Discovery and optimization of efficacious neutral 4-amino-6-biphenyl-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5-one diacylglycerol acyl transferase-1 (DGAT1) inhibitors†
Frederick W.
Goldberg
*,
Alan M.
Birch
,
Andrew G.
Leach
,
Sam D.
Groombridge
,
Wendy L.
Snelson
,
Pablo Morentin
Gutierrez
,
Clare D.
Hammond
,
Susan
Birtles
and
Linda K.
Buckett
AstraZeneca R&D, Mereside, Alderley Park, Macclesfield, Cheshire, SK10 4TG, UK. E-mail: frederick.goldberg@astrazeneca.com
First published on 1st October 2012
Abstract
A series of neutral DGAT1 inhibitors with good potency and pharmacokinetics (PK) has been designed by modification of an acidic startpoint. This was achieved by selecting the acid with the highest ligand lipophilicity efficiency (LLE) and replacing the acid with neutral isosteres. PK properties (Fabs) were then improved by removing the sidechain to reduce molecular weight and polar surface area (PSA). Compound 13 has shown good cross-species PK, with pre-clinical efficacy and PK/PD relationships comparable to those previously described for acidic inhibitors.
Introduction
Diacylglycerol acyl transferase-1 (DGAT1) is a target of great interest to the pharmaceutical industry, as inhibiting DGAT1 could be a suitable mechanism for treating diabetes, obesity and other diseases that constitute metabolic syndrome.1 Small molecule inhibitors of DGAT1 (Fig. 1) have progressed into the clinic from Novartis (LCQ-908, phase II), Pfizer (PF-04620110, phase I – discontinued)2 and AstraZeneca (AZD7687, phase I – discontinued, structure undisclosed) and preclinical compounds have been disclosed by Roche (RO-6036),3 Abbott (A-922500)4 and AstraZeneca (AZD3988),5 amongst others.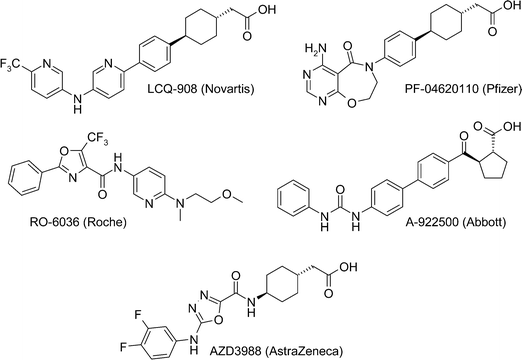 | ||
| Fig. 1 Structures of reported DGAT1 inhibitors. | ||
A recurring theme of the published DGAT1 inhibitors is the presence of a carboxylic acid moiety. In general, addition of a carboxylic acid has been used to combine good potency with good physical and pharmacokinetic (PK) properties, as we found with the recently published discovery of AZD3988.5 However, the carboxylic acid moiety can be associated with a risk of idiosyncratic toxicity in the clinic, from the formation of the corresponding acyl glucuronide.6 Despite the prevalence of acidic DGAT series, neutral DGAT inhibitors such as RO-6036 are known. As there was both rationale and precedent for discovering neutral DGAT inhibitors we initiated a program to use in-house biphenyl acidic compounds such as 1 (Fig. 2) as startpoints for the discovery of a neutral series.
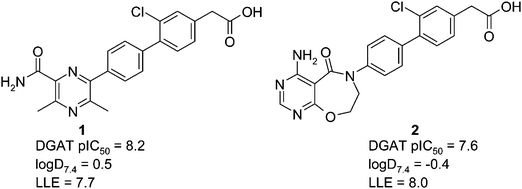 | ||
| Fig. 2 Structures of acidic startpoints. | ||
One concern we had at the outset of the program was that removing the carboxylic acid would be expected to increase log D and decrease solubility. Acid 1 is very potent (pIC50 = 8.2) with a low log D7.4 (0.5), although we were still concerned that using 1 as a startpoint for a neutral series would lead to a compound with high lipophilicity that would be difficult to optimise to a clinical candidate. We therefore synthesised and profiled 2, as the prediction was that this would give us a lower log D startpoint, and there was precedent of the 4-amino-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5-one group for DGAT inhibition.2 Pleasingly, 2 was potent (DGAT pIC50 = 7.6) at very low log D7.4 (−0.4). Although 2 is less potent than 1, it has an exceptionally high ligand lipophilicity efficiency (LLE, defined in this article as pIC50 − log D7.4) of 8.0, and was therefore our preferred startpoint for developing a neutral series.
Initially the analogous primary amide was designed, as the primary amide is a conservative isosteric neutral replacement for an acid. Thus 3 was equipotent to 2, with a log D7.4 that was in the desirable range for an oral drug (Table 1), although as expected LLE decreases as a consequence of the increase in log D. Amide 3 also retained good solubility and improved free fraction in human plasma protein binding (PPB), but at the cost of reduced in vitro metabolic stability. Nevertheless 3 was taken forward as a neutral lead compound for further optimisation.

Chemistry
Synthesis of the initial hits 2 and 3 started from methyl 2-(3-chloro-4-hydroxyphenyl)acetate 4 (Scheme 1), which was converted to triflate 5 with PhN(Tf)2 and then converted to the boronate ester 6. Suzuki coupling of 6 with bromide 7 gave the biaryl ester 8, which could be saponified to give acid 2. This was then converted with a HATU coupling to the primary amide 3.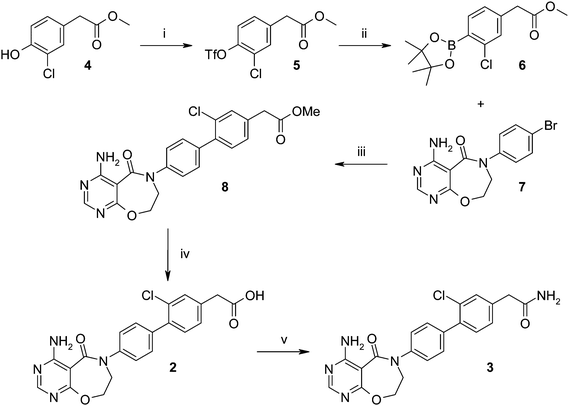 | ||
| Scheme 1 Synthesis of initial hits 2 and 3. Reagents and conditions: (i) PhN(Tf)2, Na2CO3, THF, 69%; (ii) bis(pinacolato)diboron, CH3CO2K, PdCl2(dppf)-DCM adduct, 1,4-dioxane, 42%; (iii) PdCl2(dppf)-DCM adduct, K3PO4, DME, MeOH, H2O, 46%; (iv) LiOH, 1,4-dioxane, H2O, 72%; (v) NH4Cl, HATU, i-Pr2NEt, DMF, 53%. | ||
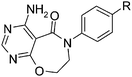
The 4-amino-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5-one core 7 was prepared using an analogous route to similar intermediates that have been previously reported by Pfizer.2 Thus 4-bromoaniline was alkylated with (2-bromoethoxy)(tert-butyl)dimethylsilane 9 (Scheme 2) to give intermediate 10, which was then acylated with 4,6-dichloropyrimidine-5-carbonyl chloride to give 11. Compound 11 was deprotected with HCl and the resulting alcohol was cyclised under basic conditions to give the desired ring system 12. The resulting chloropyrimidine was then displaced with ammonia to give 7, which could be coupled up with a range of boronate esters to give the desired biaryl target compounds, e.g. with 2-fluorobenzeneboronic acid as shown to give 13.
![Synthesis of 4-amino-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5-on core 7 and compound 13. Reagents and conditions: (i) Na2CO3, DMF, 51%; (ii) 4,6-dichloropyrimidine-5-carbonyl chloride, Et3N, THF, 106%; (iii) HCl, MeOH, 104%; (iv) Et3N, MeCN, 76%; (v) NH3, 1,4-dioxane, 89%; (vi) 2-fluorobenzeneboronic acid, PdCl2(dppf)-DCM adduct, K3PO4, DME, MeOH, H2O, 47%.](/image/article/2013/MD/c2md20231j/c2md20231j-s2.gif) | ||
| Scheme 2 Synthesis of 4-amino-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5-on core 7 and compound 13. Reagents and conditions: (i) Na2CO3, DMF, 51%; (ii) 4,6-dichloropyrimidine-5-carbonyl chloride, Et3N, THF, 106%; (iii) HCl, MeOH, 104%; (iv) Et3N, MeCN, 76%; (v) NH3, 1,4-dioxane, 89%; (vi) 2-fluorobenzeneboronic acid, PdCl2(dppf)-DCM adduct, K3PO4, DME, MeOH, H2O, 47%. | ||
Results and discussion
We initially aimed to improve the metabolic stability of 3 by varying the amide, focussing on groups with additional polarity to keep log D7.4 low (<2). Compounds 14, 15 and 16 (Table 3) incorporate secondary hydroxyl or amide functionality (designed for solubility and low metabolic liability) and were potent, although only 14 was more potent than the primary amide 3. We also investigated other polar functionality such as reversed amides, sulfones and sulfonamides (17–21). We used the reversed amide to investigate the effects of cyclic groups (22 and 23) and spacing a base out with different lengths of linker (24, 25 and 26). These changes were all tolerated, and interestingly spacing the base out can drop log D significantly and thus improve LLE, although not surprisingly given the low log D and ionisable centre the predicted absorptions of 24, 25 and 26 as assessed in vitro by Caco-2 A–B Papp were poor.Through this approach of using isosteric polar groups to the primary amide startpoint we identified a number of potent compounds with improved in vitro metabolic stability and rat PK as shown in Table 2. However, none of them improved significantly upon the potency of the startpoint 3. Our conclusion was that the SAR was surprisingly flat; despite the diversity of compounds 3–26 there is less than a log unit variation in potency, suggesting that the acid/amide isosteres that we had focussed on may not be necessary. In addition, although the carefully designed examples succeeded in reducing clearance vs. the primary amide, the molecular weight (MW) of these compounds is relatively high (437–506) and many of them had low Fabs in rat. Consequently we decided to change our strategy and cut down the sidechain and pursue simple, small MW substituents to improve absorption.7 To our surprise, removing the sidechain altogether to give 27 (Table 2) maintained potency. log D7.4 was higher (2.9) but despite moderately high in vitro and in vivo clearance, 27 had improved Caco-2 absorption and high bioavailability in rat (65%), suggesting that removing the sidechain may improve absorption. We further explored this by examining small substituents on the ring, which suggested that a range of substituents were well tolerated at C4 position (28–32). Chloro was well tolerated at C5 (35) and chloro and cyano were moderately tolerated at C3 (33 and 34). Addition of a methyl at C4 (29) gave a particularly potent compound (pIC50 = 8.3), albeit with high log D7.4 (3.5), low solubility and high clearance.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | R | DGAT pIC50 | log D7.4 | Solubilitya (μM) | Hu PPB %free | Rat heps CLint (μL min−1 × 106) | Rat F%b | Rat CLc mL min−1 kg−1 | Caco-2 A to B Papp (×10−6 cm s−1) |
| a Solubility performed under thermodynamic conditions using semicrystalline or crystalline solid. b Oral bioavailability observed when dosing compound as a low dose (2.3–6.4 μmol kg−1) solution to male Han Wistar rats. c Intravenous clearance in male Han Wistar rats when dosing compound as a low dose (1.4–2.9 μmol kg−1) solution to male Han Wistar rats. | |||||||||
| 3 |
|
7.5 | 1.3 | 201 | 17.2 | 35 | |||
| 14 |
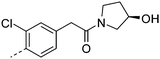
|
7.7 | 1.1 | 41 | 24.1 | <4.6 | 15 | 12 | 1.1 |
| 15 |

|
7.4 | 1.1 | 1 | 22.3 | <3.1 | |||
| 16 |
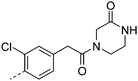
|
7.3 | 0.9 | 34 | 30.2 | <4.6 | 3 | 25 | |
| 17 |
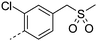
|
7.4 | 21.2 | <2.8 | 61 | 5 | |||
| 18 |

|
7.3 | 1.9 | 6 | 9.4 | 11 | 19.7 | ||
| 19 |
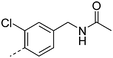
|
7.3 | 1.8 | 4 | 13.3 | <2 | 100 | 7 | 14.5 |
| 20 |
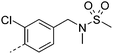
|
7.5 | 2.1 | 1 | 8.6 | 5.9 | 28 | 5 | 18.8 |
| 21 |
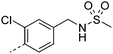
|
7.5 | 1.8 | 2 | 12.2 | 4.1 | 21 | 3 | 11.3 |
| 22 |
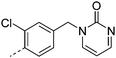
|
7.1 | 0.9 | 148 | 22.5 | 18.4 | 2.0 | ||
| 23 |
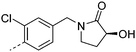
|
7.2 | 1.1 | 19 | 12.3 | <2 | 6.4 | ||
| 24 |
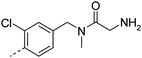
|
7.4 | 0.1 | 200 | >49 | 4.9 | 0.2 | ||
| 25 |

|
7.3 | −0.5 | 237 | 48.0 | <2 | 0.3 | ||
| 26 |
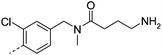
|
7.3 | −1 | 280 | 42.0 | <2 | 0.2 | ||
| 27 |
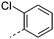
|
7.8 | 2.9 | 5 | 3.6 | 11.1 | 65 | 21 | 32.5 |
| 28 |
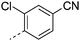
|
7.3 | 2.4 | NV | 9.3 | 4.8 | 48.2 | ||
| 29 |
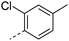
|
8.3 | 3.5 | 7 | 1.2 | 53.2 | 14 | 33 | 16.6 |
| 30 |

|
7.6 | 16.8 | ||||||
| 31 |
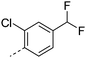
|
7.7 | 3.2 | <1 | 2.2 | 9.1 | 150 | 16 | |
| 32 |
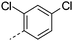
|
8.0 | >3.9 | 1 | 0.7 | 6.4 | 87 | 6 | |
| 33 |
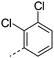
|
7.1 | 3.8 | <1 | 1.2 | 9.3 | 150 | 24 | |
| 34 |
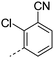
|
6.5 | 2.5 | <1 | 5.9 | 4.6 | 54.8 | ||
| 35 |
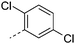
|
7.8 | 3.7 | 1 | 1.2 | 9.5 | 110 | 12 | 16.2 |
Having identified new lower molecular weight neutral startpoints such as 27, we then aimed to identify structural changes elsewhere on the scaffold that might maintain or improve potency and improve properties. An interesting feature of the biphenyl core is that ortho (2,2′) substituents would be expected to effect the conformational preference of the biphenyl (i.e. the dihedral angle between the phenyl rings), as well as placing the ortho group into a position where it may improve binding. Thus removing the chlorine to give the unsubstituted biphenyl significantly decreases potency (Table 3, cf.36 and 27). Other ortho substituents and 2,6 or 2,2′-bis-ortho substituents all improved potency relative to 36 as shown in Table 3. The 2-chloro-2′-fluoro compound 39 was the most potent (pIC50 = 8.0), although it demonstrated poor bioavailability (rat F = 18%). Ortho-fluoro compound 13 combined good potency (pIC50 = 7.4) and attractive PK properties (rat F = 93%, iv CL = 7) and thus was taken forward to mouse PK and PK/PD studies (vide infra). It is also noteworthy that replacing the phenyl ring with a pyridine reduced potency (cf.42 and 27) but also reduced log D7.4 dramatically and consequently pyridine 42 has good LLE for a neutral DGAT inhibitor (pIC50 − log D7.4 = 5.6) and excellent physical properties (solubility = 287 μM, Hu PPB = 18% free). All compounds in Table 3 showed good in vitro absorption (Caco2 A–B Papp > 10 × 10−6 cm s−1, data not shown), in marked contrast to early lead compounds such as 14 (Caco2 A–B Papp = 1.1 × 10−6 cm s−1, Table 2), presumably due to the lower MW and polar surface area of these simplified structures.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cpd | A | X | Y | Z | DGAT pIC50 | log D7.4 | Solubility (μM) | Hu PPB % free | Rat F % | Rat IV CL (mL min−1 kg−1) |
| 36 | C | H | H | H | 6.6 | 2.4 | 3 | 9.6 | ||
| 27 | C | Cl | H | H | 7.8 | 2.9 | 5 | 3.6 | 65 | 21 |
| 37 | C | Me | H | H | 7.4 | 2.7 | 6 | 6.9 | ||
| 38 | C | F | F | H | 7.8 | 2.5 | 140 | 9.5 | ||
| 39 | C | Cl | H | F | 8.0 | 3.0 | 3 | 5.6 | 14 | 18 |
| 40 | C | Cl | Me | H | 7.7 | 3.4 | 1 | 4.1 | ||
| 41 | C | F | F | F | 7.8 | 2.9 | 77 | 4.0 | ||
| 42 | N | Cl | — | H | 7.1 | 1.5 | 287 | 18.2 | 68 | 20 |
| 43 | C | Cl | CN | H | 7.5 | 2.1 | 20.6 | |||
| 13 | C | F | H | H | 7.4 | 2.8 | 12 | 8.0 | 93 | 7 |
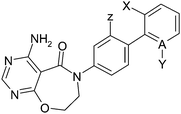
As there is an embedded biphenyl aniline in these compounds, we investigated the potential for release of Ames positive fragments. We did not observe the release of the corresponding aniline in either in vitro or in vivo rat metabolite identification (Met ID) studies, but nevertheless we assessed the Ames mutagenicity of the corresponding biphenyl fragments to assess this risk – this work has been published separately.8 We found that electron withdrawing groups, particularly F or Cl, at the position meta to the amino group (Z) in the aminobiphenyl fragment decrease the likelihood of the aniline being active in the Ames test. The meta fluorine alone is insufficient to eliminate activity. The addition of further electron withdrawing groups such as in 39 or 41 leads to aminobiphenyl fragments that are not active in the Ames test. By contrast, the aminobiphenyl embedded in 13 is found to be active in the Ames test. To further profile compound 13 we tested it for enzyme activity against ACAT1 and DGAT2, but it was found to be inactive (>100 μM) in both assays.
PK and PD of compound 13
We have previously described acidic DGAT1 inhibitors which had attractive PK profiles in pre-clinical species.5,9 Our goal in this programme was to identify neutral compounds which had similarly high bioavailability and low clearance in pre-clinical species. In addition we sought a compound which would give dose-proportional exposure at high doses in order to allow pre-clinical toxicity studies to be performed with high exposure margins compared to the IC50. Compound 13 was selected for full pre-clinical PK profiling at low dose (Table 4), where it gave good bioavailability in mouse, rat and dog. It's also noteworthy that 13 was stable to human hepatocytes (CLint < 2 μL min−1 × 106).| Species | CLp (mL min−1 kg−1) | V dss (L Kg−1) | iv t1/2 (h) | po t1/2 (h) | po Cmax (μM) | Bioavailability (%) |
|---|---|---|---|---|---|---|
a Compound was dosed iv at 2 mg kg−1 (mouse), 1 mg kg−1 (rat) and 0.5 mg kg−1 (dog) and po at 2 mg kg−1 (mouse), at 2 mg kg−1 (rat) and 0.5 mg kg−1 (dog). Vehicle for all studies was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95 DMSO:hydroxypropyl betacyclodextrin. :95 DMSO:hydroxypropyl betacyclodextrin.
|
||||||
| Mouse | 2.1 | 0.6 | 3.5 | 3.1 | 10.2 | >100 |
| Rat | 7.3 | 1.9 | 6.7 | 3.3 | 3.1 | 93 |
| Dog | 14.8 | 2.4 | 1.2 | 1.7 | 0.4 | 76 |
To investigate exposure at high dose compound 13 was given to mice at 300 mg kg−1 in a suspension formulation with polyvinylpyrrolidone–dioctyl sodium sulfosuccinate (PVT–AOT). PVT–AOT is known to stabilise drug nanoparticles, and is considered suitable to use for efficacy/toxicity studies as both PVT and AOT are common pharmaceutical excipients and are listed on the FDA inactive ingredients guide. Excellent exposure was achieved (Fig. 3) with free drug concentrations (adjusted for mouse plasma protein binding) >1000 fold above the compound's human DGAT1 IC50.
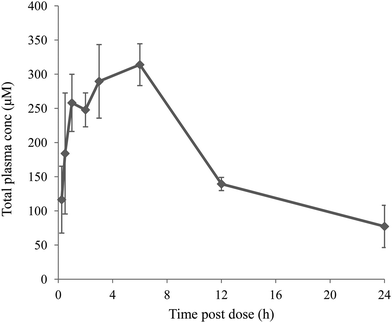 | ||
| Fig. 3 Mean total plasma concentrations following po dosing of compound 13 to 2 male CD1 mice (300 mg kg−1 suspension in PVT–AOT), with error bars showing standard deviation. | ||
Based on this favourable PK profile 13 was tested in two pharmacodynamic models in rats: the adipose triacylglycerol (TAG) synthesis test and the oral lipid tolerance test (OLTT). Methods for both these tests can be found in a previous communication.9Fig. 4 shows the relationship between adipose TAG synthesis (expressed as TAG:DAG ratio in adipose tissue) and free compound levels in plasma in the rat. A direct response PK/PD model (Emax sigmoidal model, PK/PD parameters for the model fit are given in the ESI†) was used to successfully fit the PK/PD data showing a clear correlation between free compound levels in plasma and an effect in the adipose TAG/DAG ratio. The in vivo IC50 (7.1 nM) in the adipose tissue assay is in very good agreement with the in vitro IC50 (7.7 nM).
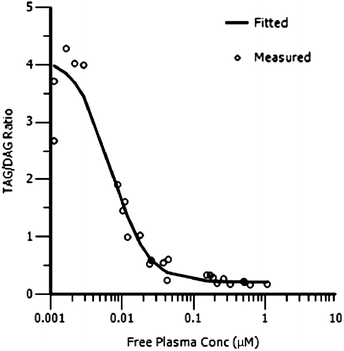 | ||
| Fig. 4 PK/PD analysis of rat adipose TAG synthesis assay (adipose tissue TAG:DAG ratio vs. free compound concentration in plasma) for compound 13. | ||
Fig. 5 shows the relationship between plasma triglycerides and free compound levels in plasma in the rat OLTT assay. A direct response PK/PD model (Emax sigmoidal model, PK/PD parameters for the model fit are given in the ESI†) was used to successfully fit the PK/PD data showing a clear correlation between compound levels in plasma and an effect in the OLTT. As for the adipose TAG synthesis assay, the in vivo IC50 (12.7 nM) in OLTT assay is in very good agreement with the in vitro IC50 (7.7 nM).
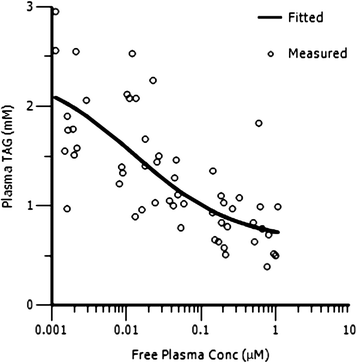 | ||
| Fig. 5 PK/PD analysis of rat OLTT (plasma triglycerides vs. free compound concentrations in plasma) for compound 13. | ||
Conclusions
In summary, we have successfully demonstrated that acidic startpoints can be modified into neutral DGAT inhibitors, which combine good potency, solubility and PK properties despite the absence of an ionisable group. This was achieved by selecting an acidic startpoint with high LLE (pIC50 − log D) and converting the acid to a neutral amide. We then found that removal of the amide to simplify the structure and reduce MW led to improved PK properties whilst (unexpectedly) maintaining potency. 13 was profiled in two different in vivo models, demonstrating similar relationships between plasma concentrations and pharmacodynamic effects to those previously reported for acidic compounds.Experimental section
General
All solvents and chemicals used were reagent grade. Flash column chromatography was carried out using prepacked silica cartridges from Redisep, Biotage, or Crawford and eluted using an Isco Companion system. Purity and characterization of compounds were established by a combination of liquid chromatography-mass spectroscopy (LC-MS) and NMR analytical techniques and was >95% for all compounds. 1H NMR were recorded on a Bruker Avance DPX400 (400 MHz) and were determined in CDCl3 or DMSO-d6. 13C NMR spectra were recorded at 101 or 175 MHz. Chemical shifts are reported in ppm relative to tetramethylsilane (TMS) (0 ppm) or solvent peaks as the internal reference. Merck precoated thin layer chromatography (TLC) plates (silica gel 60 F254, 0.25 mm, art. 5715) were used for TLC analysis. Preparative HPLC was performed on C18 reversed-phase silica on a Waters or Phenomenex preparative reversed-phase column using decreasingly polar mixtures of water (containing 1% formic acid or 1% aq. NH4OH) and MeCN. All reactions were performed under nitrogen unless otherwise stated. All in vivo experiments were performed in compliance with the relevant laws and institutional guidelines.Synthesis of key compounds 2, 3 and 13 (see supporting info for synthesis of other compounds)
Methyl 2-(3-chloro-4-(trifluoromethylsulfonyloxy)phenyl)acetate 5. Methyl 2-(3-chloro-4-hydroxyphenyl)acetate 4 (1.14 g, 5.68 mmol), 1,1,1-trifluoro-N-phenyl-N-(trifluoromethylsulfonyl)methanesulfonamide (2.03 g, 5.68 mmol) and potassium carbonate (2.36 g, 17.1 mmol) were suspended in THF (10 mL) and sealed into a microwave tube. The reaction was heated at 120 °C for 6 min and cooled to rt. The suspension was filtered, then the solid was washed with EtOAc (20 mL) and the filtrate was evaporated to afford crude product. The crude product was purified by flash silica chromatography, elution gradient 0–10% EtOAc in isohexane, to afford the title compound 5 (1.31 g, 69%) as a colourless oil; 1H NMR (400 MHz, CDCl3) 3.63 (2H, s), 3.73 (3H, s), 7.25–7.28 (1H, m), 7.31 (1H, d), 7.47 (1H, s); [M − H]− = 331. Methyl 2-(3-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acetate6. To a degassed solution of 5 (14.6 g, 37.2 mmol) in 1,4-dioxane (300 mL) was added potassium acetate (10.2 g, 104.2 mmol), bis(pinacolato)diboron (11.8 g, 46.5 mmol) and PdCl2(dppf)-DCM adduct (1.82 g, 2.23 mmol). The suspension was degassed and then heated at reflux overnight before being allowed to cool to rt. The reaction mixture was filtered through a silica pad and washed through with EtOAc, then evaporated. The residue was partitioned between EtOAc and water, and the organic layer was separated and washed with sat. brine and evaporated. The resulting crude product was purified by flash silica chromatography, elution gradient 0–50% EtOAc in isohexane to afford the title compound 6 (4.89 g, 42%) as a brown oil; 1H NMR (400 MHz, DMSO) 1.30 (12H, s), 3.61 (3H, s), 3.73 (2H, s), 7.21–7.23 (1H, m), 7.34 (1H, d), 7.58 (1H, d). 4-bromo-N-[2-[tert-butyl(dimethyl)silyl]oxyethyl]aniline10. (2-Bromoethoxy)(tert-butyl)dimethylsilane (8.61 mL, 40.1 mmol) was added dropwise to 4-bromoaniline (6.28 g, 36.5 mmol) and sodium carbonate (7.73 g, 73.0 mmol) in DMF (60 mL) at rt over a period of 3 min. The resulting suspension was stirred at 60 °C for 3 days. The reaction was partially concentrated and added dropwise to cold water (500 mL), then extracted twice with EtOAc. The combined organic layers were washed with sat. brine, dried (MgSO4) and evaporated. The resulting crude product was purified by flash silica chromatography, elution gradient 5–100% DCM in isohexane, to afford the title compound 10 (6.23 g, 52%) as an orange oil; 1H NMR (400 MHz, DMSO) 0.00 (6H, s), 0.83 (9H, s), 3.07–3.12 (2H, m), 3.66 (2H, t), 5.69 (1H, t), 6.52 (2H, d), 7.15 (2H, d); m/z MH+ = 330, 332. N-(4-bromophenyl)-N-[2-[tert-butyl(dimethyl)silyl]oxyethyl]-4,6-dichloro-pyrimidine-5-carboxamide11. 4,6-Dichloropyrimidine-5-carbonyl chloride (2.53 g, 11.95 mmol) as a solution in THF (15 mL) was added dropwise to 10 (3.76 g, 11.38 mmol) and triethylamine (1.71 mL, 12.29 mmol) in THF (30 mL) at 0 °C over a period of 1 min. The resulting solution was stirred at 0 °C for 2 h. The reaction mixture was evaporated to dryness and redissolved in EtOAc, then washed sequentially with water and sat. brine. The organic layer was dried over Na2SO4, filtered and evaporated to afford the title compound 11 (6.07 g, 106%) as an orange oil which was used without further purification; 1H NMR (400 MHz, DMSO) 0.00 (6H, s), 0.81 (9H, s), 3.77 (2H, t), 3.97 (2H, t), 7.33 (2H, d), 7.54 (2H, d), 8.84 (1H, s). N-(4-Bromophenyl)-4,6-dichloro-N-(2-hydroxyethyl)pyrimidine-5-carboxamide. N-(4-Bromophenyl)-N-(2-(tert-butyldimethylsilyloxy)ethyl)-4,6-dichloropyrimidine-5-carboxamide (6.06 g, 11.99 mmol) in methanol (8 mL) was added in one portion to conc. aq. HCl (1.55 mL) in methanol (50 mL) at rt. The resulting solution was stirred at rt for 30 min. The reaction mixture was evaporated to dryness and redissolved in EtOAc, then washed sequentially with sat. aq. NaHCO3 and sat. brine. The organic layer was dried over Na2SO4, filtered and evaporated. The resulting crude product was purified by flash silica chromatography, elution gradient 10–90% EtOAc in isohexane, to afford the title compound (4.88 g, 104%) as a colourless oil; 1H NMR (400 MHz, DMSO) 3.57–3.62 (2H, m), 3.90 (2H, t), 4.84 (1H, t), 7.37 (2H, d), 7.55 (2H, d), 8.83 (1H, s); m/z MH+ = 392. 6-(4-Bromophenyl)-4-chloro-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5(6H)-one12. Triethylamine (9.23 mL, 66.39 mmol) was added in one portion to N-(4-bromophenyl)-4,6-dichloro-N-(2-hydroxyethyl)pyrimidine-5-carboxamide (6.83 g, 17.5 mmol) in acetonitrile (78 mL) at rt. The resulting solution was stirred at 80 °C for 6 h. The reaction mixture was evaporated to dryness and redissolved in EtOAc, then washed with water and sat. brine. The organic layer was dried over Na2SO4, filtered and evaporated. The resulting crude solid was triturated with MeOH (50 mL) then washed with MeOH (25 mL) and ether (50 mL) and dried under vacuum to give the title compound 12 (4.73 g, 76%) as a white solid; 1H NMR (400 MHz, DMSO) 4.23 (2H, t), 4.79 (2H, t), 7.49 (2H, d), 7.73 (2H, d), 8.89 (1H, s); m/z MH+ = 354, 356. 4-Amino-6-(4-bromophenyl)-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5(6H)-one7. 0.5 M ammonia in 1,4-dioxane (300 mL) was added in one portion 12 (6.59 g, 14.87 mmol) at rt. The resulting solution was stirred at 55 °C for 3 days. The reaction mixture was allowed to cool and a precipitate was collected by filtration, washed with water and methanol and dried under vacuum to afford the title compound 7 (4.44 g, 89%) as a white solid; 1H NMR (400 MHz, DMSO) 3.95–4.00 (2H, m), 4.57–4.62 (2H, m), 7.34–7.38 (2H, m), 7.57–7.63 (4H, m), 8.16 (1H, s); m/z MH+ = 335, 337. Methyl 2-(4′-(4-amino-5-oxo-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-6(5H)-yl)-2-chlorobiphenyl-4-yl)acetate8. 4-Amino-6-(4-bromophenyl)-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5(6H)-one 7 (214 mg, 0.64 mmol), methyl 2-(3-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acetate 6 (198 mg, 0.64 mmol) and tripotassium phosphate (163 mg, 0.77 mmol) were suspended in DME (4 mL), methanol (2 mL) and water (1 mL) and sealed into a microwave tube. The mixture was degassed under vacuum and the atmosphere replaced with nitrogen. (1,1′-Bis(diphenylphosphino)ferrocene)-dichloropalladium(II) (DCM adduct) (26 mg, 0.03 mmol) was added and the reaction was heated at 110 °C for 30 min and cooled to rt. The reaction mixture was evaporated to dryness, redissolved in DCM and washed with water. The organic layer was dried over Na2SO4, filtered and evaporated. The resulting crude product was purified by flash silica chromatography, elution gradient 10–100% EtOAc in iso-hexane then 0–30% MeOH in EtOAc to afford the title compound 8 (129 mg, 46%) as a white solid; 1H NMR (400 MHz, DMSO) 3.65 (3H, s), 3.78 (2H, s), 4.05 (2H, t), 4.63 (2H, t), 7.32–7.35 (1H, m), 7.36–7.39 (2H, m), 7.41–7.44 (1H, m), 7.49–7.52 (3H, m), 7.62 (2H, s), 8.18 (1H, s); m/z MH+ = 439. 2-(4′-(4-Amino-5-oxo-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-6(5H)-yl)-2-chlorobiphenyl-4-yl)acetic acid2. 1 M aq. LiOH (0.86 mL, 0.86 mmol) was added to methyl 2-(4′-(4-amino-5-oxo-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-6(5H)-yl)-2-chlorobiphenyl-4-yl)acetate 8 (126 mg, 0.29 mmol) in 1,4-dioxane (2.23 mL) and water (0.74 mL) at rt. The resulting solution was stirred at 50 °C for 45 min. The mixture was cooled to rt and the pH adjusted to 3–4 using 2 M aq. HCl to give a thick precipitate. The resulting mixture was evaporated to remove the organic solvent and the residual suspension was diluted with water (10 mL) and stirred vigorously for 1 h. The resulting solid was filtered off and washed with water and ether and dried under vacuum to give a white solid which was triturated with MeOH to afford the title compound 2 (89 mg, 73%) as a white solid; 1H NMR (400 MHz, DMSO) 3.73 (2H, s), 4.12 (2H, t), 4.70 (2H, t), 7.37–7.40 (1H, m), 7.44–7.46 (1H, m), 7.53–7.57 (5H, m), 7.69 (2H, s), 8.24 (1H, s), 12.51 (1H, s); m/z MH+ = 425. 2-(4′-(4-Amino-5-oxo-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-6(5H)-yl)-2-chlorobiphenyl-4-yl)acetamide3. HATU (175 mg, 0.46 mmol) was added to 2-(4′-(4-amino-5-oxo-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-6(5H)-yl)-2-chlorobiphenyl-4-yl)acetic acid 2 (150 mg, 0.35 mmol), ammonium hydrochloride (23 mg, 0.42 mmol) and N,N-diisopropylethylamine (0.151 mL, 0.88 mmol) in DMF (2 mL). The reaction mixture was stirred at rt for 2 h, then was concentrated and diluted with 1:1 mix of THF:EtOAc (100 mL), and washed sequentially with water and sat. brine. The organic layer was dried over Na2SO4, filtered and evaporated, and the resulting crude product was triturated with EtOAc. The resulting solid was washed with water and dried under vacuum to afford the title compound 3 (79 mg, 53%) as an off white solid; 1H NMR (400 MHz, DMSO) 3.44 (2H, s), 4.05 (2H, t), 4.63 (2H, t), 6.94 (1H, s), 7.29–7.32 (1H, m), 7.37 (1H, d), 7.46–7.50 (5H, m), 7.52 (1H, s), 7.64 (2H, s), 8.18 (1H, s); 13C NMR (101 MHz, DMSO) 41.20, 49.06, 73.11, 96.33, 126.11 (2C), 128.31, 129.71 (2C), 130.30, 130.75, 131.10, 136.81, 136.96, 137.94, 141.99, 159.15, 165.25, 165.68, 166.01, 171.52; m/z MH+ = 424; HRMS (ESI) calc. for C21H19O3N5Cl(MH+) 424.1171, found 424.1173. 4-Amino-6-(2′-fluorobiphenyl-4-yl)-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5(6H)-one13. 4-Amino-6-(4-bromophenyl)-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5(6H)-one 7 (0.25 g, 0.75 mmol), 2-fluorobenzeneboronic acid (0.136 g, 0.97 mmol), PdCl2(dppf)-DCM adduct (0.030 g, 0.04 mmol) and tripotassium phosphate (0.190 g, 0.90 mmol) were suspended in DME (3 mL), methanol (1.5 mL) and water (0.7 mL) and sealed into a microwave tube. The mixture was degassed under vacuum and the atmosphere replaced with nitrogen. The reaction was heated at 110 °C for 40 min in the microwave reactor and cooled to rt. The reaction mixture was evaporated to dryness and re-dissolved in methyl THF, and washed sequentially with water and sat. brine. The resulting solid was isolated and stirred with with MeOH/DCM, then filtered. The filtrate was combined with the organic layer from above, filtered through a phase separation funnel and evaporated. The resulting crude product was purified by preparative HPLC to afford the title compound 13 (0.123 g, 47%) as a white solid; 1H NMR (400 MHz, CDCl3) 4.08 (2H, dd), 4.70–4.75 (2H, m), 5.64 (1H, s), 7.13–7.24 (2H, m), 7.31–7.40 (3H, m), 7.44 (1H, td), 7.61–7.68 (2H, m), 8.09–8.26 (1H, m), 8.30 (1H, s); 13C NMR (101 MHz, DMSO) 49.1, 73.0, 96.4, 116.1, 124.9, 126.5, 126.5, 127.5, 129.3, 129.3, 129.6, 130.7, 133.3, 142.2, 159.1, 159.3, 165.3, 165.8, 166.1; m/z MH+ = 351; HRMS (ESI) calc. for C19H16O2N4F(MH+) 351.1252, found 351.1249.
Acknowledgements
The authors would like to thank Usha Chauhan for generating high quality enzyme data and Andy Turnbull for all his support and leadership of the project.Notes and references
- A. M. Birch, L. K. Buckett and A. V. Turnbull, Curr. Opin. Drug Discovery Dev., 2010, 13, 489 Search PubMed.
- R. L. Dow, J.-C. Li, M. P. Pence, E. M. Gibbs, J. L. LaPerle, J. Litchfield, D. W. Piotrowski, M. J. Munchhof, T. B. Manion, W. J. Zavadoski, G. S. Walker, R. K. McPherson, S. Tapley, E. Sugarman, A. Guzman-Perez and P. DaSilva-Jardine, ACS Med. Chem. Lett., 2011, 2, 407 Search PubMed.
- W. Yuna, M. Ahmada, Y. Chena, P. Gillespiea, K. Conde-Knapea, S. Kazmera, S. Lia, Y. Qiana, R. Taubb, S. J. Wertheimera, T. Whittarda and D. Bolin, Bioorg. Med. Chem. Lett., 2011, 21, 7205 Search PubMed.
- G. Zhao, A. J. Souers, M. Voorbach, H. D. Falls, B. Droz, S. Brodjian, Y. Y. Lau, R. R. Iyengar, J. Gao, A. S. Judd, S. H. Wagaw, M. M. Ravn, K. M. Engstrom, J. K. Lynch, M. M. Mulhern, J. Freeman, B. D. Dayton, X. Wang, N. Grihalde, D. Fry, D. W. A. Beno, K. C. Marsh, Z. Su, G. J. Diaz, C. A. Collins, H. Sham, R. M. Reilly, M. E. Brune and P. R. Kym, J. Med. Chem., 2008, 51, 380 Search PubMed.
- W. McCoull, M. S. Addie, A. M. Birch, S. Birtles, L. K. Buckett, R. J. Butlin, S. S. Bowker, S. Boyd, S. Chapman, R. D. M. Davies, C. S. Donald, C. P. Green, C. Jenner, P. D. Kemmitt, A. G. Leach, G. C. Moody, P. M. Gutierrez, N. J. Newcombe, T. Nowak, M. J. Packer, A. T. Plowright, J. Revill, P. Schofield, C. Sheldon, S. Stokes, A. V. Turnbull, S. J. Y. Wang, D. P. Whalley and J. M. Wood, Bioorg. Med. Chem. Lett., 2012, 22, 3873 CrossRef CAS.
- (a) S. L. Regan, J. L. Maggs, T. G. Hammond, C. Lambert, D. P. Williams and B. K. Park, Biopharm. Drug Dispos., 2010, 31, 367 CrossRef CAS; (b) U. A. Boelsterli and V. Ramirez-Alcantara, Curr. Drug Metab., 2011, 12, 245 Search PubMed; (c) A. V. Stachulski, J. R. Harding, J. C. Lindon, J. L. Maggs, B. K. Park and I. D. Wilson, J. Med. Chem., 2006, 49, 6931 CrossRef CAS.
- M. J. Waring, Bioorg. Med. Chem. Lett., 2009, 19, 2844 CrossRef CAS.
- A. M. Birch, S. Groombridge, R. Law, A. G. Leach, C. D. Mee and C. Schramm, J. Med. Chem., 2012, 55, 3923 Search PubMed.
- A. M. Birch, S. Birtles, L. K. Buckett, P. D. Kemmitt, G. J. Smith, T. J. D. Smith, A. V. Turnbull and S. J. Y. Wang, J. Med. Chem., 2009, 52, 1558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols and references thereof are given. See DOI: 10.1039/c2md20231j |
| This journal is © The Royal Society of Chemistry 2013 |