 Open Access Article
Open Access ArticlePseudopeptides with a centrally positioned alkene-based disulphide bridge mimetic stimulate kallikrein-related peptidase 3 activity†
Kristian
Meinander
a,
Janne
Weisell
b,
Miikka
Pakkala
b,
Andrew C.
Tadd
a,
Can
Hekim
c,
Roope
Kallionpää
a,
Kim
Widell
a,
Ulf-Håkan
Stenman
c,
Hannu
Koistinen
c,
Ale
Närvänen
b,
Jouko
Vepsäläinen
b,
Kristina
Luthman
d and
Erik A. A.
Wallén
*a
aDivision of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Helsinki, Helsinki, Finland. E-mail: erik.wallen@helsinki.fi
bSchool of Pharmacy, Faculty of Health Sciences, University of Eastern Finland and Biocenter Kuopio, Kuopio, Finland
cDepartment of Clinical Chemistry, University of Helsinki and Helsinki University Central Hospital, Helsinki, Finland
dMedicinal Chemistry, Department of Chemistry and Molecular Biology, University of Gothenburg, Gothenburg, Sweden
First published on 14th January 2013
Abstract
Pseudopeptides based on the kallikrein-related peptidase 3 (KLK3) activating bicyclic peptide “C-4” comprising hydrocarbon-based disulphide bridge mimetics have been synthesized. After investigating different synthetic approaches, the pseudopeptides were successfully cyclized from two L-allylglycine side chains via an alkene ring-closing metathesis reaction during the peptide synthesis. The alkene-linker was formed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z isomer ratio. The resulting pseudopeptides were almost as potent as the parent peptide, increasing the activity of KLK3 over four-fold at 200 μg ml−1 (130–140 μM) concentrations.
:1 E/Z isomer ratio. The resulting pseudopeptides were almost as potent as the parent peptide, increasing the activity of KLK3 over four-fold at 200 μg ml−1 (130–140 μM) concentrations.
Introduction
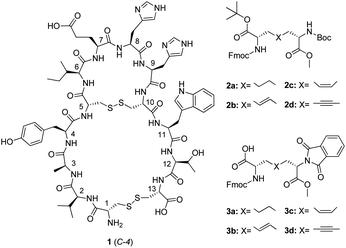
The human kallikrein-related peptidases, a family of 15 closely related serine proteases (KLK1–KLK15), are attracting increased attention due to their role as biomarkers for screening, diagnosis, and monitoring of various cancers.1 Among these KLK3, also known as prostate-specific antigen (PSA), is the most studied. It has been shown to be highly prostate specific and there is growing evidence supporting its use for targeted treatment and selective imaging of prostate cancer.2,3 In prostate cancer, the KLK3 expression is reduced and more aggressive tumours have lower KLK3 levels than less aggressive ones.4–6 At the same time the levels of KLK3 in blood are elevated due to increased leakage from the prostate, however, in blood circulation the majority of KLK3 is rapidly inactivated by endogenous protease inhibitors.7 KLK3 exerts an antiangiogenetic activity that is related to its enzymatic activity.8–10 It has therefore been suggested that tumour angiogenesis and thus prostate cancer growth might be controlled by increasing the proteolytic activity of KLK3.9,11Peptide 1 (C-4), developed by Stenman and coworkers, is so far the most potent KLK3 activating compound,12 increasing the proteolytic activity of KLK3 several-fold at micromolar concentrations (Table 3). Interestingly, all known potent compounds having this unique biological activity are peptides.11Peptide1 is likely to bind near the active site of KLK3, as the binding could be inhibited by monoclonal antibodies binding close to the active site.12 Recent studies demonstrate that peptide1 enhances the antiangiogenic activity of KLK3 in an endothelial cell tube formation assay.13
Peptide 1 consists of 13 amino acids and is cyclized by two disulphide bridges, one between Cys-1 and Cys-13, and one between Cys-5 and Cys-10. The side chains of Tyr-4, Ile-6 and Trp-11 have previously been identified to be important for their biological activity.14 As these amino acid residues are directly attached to the central disulphide bridge, this part of the peptide was considered to be most important for the development of pseudopeptides of 1. In vivo stable replacements of the central disulphide bridge would result in pseudopeptides with new molecular backbones.
We have earlier reported the successful synthesis of a series of orthogonally protected disulphide bridge mimetics (2a–d),15 and in the present study our aim was to synthesize pseudopeptides of peptide1 with these mimetics as replacements for the centrally positioned disulphide bridge. As described earlier, 2a–d can easily be converted to 3a–d, which have a more suitable combination of protecting groups for solid phase peptide synthesis using Fmoc-chemistry.
An additional objective was to identify all important side chains in 1 using multiple alanine replacements. In our earlier single alanine replacement study the importance of other amino acid side chains except Tyr-4, Ile-6 and Trp-11 was not fully determined.14 As the pseudopeptides are cyclized in a way that gives them a new backbone, we wanted to clarify if some parts of the original peptide structure could be simplified without decreasing the biological activity.
Results and discussion
Identification of all important amino acid side chains in peptide1
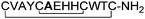
We started the current study by performing simultaneous substitutions of more than one amino acid residue with alanine on peptide1. The peptides were synthesized on a peptide synthesizer using Fmoc-chemistry on Rink AM resin, resulting in peptides with a C-terminal amide instead of the C-terminal carboxylic acid. This difference has however no significant influence on the biological activity (unpublished data). The effect of the peptides on the proteolytic activity of KLK3 was tested in vitro using a chromogenic peptide substrate according to a published procedure (Table 1).16| Peptide | Structure | KLK3 proteolytic activity [%] |
|---|---|---|
| 4 |

|
226 |
| 5 |

|
476 |
| 6 |

|
239 |
| 7 |

|
165 |
| 8 |

|
84 |
| 9 |
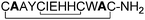
|
127 |
| 10 |

|
583 |
| 11 |

|
436 |
| 12 |
|
299 |
| 13 |

|
152 |
Although the original single alanine replacement study indicated that residues Glu-7, His-8 and His-9 could all be replaced with alanines, His-9 was found to be necessary for retaining full activity when two or three of these residues were replaced with alanines at the same time (peptides4–7). Furthermore, replacing Cys-5 and Cys-10 with alanines in combination with replacing Ile-6 and His-9 with cysteines to form the centrally positioned disulphide bridge resulted in an inactive peptide (peptide8). Interestingly, Val-2 and Thr-12 could not be replaced simultaneously with alanines (peptide9). Thus, the amino acids directly connected to the cysteine residues of both disulphide bridges seem to have a significant influence on the biological activity of 1.
The identified important side chains can either have direct interactions with the binding site on KLK3 or have a critical influence on the biologically active conformation of the peptide. According to a recent report, the binding site of 1 was suggested to be located in the kallikrein loop region on KLK3,17 which makes the analysis of the binding site more challenging due to the flexibility of the loop region.
Ile-6 was also replaced with different non-polar amino acids (peptides10–13). Interestingly, this residue could be replaced with phenylalanine and leucine, which both have lipophilic side chains of similar size to that of isoleucine. However, considerably smaller amino acids such as glycine or alanine were not successful replacements.
Synthesis and evaluation of the pseudopeptides
The pseudopeptides were designed based on our previously reported disulphide bridge mimetics 2a–d/3a–d.15 The three most potent peptides, original peptide1 and analogues 5 and 10, were used as starting peptides in this part of the study. Peptide5 was of primary interest due to its lower molecular weight. The pseudopeptides were synthesized manually using Fmoc-chemistry on Cys(Acm)-preloaded Wang resin.Our initial strategy was to use the orthogonally protected disulphide bridge mimetics 3a–d directly in solid phase peptide synthesis. However, an initial study with Fmoc-chemistry on Wang resin with mimetic 3b failed due to problems in the selective removal of the methyl ester and/or phthalimide protecting groups from the resin-bound intermediate 14 during peptide synthesis.
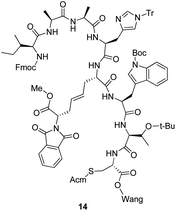
We also synthesized the small ring 18 with the intention of using it as an Fmoc-protected building block in solid phase peptide synthesis after hydrolysis of the methyl ester. Linear tetrapeptide15 was straightforwardly accessed in solution starting from Boc-Ile, coupling each residue as an amino acid methyl ester and subsequently hydrolyzing the ester. The Boc and the tert-butyl ester groups of 2b were removed with TFA before coupling, resulting in 16. The risk for racemization in the coupling steps was suppressed by using EDC·HCl with HOBt and DIPEA as coupling reagents. After selectively removing the Boc-group from 17 with 1.75 M HCl in EtOAc at room temperature for 1 h followed by cyclization with TBTU, HOBt and DIPEA, 18 was obtained in good yield (42% over two steps). Although methyl ester groups can generally be selectively removed in the presence of Fmoc groups using Me3SnOH in 1,2-dichloroethane (DCE), all our attempts to perform this reaction on 18 failed. This might be due to the steric hindrance around the ester group, from the neighbouring Trt- and Fmoc-groups (Scheme 1).
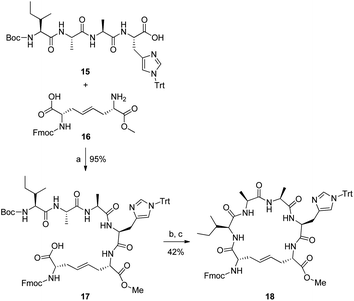 | ||
| Scheme 1 (a) EDC·HCl, HOBt, DIPEA/MeCN, 0 °C; (b) 1.75 M HCl/EtOAc; (c) TBTU, HOBt, DIPEA/DCM, 0 °C. | ||
After the pioneering work by Miller and Grubbs,18 ring-closing alkene metathesis reactions on peptides have been successfully reported for a wide variety of peptides.19 This inspired us to attempt this synthetic strategy also for our pseudopeptides. However, the drawbacks with this strategy are that only the alkene-based mimetics can be accessed and that the E/Z ratio of the formed alkenes cannot be controlled. Peptide19 was first synthesized using Fmoc-chemistry on Wang resin with L-allylglycine residues as replacements for the cysteines in positions 5 and 10. The ring-closing alkene metathesis reaction was first attempted for cleaved 19 and resin-bound 20a and 20b, but all attempts were unsuccessful as the MS spectra did not reveal even trace amounts of the cyclized peptide independent of the conditions used (Table 2). Thus, as unreacted starting material was the main component in all unsuccessful reactions, the full length peptide was unreactive in the ring-closing metathesis reaction.
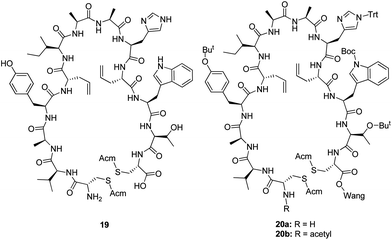
| Starting material | Catalysta | Additiveb | Solventc | Temperatured [°C] | Time [h] | Producte |
|---|---|---|---|---|---|---|
| a HG2, Hoveyda–Grubbs 2nd generation catalyst; G2, Grubbs 2nd generation catalyst. b DMF, N,N-dimethylformamide. c TFE, trifluoroethanol; DCM, dichloromethane; DCE, 1,2-dichloroethane. d r.t., room temperature; MW, microwave heating. e Analyzed by MS, in the case of high conversion the product was the main component and the main other component was a trace amount of the starting material. | ||||||
| 19 | HG2 | TFE:DCM 4:1 |
r.t. | 24 | Not detected | |
| 20a | HG2 | TFE:DCM 4:1 |
r.t. | 48 | Not detected | |
| 20b | HG2 | DCM | r.t. | 24 | Not detected | |
| 20b | HG2 | TFE:DCM 4:1 |
60 | 1 | Not detected | |
| 20b | HG2 | DCM | 60 | 1 | Not detected | |
| 20b | HG2 | DCE | 60 | 20 | Not detected | |
| 20b | G2 | DCE | 60 | 20 | Not detected | |
| 20b | HG2 | LiCl/DMF | DCE | 100 (MW) | 1 | Not detected |
| 20b | G2 | LiCl/DMF | DCE | 100 (MW) | 1 | Not detected |
| 21 | HG2 | DCE | r.t. | 48 | Not detected | |
| 21 | HG2 | LiCl/DMF | DCE | 100 (MW) | 1 | High conversion |
| 22 | HG2 | LiCl/DMF | DCE | 100 (MW) | 1 | High conversion |
| 23 | HG2 | LiCl/DMF | DCE | 100 (MW) | 1 | High conversion |
We refined our strategy by stopping the peptide synthesis after attachment of the second Fmoc-L-allylglycine, resulting in intermediate 21. An initial attempt to perform the ring-closing metathesis on 21 using the Hoveyda–Grubbs 2nd generation catalyst (HG2) in DCE at room temperature for 48 h was unsuccessful, but in later attempts the reaction was found to be highly successful when using LiCl in DMF as an additive and performing the reaction under microwave heating at 100 °C for 60 min (Table 2). The only impurity was a trace amount of uncyclized starting material. Intermediates 22 and 23 were also cyclized successfully using the same methodology. The addition of LiCl in DMF in combination with microwave heating to enable/improve cyclization of peptides with two L-allylglycine residues on Wang resin by ring-closing metathesis reaction has also been presented previously by other groups.20 Pre-treatment with LiCl in DMF has also been reported to enable a similar cyclization of peptides with thermal heating (Scheme 2).21
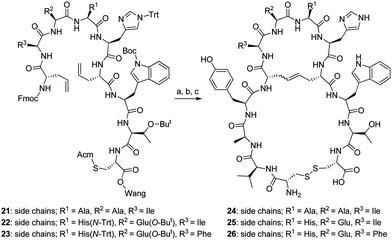 | ||
| Scheme 2 (a) HG2, 0.4 M LiCl in DMF (10% (v/v))/DCE, 100 °C, MW; (b) (i) solid-phase peptide synthesis (coupling of Tyr(tBu), Ala, Val and Cys(Acm)), (ii) reagent K (cleavage from resin); (c) I2, HCl/AcOH, H2O. | ||
There are several reported NMR spectroscopy methods for studying the E/Z ratio of the formed alkene.22,23 Product 27 (the cleaved ring-closing alkene metathesis product of 22) was studied by NMR spectroscopy according to the method reported by Bhattacharya et al.22 The sample was purified by preparative HPLC. The NMR spectra from the crude and purified ring-closing alkene metathesis product showed that the E/Z ratio was approximately 1:1.
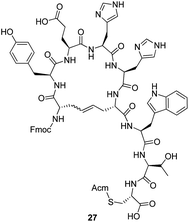
As the only impurity was a trace amount of uncyclized starting material in all three cyclizations, peptide synthesis was continued on a solid support with addition of the remaining four amino acids (Tyr-4, Ala-3, Val-2 and Cys-1). The pseudopeptides were cleaved from the resin, and the Acm-protected cysteines were then deprotected and cyclized with iodine in acetic acid in the presence of aqueous HCl. The yield of the crude pseudopeptides 24, 25 and 26 were 37%, 65% and 81%, respectively. After purification by preparative HPLC to over 90% purity, the total yield for 24, 25 and 26 went down to 1%, 3% and 6%, respectively. To confirm that the purification procedure had not changed the E/Z isomer ratio, the NMR spectra of the purified pseudopeptide 26 showed that the E/Z isomer ratio was unchanged. In biological testing, the pseudopeptides 24, 25 and 26 increased the activity of KLK3 to 225%, 189% and 223%, respectively, of that of KLK3 alone at a concentration 20 μg ml−1 (Table 3). The original peptide1 increased the activity to 625% at the same concentration. Increasing the concentration of pseudopeptides 24, 25 and 26 showed that the stimulating effect approached the level of peptide1 at higher concentrations (Fig. 1). At 200 μg ml−1 concentration they stimulated the KLK3 activity about four-fold.
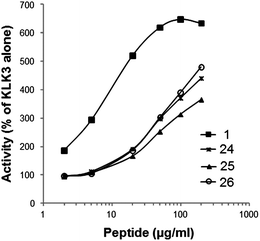 | ||
| Fig. 1 Effect on the proteolytic activity of KLK3 by peptide1 and the synthesized pseudopeptides 24–26 at different concentrations. | ||
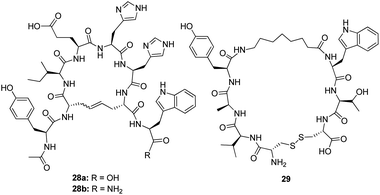
In order to test if pseudopeptides with approximately half the size of the original peptide can increase the activity of KLK3, we synthesized pseudopeptides 28a, 28b and 29. Pseudopeptides 28a and 28b were synthesized according to the previously described procedure with the exception that 28a was synthesized on Trp(Boc)-preloaded Wang resin and 28b on Rink amide resin. Pseudopeptide 29 has 8-aminooctanoic acid as a replacement of the disulphide bridge and the small ring, and this linker was introduced in the peptide synthesis as an Fmoc-protected amino acid. The yield of the crude pseudopeptides 28a, 28b and 29 were 91%, 29% and 52%, respectively. After purification by preparative HPLC to over 90% purity, the total yield for 28a, 28b and 29 went down to 7%, 3% and 3%, respectively. None of these smaller pseudopeptides increased the activity of KLK3 at a concentration of 20 μg ml−1 (data not shown), which clearly indicates that neither ring alone is biologically active.
Conclusions
Based on the multiple alanine replacement study, we succeeded in identifying the important amino acid side chains in peptide1. The only non-important side chains were Glu-7 and His-8, as these could simultaneously be replaced with alanines. The multiple alanine replacement showed that most amino acid side chains are important and the size of the peptide will therefore be difficult to reduce. Furthermore, Ile-6 could be replaced with leucine and phenylalanine, which are similar in size and non-polar character.We have developed a general synthetic route towards the pseudopeptides with a centrally positioned alkene-based disulphide bridge mimetic. However, the alkene was formed as a 1:1 mixture of the E and Z isomers, and the two isomers could not be separated. The same amino acid replacements that were allowed for peptide1 were also allowed for the pseudopeptides. The most potent pseudopeptides (24 and 26) increased the activity of KLK3 to over 200% (doubling the activity) at a concentration of 20 μg ml−1 (13–14 μM) and they were almost as potent as the parent peptide at higher concentrations, increasing the activity of KLK3 over four-fold at a concentration of 200 μg ml−1 (130–140 μM). As the pseudopeptides have a new backbone consisting of the alkene-linker, we also synthesized pseudopeptides corresponding to either the small or the large ring on peptide1. These smaller pseudopeptides did not stimulate the KLK3 activity which indicates that it will be difficult to drastically decrease the size of the pseudopeptides.
Acknowledgements
This work was supported by the Academy of Finland (grants 126969, 135937 and 140929), Graduate School of Organic Chemistry and Chemical Biology, Research Funds of the University of Helsinki, Helsinki University Central Hospital, Finnish Cancer Foundation, Sigrid Juselius Foundation, Finska Läkaresällskapet, Magnus Ehrnrooth foundation, and The Swedish Research Council (grant 2010-4846). We thank Dr Timo Mauriala, Mr Miikka Olin and Ms Marianne Niemelä for technical assistance.Notes and references
- M. G. Lawrence, J. Lai and J. A. Clements, Endocr. Rev., 2010, 31, 407–446 CrossRef CAS.
- A. M. LeBeau, M. Kostova, C. S. Craik and S. R. Denmeade, Biol. Chem., 2010, 391, 333–343 CrossRef CAS.
- D. Ulmert, M. J. Evans, J. P. Holland, S. L. Rice, J. Wongvipat, K. Pettersson, P. A. Abrahamsson, P. T. Scardino, S. M. Larson, H. Lilja, J. S. Lewis and C. L. Sawyers, Cancer Discovery, 2012, 2, 320–327 CrossRef CAS.
- R. Stege, M. Grande, K. Carlstrom, B. Tribukait and A. Pousette, Clin. Cancer Res., 2000, 6, 160–165 CAS.
- P. A. Abrahamsson, H. Lilja, S. Falkmer and L. B. Wadstrom, Prostate, 1988, 12, 39–46 CrossRef CAS.
- A. Paju, K. Hotakainen, Y. Cao, T. Laurila, V. Gadaleanu, A. Hemminki, U.-H. Stenman and A. Bjartell, Eur. Urol., 2007, 52, 1670–1679 CrossRef CAS.
- U.-H. Stenman, J. Leinonen, W. M. Zhang and P. Finne, Semin. Cancer Biol., 1999, 9, 83–93 CrossRef CAS.
- J. M. Mattsson, L. Valmu, P. Laakkonen, U.-H. Stenman and H. Koistinen, Prostate, 2008, 68, 945–954 CrossRef CAS.
- A. H. Fortier, B. J. Nelson, D. K. Grella and J. W. Holaday, J. Natl. Cancer Inst., 1999, 91, 1635–1640 CrossRef CAS.
- H. H. Heidtmann, D. M. Nettelbeck, A. Mingels, R. Jager, H. G. Welker and R. E. Kontermann, Br. J. Cancer, 1999, 81, 1269–1273 CrossRef CAS.
- H. Koistinen, A. Närvänen, M. Pakkala, C. Hekim, J. M. Mattsson, L. Zhu, P. Laakkonen and U.-H. Stenman, Biol. Chem., 2008, 389, 633–642 CrossRef CAS.
- P. Wu, J. Leinonen, E. Koivunen, H. Lankinen and U.-H. Stenman, Eur. J. Biochem., 2000, 267, 6212–6220 CrossRef CAS.
- J. M. Mattsson, A. Närvänen, U.-H. Stenman and H. Koistinen, Prostate, 2012, 72, 1588–1594 CrossRef CAS.
- M. Pakkala, A. Jylhäsalmi, P. Wu, J. Leinonen, U.-H. Stenman, H. Santa, J. Vepsäläinen, M. Peräkylä and A. Närvänen, J. Pept. Sci., 2004, 10, 439–447 CrossRef CAS.
- A. C. Tadd, K. Meinander, K. Luthman and E. A. A. Wallén, J. Org. Chem., 2011, 76, 673–675 CrossRef CAS.
- M. Pakkala, J. Weisell, C. Hekim, J. Vepsäläinen, E. A. A. Wallén, U.-H. Stenman, H. Koistinen and A. Närvänen, Amino Acids, 2010, 39, 233–242 CrossRef CAS.
- H. H. Härkönen, J. M. Mattsson, J. A. Määttä, U.-H. Stenman, H. Koistinen, S. Matero, B. Windshügel, A. Poso and M. Lahtela-Kakkonen, ChemMedChem, 2011, 6, 2170–2178 CrossRef.
- S. J. Miller and R. H. Grubbs, J. Am. Chem. Soc., 1995, 117, 5855–5856 CrossRef CAS.
- Ø. Jacobsen, J. Klaveness and P. Rongved, Molecules, 2010, 15, 6638–6677 CrossRef.
- J. Illesinghe, C. X. Guo, R. Garland, A. Ahmed, B. van Lierop, J. Elaridi, W. R. Jackson and A. J. Robinson, Chem. Commun., 2009, 295–297 RSC.
- D. J. Derksen, J. L. Stymiest and J. C. Vederas, J. Am. Chem. Soc., 2006, 128, 14252–14253 CrossRef CAS.
- S. Bhattacharya, H. Zhang, D. Cowburn and A. K. Debnath, Biopolymers, 2012, 97, 253–264 CrossRef CAS.
- J. L. Stymiest, B. F. Mitchell, S. Wong and J. C. Vederas, Org. Lett., 2003, 5, 47–49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3md20292e |
| This journal is © The Royal Society of Chemistry 2013 |