Mechanistic insight into inhibition of two-component system signaling†
Samson
Francis
a,
Kaelyn E.
Wilke
a,
Douglas E.
Brown
a and
Erin E.
Carlson
*ab
aDepartment of Chemistry, Indiana University, 800 E. Kirkwood Avenue, Bloomington, Indiana, USA. E-mail: carlsone@indiana.edu; Tel: +1 812-855-3665
bDepartment of Molecular and Cellular Biochemistry, Indiana University, 212 S. Hawthorne Drive, Bloomington, Indiana, USA
First published on 21st November 2012
Abstract
Two-component signal transduction systems (TCSs) are commonly used by bacteria to couple environmental stimuli to adaptive responses. Targeting the highly conserved kinase domain in these systems represents a promising strategy for the design of a broad-spectrum antibiotic; however, development of such compounds has been marred by an incomplete understanding of the conserved binding features within the active site that could be exploited in molecule design. Consequently, a large percentage of the available TCS inhibitors demonstrate poor target specificity and act via multiple mechanisms, with aggregation of the kinase being the most notable. In order to elucidate the mode of action of some of these compounds, molecular modeling was employed to dock a suite of molecules into the ATP-binding domain of several histidine kinases. This effort revealed a key structural feature of the domain that is likely interacting with several known inhibitors and is also highly conserved. Furthermore, generation of several simplified scaffolds derived from a reported inhibitor and characterization of these compounds using activity assays, protein aggregation studies and saturation transfer differential (STD) NMR suggests that targeting of this protein feature may provide a basis for the design of ATP-competitive compounds.
Introduction
Two-component systems (TCSs) are signal transduction pathways that are ubiquitous in bacterial systems.1,2 Consisting of a histidine kinase (HK) and a response regulator (RR), these systems regulate diverse processes ranging from chemotaxis3 to virulence.4 Extracellular events activate HKs, inducing autophosphorylation of a conserved histidine (His) residue (Fig. 1). The phosphate group is subsequently transferred to an aspartate (Asp) on a cognate RR, eliciting an adaptive response such as quorum sensing,5 antibiotic resistance,6 or the production of virulence factors in pathogenic bacteria.7 Furthermore, the essentiality of some of these systems in the promotion of virulence traits8,9 and activation of resistance mechanisms10 makes them attractive drug targets.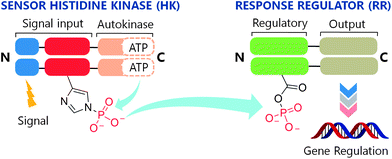 | ||
| Fig. 1 The two-component system signaling (TCS) cascade. Upon detection of an appropriate signal, autophosphorylation occurs at a conserved His residue of the HK, followed by phosphoryl group transfer to an Asp residue of the RR. A typical function for the RR is gene regulation. | ||
To date, although ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TCS proteins have been identified from genomic sequences, most have not been characterized.11 However, with the availability of X-ray crystal structures for a variety of HKs12–14 and RRs,15,16 and recently an HK–RR complex,17 much knowledge has been gained about the structural features that govern functionality in this family of signaling proteins. As a result, several sites have been identified for the action of chemical compounds to disrupt the intracellular signaling activities of these systems. Intervention by such a compound could occur at key junctions in the cascade (Fig. 1) including receipt of the activation signal, binding of ATP, autophosphorylation of the HK (HK∼P), dephosphorylation of HK∼P, phosphotransfer from HK∼P to the RR, and binding of phosphorylated RR (RR∼P) to the gene promoter.
000 TCS proteins have been identified from genomic sequences, most have not been characterized.11 However, with the availability of X-ray crystal structures for a variety of HKs12–14 and RRs,15,16 and recently an HK–RR complex,17 much knowledge has been gained about the structural features that govern functionality in this family of signaling proteins. As a result, several sites have been identified for the action of chemical compounds to disrupt the intracellular signaling activities of these systems. Intervention by such a compound could occur at key junctions in the cascade (Fig. 1) including receipt of the activation signal, binding of ATP, autophosphorylation of the HK (HK∼P), dephosphorylation of HK∼P, phosphotransfer from HK∼P to the RR, and binding of phosphorylated RR (RR∼P) to the gene promoter.
The search for inhibitors capable of interrupting TCS signal transduction has yielded several classes of synthetic compounds through a combination of high-throughput screening,18 rational design and structure-based design.19 While these initiatives have generated large libraries of inhibitors, examples of which are shown in Fig. 2, the mechanisms of action of most of these agents remain poorly understood. For example, RWJ-49815 (30; Fig. 3), a representative HK inhibitor, was initially reported to exert its inhibitory activity by targeting the catalytic and ATP-binding domain (CA) of several HKs;20 however, subsequent studies have shown that this compound inhibits HK autokinase activity non-specifically.21 In fact, 30, along with a large majority of compounds reported to inhibit TCS activity, do so through aggregation of the protein.22
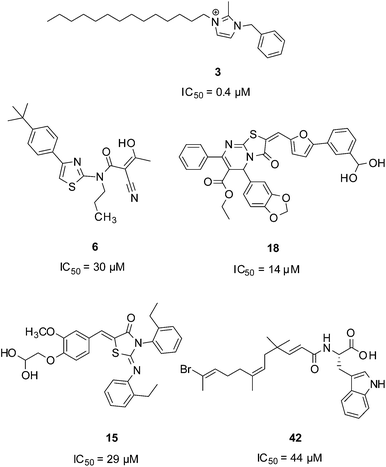 | ||
| Fig. 2 Molecules reported to inhibit TCS activity. | ||
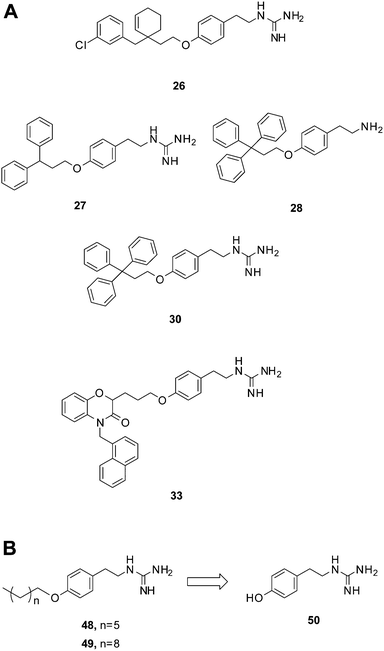 | ||
| Fig. 3 Compounds bearing guanidine moieties. (A) Subset of compounds predicted to interact with an active site Asp using molecular modeling. (B) Simplified variants of compounds 26, 27, 30 and 33. Further deconstruction of these compounds resulted in the fragment-like compound 50. | ||
Clearly, attaining specificity in the development of compounds capable of disrupting TCS signaling remains a challenging task. In recent years, there has been a heightened interest in deactivating TCS transduction by targeting the CA domain of the HK.22,23 Similar to other classes of kinases, the catalytic core within HKs has been reported to exhibit a high degree of homology11,24 in both Gram-positive and Gram-negative bacteria. Exploiting such a feature could yield an inhibitor with the potential to handicap multiple TCSs in a single pathogen, perhaps providing an additional avenue towards addressing antimicrobial drug resistance. Therefore, a better understanding of the interactions that take place between the CA domain and possible ligands is paramount. Herein, we report a chemical scaffold with the potential to interact with a conserved structural feature in the CA of HKs. This scaffold was identified, optimized and validated through a combination of molecular modeling, kinase inhibition and aggregation experiments, and ligand-observed NMR studies.
Results and discussion
To examine the probable ways in which previously reported inhibitors may be interacting with an HK, we sourced a diverse set of compounds with reported activity against TCS signaling from the literature. In all, forty-five inhibitors with IC50 values in the range of 0.4 μM to 1000 μM were identified and added to a virtual library for screening (compounds 1–38, 41–47; Table S1†).18,20,23,25–38 This library was appended with a subset of 1000 randomly selected drug-like compounds from the ZINC small-molecule database to expand library diversity and enhance screening for actives.39,40 In the selection of suitable receptors from the RSCB Protein Data Bank (PDB),41 preference was given to those that had been co-crystallized with a nucleotide and/or ligand. These included the virulence sensor PhoQ from Salmonella typhimurium (PDB: 3CGY)42 and Escherichia coli (PDB: 1ID0),14 the chemotaxis HK CheA (PDB: 1I58)43 and the kinase HK853 (PDB: 2C2A),13 both from Thermotoga maritima.The combined 1045 compound set was docked into the active sites of the selected receptors using the Surflex-Dock module44 of SYBYL.45 The fragments-constraints feature of Surflex was implemented and utilized a portion of the co-crystallized ligand as a guide during docking (Fig. S1†). In order to gauge docking accuracy, the ATP analogue AMP-PNP (5), was also included in the compound database. This nucleotide derivative ranked among the top 0.5% of all top-scoring compounds for one of the receptors, PhoQ (PDB: 1ID0), with the predicted binding pose of its adenine moiety closely matching that observed crystallographically (Fig. S2†). This ‘positive control’ helped to verify the docking and scoring parameters.
A notable aspect of many of the compounds in the inhibitor subset, particularly those in the top 10% (as surveyed across all utilized receptors), was the manner in which they interacted with a deeply buried Asp residue in the receptor active sites (Fig. S3†). Hydrogen bonding interactions have been observed between a phenolic hydrogen in radicicol and the analogous Asp in the HK PhoQ from Salmonella typhimurium in a co-crystallized structure.42 We were particularly drawn to a series of high-scoring (0.1–7.2% across all receptors) guanidine-bearing compounds (26, 27, 30, 33; Fig. 3A), that when docked, were predicted to interact with this active site Asp through a salt-bridge (Fig. S4†). For instance, a representative docked pose of the diphenyl compound 27 reveals that a strong interaction is established between this molecule's guanidine-like head group and Asp411 within the active site of HK853 (Fig. 4). Intriguingly, compound 28 (Fig. 3A), a close derivative of 30 that lacks the guanidine moiety and instead displays an amine, did not score as well and ranked in the range of 11.5–35% across all four receptors. Other noteworthy interactions include a π–π stacking between the aromatic ring of 27 with Tyr384, as well as other complementary hydrophobic interactions that occur on the outskirts of the binding site.
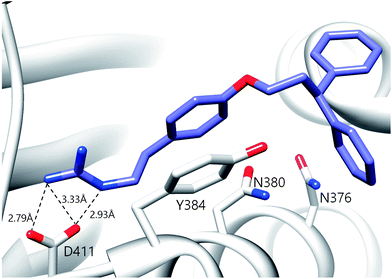 | ||
| Fig. 4 Predicted binding pose of compound 27 in the active site of HK853. The guanidine moiety of 27 forms a salt-bridge with D411. The ligand is also projected to participate in π–π stacking interactions with Y384. | ||
Previous active site mapping efforts indicated that this Asp residue, which is present in the G1 box, is conserved across HKs and is involved in ATP binding.46,47 Using the multiple sequence alignment (MSA) of 150 HK homologs (Fig. S5†)48 rendered onto the crystal structure of HK853 from Thermotoga maritima (Fig. 5), we generated a map of the highly conserved residues in the ATP-binding domain. This deeply situated Asp residue, along with a pair of distal active-site residues, Asn380 and Leu446, play key roles in the binding of the nucleotide to the CA domain (Asn in N Box, Leu in G2 box; Fig. 5, inset) and may provide viable contacts for appropriately designed ATP-competitive inhibitors.
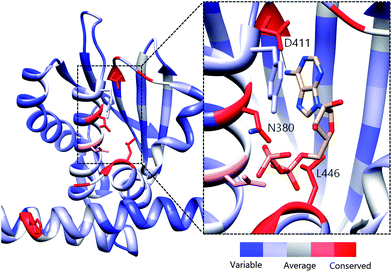 | ||
| Fig. 5 Evolutionary profile of active site residues in HKs. Derived from the sequence alignment of 150 HK homologs and rendered onto HK853 from Thermotoga maritima. Red colored residues/areas indicate high degrees of evolutionary conservation while blue regions indicate variations in conservation. Inset: Co-crystallized structure of ADP in the CA domain of HK853 (ref. 13) illustrating residues that are involved in binding of the nucleotide. The measured distance between the exocylic N of ADP and D411 is 2.83 Å. | ||
Salt-bridge mediated interactions between guanidine-like motifs and Asp residues situated in an active site have been observed before in serine proteases, thrombin,24 and even utilized successfully as starting points for inhibitor design,49 prompting us to further investigate this scaffold. However, the compound that was reported to be the most potent of this collection, 30, has also been shown to elicit protein aggregation as part of its mechanism of action.21,22 Shoichet and co-workers reported the triphenyl-bearing compound, clotrimazole, as also being a strong protein aggregator, suggesting that large hydrophobic moieties are likely involved in this undesirable outcome.50 Therefore, we postulated that more simplified variants of compounds 26, 27, 30 and 33 devoid of the rigid, hydrophobic rings would be better suited for study, with the goal of identifying a scaffold that does not cause protein aggregation (48, 49; Fig. 3B).
We used our previously disclosed assay, which utilizes the fluorescent nucleotide BODIPY-FL-ATP-γS,51 to evaluate the activity of HK853 from Thermotoga maritima upon subjection to the designed molecules. Inhibition of HK autophosphorylation was observed with both 48 and 49 and the commercially available HK inhibitor, NH-125 (3, Fig. 2 and S9†). The activity of compounds 48 and 49 was modest, with inhibition seen starting from 600 μM to 1 mM (Fig. S6†). Competition was observed between BODIPY-FL-ATP-γS and the test compounds (Fig. S7†), suggesting that either the lead molecules also bind in the active site or that they aggregate the protein reversibly. When subjected to in-gel aggregation studies (Fig. S8†), we confirmed that even the simplified scaffolds were still operating in a non-specific fashion by inducing aggregation of the protein, a phenomenon that was easily reversed by addition of the detergent Triton X-100. Examination of NH125 (3) in these assays also revealed that protein aggregates appear at concentrations where it inhibits autophosphorylation (Fig. 6A and S8†). We postulated that the aggregation potential of these compounds, all three of which contain a long alkyl tail, could be attributed to this moiety. Therefore, to further the study, we pursued the variant, 50 (Fig. 3B), that lacked the alkyl chain. Given the small size of this compound, we were unsurprised that even at concentrations nearing ∼10 mM, inhibition of autophosphorylation by compound 50 was not detected in our activity assay (Fig. S10†). Gel-based aggregation studies, however, using 50 confirmed that this compound did not induce aggregation of HK853 (Fig. 6B) suggesting that if this component does bind to the protein, it could provide a viable starting point for the design of an ATP-competitive inhibitor (Fig. S11 and S12†). The fragment-like nature of compound 50 presented significant challenges in assessing its potential interactions with a protein receptor using standard biological assays. Therefore, we utilized a ligand-observed NMR spectroscopy technique, saturation transfer differential (STD),52 to characterize the interactions between 50 and the receptor HK853 (Fig. 7). STD-NMR has become an invaluable tool in characterizing ligand–receptor complexes and has seen great utility in fragment-based drug discovery. Based on the nuclear-Overhauser effect, STD-NMR relies upon the selective saturation of the receptor in order to elicit resonances from any interacting ligands, distinguishing between binders and non-binders. In the presence of protein (HK853), resonances corresponding to compound 50 were observed in the STD-NMR spectrum, confirming a ligand–protein association (compare Fig. 7A and B). By utilizing the STD initial growth rates approach,53 the Kd for compound 50 was determined (Fig. 8) to be 2.41 (±0.25) mM, affording it a ligand efficiency (LE) of 0.27. If the guanidine-bearing group of 50 binds specifically in the ATP-binding site, adenosine diphosphate (ADP), the kinase reaction product, would disrupt this interaction. Thus, STD-NMR was repeated with protein and an equimolar mixture of ADP and 50. This yielded resonances corresponding only to those of the nucleotide indicating a competitive relationship between ligand 50 and ADP (compare Fig. 7C and D). Additionally, mutation of the active-site Asp residue of the protein to Ala resulted in significant attenuation of ligand 50's affinity for HK853 as judged by the diminishment of its resonances in the STD-NMR spectrum, which demonstrated the importance of this residue in ligand binding (compare Fig. 7B and E). We also examined the ability of tyramine, an analogous structure that contains an amine instead of a guanidine, to interact with the histidine kinase. Extremely weak ligand signals were observed, indicating that little to no binding was taking place (compare Fig. 7F and G). These data are consistent with the previous finding that the guanidine-functionalized compound 30 is 10-fold more potent than the amine-containing analogue (28; Table S1†). Collectively, our results are strongly suggestive that the simplified fragment 50 is binding in the nucleotide-binding domain of HK853 and is likely interacting with the protein via a salt-bridge with Asp411 in the active site.
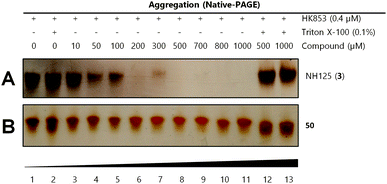 | ||
| Fig. 6 Gel-based aggregation studies using native-PAGE. (A) NH125 (3) induces aggregation of the protein starting at concentrations as low as 50 μM as evidenced by the disappearance of protein bands and (B) compound 50 does not aggregate HK853 even at concentrations as high as 1 mM. | ||
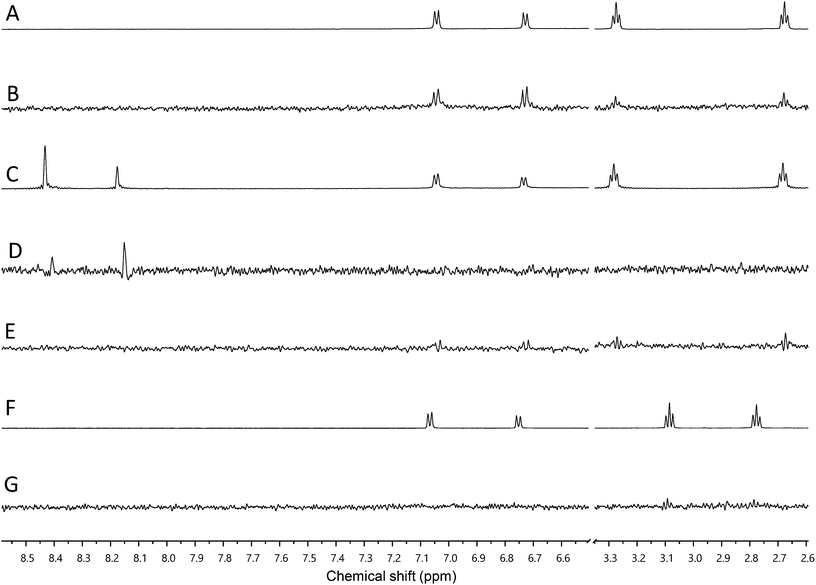 | ||
| Fig. 7 Assessment of binding using saturation transfer differential (STD) experiments (A) 1H spectrum of 50 and HK853, (B) 1H STD-NMR spectrum of 50 and HK853, (C) 1H NMR spectrum of 50, ADP and HK853, (D) 1H STD-NMR spectrum of 50, ADP and HK853, (E) 1H STD-NMR spectrum of 50 and mutated (D411A) HK853, (F) 1H spectrum of tyramine and HK853 and (G) 1H STD-NMR spectrum of tyramine and HK853. | ||
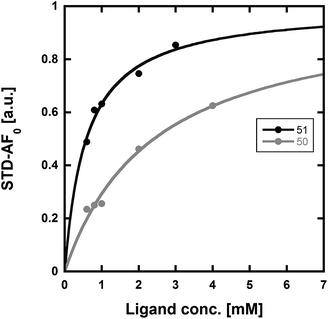 | ||
| Fig. 8 Determination of the Kd values of compounds 50 and 51 using STD-NMR. Using the initial growth rates approach53 and mathematical fitting to a Langmuir isotherm, the Kd values of 50 and 51 were found to be 2.41 (±0.25) mM and 0.58 (±0.06) mM, respectively. | ||
With fragment 50 as a starting point, we sought to determine if a molecule with increased affinity could be designed without generation of a compound that causes protein aggregation. Our modeling studies suggest that 50 is mimicking the adenosine portion of ATP. We anticipated that appending fragment 50 with a phosphate mimic, such as a sulfonyl group, could increase compound potency. Structural data indicates that the phosphate of ADP interacts with a highly conserved residue, N380 (Fig. 5), which could also be expected to bond to a sulfonyl moiety. Accordingly, we synthesized the modified fragment 51 (Fig. 9A) and proceeded to assess both its inhibitory and binding potentials using our activity-based assay and STD-NMR, respectively. Indeed, the evolved fragment 51 was found to not only inhibit autophosphorylation of HK853 (Fig. S13†) without inducing aggregation of the protein (Fig. S14†), but to also register a binding event to the receptor that is competitive with ADP (compare Fig. 9B and C). NMR Ranking experiments based on the STD factor values (fSTD)54 of ADP, tyramine, 50 and 51 (Table 1) reflected the increased affinity of 51 for HK853 [Kd = 0.58 (±0.06) mM; Fig. 8]. These data also indicate, as previously discerned, that binding of the guanidine-containing fragments is tighter than the related compound, tyramine, that is instead functionalized with an amine, establishing the importance of this group.
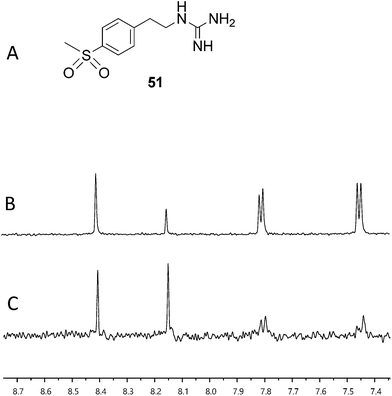 | ||
| Fig. 9 Assessment of binding using saturation transfer differential (STD) experiments (A) compound 51, (B) 1H spectrum of 51, ADP and HK853 and (C) 1H STD-NMR spectrum of 51, ADP and HK853. | ||
Conclusions
The catalytic and ATP-binding domains of bacterial HKs represent an ideal target for the action of new antibiotics. Although challenging, the design of ATP-competitive molecules specific for this site can likely be achieved through a better understanding of its structural features. Using a combination of molecular docking and sequence alignments, we have identified that the conserved Asp residue in the active site of HKs may interact with a portion of the compounds reported to have TCS inhibitory activities. Also, by deconstructing a series of known aggregators, we have identified a fragment (50) that likely binds with this residue, providing a basis for the design of selective HK inhibitors. Elaboration of this fragment into the sulfonyl 51, which may interact similarly to AMP, resulted in the identification of a compound with increased affinity. Our studies suggest that rational design is a viable avenue for the generation of potent inhibitors that specifically preclude autophosphorylation in this family of proteins.Experimental section
Synthetic methods
:0.6 hexanes:EtOAc) to afford the Boc-protected derivative of 48 (9.7 mg, 0.020 mmol, 70% yield) which then underwent a 2 h deprotection step in a stirred solution of 1 mL dichloromethane and 1 mL TFA. Evaporation of the solvents in vacuo followed by chromatography through a plug of neutral alumina (0.1:2 MeOH:CHCl3) afforded 48 (5.1 mg, 0.018 mmol, 91% yield). 1H NMR (400 MHz, CD3OD) δ 7.16 (d, J = 8.3 Hz, 2H), 6.87 (d, J = 8.3 Hz, 2H), 3.94 (t, J = 6.4 Hz, 2H), 3.41 (t, J = 7.0 Hz, 2H), 2.81 (t, J = 6.9 Hz, 2H), 1.82–1.71 (m, 2H), 1.51–1.43 (m, 2H), 1.41–1.32 (m, 6H), 0.91 (t, J = 6.1 Hz, 3H). 13C NMR δ, 15.0, 24.3, 27.8, 30.8, 31.1, 33.6, 35.8, 44.5, 69.7, 116.4, 131.4, 160.2 MS (m/z): [M + H]+ calcd for C16H27N3O 278.2232; found 278.2220.
:1 hexanes:EtOAc) to afford 52, the protected variant of 50 (0.22 g, 0.58 mmol, 79% yield). 1H NMR (400 MHz, CD2Cl2) δ 7.00 (d, J = 8.4 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 3.53 (t, J = 7.1 Hz, 2H), 2.75 (t, J = 7.3 Hz, 2H), 1.48 (s, 9H), 1.43 (s, 9H). Compound 52 (0.10 g, 0.26 mmol) was taken up in 1 mL dichloromethane and 1 mL trifluoroacetic acid and the reaction mixture was stirred for 2 h at r.t. after which the solvents were removed in vacuo. Purification of the crude material was accomplished by passage through a plug of neutral alumina (0.1:2 MeOH:CHCl3) to afford compound 50 (0.046 g, 0.256 mmol, 97% yield). 1H NMR (400 MHz, CD3OD) δ 7.04 (d, J = 8.1 Hz, 2H), 6.71 (d, J = 8.1 Hz, 2H), 3.36 (t, J = 7.0 Hz, 2H), 2.75 (t, J = 7.0 Hz, 2H). 13C NMR δ 33.7, 42.5, 115.0, 129.4, 155.9 MS (m/z): [M + H]+ calcd for C9H14N3O 180.1137; found 180.1133.
Biochemical methods
Computational methods
Acknowledgements
We thank D. Ma for assistance with the NMR spectrometers, C. Dann and T. Stone for helpful discussions and F. Hardin, Jr for generating HK853 protein. This work was supported by NIH R00GM82983, NIH DP2OD008592, a Pew Biomedical Scholar Award (E.E.C.), and an Indiana University Quantitative and Chemical Biology Training Fellowship (K.E.W.).Notes and references
- A. M. Stock, V. L. Robinson and P. N. Goudreau, Annu. Rev. Biochem., 2000, 69, 183–215 CrossRef CAS.
- L. A. Egger, H. Park and M. Inouye, Genes Cells, 1997, 2, 167–184 CAS.
- A. Stock, T. Chen, D. Welsh and J. Stock, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 1403–1407 CrossRef CAS.
- S. J. Clough, K. E. Lee, M. A. Schell and T. P. Denny, J. Bacteriol., 1997, 179, 3639–3648 CAS.
- L. C. M. Antunes, R. B. R. Ferreira, M. M. C. Buckner and B. B. Finlay, Microbiology, 2010, 156, 2271–2282 CrossRef CAS.
- E. Guenzi, A.-M. Gasc, M. A. Sicard and R. Hakenbeck, Mol. Microbiol., 1994, 12, 505–515 CrossRef CAS.
- G. K. Paterson, C. E. Blue and T. J. Mitchell, J. Med. Microbiol., 2006, 55, 355–363 CrossRef CAS.
- D. Beier and R. Gross, Curr. Opin. Microbiol., 2006, 9, 143–152 CrossRef CAS.
- N. C. Reading, D. A. Rasko, A. G. Torres and V. Sperandio, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5889–5894 CrossRef CAS.
- G. A. Cangelosi, J. S. Do, R. Freeman, J. G. Bennett, M. Semret and M. A. Behr, Antimicrob. Agents Chemother., 2006, 50, 461–468 CrossRef CAS.
- R. Gao and A. M. Stock, Annu. Rev. Microbiol., 2009, 63, 133–154 CrossRef CAS.
- M. J. Bick, V. Lamour, K. R. Rajashankar, Y. Gordiyenko, C. V. Robinson and S. A. Darst, J. Mol. Biol., 2009, 386, 163–177 CrossRef CAS.
- A. Marina, C. D. Waldburger and W. A. Hendrickson, EMBO J., 2005, 24, 4247–4259 CrossRef CAS.
- A. Marina, C. Mott, A. Auyzenberg, W. A. Hendrickson and C. D. Waldburger, J. Biol. Chem., 2001, 276, 41182–41190 CrossRef CAS.
- A. K. Park, J. H. Moon, K. S. Lee and Y. M. Chi, Biochem. Biophys. Res. Commun., 2012, 421, 403–407 CrossRef CAS.
- S. Yamada, H. Sugimoto, M. Kobayashi, A. Ohno, H. Nakamura and Y. Shiro, Structure, 2009, 17, 1333–1344 CrossRef CAS.
- P. Casino, V. Rubio and A. Marina, Cell, 2009, 139, 325–336 CrossRef CAS.
- Z. Qin, J. Zhang, B. Xu, L. Chen, Y. Wu, X. Yang, X. Shen, S. Molin, A. Danchin, H. Jiang and D. Qu, BMC Microbiol., 2006, 6, 96 CrossRef.
- Y. T. Tang, R. Gao, J. J. Havranek, E. A. Groisman, A. M. Stock and G. R. Marshall, Chem. Biol. Drug Des., 2012, 79, 1007–1017 CAS.
- J. F. Barrett, R. M. Goldschmidt, L. E. Lawrence, B. Foleno, R. Chen, J. P. Demers, S. Johnson, R. Kanojia, J. Fernandez, J. Bernstein, L. Licata, A. Donetz, S. Huang, D. J. Hlasta, M. J. Macielag, K. Ohemeng, R. Frechette, M. B. Frosco, D. H. Klaubert, J. M. Whiteley, L. Wang and J. A. Hoch, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5317–5322 CrossRef CAS.
- E. C. Theodorou, M. C. Theodorou and D. A. Kyriakidis, Cell. Signalling, 2011, 23, 1327–1337 CrossRef CAS.
- K. Stephenson, Y. Yamaguchi and J. A. Hoch, J. Biol. Chem., 2000, 275, 38900–38904 CrossRef CAS.
- R. Gilmour, J. E. Foster, Q. Sheng, J. R. McClain, A. Riley, P.-M. Sun, W.-L. Ng, D. Yan, T. I. Nicas, K. Henry and M. E. Winkler, J. Bacteriol., 2005, 187, 8196–8200 CrossRef CAS.
- V. Škedelj, T. Tomašić, L. P. Mašič and A. Zega, J. Med. Chem., 2011, 54, 915–929 CrossRef.
- J. E. Foster, Q. Sheng, J. R. McClain, M. Bures, T. I. Nicas, K. Henry, M. E. Winkler and R. Gilmour, Microbiology, 2004, 150, 885–896 CrossRef CAS.
- E. Furuta, K. Yamamoto, D. Tatebe, K. Watabe, T. Kitayama and R. Utsumi, FEBS Lett., 2005, 579, 2065–2070 CrossRef CAS.
- J. J. Hilliard, R. M. Goldschmidt, L. Licata, E. Z. Baum and K. Bush, Antimicrob. Agents Chemother., 1999, 43, 1693–1699 CAS.
- D. J. Hlasta, J. P. Demers, B. D. Foleno, S. A. Fraga-Spano, J. Guan, J. J. Hilliard, M. J. Macielag, K. A. Ohemeng, C. M. Sheppard, Z. Sui, G. C. Webb, M. A. Weidner-Wells, H. Werblood and J. F. Barrett, Bioorg. Med. Chem. Lett., 1998, 8, 1923–1928 CrossRef CAS.
- R. M. Kanojia, W. Murray, J. Bernstein, J. Fernandez, B. D. Foleno, H. Krause, L. Lawrence, G. Webb and J. F. Barrett, Bioorg. Med. Chem. Lett., 1999, 9, 2947–2952 CrossRef CAS.
- T. Kitayama, R. Iwabuchi, S. Minagawa, S. Sawada, R. Okumura, K. Hoshino, J. Cappiello and R. Utsumi, Bioorg. Med. Chem. Lett., 2007, 17, 1098–1101 CrossRef CAS.
- N. Li, F. Wang, S. Niu, J. Cao, K. Wu, Y. Li, N. Yin, X. Zhang, W. Zhu and Y. Yin, BMC Microbiol., 2009, 9, 129 CrossRef.
- A. Okada, Y. Gotoh, T. Watanabe, E. Furuta, K. Yamamoto and R. Utsumi, Methods Enzymol., 2007, 422, 386–395 CAS.
- Z. Qin, B. Lee, L. Yang, J. Zhang, X. Yang, D. Qu, H. Jiang and S. Molin, FEMS Microbiol. Lett., 2007, 273, 149–156 CrossRef CAS.
- H. Ramström, M. Bourotte, C. Philippe, M. Schmitt, J. Haiech and J.-J. Bourguignon, J. Med. Chem., 2004, 47, 2264–2275 CrossRef.
- S. Roychoudhury, S. E. Blondelle, S. M. Collins, M. C. Davis, H. D. McKeever, R. A. Houghten and C. N. Parker, Mol. Diversity, 1998, 4, 173–182 CrossRef CAS.
- S. Roychoudhury, N. A. Zielinski, A. J. Ninfa, N. E. Allen, L. N. Jungheim, T. I. Nicas and A. M. Chakrabarty, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 965–969 CrossRef CAS.
- Z. Sui, J. Guan, D. J. Hlasta, M. J. Macielag, B. D. Foleno, R. M. Goldschmidt, M. J. Loeloff, G. C. Webb and J. F. Barrett, Bioorg. Med. Chem. Lett., 1998, 8, 1929–1934 CrossRef CAS.
- S. J. Trew, S. K. Wrigley, L. Pairet, J. Sohal, P. Shanu-Wilson, M. A. Hayes, S. M. Martin, R. N. Manohar, M. I. Chicarelli-Robinson, D. A. Kau, C. V. Byrne, E. M. Wellington, J. M. Moloney, J. Howard, D. Hupe and E. R. Olson, J. Antibiot., 2000, 53, 1–11 CrossRef CAS.
- T. A. Pham and A. N. Jain, J. Med. Chem., 2005, 49, 5856–5868 CrossRef.
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2004, 45, 177–182 CrossRef.
- RSCB Protein Data Bank, http://www.pdb.org.
- M. T. Guarnieri, L. Zhang, J. Shen and R. Zhao, J. Mol. Biol., 2008, 379, 82–93 CrossRef CAS.
- A. M. Bilwes, C. M. Quezada, L. R. Croal, B. R. Crane and M. I. Simon, Nat. Struct. Biol., 2001, 8, 353–360 CrossRef CAS.
- A. Jain, J. Comput.-Aided Mol. Des., 2007, 21, 281–306 CrossRef CAS.
- T. Associates, Certara, St. Louis, MI, 2012.
- A. Hirschman, M. Boukhvalova, R. VanBruggen, A. J. Wolfe and R. C. Stewart, Biochemistry, 2001, 40, 13876–13887 CrossRef CAS.
- A. M. Bilwes, C. M. Quezada, L. R. Croal, B. R. Crane and M. I. Simon, Nat. Struct. Mol. Biol., 2001, 8, 353–360 CAS.
- H. Ashkenazy, E. Erez, E. Martz, T. Pupko and N. Ben-Tal, Nucleic Acids Res., 2010, 38, W529–W533 CrossRef CAS.
- J. A. Olsen, D. W. Banner, P. Seiler, U. Obst Sander, A. D'Arcy, M. Stihle, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 2507–2511 CrossRef CAS.
- J. Seidler, S. L. McGovern, T. N. Doman and B. K. Shoichet, J. Med. Chem., 2003, 46, 4477–4486 CrossRef CAS.
- K. E. Wilke, S. Francis and E. E. Carlson, J. Am. Chem. Soc., 2012, 134, 9150–9153 CrossRef CAS.
- M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108–6117 CrossRef CAS.
- J. Angulo, P. M. Enríquez-Navas and P. M. Nieto, Chem.–Eur. J., 2010, 16, 7803–7812 CrossRef CAS.
- S. Barelier, J. Pons, K. Gehring, J.-M. Lancelin and I. Krimm, J. Med. Chem., 2010, 53, 5256–5266 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and tables are provided depicting the library of known inhibitors, additional modeling studies, kinase activity assays, protein aggregation studies, sequence alignments and experimental methods. See DOI: 10.1039/c2md20308a |
| This journal is © The Royal Society of Chemistry 2013 |