Experimental validation of in silico target predictions on synergistic protein targets†
Isidro
Cortes-Ciriano‡
a,
Alexios
Koutsoukas
b,
Olga
Abian‡
ac,
Robert C.
Glen
b,
Adrian
Velazquez-Campoy‡
*ad and
Andreas
Bender‡
*b
aInstitute of Biocomputation and Physics of Complex Systems (BIFI), Unidad Asociada IQFR-CSIC-BIFI, and Department of Biochemistry and Molecular and Cellular Biology, Universidad de Zaragoza, Zaragoza, Spain. E-mail: ab454@cam.ac.uk; adrianvc@unizar.es
bUnilever Centre for Molecular Science Informatics, Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
cAragon Health Sciences Institute (I+CS), Zaragoza, Spain
dFundacion ARAID, Diputacion General de Aragon, Spain
First published on 23rd November 2012
Abstract
Two relatively recent trends have become apparent in current early stage drug discovery settings: firstly, a revival of phenotypic screening strategies and secondly, the increasing acceptance that some drugs work by modulating multiple targets in parallel (‘multi-target drugs’). The work presented here combines both those aspects by integrating experimental phenotypic screening for cytotoxic compounds with an experimental validation of individual protein targets predicted in silico. In this first step of this work, in silico target predictions for a dataset comprising cytotoxic compounds showed an enrichment of enzymes involved in cell cycle progression (such as Topoisomerase I, Bcl-X and Protein Kinase C alpha) as well as in the defense against xenobiotic compounds (such as P-gp 1 and the CYPs). Ten compounds predicted to be active on each of two of the enriched targets, P-glycoprotein 1 and Topoisomerase I, were tested in vitro to validate (or invalidate) the predicted mode of action. Hoechst 33342 dye uptake, P-gp ATPase activity and Topoisomerase I DNA relaxation assays were able to identify two inhibitors of P-gp with IC50 values of 37 ± 5 and 28 ± 2 μM, respectively (comparable to the activity of Verapamil of 12 μM measured with the same assay) as well as five moderate inhibitors of Topoisomerase I. Furthermore, we also screened combinations of compounds with different modes of action to evaluate possible synergistic effects. When evaluating compound synergies, four of the five compounds exhibit synergistic effects in HeLa cell cultures in the presence of the two P-gp inhibitors identified (two independent samples t-test, p < 0.01). Hence, this appears to be one of the first studies where multiple aspects of compound action as predicted by in silico models are prospectively validated, namely phenotypic effect as well as on-target activities, and where synergies between compound combinations could also be experimentally confirmed.
Introduction
Historically, drug discovery has been performed phenotypically, and in many cases even at the organism level.1,2 Since the 1980s, while target-based approaches (together with developments such as combinatorial chemistry and high-throughput screening) have gained considerable attention, recent studies show that phenotypic screening is still one of the main sources of ‘first in class’ drugs (i.e. those with a new mode of action; 27 out of 45 drugs approved by the FDA between 1999 and 2008).3Both target-based1 and phenotypic-based screening2 are based on different but complementary principles (see Fig. 1). Target-based drug discovery makes it necessary to first identify biochemical factor(s) mediating disease biology,4 and in a second step high-affinity modulators of those proteins are identified in the hope that this modulation will reverse the diseased state back to the healthy state.5,6 While this approach is very amenable to high-throughput techniques, in this case no network information (neither of intra- nor inter-cellular networks) is taken into account, and in addition no information concerning ADME/Tox properties or the modulation of additional targets can be gathered. Hence, while a precisely defined area of chemical space is sampled in target-based screens (with the hope that this particular activity will be important in later stages), the information that can be gathered about a compound (in particular from the efficacy standpoint) is rather limited.
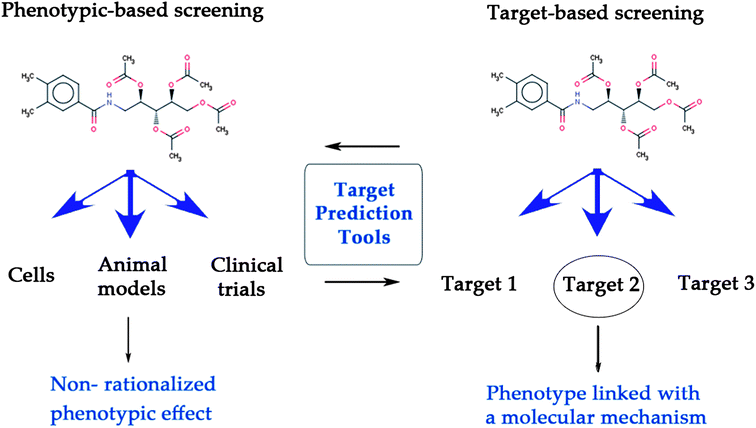 | ||
| Fig. 1 Target prediction tools bridge the gap between target-based and phenotypic screening, leading to the linkage of a phenotype with its underlying mode-of-action. While phenotypic screening can assess the efficacy of compounds at a physiological level (left), target-based screening determines whether a compound interacts with a target or not (right). | ||
On the other hand, phenotypic screening evaluates the response of a biological system to the application of compounds at more complex biological levels, from cell cultures to whole organisms.2,7,8 Here, an efficacy assessment of chemical substances ‘closer to disease biology’ is obtained, hence facilitating the collection of bioactivity information in a physiologically more relevant context. (In turn it should be said that phenotypic screens generally deliver more noisy data than biochemical screens also due to the underlying biology, and that often high-dimensional readouts are obtained which are more difficult to interpret.) However although the phenotypic readout is often thought to be more predictive for efficacy in man,9–11 this approach does not permit the identification of the mode of action (MoA) of the compounds exhibiting bioactivity.4 Still, when combined with subsequent target elucidation techniques phenotypic screening followed by identification of the target can be seen as a commonly applied and feasible screening strategy in early stage drug discovery.12,13
Given that phenotypic screening seems to be more successful in identifying compounds with novel modes of action, and the fact that target deconvolution is one of the hurdles when using this approach, it appears that additional help in the target deconvolution area would be of tremendous practical value. One recent area of development, sparked by the amount of bioactivity data available publicly, has been the prediction of compound bioactivities using in silico approaches. Target prediction algorithms are based upon either the ligand or the protein side,14 and in the current work we will focus on utilizing the wealth of public bioactivity information15 for the development of ligand-based target prediction models. Data mining techniques in the target prediction area are an application of the ‘chemogenomics principle’,16,17 which analyzes (and predicts) the relationships between protein and ligand space on a large scale.18 As an example of machine learning algorithms employed in this area, multiple-category Naïve Bayes based approaches were firstly implemented by Nidhi et al.,19,20 with more recent approaches (with roughly comparable performance) being e.g. the Parzen–Rosenblatt Window algorithm.21
The application of in silico target prediction algorithms has also led to the possibility of completing compound–target bioactivity profiles in a more comprehensive manner than before, enabling the prediction of broad bioactivity profiles against large numbers of targets, now also known as the polypharmacology that many compounds are thought to exhibit to achieve the observed clinical effects.22,23 In other words, the conventional lock-and-key paradigm,24 which states that a compound exerts its activity via a unique protein target relevant for this particular disease, has been extended, and in line with this thinking it was recently established that drugs modulate on average not only one, but around six targets (though of course the distribution between targets contribution to the desired action and those that are detrimental or toxic are often not known).25
Consistent with the notion that cross-reactivity and side-effects stem from the modulation of additional targets (‘antitargets’),23,26–29 which is an important cause of attrition in drug development,22,28 Lounkine et al.30 conducted the largest prospective evaluation of in silico target prediction to date for 656 known drugs approved for human use on 73 protein targets (focusing on undesired off-targets). Applying the similarity ensemble approach (SEA),31 adverse drug reactions were linked to predicted off-target interactions. Biochemical assays confirmed about half of the predictions at in many cases pharmacologically significant concentrations (1 nM to 30 μM). Due to the large number of classes and relatively low random hit rates this represents a significant enrichment of active compounds at the anti-targets considered in the study.
As one of the earliest examples of in silico target prediction methods, the PASS (‘Prediction of Activity Spectra for Substances’) method was applied to 130 of the 200 best-selling medicines on the market, and it was found that their known bioactivity spectrum was in accordance with the predicted bioactivity classes in 93.2% of the cases.32 Moreover, new cognition enhancers in animal models were discovered by the application of the algorithm to novel chemical structures.33
Also, a vast number of traditional medicines, such as from traditional Chinese and ayurvedic medicine, potentially reverse diseased phenotypes back to the healthy state (with the limitation being that the number of statistically sound clinical studies has only recently reached an acceptable level of stringency).34 However, in the case of traditional medicines the identification of the active principle from natural products (NP) responsible for the desired phenotype is necessary, but often not trivial. In order to alleviate this situation,35 Schuster et al. applied pharmacophore ensembles to the target prediction of entities from Traditional Chinese Medicine (TCM), thereby successfully elucidating the involvement of four targets in the antiinflammatory effects of those compounds.36
Of particular relevance for the work presented here is the application of Flachner et al.37 to compound target profiling in combination with high-throughput cytotoxicity screening in the context of cancer chemotherapy. A differential in vitro and in vivo screening strategy (DIVISS) identified 115 enriched targets for compounds selectively cytotoxic to tumor cells. Among these targets 58% were enzymes, while this percentage fell to 37% for compounds exhibiting cytotoxicity to either only healthy or both cell lines. While we do not compare healthy and cancer cells in the current work, the calculation of target enrichments will also be encountered later, with prospective predictions being tested both phenotypically and in biochemical assays as outlined below.
In this work, we focus on both the phenotypic and target-based aspects of identifying novel bioactive compounds. More precisely, we present a practical application of the Laplacian-modified Naïve Bayes classifier proposed by Nigsch et al.21 for cytotoxicity-related target prediction (Fig. 2). By comparing the targets for a cytotoxicity dataset to a background distribution, we calculated which targets were enriched among compounds cytotoxic to HeLa cells. In order to validate this prediction, ten compounds from an external library (HitFinder)38 predicted to be active on two of the enriched targets, P-glycoprotein 1 and Topoisomerase I, were selected and experimentally validated with respect to their bioactivity (and, hence, to find supporting evidence for their putative modes of action). Furthermore, given that compounds active against both targets can likely contribute to cytotoxicity, we also explored the possibility of synergism between these by applying both compounds in parallel when evaluating cytotoxicity. This is of particular relevance given that efflux pumps such as P-gp are thought to decrease efficacy of e.g. anti-cancer drugs, so that blocking this protein while at the same time applying the Topoisomerase inhibitor should increase efficacy over applying the latter by itself.39 Hence, both aspects discussed above – the importance of considering both phenotypic and protein bioactivities in parallel as well as the area of polypharmacology – will be considered in the experimental aspects of this study.
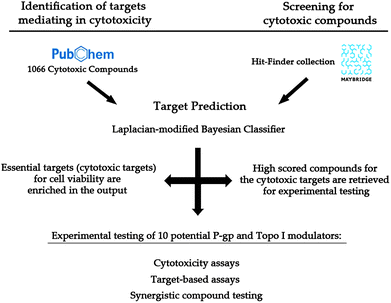 | ||
| Fig. 2 Workflow followed to experimentally evaluate the applicability of an in silico target prediction for cytotoxic compounds on synergistic protein targets (P-glycoprotein 1 and Topoisomerase I). Firstly, 1066 compounds cytotoxic to HeLa cells were retrieved from PubChem Bioassay (ESI, Table S4†) and their targets were predicted (left side). 18 enriched targets were used in further assessment (Table S1†). Secondly, the target prediction algorithm was applied to a supplier library (right). Ten compounds predicted to interact with those targets selected in the first step were experimentally tested separately and in combination (bottom). | ||
Results and discussion
The enrichment analysis of protein targets predicted for 1066 compounds cytotoxic to HeLa cells retrieved a list of 18 protein targets which could mechanistically be involved in this effect with a certain confidence (as discussed in the model validation – Materials and methods –; 2.01% of the training dataset, see Table S1†). Among the enriched targets, enzymes crucial for the viability of the cell as well as those important for the defense against xenobiotics (P-glycoprotein 1 and CYPs)40 were widely represented (13.41% and 10.98% respectively). Topoisomerase I was repeatedly predicted as a target for cytotoxic compounds (6.66%), owing to its relevance for DNA replication.39 This was also the case for Protein Kinase Cα (PKCα; 17.45%) and DNA-dependent Protein Kinase (DNA-PK; 6.75%). The role of PKCα in key cellular processes such as apoptosis, differentiation or proliferation has been exhaustively demonstrated41 and DNA-PK has been implicated in DNA double-strand break (DSB) repair.42Given the enrichment of the above targets among compounds cyototoxic to HeLa cells we now hypothesized a causal link between the two – cytotoxicity and targets involved – which was evaluated experimentally.
For this purpose, the target prediction model was employed to select the highest-scoring compounds from the HitFinder library38 for two of the enriched targets for which assays were available, namely human Topoisomerase I (compounds Topo 1–Topo 5) and P-glycoprotein 1 (compounds P-gp 1–P-gp 5), for experimental validation. Although not being the most enriched targets, both the relevance of P-glycoprotein 1 and Topoisomerase I for chemotherapy and their spatially distinct localization, plasma membrane and nucleus respectively, make them suitable for the assessment of compound synergism. Furthermore, compounds Topo 1, Topo 3 and Topo 5 were also predicted to interact with P-gp, with scores of 3.20, 3.83 and 5.40 respectively, therefore allowing validation of diverse target–compound interactions for unique chemical structures.
The structures of the selected compounds are shown in Fig. 3 and 4 for Topoisomerase I and P-gp, respectively. Visual inspection and similarity analyses of the 5 nearest neighbors between selected compounds and those present in the cytotoxicity dataset (using the Tanimoto coefficient and OpenBabel FP2 fingerprints) between the ten selected compounds and the cytotoxicity dataset showed relatively diverse chemistry (Table S2†) with nearest-neighbour Tanimoto coefficients ranging from 0.19 to a maximum 0.68. By chance, compound Topo 2 identified by our model had already been described as cyototoxic to several cancer cell lines by Kolocouris et al.43 (ESI, Table S2†), which is why it was also present in the cytotoxicity dataset extracted from the PubChem BioAssay.44 We were not overly concerned with this compound overlapping between both sets since this in effect confirmed the validity of our data modeling pipeline.
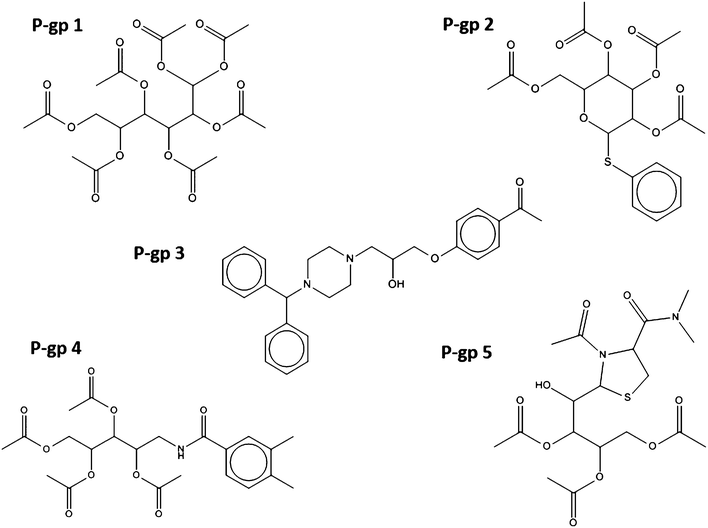 | ||
| Fig. 3 Chemical structure of the five compounds selected according to their predicted activity against P-glycoprotein 1. Virtually all compounds cover different chemical classes, ensuring that sufficient chemical space is sampled in subsequent biochemical and phenotypic assays. | ||
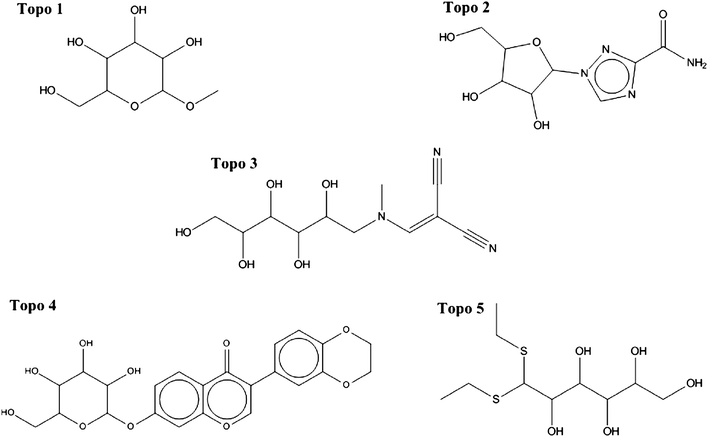 | ||
| Fig. 4 Chemical structure of the five selected compounds targeting Topoisomerase I. Considerable diversity is also apparent. In addition, the differences between structures shown here and known Topoisomerase inhibitors from CHEMBL (v10) were evaluated by a more detailed similarity analysis (Table S2†). | ||
In order to establish the novelty of our compounds selected for Topoisomerase I we compared the compounds to those present in ChEMBL v10. While compound Topo 1 had close analogues in the database, all other compounds selected were structurally novel (Fig. S1†). Furthermore, only compound P-gp 3 exhibits Tanimoto coefficients higher than 0.5 when performing a similarity analysis of our compound selected against P-gp to the P-gp inhibitors database described by Chen et al. (Fig. S2†).45 Both bioactivities are also orthogonal in ligand space, as indicated by low overall similarity of the ligands (Tanimoto coefficient is 0.4), except for compound pairs Topo 1/P-gp 1 and P-gp 4/P-gp 5 (Fig. S3†). The Maximum Common Subgraph (MCS) of the ten selected compounds and both the P-glycoprotein 1 and Topoisomerase I databases are depicted in Fig. S4 and S5† respectively.46
In order to lend support for the involvement of P-gp in cytotoxicity against HeLa cells firstly biochemical assays against this protein were conducted. With the assay employed here47,48 all five compounds tested were found to inhibit P-glycoprotein 1 in a statistically significant way (p < 0.05, ESI, Fig. S6†). In particular, P-gp 2 (IC50 = 37 ± 5 μM, ESI, Fig. S7†) and P-gp 4 (IC50 = 28 ± 2 μM, ESI, Fig. S7†) showed higher activity, being only slightly less potent than the well-established P-gp inhibitor Verapamil (IC50 = 12 μM) when measured with the same assay.47,48 Also in cellular assays both compounds showed activity in reducing the uptake of Hoechst 33342 in HeLa cell cultures, a process which is known to be mediated by P-glycoprotein 1,49 with IC50 values of 15.38 μM and 15.50 μM, respectively (see Fig. 5). Given that many of the P-gp inhibitors contained in ChEMBL are also active in the low micromolar range, the activities discovered are consistent with the activities of inhibitors used to generate the target prediction in the first place.
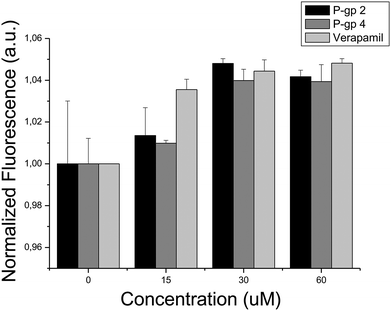 | ||
| Fig. 5 Hoechst 33342 uptake of HeLa cells in the presence of: (i) compound P-gp 2, (ii) compound P-gp 4, and (iii) Verapamil. The fluorescence signal increase reflects the intracellular accumulation of dye due to the inhibitory action of either compounds targeting P-gp or Verapamil. According to the IC50 values of compounds P-gp 2 and P-gp 4 (IC50 = 15.38 μM and 15.50 μM, respectively), Hoechst 33342 is accumulated within the cells as compound concentration increases, being the effect comparable to that induced by the presence of Verapamil (IC50 = 1.7 μM). | ||
When evaluating bioactivity for the second enriched target for which experimental validations were performed, biochemical assays confirmed activity of two out of the five compounds selected for Topoisomerase I, where it was found that compounds Topo 2 and Topo 3 possess IC50 values of 65.88 μM and 47.53 μM, respectively (see Fig. 6). On the contrary, bacterial proliferation was not affected at a concentration of 200 μM by the presence of either of those compounds, suggesting inhibition of human Topoisomerase I selectively, while not compromising bacterial Topoisomerase activity.
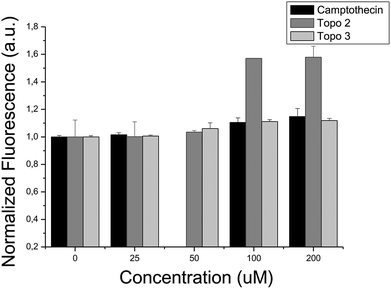 | ||
| Fig. 6 In vitro interaction between Topoisomerase I and: (i) camptothecin, (ii) compound Topo 2 and (iii) compound Topo 3. An increase in the fluorescence signal reflects the formation of DNA triplex and the presence of supercoiled DNA, inversely proportional to the Topoisomerase activity. Compounds Topo 2 and Topo 3 inhibit Topoisomerase activity, exhibiting IC50 values of 65.88 μM and 47.53 μM, respectively. | ||
As we have seen above, 7 out of the 10 compounds selected showed bioactivity against the target they were selected for. Given that the selection of compounds was guided by the putative involvement of the target in cytotoxicity, we now evaluated those compounds – separately and in combination – in cytotoxicity assays. It was found that the most active compound identified, P-gp 3 (tested at 120 μM), caused a considerable drop in cell viability to 37% (p < 0.01, Fig. 7) while P-gp 2 exhibited moderate, but not statistically significant cytotoxicity at the same concentration (showing a cell viability drop from 100% to 87%, p > 0.05). Compounds P-gp 1, P-gp 4 and P-gp 5 were not cytotoxic at the maximum concentration employed (120 μM). Among the Topoisomerase inhibitors Topo 4 was the most active compound (cell viability reduction to 82%, p < 0.01) at a concentration of 120 μM while Topo 1–Topo 3 and Topo 5 were not cytotoxic at 120 μM although a compound–target interaction was confirmed in the corresponding target-based assay. Many reasons are possible for inactivity, ranging from cell permeability to insufficient activity to cause cellular effects. In all MTT cytotoxicity assays, a transient increase of the reduction of MTT by living cells appears at low compound concentrations.50 Although we cannot determine the reasons for this phenomenon here, Weyermann et al.51 reported the same effect when measuring the reduction of MTT by mouse fibroblasts after the addition of sodium azide.
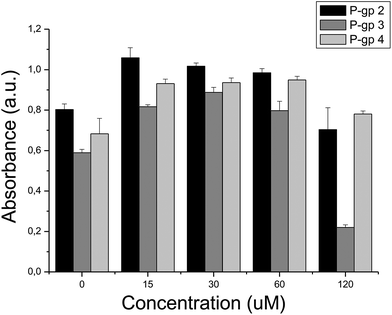 | ||
| Fig. 7 Cell viability measured in the presence of compounds: (i) P-gp 2, (ii) P-gp 3, (iii) P-gp 4. The intensity of the signal is directly proportional to cell viability. At the maximum compound concentration tested – 120 μM – cell viability decreases in the presence of compounds P-gp 2 and P-gp 3, while it is not compromised in the presence of compound P-gp 4. | ||
We further tested the hypothesis that applying inhibitors of Topoisomerase I, while blocking the efflux pump P-gp in parallel, increases the activity over inhibiting Topoisomerase I alone. In line with this hypothesis, indeed several synergistic effects between compounds from both activity classes could be found (see Fig. 8 and also Fig. 9). The presence of compound P-gp 4 (50 μM) maintained the previously indicated cytotoxicity levels of Topo 1, while significantly increasing cytotoxicity of Topo 2 (p < 0.01) and Topo 3–Topo 5 (all p < 0.01; Fig. 9 and ESI, Table S3†). In agreement with the relative IC50 values of P-gp 2 and P-gp 4, comparable combined cytotoxic effects were observed when compound P-gp 2 was added to the above compounds (all p < 0.01), interestingly this time with the exception of Topo 4 (with p = 0.28).
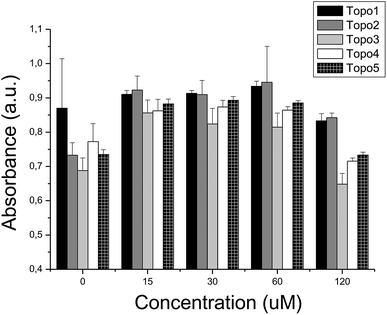 | ||
| Fig. 8 Combined cytotoxicity evaluation in the presence of compound P-gp 4 (50 μM) and compounds Topo: (i) 1, (ii) 2, (iii) 3, (iv) 4 and (v) 5 at 120 μM. The presence of a synergistic effect of compound P-gp 4 and compounds Topo 2, Topo 3, Topo 4 and Topo 5 may suggest a causal link between the inhibition of P-gp and an increase of the inhibition of the Topoisomerase activity. | ||
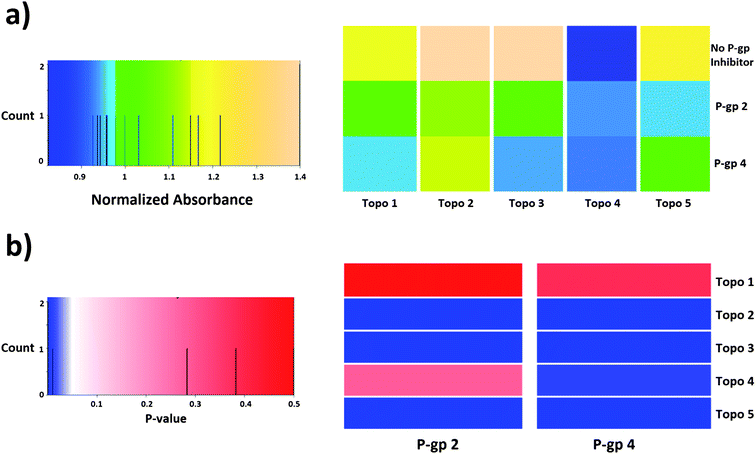 | ||
| Fig. 9 Heatmaps representing (a) normalized absorbance values obtained in the cytotoxicity assay and (b) p-values of the analysis of compound synergism in HeLa cells (t-test of independent samples). According to the statistical analysis, compound synergies could be established in 7 out of the 10 cases presented here (out of a total of 25 combinations). Compound P-gp 4 enhances the cytotoxic effect of compounds Topo 2, Topo 3, Topo 4 and Topo 5, while compound P-gp 2 produces a similar effect except for Topo 4. | ||
One remark regarding the activities of the compounds discovered here should also be made: while for single-compound drug discovery usually nanomolar binders are required to achieve efficacy in biological systems, it is thought that in the case of combination therapy multiple weak interactions are able to achieve comparable effects, which are able to stabilize the cellular system in the desired (usually healthy) state, and prevent it from shifting to an alternative resistant equilibrium.52 Therefore, compounds eliciting a desired inhibitory effect with relatively high IC50 values, as is the case here, should not be discarded beforehand in case multiple modulations of biological systems are performed in parallel.
In the case of compounds predicted to inhibit P-gp, high cytotoxicity was not expected owing to its cellular localization and role (loss of P-gp function does not necessarily compromise cell viability in the context of aseptic laboratory cell cultures), except for those cases where compounds for both targets were present (synergy evaluation).
Furthermore, apparently neither compound P-gp 2 nor compound P-gp 4 induced apoptosis, a hallmark of DNA damage,53 with the morphology of the cells and their extracellular membranes similar to that of non-treated cells (ESI, Fig. S8†). The absence of apoptotic cells suggests that compounds inhibiting P-gp do not interact with either apoptosis related proteins or with the DNA to a remarkable extent (which concurs with the five top ranked predicted targets for P-gp 2 and P-gp 4, namely membrane channels). As previously shown by Zheng et al.,54 compound cytotoxicity is in some cases responsible for transport inhibition when in vitro screening membrane channel inhibitors and therefore, a source of false positives in drug-transporter inhibition assays. The ability of compounds predicted to inhibit P-gp to compromise its activity was measured via biochemical assays and Hoechst 33342 dye uptake, while not compromising cell viability in a comparable manner in cell-based assays, let us conclude that the inhibition of P-gp is not causing cytotoxicity in the cell line tested.54
Conclusions
In this work we have illustrated the combination of phenotypic screening and in silico target prediction for both mode-of-action analysis and the evaluation of synergistic effects. As a result, two modulators of the human P-glycoprotein 1 with IC50 values of 37 ± 5 and 28 ± 2 μM, respectively, have been identified which are comparable to that of Verapamil with 12 μM when measured with the same assay. Furthermore, we identified five moderate affinity Topoisomerase I inhibitors via a prospective validation of both target-based and phenotypic screens. The validation of synergistic combinations of compounds was partially successful (and where successful statistically significant), namely for compound P-gp 2 in combination with compounds Topo 2, Topo 3 and Topo 5, and compound P-gp 4 in combination with compounds Topo 2, Topo 3, Topo 4 and Topo 5.Hence it appears that in silico target prediction still has more applications than obvious at first sight, and the rational selection of compound combinations to arrive at the desired effect – as illustrated here for cytotoxicity – is also likely to deserve more attention in the future.
Materials and methods
Laplacian-modified Naïve Bayesian classifier
The Laplacian-modified Naïve Bayesian classifier described by Nigsch et al.21 was implemented which is based on Bayes' theorem:with A being the event for which a compound showing activity A and B the event for which an array of n molecular features fi is present concurrently in that compound:
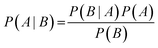
| P(B) = P(f1 ∩ f2 ∩…∩ fn) |
Two events are independent if:
| P(A ∩ B) = P(A)P(B) |
Given a number m of independent events, Am:
| P(A1 ∩ A2 ∩…∩ Am) = P(A1)P(A2)…P(Am) |
As a result, in the case of a compound containing n features, the conditional probability of the event of that compound belonging to a concrete class is defined by:
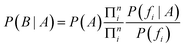
We can now consider Ci and Ti, the number of occurrences of feature fi in compounds in a class and in the total dataset, respectively. Thus, the uncorrected probability of a compound of a class, A, if feature fi were present is:
| P(A|fi) = Ci/Ti |
In cases where Ti numbers are small, e.g. if fi is rarely represented within the dataset, the estimate deduced would be an overestimate. To illustrate this fact, imagine a feature occurring only a few times in the dataset, belonging to all the compounds in a class. Consequently, P(A|fi) = 1. Nonetheless, the corrected conditional probability, P′(A|fi), attenuates this problem by introducing additional virtual samples, D:
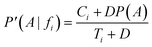
If Ci→0 and Ti→0, in cases where fi is barely present, the corrected result arises:
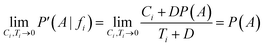
Now, considering the Laplacian correction D = P(A)−1:
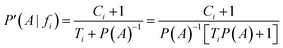
Dividing by P(A) results in the relative probability estimate, Prel:
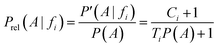
On the basis of the previous equations, the probability of a compound belonging to a given class, Pclass, bearing a set of features fi, is determined from the relative estimates:
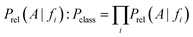
The need for interpretable results and to avoid the very small numbers yielded by the algorithm motivates the use of logarithms. Consequently, a combined score S is the final output:
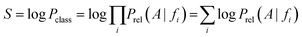
Targets are ranked by this score in descending order and recall and precision values can be assigned to every score. When using rank-based cut-offs, a recall of 83% is achieved if the nine top-ranked targets (corresponding to 1% of the total number of targets) are considered as positive predictions. When using a score cut-off of 30 on the other hand, a precision of 31.43% and a recall of 54.23% are achieved. Here a score cut-off of 35 was used, where it was found that recall was equaling precision at a value of around 72%.
Datasets, molecular descriptors and fingerprints
1066 compounds cytotoxic for HeLa cells were retrieved from PubChem Bioassay (ESI, Table S4†). The training dataset for the Laplacian-modified Naïve Bayesian classifier was extracted from CHEMBL version 10, containing 105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 946 compounds with reported activities (Ki/Kd/IC50/EC50) equal or better than 10 μM covering 894 human protein targets with a confidence score of 8 or 9 (provided in CHEMBL as a measure of the confidence of the assay-to-target relationship) in 155259 ligand–target instances. Experimentally tested compounds were identified in the Maybridge HitFinder library.38
946 compounds with reported activities (Ki/Kd/IC50/EC50) equal or better than 10 μM covering 894 human protein targets with a confidence score of 8 or 9 (provided in CHEMBL as a measure of the confidence of the assay-to-target relationship) in 155259 ligand–target instances. Experimentally tested compounds were identified in the Maybridge HitFinder library.38
MOLPRINT 2D55 fingerprints were calculated using Open Babel version 2.3.1 (GNU Free Documentation License 1.2.).56 MOLPRINT 2D fingerprints were considered appropriate for the purpose of this work given their previously reported performance in linking the chemical structure and bioactivity against protein targets.21
Cell culture
HeLa cells were grown at 37 °C and 5% CO2 in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), glutamine/penicillin/streptavidin (Invitrogen), and non-essential amino acids (Promega). Confluent cultures were switched to supplemented DMEM medium, except for cytotoxicity and Hoechst 33342 uptake assays, where no supplemented DMEM medium was applied.Human P-glycoprotein 1 enzymatic assay
The effect of potential P-glycoprotein modulators was assessed with the luminescent P-glycoprotein ATPase assay Pgp-Glo™ Assay Systems (Promega). Verapamil and sodium orthovanadate were employed as controls, with the former increasing the consumption of ATP by P-gp, while the latter producing the opposite effect. According to the commercial kit,47,48 the luminescence signal is proportional to the concentration of remaining ATP after P-gp-mediated substrate transfer activity, which is in turn inversely proportional to the activity of P-gp (since P-gp consumes ATP when externalizing substrates). IC50 values were estimated via data fitting using the Hill equation as follows:where L0 and Lmax determine the luminescence values at zero and infinite compound concentration, respectively, [C] is the compound concentration, and n is the Hill coefficient.
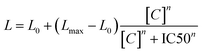
Concerning the level of expression of P-gp in HeLa cell lines, the scientific literature appears controversial. de Inés et al.57 stated that no expression of P-gp is found in HeLa cells, according to Daoud et al.58 and Zhao et al.59 Nonetheless, the MDR1 (codifying for P-gp 1) mRNA levels in HeLa cells were estimated to be 0.30 (105 copies per RNA μg) by Yang et al.60via semi-quantitative PCR, being determined a MDR1/GADPH mRNA ratio of 4.22 ± 0.35 by Komoto et al.61 With respect to P-gp expression in HeLa cells, Zhang et al.,62 calculated an approximate number of 8.47 × 106 P-gp molecules per cell. Therefore, we assume P-gp expression in wild type HeLa cells.
Human Topoisomerase 1 enzymatic assay63
A human Topoisomerase I Microplate Assay Kit (purchased from Inspiralis) was used as a means to evaluate the interaction between human Topoisomerase I and compounds predicted to inhibit its activity with camptothecin as a positive control. In this assay, the fluorescence signal reflects the ability for DNA triplex formation, which is promoted by the presence of supercoiled DNA, which is inversely proportional to the DNA-relaxing Topoisomerase activity.In vivo Hoechst 33342 uptake
An in-house protocol (inspired by Müller et al.)64 was designed to measure the uptake of the nuclear dye Hoechst 33342 (Sigma) in the presence of compounds targeting P-gp 1 with Verapamil as a positive control. Cells were incubated in triplicate for 30 minutes with both compound and Hoechst 33342 at various concentrations (4.4 μM, 2.2 μM and 1.1 μM). New DMEM medium was added and cells were incubated for two additional days. Subsequently, the medium was switched and fluorescence was measured in a Synergy HT Multi-Mode Microplate Reader (Biotek Instruments). Fluorescence microscopy images were generated with a Leica DMI 6000B fluorescence microscope. The fluorescence signal reflects the intracellular dye concentration. Inhibitory action of either Verapamil or compounds targeting P-gp (substrates of P-gp act as P-gp inhibitors regarding other potential transported molecules by blocking access to P-gp) would lead to an increased fluorescence signal.Cytotoxicity assays
CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) was employed to obtain dose–response curves for cytotoxicity evaluation of compounds selected for their predicted inhibitory activity against both P-gp and Topoisomerase on HeLa cell cultures. Cells were seeded in 96-well microplates at an initial cell concentration of around 5000 cells per microwell (100 μL). Subsequently, the cells were incubated for 24 hours to allow attachment. Compounds were added in triplicate to the cells, which were incubated for three days. Then, CellTiter solution was added after the removal of medium. Absorbance at 490 nm was measured 4 hours later in a Synergy HT Multi-Mode Microplate Reader (Biotek Instruments).Statistics analysis of compound synergy in HeLa cells
Normalized absorbance values of the MTT assay for (i) compounds Topo 1–5 at 120 μM and (ii) compounds Topo 1–5 at 120 μM in the presence of either P-gp 2 or P-gp 4 at 50 μM were statistically compared to evaluate compound synergism in cell-based assays (HeLa cells). Given the design of the experiments samples were considered as independent. Normality (Shapiro–Wilk) and homoscedasticity (Levene's test) were evaluated. On this basis, a two-tailed t-test of independent samples (α = 0.05) was applied (see Table S3†). This test was also applied to evaluate the statistical significance of the cytotoxicity and P-gp ATPase activity assays.Escherichia coli proliferation assay
Escherichia coli were grown on LB agar Petri plates for 24 hours with compounds targeting human Topoisomerase I at 200 μM. The effect of the inhibition was measured qualitatively by visual inspection and counting bacterial colonies.Abbreviations
| ADME | Absorption–distribution–metabolism–excretion; |
| DMEM | Dulbecco's modified Eagle's medium; |
| FBS | Fetal bovine serum; |
| IC50 | Half maximal inhibitory concentration; |
| LB | Luria-Bertani medium; |
| MDR | Multidrug resistance; |
| MoA | Mode of action; |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; |
| P-gp | P-glycoprotein 1; |
| SAR | Structure–activity relationship; |
| Tc | Tanimoto coefficient; |
| Topo | Topoisomerase I. |
Acknowledgements
AB and AK thank Unilever for funding. This work was supported by Spanish Ministerio de Ciencia e Innovación (BFU2010-19451 to A.V.-C.), Miguel Servet Program from Instituto de Salud Carlos III (CP07/00289 to O.A.), Fondo de Investigaciones Sanitarias (PI10/00186 to O.A.).References
- D. Brown, Drug Discovery Today, 2007, 12, 1007–1012 CrossRef CAS.
- Y. Feng, T. J. Mitchison, A. Bender, D. W. Young and J. A. Tallarico, Nat. Rev. Drug Discovery, 2009, 8, 567–578 CrossRef CAS.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS.
- C. P. Hart, Drug Discovery Today, 2005, 10, 513–519 CrossRef CAS.
- M. J. Allen and A. H. Carey, Drug Discovery Today, 2004, 3, 183–190 CrossRef CAS.
- A. D. Roses, D. K. Burns, S. Chissoe, L. Middleton and P. St. Jean, Drug Discovery Today, 2005, 10, 177–189 CrossRef.
- V. C. Abraham, D. L. Taylor and J. R. Haskins, Trends Biotechnol., 2004, 22, 15–22 CrossRef CAS.
- K. Korn and E. Krausz, Curr. Opin. Chem. Biol., 2007, 11, 503–510 CrossRef CAS.
- A. G. Reaume, Drug Discovery Today: Ther. Strategies, 2011, 8, 85–88 CrossRef.
- D. W. Young, A. Bender, J. Hoyt, E. McWhinnie, G. W. Chirn, C. Y. Tao, J. A. Tallarico, M. Labow, J. L. Jenkins, T. J. Mitchison and Y. Feng, Nat. Chem. Biol., 2008, 4, 59 CrossRef CAS.
- P. A. Clemons, Curr. Opin. Chem. Biol., 2004, 8, 334–338 CrossRef CAS.
- C. J. O'Connor, L. Laraia and D. R. Spring, Chem. Soc. Rev., 2011, 40, 4332–4345 RSC.
- D. R. Spring, Chem. Soc. Rev., 2005, 34, 472–482 RSC.
- A. Koutsoukas, B. Simms, J. Kirchmair, P. J. Bond, A. V. Whitmore, S. Zimmer, M. P. Young, J. L. Jenkins, M. Glick, R. C. Glen and A. Bender, J. Proteomics, 2011, 74, 2554–2574 CrossRef CAS.
- A. Bender, Nat. Chem. Biol., 2010, 6, 309 CrossRef CAS.
- T. Klabunde, Br. J. Pharmacol., 2007, 152, 5–7 CrossRef CAS.
- M. Bredel and E. Jacoby, Nat. Rev. Genet., 2004, 5, 262–275 CrossRef CAS.
- A. R. Leach, V. J. Gillet, R. A. Lewis and R. Taylor, J. Med. Chem., 2010, 53, 539–558 CrossRef CAS.
- A. Bender, H. Y. Mussa, R. C. Glen and S. Reiling, J. Chem. Inf. Comput. Sci., 2004, 44, 170–178 CrossRef CAS.
- Nidhi, M. Glick, J. W. Davies and J. L. Jenkins, J. Chem. Inf. Model., 2006, 46, 1124–1133 CrossRef CAS.
- F. Nigsch, A. Bender, J. L. Jenkins and J. B. Mitchell, J. Chem. Inf. Model., 2008, 48, 2313–2325 CrossRef CAS.
- A. L. Hopkins, Nat. Biotechnol., 2007, 25, 1110–1111 CrossRef CAS.
- S. K. Mencher and L. G. Wang, BMC Clin. Pharmacol., 2005, 5, 3 CrossRef.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985–2993 CrossRef CAS.
- J. Mestres, E. Gregori-Puigjane, S. Valverde and R. V. Sole, Mol. BioSyst., 2009, 5, 1051–1057 RSC.
- M. Liu, Y. Wu, Y. Chen, J. Sun, Z. Zhao, X. W. Chen, M. E. Matheny and H. Xu, J. Am. Med. Informat. Assoc., 2012, 19, 28–35 CrossRef.
- T. Klabunde and A. Evers, ChemBioChem, 2005, 6, 876–889 CrossRef CAS.
- I. Kola and J. Landis, Nat. Rev. Drug Discovery, 2004, 3, 711–715 CrossRef CAS.
- R. Morphy, J. Med. Chem., 2010, 53, 1413–1437 CrossRef CAS.
- E. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J. Hamon, J. L. Jenkins, P. Lavan, E. Weber, A. K. Doak, S. Côté, B. K. Shoichet and L. Urban, Nature, 2012, 486, 361–367 CAS.
- M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin and B. K. Shoichet, Nat. Biotechnol., 2007, 25, 197–206 CrossRef CAS.
- V. Poroikov, D. Akimov, E. Shabelnikova and D. Filimonov, SAR QSAR Environ. Res., 2001, 12, 327–344 CrossRef CAS.
- A. A. Geronikaki, J. C. Dearden, D. Filimonov, I. Galaeva, T. L. Garibova, T. Gloriozova, V. Krajneva, A. Lagunin, F. Z. Macaev, G. Molodavkin, V. V. Poroikov, S. I. Pogrebnoi, F. Shepeli, T. A. Voronina, M. Tsitlakidou and L. Vlad, J. Med. Chem., 2004, 47, 2870–2876 CrossRef CAS.
- M. S. Butler, Nat. Prod. Rep., 2008, 25, 475–516 RSC.
- D. Schuster, Drug Discovery Today: Technol., 2010, 7, 205–2011 CrossRef.
- T. M. Ehrman, D. J. Barlow and P. J. Hylands, Bioorg. Med. Chem., 2010, 18, 2204–2218 CrossRef CAS.
- B. Flachner, Z. Lörincz, A. Carotti, O. Nicolotti, P. Kuchipudi, N. Remez, F. Sanz, J. Tóvári, M. J. Szabó, B. Bertók, S. Cseh, J. Mestres and G. Dormán, PLoS One, 2012, 7, e35582 CAS.
- Maybridge HitFinder Library version 11 (14400 compounds, accessed 8 Nov 2011).
- R. B. Ewesuedo and M. J. Ratain, Oncologist, 1997, 2, 359–364 CAS.
- M. Ingelman-Sundberg, Toxicology, 2002, 181, 447–452 CrossRef.
- O. Leontieva and J. D. Black, J. Biol. Chem., 2004, 279, 5788–5801 CrossRef CAS.
- T. Helleday, J. Lo, D. C. van Gent and B. P. Engelward, DNA Repair, 2007, 7, 923–935 CrossRef.
- A. Kolocouris, K. Dimas, C. Pannecouque, M. Witvrouw, G. B. Foscolos, G. Stamatiou, G. Fytas, G. Zoidis, N. Kolocouris, G. Andrei, R. Snoeck and E. De Clercq, Bioorg. Med. Chem. Lett., 2002, 5, 723–727 CrossRef.
- Y. Wang, J. Xiao, T. O. Suzek, J. Zhang, J. Wang, Z. Zhou, L. Han, K. Karapetyan, S. Dracheva, B. A. Shoemaker, E. Bolton, A. Gindulyte and S. H. Bryant, Nucleic Acids Res., 2012, 40, 400–412 CrossRef.
- L. Chen, Y. Li, Q. Zhao, H. Peng and T. Hou, Mol. Pharmacol., 2011, 8, 889–900 CrossRef CAS.
- S. A. Rahman, M. Bashton, G. Holliday, R. Schrader and J. Thornton, J. Cheminf., 2009, 1, 12 Search PubMed.
- S. L. Mercer, C. W. Cunningham, H. Hassan, N. D. Eddington and A. Coop, Bioorg. Med. Chem. Lett., 2007, 5, 1160–1162 CrossRef.
- J. Rautio, J. E. Humphreys, L. O. Webster, A. Balakrishnan, J. P. Keogh, J. R. Kunta, C. J. Serabjit-Singh and J. W. Polli, Drug Metab. Dispos., 2006, 34, 786–792 CrossRef CAS.
- A. B. Shapiro, A. B. Corder and V. Ling, Eur. J. Biochem., 1997, 250, 115–121 CAS.
- I. Isobe, M. Michikawa and K. Yanagisawa, Neurosci. Lett., 1999, 266, 129–132 CrossRef CAS.
- J. Weyermann, D. Lochmann and A. Zimmer, Int. J. Pharm., 2005, 288, 369–376 CrossRef CAS.
- A. A. Borisy, P. J. Elliott, N. W. Hurst, M. S. Lee, J. Lehar, E. R. Price, G. Serbedzija, G. R. Zimmermann, M. A. Foley, B. R. Stockwell and C. T. Keith, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7977–7982 CrossRef CAS.
- O. Sordet, Q. A. Khan, K. W. Kohn and Y. Pommier, Curr. Med. Chem. Anti Canc. Agents, 2003, 3, 271–290 CrossRef CAS.
- X. Zheng, L. Diao, S. Ekins and J. E. Polli, Biochem. Pharmacol., 2010, 7, 1087 CrossRef.
- A. Bender, H. Y. Mussa, R. C. Glen and S. Reiling, J. Chem. Inf. Comput. Sci., 2004, 44, 1708–1718 CrossRef CAS.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 CAS.
- C. de Inés, V. H. Argandoña, J. Rovirosa, A. San-Martín, A. R. Díaz-Marrero, M. Cueto and A. González-Coloma, Z. Naturforsch., C: J. Biosci., 2004, 5, 339–344 Search PubMed.
- R. Daoud, C. Kast, P. Gros and E. Georges, Biochemistry, 2000, 50, 15344–15352 CrossRef.
- J. Y. Zhao, N. Savaraj, R. Song, W. Priebe and M. T. Kuo, Anticancer Res., 1994, 5, 1735–1742 Search PubMed.
- Z. Yang, E. L. Woodahl, X. Y. Wang, T. Bui, D. D. Shen and R. J. Ho, BioTechniques, 2002, 1, 196–200 Search PubMed.
- C. Komoto, T. Nakamura, M. Yamamori, N. Ohmoto, H. Kobayashi, A. Kuwahara, K. Nishiguchi, K. Takara, Y. Tanigawara, N. Okamura, K. Okumura and T. Sakaeda, Kobe J. Med. Sci., 2008, 6, 355–363 Search PubMed.
- J. J. Zhang, F. F. Cheng, T. T. Zheng and J. J. Zhu, Anal. Chem., 2010, 9, 3547–3555 CrossRef.
- A. Maxwell, N. P. Burton and N. O'Hagan, Nucleic Acids Res., 2006, 34, e104 CrossRef.
- H. Müller, W. Klinkhammer, C. Globisch, M. U. Kassack, I. K. Pajeva and M. Wiese, Bioorg. Med. Chem., 2007, 15, 7470–7479 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2md20286g |
| ‡ A.B. and A.V.-C. designed the study. O.A. assisted in experimental protocols and provided reagents and cell lines. I.C.-C. performed experiments and analyzed the experimental data. I.C.-C., A.B. and A.V.-C. wrote the paper. |
| This journal is © The Royal Society of Chemistry 2013 |