 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
6-Alkyl-, 6-aryl- or 6-hetaryl-7-deazapurine ribonucleosides as inhibitors of human or MTB adenosine kinase and potential antimycobacterial agents†
Pavla Perlíková‡a, Petr Konečný‡b, Petr Nauša, Jan Snášela, Ivan Votrubaa, Petr Džubákb, Iva Pichováa, Marián Hajdúchb and Michal Hocek*ac
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Gilead Sciences & IOCB Research Center, Flemingovo nam. 2, CZ-16610 Prague 6, Czech Republic. E-mail: hocek@uochb.cas.cz; Tel: +420 220183324
bInstitute of Molecular and Translational Medicine, Laboratory of Experimental Medicine, Palacky University and University Hospital in Olomouc, Faculty of Medicine and Dentistry, Hněvotínská 5, CZ-775 15 Olomouc, Czech Republic
cDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, CZ-12843 Prague 2, Czech Republic
First published on 17th September 2013
Abstract
Title 6-alkyl-, 6-aryl- and 6-hetaryl-7-deazapurine ribonucleosides previously known as nanomolar cytostatics were found to be potent inhibitors of either human or mycobacterial (MTB) adenosine kinase (ADK). Several new derivatives bearing bulky substituents at position 6 were non-cytotoxic but selectively inhibited MTB ADK. However, most of the nucleosides (ADK inhibitors) as well as their octadecylphosphate prodrugs were inactive in the whole cell assay of inhibition of Mycobacterium bovis growth. 6-Methyl-7-deazapurine ribonucleoside was found to be a potent antimycobacterial agent.
Introduction
Modified purine nucleoside derivatives and analogs display a wide range of biological activities. Their antiviral1 and antitumor2 properties are particularly important and used in clinical therapeutics. However, purine derivatives3 and nucleosides4 also are potent antimycobacterial agents, i.e. for treatment of Mycobacterium tuberculosis (MTB). Out of the variety of possible target enzymes from the purine salvage pathway in MTB,5 adenosine kinase (ADK)6 is considered as a promising target for drug development since it is structurally very different from the human ADK.7Recently, we have discovered 6-hetaryl-7-deazapurine ribonucleosides 1–3 with nanomolar cytostatic activities towards a wide panel of leukemia and cancer cell-lines.8 Their cycloSal-phosphate9 and phosphoramidate prodrugs10 were less active due to increased efflux from the cells. However, cycloSal-phosphates were also found9 to be potent inhibitors of human and moderate inhibitors of MTB ADKs. Since also other 7-deazapurine nucleosides are known as inhibitors of ADKs,11 we have revisited the whole class of 7-deazapurine nucleosides with diverse aryl and hetaryl groups at position 6 and systematically studied their activity toward human and MTB ADKs.
Results and discussion
The synthesis of a series of nineteen 6-alkyl, 6-aryl- and 6-hetaryl-7-deazapurine ribonucleosides 1b–t, nine 7-fluoro derivatives 2e, j–m, o–r and six 7-chloro derivatives 3e, g, j–m has been reported earlier,8 as well as the 6-methyl derivative 1a (ref. 12) (Chart 1). Most of these derivatives exhibited strong cytostatic or cytotoxic activities.8,12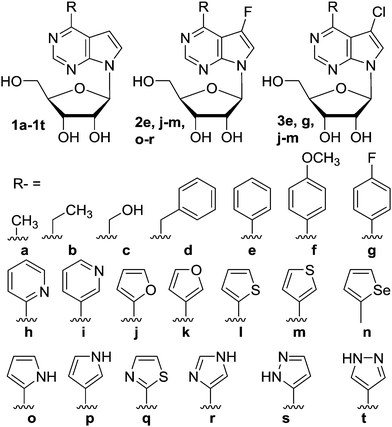 | ||
| Chart 1 Structures of previously known nucleosides 1a–t. | ||
In order to get less cytotoxic derivatives, we have extended the series by synthesis of other six derivatives bearing bulky (het)aryl groups at position 6 (Scheme 1). The dibenzofuryl and benzofuryl derivatives 1u and v were prepared by the Suzuki coupling of isopropylidene-protected nucleoside 4 followed by deprotection. The other derivatives 1w–z were synthesized by direct aqueous Suzuki coupling of 6-chloro-7-deazapurine ribonucleoside with the corresponding hetarylboronic acid in the presence of Pd(OAc)2, triphenylphosphine-3,3′,3′′-trisulfonate (TPPTS) and Na2CO3. In the case of indole derivative 1x, the coupling was performed with the Boc-protected indole-2-boronic acid and the TFA treatment was used for deprotection. In all cases, the products were obtained in good 71–85% yields.
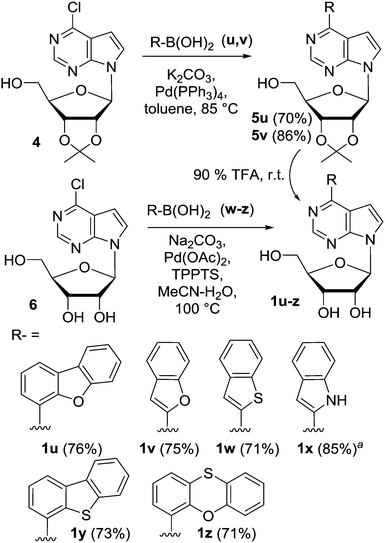 | ||
| Scheme 1 Synthesis of nucleosides 1u–z. aN-Boc-protected indole-2-boronic acid was used and the Boc was cleaved off by 90% TFA at rt; the yield is given over two steps. | ||
A synthetic path to 6-(het)aryl-7-deazapurine ribonucleoside-5′-octadecylphosphates, as potential lipophilic phosphate prodrugs, was developed starting from isopropylidene-protected ribonucleoside 4. An octadecylphosphate group was attached by reaction with octadecylphosphate in the presence of 2,4,6-trimethylbenzene-1-sulphonyl chloride (MtsCl) in pyridine. Nucleoside-5′-octadecylphosphate 7 was obtained in 58% yield. Deprotection by treatment with 90% aqueous TFA provided free nucleoside-5′-octadecylphosphate 8 (94%), which was used as a starting material for a series of Stille and Suzuki cross-coupling reactions. Stille cross-coupling reactions with hetaryltributylstannanes were performed in the presence of PdCl2(PPh3)2 in DMF at 105 °C. As 5′-octadecylphosphate 8 is insoluble in toluene, standard conditions for the Suzuki cross-coupling reaction had to be slightly modified. The reactions with (het)arylboronic acids were performed in the presence of potassium carbonate and Pd(PPh3)4 in DMF/water (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 105 °C. 6-(Het)aryl-7-deazapurine ribonucleoside-5′-octadecylphosphates 9e, j–m and v were obtained in 62–85% yields (Scheme 2).
:1) at 105 °C. 6-(Het)aryl-7-deazapurine ribonucleoside-5′-octadecylphosphates 9e, j–m and v were obtained in 62–85% yields (Scheme 2).
 | ||
| Scheme 2 Reagents and conditions: (i) C18H37OP(O)(OH)2 (1.5 equiv), MtsCl (6 equiv), pyridine, rt; (ii) 90% TFA, rt; (iii) R-SnBu3 (1.5 equiv), PdCl2(PPh3)2 (0.05 equiv)/DMF, 105 °C; (iv) R–B(OH)2 (1.5 equiv), K2CO3 (2 equiv), Pd(PPh3)4 (0.05 equiv), DMF/H2O (8:1), 105 °C. | ||
All the title nucleosides 1a–z, 2e, j–m, o–r, 3e, g, j–m and nucleotides 9e, j–m, v were tested for the inhibition of human and MTB ADKs (for cloning expression and purification of these enzymes, see ref. 9) and most of them also for substrate activity to the kinases, i.e. for phosphorylation. The results were correlated with their in vitro cytotoxicity (MTT) against non-malignant BJ and MRC-5 human fibroblast cell lines. The cytotoxicity strongly depended on the bulkiness of the substituent at position 6. Most active were derivatives bearing five-membered heterocycles, whereas derivatives bearing bulky aryl groups were generally not cytotoxic, which was consistent with previously published8 cytostatic and cytotoxic activities on leukemia and solid tumor cell lines. Most of the nucleosides did not show significant inhibition to human ADK with the exception of bulky derivatives 1u–z, which showed inhibition with micromolar IC50 values. On the other hand, most of the nucleosides were moderate to good substrates for the human ADK and were readily phosphorylated to nucleoside 5′-O-monophosphates, which is a necessary step in their activation for eventual inhibition of RNA synthesis in their cytostatic or cytotoxic effect.8 On the other hand, none of the compounds was found to be a substrate for MTB ADK and most compounds efficiently inhibited this enzyme. While the alkyl-substituted derivatives 1a–d were weak inhibitors of MTB ADK, all the aryl- and hetaryl-substituted 7-deazapurine ribonucleosides (1e–z), including 7-fluoro- (2) and 7-chloro-derivatives (3) were strong inhibitors of this enzyme with submicromolar to low nanomolar IC50 values. Most of the derivatives bearing phenyl and five-membered hetaryl groups at position 6 (1e–i, k–t, 2e, k, m–r and 3e, g, m) were selective inhibitors of the MTB ADK and did not significantly inhibit the human enzyme (but were strongly cytotoxic). 6-Furyl derivatives 1j and 2j and the thienyl derivative 2l were less selective, inhibited both enzymes and were cytotoxic. The derivatives bearing bulky aryl groups 1u–z inhibited the MTB ADK in low nanomolar concentrations while the inhibition of human enzyme was observed at micromolar concentrations, and so the selectivity index was 2–3 orders of magnitude. These bulky derivatives were not cytotoxic. The octadecylphosphate prodrugs 9 were moderate inhibitors of the human ADK and inactive against MTB enzyme.
All the derivatives were also tested for in vivo inhibition of Mycobacterium bovis growth (Table 1). From all tested nucleosides only compound 1a displayed very significant antimycobacterial activity (IC50 = 0.3 μM) but showed the highest cytotoxicity. This derivative, however, only weakly inhibited in vitro MTB ADK (IC50 = 8.8 μM) which may indicate that the mode of antimycobacterial activity of this compound is independent of MTB ADK and rather suggests a more general cytotoxic mechanism. Two 6-(imidazolyl)deazapurine nucleosides 1r and 2r exerted moderate antimycobacterial activity (IC50 = 15.6 and 11.7 μM, respectively) accompanied by preferential inhibition of MTB ADK and low cytotoxicity, whereas all other nucleosides were virtually inactive. The octadecylphosphate prodrugs 9e, j–m and v, which were designed as lipophilic derivatives with increased penetration through the mycobacterial cell wall, did not show any antimycobacterial activity either.
| Compd | BJa CC50 (μM) | MRC-5 CC50 (μM) | ADK substrateb (%) | ADK inhibition IC50 (μM) | Mycobacterium bovisc IC50 (μM) | |
|---|---|---|---|---|---|---|
| Human | Human | MTB | ||||
| a Cytotoxicity (MTT test) in BJ and MRC-5 fibroblasts.b ADK substrate activity, conversion to 5′-phosphate (%).c 50% growth inhibitory concentration of in vitro cultivated Mycobacterium bovis BCG. | ||||||
| 1a | 0.098 | 0.081 | 54 | >10 | 8.8 ± 0.03 | 0.30 |
| 1b | 2.70 | 50 | n.d. | >20 | 8.1 ± 0.9 | 88.84 |
| 1c | 0.34 | 0.28 | 26 | >10 | >5.00 | 32.61 |
| 1d | 36.92 | >50 | >20 | >20 | >100 | |
| 1e | 44.15 | >50 | n.d. | >10 | 0.12 ± 0.03 | 75.16 |
| 1f | >50 | >50 | n.d. | >10 | 0.058 ± 0.003 | 56.88 |
| 1g | >50 | >50 | 17 | >10 | 0.08 ± 0.008 | 93.82 |
| 1h | 1.74 | >50 | 39 | >10 | 0.12 ± 0.02 | 38.22 |
| 1i | 50 | >50 | n.d. | >10 | 1.00 ± 0.11 | >100 |
| 1j | 0.23 | 11.83 | 56 | 1.30 ± 0.25 | 0.32 ± 0.05 | >100 |
| 1k | 1.20 | 45.38 | 32 | >10 | 0.32 ± 0.04 | 83.67 |
| 1l | 0.31 | >50 | n.d. | >5 | 0.046 ± 0.003 | >100 |
| 1m | 21.33 | >50 | 56 | >10 | 0.195 ± 0.05 | 81.00 |
| 1n | 1.52 | >50 | 43 | >10 | 0.030 ± 0.005 | >100 |
| 1o | >50 | >50 | 40 | >10 | 0.073 ± 0.007 | 56.44 |
| 1p | 1.87 | 40.15 | 70 | >10 | 0.30 ± 0.04 | 98.23 |
| 1q | 0.26 | 37.00 | 41 | >20 | 0.023 ± 0.003 | 80.53 |
| 1r | 0.21 | >50 | 28 | >10 | 0.78 ± 0.10 | 15.55 |
| 1s | 43.93 | 1.16 | 31 | >20 | 0.059 ± 0.005 | 91.10 |
| 1t | 42.13 | >50 | 7 | >20 | 0.67 ± 0.10 | >100 |
| 1u | >50 | >50 | n.d. | 5.26 ± 0.66 | 2.35 ± 0.35 | >100 |
| 1v | >50 | >50 | n.d. | 2.10 ± 0.03 | 0.0145 ± 0.001 | >100 |
| 1w | >50 | >50 | n.d. | 0.30 ± 0.02 | 0.0075 ± 0.0007 | >100 |
| 1x | >50 | >50 | n.d. | 8.35 ± 0.75 | 0.04 ± 0.005 | >100 |
| 1y | >50 | 43.76 | n.d. | 15.4 ± 1.2 | 0.15 ± 0.007 | >100 |
| 1z | >50 | >50 | n.d. | 3.07 ± 0.40 | 1.46 ± 0.07 | >100 |
| 2e | >50 | >50 | 17 | >10 | 0.20 ± 0.05 | 74.81 |
| 2j | 0.31 | 29.04 | 50 | 1.30 ± 0.25 | 0.19 ± 0.10 | 73.20 |
| 2k | 0.35 | 15.38 | n.d. | >10 | 0.15 ± 0.05 | 91.19 |
| 2l | 0.17 | 47.21 | 52 | 1.10 ± 0.20 | 0.035 ± 0.005 | >100 |
| 2m | 0.62 | 50 | 51 | >10 | 0.063 ± 0.003 | >100 |
| 2o | 2.99 | 44.45 | 76 | >10 | 0.070 ± 0.010 | 45.78 |
| 2p | 2.02 | 48.94 | 97 | >10 | 0.40 ± 0.06 | >100 |
| 2q | 0.63 | >50 | 75 | >20 | 0.028 ± 0.007 | 68.99 |
| 2r | >50 | >50 | 11 | >20 | 1.00 ± 0.12 | 11.73 |
| 3e | 44.49 | >50 | 29 | >10 | 0.22 ± 0.05 | >100 |
| 3g | 49.52 | >50 | n.d | >20 | 0.40 ± 0.02 | 89.63 |
| 3j | 37.25 | >50 | 77 | 0.29 ± 0.03 | 0.11 ± 0.01 | 89.23 |
| 3k | 21.62 | >50 | 100 | 3.4 | 0.30 ± 0.03 | 97.10 |
| 3l | 4.97 | >50 | 100 | 1.20 ± 0.20 | 0.07 ± 0.007 | 96.38 |
| 3m | 1.72 | >50 | 97 | >10 | 0.25 ± 0.03 | 99.57 |
| 9e | >50 | >50 | — | 4.40 ± 0.10 | >20 | >100 |
| 9j | >50 | 45.26 | — | 2.4 ± 0.11 | >20 | >100 |
| 9k | 44.20 | >50 | — | 5.15 ± 0.35 | >20 | 56.56 |
| 9l | n.d. | n.d. | — | 1.97 ± 0.16 | >20 | n.d. |
| 9m | >50 | >50 | — | 6.35 ± 0.34 | >20 | 82.78 |
| 9v | >50 | >50 | — | 2.9 ± 0.30 | >20 | 93.39 |
Conclusions
It can be concluded that the 6-(het)aryl-7-deazapurine ribonucleosides are strong and mostly selective inhibitors of MTB ADK but not the human ADK. Nonetheless, they showed only limited potential to inhibit growth of mycobacteria. One reason could be their poor penetration through the mycobacterial cell wall. Alternatively, the mycobacterial ADK may not be a suitable target for therapy since AMP also can be biosynthesized by the salvage pathway from adenine utilizing adenine phosphoribosyl transferase or by a reaction sequence from IMP.5Acknowledgements
This work was supported by the institutional support from the Academy of Sciences of the Czech Republic (RVO: 61388963), a grant of the Czech Science Foundation (P207/11/0344), EU-PF7 SysteMtb Collaborative Project no. 241587, and by Gilead Sciences, Inc. Infrastructural part of this project (Institute of Molecular and Translational Medicine) was supported by the Operational Programme Research and Development for Innovations (project CZ.1.05/2.1.00/01.0030). We thank Mrs Dagmar Grundová for excellent technical assistance.Notes and references
- Reviews: (a) E. De Clercq, J. Med. Chem., 2010, 53, 1438–1450 CrossRef CAS PubMed; (b) E. De Clercq, Nucleosides, Nucleotides Nucleic Acids, 2012, 31, 339–352 CrossRef CAS PubMed.
- Reviews: (a) W. B. Parker, J. A. Secrist, III and W. R. Waud, Curr. Opin. Invest. Drugs, 2004, 5, 592–596 CAS; (b) C. M. Galmarini, F. Popowycz and B. Joseph, Curr. Med. Chem., 2008, 15, 1072–1082 CrossRef CAS; (c) W. B. Parker, Chem. Rev., 2009, 109, 2880–2893 CrossRef CAS PubMed.
- Examples: (a) A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, Bioorg. Med. Chem. Lett., 2001, 11, 1675–1678 CrossRef CAS; (b) A. K. Pathak, V. Pathak, L. E. Seitz, W. J. Suling and R. C. Reynolds, J. Med. Chem., 2004, 47, 273–276 CrossRef CAS PubMed; (c) L.-L. Gundersen, J. Nissen-Meyer and B. Spilsberg, J. Med. Chem., 2002, 45, 1383–1386 CrossRef CAS PubMed; (d) A. K. Bakkestuen, L.-L. Gundersen and B. T. Utenova, J. Med. Chem., 2005, 48, 2710–2723 CrossRef CAS PubMed; (e) M. Braendvang and L.-L. Gundersen, Bioorg. Med. Chem., 2005, 13, 6360–6373 CrossRef CAS PubMed.
- Reviews: (a) M. C. Long and W. B. Parker, Biochem. Pharmacol., 2006, 71, 1671–1682 CrossRef CAS PubMed; (b) M. C. Long, S. C. Shaddix, O. Moukha-Chafiq, J. A. Maddry, L. Nagy and W. B. Parker, Biochem. Pharmacol., 2008, 75, 1588–1600 CrossRef CAS PubMed.
- Reviews: (a) W. B. Parker and M. C. Long, Curr. Pharm. Des., 2007, 13, 599–608 CrossRef CAS; (b) R. G. Ducati, A. Breda, A. Basso, L. A. Basso and D. S. Santos, Curr. Med. Chem., 2011, 18, 1258–1275 CrossRef CAS.
- M. C. M. Reddy, S. K. Palaninathan, N. D. Shetty, J. L. Owen, M. D. Watson and J. C. Sacchettini, J. Biol. Chem., 2007, 282, 27334–27342 CrossRef CAS PubMed.
- S. W. Muchmore, R. A. Smith, A. O. Stewart, M. D. Cowart, A. Gomtsyan, M. A. Matulenko, H. Yu, J. M. Severin, S. S. Bhagwat, C.-H. Lee, E. A. Kowaluk, M. F. Jarvis and C. L. Jakob, J. Med. Chem., 2006, 49, 6726–6731 CrossRef CAS PubMed.
- P. Nauš, R. Pohl, I. Votruba, P. Džubák, M. Hajdúch, R. Ameral, G. Birkus, T. Wang, A. S. Ray, R. Mackman, T. Cihlar and M. Hocek, J. Med. Chem., 2010, 53, 460–470 CrossRef PubMed.
- P. Spáčilová, P. Nauš, R. Pohl, I. Votruba, J. Snášel, H. Zábranská, I. Pichová, R. Ameral, G. Birkuš, T. Cihlář and M. Hocek, ChemMedChem, 2010, 5, 1386–1396 CrossRef PubMed.
- P. Perlíková, R. Pohl, I. Votruba, R. Shih, G. Birkuš, T. Cihlář and M. Hocek, Bioorg. Med. Chem., 2011, 19, 229–242 CrossRef PubMed.
- Examples: (a) B. G. Ugarkar, J. M. DaRe, J. J. Kopcho, C. E. Browne, III, J. M. Schanzer, J. B. Wiesner and M. D. Erion, J. Med. Chem., 2000, 43, 2883–2893 CrossRef CAS PubMed; (b) B. G. Ugarkar, A. Castellino, J. M. DaRe, J. J. Kopcho, J. B. Wiesner, J. M. Schanzer and M. D. Erion, J. Med. Chem., 2000, 43, 2894–2905 CrossRef CAS PubMed; (c) B. G. Ugarkar, A. J. Castellino, J. S. DaRe, M. Ramirez-Weinhouse, J. J. Kopcho, S. Rosengren and M. D. Erion, J. Med. Chem., 2003, 46, 4750–4760 CrossRef CAS PubMed; (d) B. C. Bookser, M. C. Matelich, K. Ollis and B. G. Ugarkar, J. Med. Chem., 2005, 48, 3389–3399 CrossRef CAS PubMed; (e) S. H. Boyer, B. G. Ugarkar, J. Solbach, J. J. Kopcho, M. C. Matelich, K. Ollis, J. E. Gomez-Galeno, R. Mendonca, M. Tsuchiya, A. Nagahisa, M. Nakane, J. B. Wiesner and M. D. Erion, J. Med. Chem., 2005, 48, 6430–6441 CrossRef CAS PubMed; (f) Y. A. Kim, A. Sharon, C. K. Chu, R. H. Rais, O. N. Al Safarjalani, F. N. M. Naguib and M. H. el Kouni, J. Med. Chem., 2008, 51, 3934–3945 CrossRef CAS PubMed.
- R. Wu, E. D. Smidansky, H. S. Oh, R. Takhampunya, R. Padmanabhan, C. E. Cameron and B. R. Peterson, J. Med. Chem., 2010, 53, 7958–7966 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental part and characterization data for all new compounds. See DOI: 10.1039/c3md00232b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |