., 2006, 84, 960–978 Search PubMed.
- S. Kissler, P. Stern, K. Takahashi, K. Hunter, L. B. Peterson and L. S. Wicker, In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes, Nat. Genet., 2006, 38, 479–483 Search PubMed.
- K. Hunter, D. Rainbow, V. Plagnol, J. A. Todd, L. B. Peterson and L. S. Wicker, Interactions between Idd5.1/Ctla4 and other type 1 diabetes genes, J. Immunol., 2007, 179, 8341–8349 Search PubMed.
- B. A. O'Brien, N. S. Archer, A. M. Simpson, F. R. Torpy and N. T. Nassif, Association of SLC11A1 promoter polymorphisms with the incidence of autoimmune and inflammatory diseases: a meta-analysis, J. Autoimmun., 2008, 31, 42–51 Search PubMed.
- O. Ates, L. Dalyan, B. Müsellim, G. Hatemi, H. Türker, G. Ongen, V. Hamuryudan and A. Topal-Sarikaya, NRAMP1 (SLC11A1) gene polymorphisms that correlate with autoimmune versus infectious disease susceptibility in tuberculosis and rheumatoid arthritis, Int. J. Immunogenet., 2009, 36, 15–19 Search PubMed.
- Y. D. Dai, I. G. Marrero, P. Gros, H. Zaghouani, L. S. Wicker and E. E. Sercarz, Slc11a1 enhances the autoimmune diabetogenic T-cell response by altering processing and presentation of pancreatic islet antigens, Diabetes, 2009, 58, 156–164 Search PubMed.
- D. Paccagnini, L. Sieswerda, V. Rosu, S. Masala, A. Pacifico, M. Gazouli, J. Ikonomopoulos, N. Ahmed, S. Zanetti and L. A. Sechi, Linking chronic infection and autoimmune diseases: Mycobacterium avium subspecies paratuberculosis, SLC11A1 polymorphisms and type-1 diabetes mellitus, PLoS One, 2009, 4, e7109 Search PubMed.
- S. Sreedharan, O. Stephansson, H. B. Schiöth and R. Fredriksson, Long evolutionary conservation and considerable tissue specificity of several atypical solute carrier transporters, Gene, 2011, 478, 11–18 Search PubMed.
- J. H. Yang, K. Downes, J. M. Howson, S. Nutland, H. E. Stevens, N. M. Walker and J. A. Todd, Evidence of association with type 1 diabetes in the SLC11A1 gene region, BMC Med. Genet., 2011, 12, 59 Search PubMed.
- T. Canhamero, B. Reines, L. C. Peters, A. Borrego, P. S. Carneiro, L. L. Albuquerque, W. H. Cabrera, O. G. Ribeiro, J. R. Jensen, N. Starobinas, O. M. Ibañez and M. De Franco, Distinct early inflammatory events during ear tissue regeneration in mice selected for high inflammation bearing Slc11a1 R and S alleles, Inflammation, 2011, 34, 303–313 Search PubMed.
- T. B. Aydemir, J. P. Liuzzi, S. McClellan and R. J. Cousins, Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-gamma expression in activated human T cells, J. Leukocyte Biol., 2009, 86, 337–348 Search PubMed.
- F. W. Heineman and R. S. Balaban, Effects of afterload and heart rate on NAD(P)H redox state in the isolated rabbit heart, Am. J. Physiol., 1993, 264, H433–H440 Search PubMed.
- M. R. Laughlin and F. W. Heineman, The relationship between phosphorylation potential and redox state in the isolated working rabbit heart, J. Mol. Cell Cardiol., 1994, 26, 1525–1536 Search PubMed.
- J. R. Pungercar, D. Caglic, M. Sajid, M. Dolinar, O. Vasiljeva, U. Pozgan, D. Turk, M. Bogyo, V. Turk and B. Turk, Autocatalytic processing of procathepsin B is triggered by proenzyme activity, FEBS J., 2009, 276, 660–668 Search PubMed.
- D. K. Perry, M. J. Smyth, H. R. Stennicke, G. S. Salvesen, P. Duriez, G. G. Poirier and Y. A. Hannun, Zinc is a potent inhibitor of the apoptotic protease, caspase-3, A novel target for zinc in the inhibition of apoptosis, J. Biol. Chem., 1997, 272, 18530–18533 CrossRef CAS.
- R. P. Guttmann and G. Johnson, Oxidative stress inhibits calpain activity in situ, J. Biol. Chem., 1998, 273, 13331–13338 Search PubMed.
- P. Kiss, A. Szabó, E. Hunyadi-Gulyás, K. F. Medzihradszky, Z. Lipinszki, M. Pál and A. Udvardy, Zn2+ -induced reversible dissociation of subunit Rpn10/p54 of the Drosophila 26 S proteasome, Biochem. J., 2005, 391, 301–310 Search PubMed.
- A. U. Chouduri, K. Mishina, T. Shimizu, M. Yamazaki, T. Tokumoto and S. Yamada, High affinity Zn2+ inhibitory site(s) for the trypsin-like peptidase of the 20S proteasome, Arch. Biochem. Biophys., 2008, 477, 113–120 Search PubMed.
- M. Chen, Q. Chen, X. W. Cheng, T. J. Lu, H. X. Liu, J. M. Jia, C. Zhang, L. Xu and Z. Q. Xiong, Zn2+ mediates ischemia-induced impairment of the ubiquitin-proteasome system in the rat hippocampus, J. Neurochem., 2009, 111, 1094–1103 Search PubMed.
- G. Arena, R. Fattorusso, G. Grasso, G. I. Grasso, C. Isernia, G. Malgieri, D. Milardi and E. Rizzarelli, Zinc(II) complexes of ubiquitin: speciation, affinity and binding features, Chemistry, 2011, 17, 11596–11603 Search PubMed.
- F. Kriegenburg, E. G. Poulsen, A. Koch, E. Krüger and R. Hartmann-Petersen, Redox control of the ubiquitin-proteasome system: from molecular mechanisms to functional significance, Antioxid. Redox Signaling, 2011, 15, 2265–2299 Search PubMed.
- Q. Wei, J. Wang, M. H. Wang, F. Yu and Z. Dong, Inhibition of apoptosis by Zn2+ in renal tubular cells following ATP depletion, Am. J. Physiol. Renal Physiol., 2004, 287, F492–F500 Search PubMed.
- I. Dolenc and M. Mihelic, Purification and primary structure determination of human lysosomal dipeptidase, Biol. Chem., 2003, 384, 317–320 Search PubMed.
- S. P. Fricker, Cysteine proteases as targets for metal-based drugs, Metallomics, 2010, 2, 366–377 RSC.
- M. M. Moran, M. A. McAlexander, T. Bíró and A. Szallasi, Transient receptor potential channels as therapeutic targets, Nat. Rev. Drug Discovery, 2011, 10, 601–620 Search PubMed.
- M. Usenovic, E. Tresse, J. R. Mazzulli, J. P. Taylor and D. Krainc, Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity, J. Neurosci., 2012, 32, 4240–4246 Search PubMed.
- M. Lang, L. Wang, Q. Fan, G. Xiao, X. Wang, Y. Zhong and B. Zhou, Genetic inhibition of solute-linked carrier 39 family transporter 1 ameliorates aβ pathology in a Drosophila model of Alzheimer's disease, PLoS Genet., 2012, 8, e1002683 Search PubMed.
- A. P. Lieberman, R. Puertollano, N. Raben, S. Slaugenhaupt, S. U. Walkley and A. Ballabio, Autophagy in lysosomal storage disorders, Autophagy, 2012, 8, 719–730 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |