Small molecules aimed at type III secretion systems to inhibit bacterial virulence
Lun K.
Tsou†
,
Paul D.
Dossa
and
Howard C.
Hang
*
Laboratory of Chemical Biology and Microbial Pathogenesis, The Rockefeller University, New York, NY 10065, USA. E-mail: hhang@mail.rockefeller.edu
First published on 14th September 2012
Abstract
The development of new anti-bacterial compounds presents a major challenge to modern medicine as bacterial strains resistant to traditional antibiotics are constantly emerging. Type III secretion systems (T3SSs) are essential for virulence mechanisms of many Gram-negative bacterial pathogens and have therefore emerged as an attractive target for small molecule anti-virulence therapeutics. This has led to several high-throughput screens in search for specific chemicals that inhibit the secretion and translocation of bacterial effectorproteins. Several classes of small molecules have now been identified from these screens and this review focuses on their discovery and discusses their potential mechanisms of action and prospects for clinical development.
Introduction
The emergence of multidrug-resistant bacterial strains and new microbial pathogens demands novel antimicrobial strategies to replenish the dwindling antibiotic arsenal.1,2 Not all bacteria are malevolent, as an increasing number of studies have been begun to reveal specific beneficial mechanisms of commensal bacteria on host immunity.3 Indeed, antibiotic disruption of bacterial communities (microbiome) can alter host immunity and exacerbate microbial infections.4 Therapeutic strategies that target bacterial virulence rather than growth have thus received considerable attention,1,2 as these approaches are proposed to selectively disarm pathogens while preserving the integrity of the host microbiome. The discovery of many bacterial virulence mechanisms including protein secretion systems, adhesins, nutrient acquisition, and associated toxins are new targets for anti-virulence strategies.1,2 Amongst the many bacterial virulence mechanisms that have been discovered, specialized protein secretion systems appear to be prime targets for small molecule inhibition of infection5–7 and are the focus of this Minireview.The highly conserved type III secretion system (T3SS) is central to the virulence of many human Gram-negative pathogens such as Salmonella, Shigella, Pseudomonas, enteropathogenic Escherichia coli (EPEC), enterohemorrhagic E. coli (EHEC), Vibrio, Yersinia, and Chlamydia.8–10 The T3SS is a transmembrane machine comprised of about 20 proteins that assemble into a needle-like complex spanning the bacterial membranes and function in a highly regulated manner to transport effectorproteins from the bacterial cytoplasm directly into host cells (Fig. 1).11–14 Construction of the T3SS requires both structural components and ancillary proteins for the assembly process.15,16 The basal body spans the inner (IM) and outer (OM) bacterial membranes.15,16 The rings that span the IM are brought to the envelope by a Sec-dependent pathway and anchored by N-terminal lipidation.8 A short rod connects the IM rings to the OM components, which belong to the secretin family of pore-forming proteins.16 The needle, with various lengths among species and strains, is a polymer of helical proteins that protrudes from the bacterial surface and delivers proteineffectors to the extra-bacterial environment.15,16 The tip of the needle is composed of a complex known as the “translocon”, which can insert itself into the host cell membranes.15 The T3SS is activated by environmental cues, and upon contact with host cells, secretes and translocates proteineffectors into the host cytoplasm (Fig. 1A).9 T3SSs thus facilitate highly coordinated and regulated secretion of specific bacterial proteineffectors during infection.17 Hundreds of effectorproteins among diverse bacterial pathogens have been identified as potential substrates of T3SSs, with as many as 30 different effectorproteins being secreted from a single pathogen.10 Several bacterial effectors can directly induce host cell death to facilitate bacterial pathogenesis (Fig. 1A).18,19 Other bacterial effectors mimic host signaling proteins and enzymes to alter cellular signaling pathways. This allows bacteria to enter and replicate within the host cells while evading detection and destruction by the host immune system (Fig. 1B).20 Depending on the bacterial pathogen, T3SSs can thus facilitate bacterial entry and replication in host cells or cause host cell death directly (Fig. 1). Even though many detailed molecular mechanisms regarding the biogenesis of T3SSs and bacterial effector functions are still under investigation, genetic and biochemical studies of many Gram-negative bacterial pathogens have revealed that effector translocation by T3SSs is crucial for successful infection.16 Despite normal in vitro growth rates, T3SS bacterial mutants cannot deliver proteineffectors into host cells, which renders the pathogens avirulent and significantly attenuated in their ability to cause disease in animals.16 Moreover, directly blocking the injection of proteineffectors into host cells through active or passive immunization using specific antibodies further supports T3SSs as key virulence mechanisms for infection.
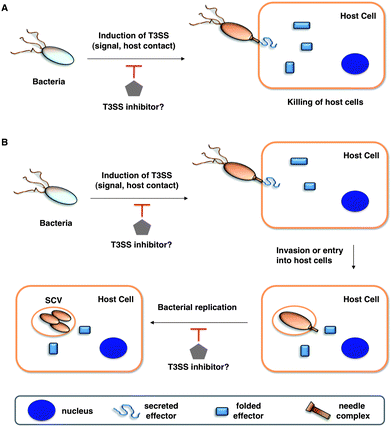 | ||
| Fig. 1 Potential anti-virulence strategies with small molecule inhibitors of T3SSs in Gram-negative bacterial pathogens. (A) T3SS effectors that kill the host cells (for example, in Pseudomonas aeruginosa with translocation of ExoU). (B) T3SSs allow entry and replication in host cells. Legends are indicated in the bottom grey box. | ||
T3SSs have therefore emerged as attractive targets for small molecule anti-virulence therapeutics and have motivated several high-throughput screens (HTS) in search for specific chemical inhibitors that inhibit the secretion and translocation of bacterial effectorproteins.5–7 As T3SSs have not been reconstituted in vitro, the search for small molecule inhibitors has largely employed cell-based assays for protein secretion coupled to sensitive and high-throughput readouts or targeted screens against the enzymatic activity of the ATPase required for protein secretion (Fig. 2). Several classes of small molecules have now been identified from these screens (Tables 1 and 2), which we will summarize below and discuss their potential mechanisms of action and prospects for clinical development.
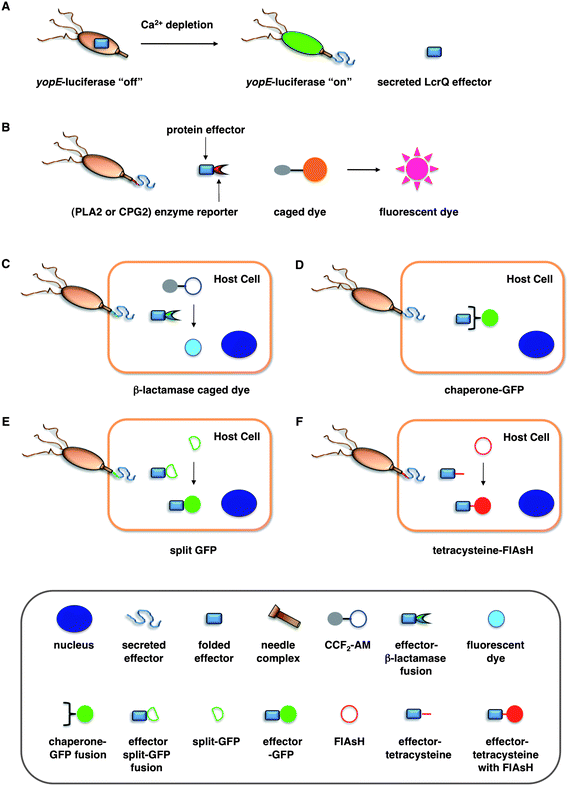 | ||
| Fig. 2 Assays for bacterial effector secretion and HTS. (A) Whole cell HTS using a Yersinia pseudotuberculosis (yopE:luxAB) strain21 that utilizes a transcriptional readout linked to secretion. (B) Whole-cell HTSs were performed using an effector-enzyme fusion where the enzymatic activity can be monitored by fluorescence.38,41 (C) A bacterial effector is fused with β-lactamase (βla) that cleaves a βla-sensitive FRET probe, CCF2-AM, in the host cells. (D) GFP-labeled chaperones were used as probes to visualize translocation of bacterial effectors by imaging effector accumulation in the cytosol of the host cells. (E) Upon T3SS effector translocation, the association of the two fragments, small 13-amino-acid 11th strand of the GFP β-barrel and the complementary fragment of the first 10 GFP strands, leads to GFP fluorescent tagging of the effector population in the host cells. (F) The fluorescein-based biarsenical dye FlAsH in the host cells allow the labeling of effectors with a genetically encoded sequence containing the tetracysteine repeat motif as the tag. Legends for C–F are indicated in the bottom grey box. | ||
| Compound | Regulation | Secretion/translocation | T3SS IC50 | Infection | Other Pathways | Ref. |
|---|---|---|---|---|---|---|
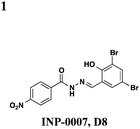
|
C. trachomatis: down-regulates transcription of late-cycle virulence genes | C. trachomatis: prevents Inc A secretion during infection | ∼15 μM | C. trachomatis: inhibits replication in HeLa | 21,26,31,35 and 38 | |
| S. typhimurium | ∼5 μM* | S. typhimurium: reduces hemolysis of erythrocytes, invasion of HeLa, and immune response in bovine ligated loop model | ||||
| Y. pseudotuberculosis, Y. pseudotuberculosis: prevents YopH translocation into HeLa | Y. pseudotuberculosis: inhibits invasion of HeLa | Y. pseudotuberculosis: inhibits motility | ||||
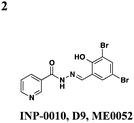
|
C. pneumoniae, S. typhimurium: down-regulates transcription of some virulence genes, EHEC: down-regulates transcription of LEE but not iron-related genes | EHEC: inhibits Tir and EspD secretion, S. typhimurium | ∼25 μM | C. trachomatis: inhibits replication in mammalian cell lines, S. typhimurium: inhibits invasion of MDCK cells and replication in macrophages, Y. pseudotuberculosis: reduces macrophage cytotoxicity | 26,27 and 32 | |
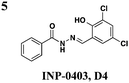
|
EHEC: down-regulates transcription of LEE but not iron-related genes and up-regulates flagellum expression | EHEC: inhibits Tir and EspD secretion | ∼25 μM | EHEC: reduces number of A/E lesions on bovine epithelial cells | 27 | |
|
S. flexneri: affects needle complex assembly | S. typhimurium, S. flexneri | C. trachomatis: inhibits replication in HeLa and McCoy cells, S. typhimurium: reduces HeLa cytotoxicity, S. flexneri: reduces invasion of HeLa and macrophage cytotoxicity | 33,34,36 and 39 | ||
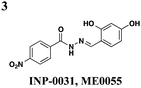
|
S. typhimurium: down-regulates transcription of invC and hilD but not hilA or effectors | S. typhimurium | ∼25 μM | S. typhimurium: reduces hemolysis of erythrocytes, invasion of HeLa, and immune response in bovine ligated loop model | S. typhimurium: effects reversed with exogenous iron | 35–37 |
| Compound | Regulation | Secretion/translocation | T3SS IC50 | Infection | Other pathways | Ref. |
|---|---|---|---|---|---|---|
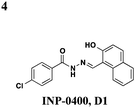
|
S. typhimurium: inhibits needle complex assembly, no effect on general transcription | S. typhimurium, Y. enterocolitica, F. tularensis | 83 μM | P. syringae: prevents HR response in tobacco leaves, S. typhimurium: reduces macrophage cytotoxicity | S. typhimurium: no effect on flagellar system or motility, P. aeruginosa: inhibits T4-dependent motility & T2S | 41–43 |
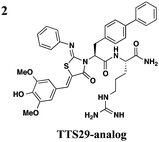
|
S. typhimurium: no effect on general transcription | S. typhimurium | 3 μM | S. typhimurum: reduces macrophage cytotoxicity | P. aeruginosa: inhibits T2S | 41–43 |
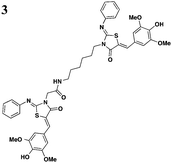
|
S. typhimurium: Inhibits secretion of SipA | 5 μM | 43 |
Salicylidene acylhydrazides
The salicylidene acylhydrazides were the first class of small molecules identified from a T3SS effector screen.21 Kauppi et al. developed a whole cell HTS using a Yersinia pseudotuberculosis reporter strain that utilizes a transcriptional readout linked to secretion (Fig. 2A).21 They engineered the luciferase luxAB gene under the control of the effectoryopE promoter. When grown at 37 °C in Ca2+-rich media, LcrQ prevents T3SS gene expression. Upon contact with a eukaryotic cell or depletion of Ca2+, LcrQ is secreted via the T3SS, thus releasing transcriptional repression of type III secretion genes such as yopE.22 Under these conditions, this strain will then express luciferase and luminescence can be used to monitor yopE expression. Using this assay, Kauppi et al. screened 9400 compounds for their ability to block expression of luciferase activity. Luminescence was measured after bacteria were incubated with compound in Ca2+-depleted media. In wells with inactive compounds, LcrQ would be secreted and luciferase would be expressed. For compounds that block T3S, LcrQ would remain in the cell and repress expression of the luciferase gene. Since this assay monitors loss of signal on a transcriptional level, follow-up assays are required to assess the activity of compounds on type III secretion directly. Compounds that affect the growth of the bacteria are excluded, and compounds with effects on transcription would also result in hits by indirectly affecting type III secretion.Three notable classes of compounds were discovered to prevent type III secretion at low micromolar concentrations without affecting growth of the bacteria: the sulfonylamino-benzanilides, salicylanilides, and salicylidene acylhydrazides (Table 1). Incubation of bacteria with each class resulted in decreased levels of secreted effectors into culture supernatant, confirming the transcriptional readout. The individual compounds have varying effects on transcription and motility, suggesting different targets and mechanisms of action. The flagellar system is related to the type III secretion machinery, so compounds that affect one could also target the other. The sulfonylamino-benzanilides did not affect motility or general transcriptional regulation of the T3SS, but only one follow-up study has been done, potentially due to their limited solubility.23 The salicylanilides have no effect on motility and likely work upstream of a transcriptional activator of the Yops, potentially by affecting two-component signaling, thus indirectly affecting type III secretion. There are other reports confirming their effect on transcription.24,25 The salicylidene acylhydrazides, specifically INP-0007 from this study, affected both secretion and motility without generally affecting transcription (Table 1, entry 1). This suggests a conserved component between the two systems as a potential target. Inhibiting secretion and motility is advantageous for an anti-virulence compound, making this class a good candidate for follow-up studies.
Since the initial discovery of the salicylidene acylhydrazides in Yersina, follow-up studies in Yersinia demonstrated INP-0007 blocks secretion in a constitutively secreting mutant and prevents bacterial invasion of HeLa cells.26 Compounds in this class have since been shown to have T3SS inhibitory activity in several Gram-negative pathogens including E. coli,27,28Chlamydia,29–34Salmonella,35–38 and Shigella,39 which results in an attenuation of infection. Various INP-0007 derivatives were shown to prevent invasion of Yersinia, Salmonella, and Shigella, disrupt the infectious cycle in Chlamydia, and decrease the number of attachment and effacement lesions in a bovine epithelial cell line when infected with E. coli.27 INP-0007 and INP-0403 were shown to inhibit T3SS-induced hemolysis in erythrocytes and decrease the secretory and inflammatory responses in vivo with a bovine intestinal ligated loop model when infected with Salmonella (Table 1, entries 1 and 5), but the caveat for drug development is that these protective effects were only observed when the bacteria were pre-treated with inhibitor.35 In Chlamydia, however, even when inhibitor was added 24 hours post-infection to HeLa cells, it still had a significant impact on bacterial replication.33
While there is variability in the exact structure of active compounds between species, there is a common core scaffold. One study used statistical molecular design (SMD) and quantitative structure–activity relationships (QSAR) on 50 compounds tested in Yersinia to make predictions about untested compounds.40 Of eight candidate compounds, they were able to correctly predict that three would be inactive, but only three of the five predicted to be active demonstrated inhibitory activity. Though there was no obvious consensus motif that confers activity, the authors found that the pKa of the phenol was the most important property in the model and that the electrostatic potential of the carbon atoms in that aromatic ring may play a key role.
Though there have been several studies using the salicylidene acylhydrazides in various bacterial species, the precise target(s) and mechanisms of action remain unclear. Since these compounds have broad-spectrum inhibitory activity, it is likely that they target something conserved among the species. It was initially proposed that the compounds act directly on the T3S machinery, and one report in Shigella suggests that incubation with INP-0400 affected needle assembly39 (Table 1, entry 4). Further support for this notion is that the T3S needle complex is related to the flagellar assembly, and motility is affected in Yersinia21 and Salmonella.36 Several reports have examined the effects of these compounds have on transcription. In Chlamydia, Wolf and co-workers found that INP-0007 down-regulates some virulence genes, and while levels of needle components are unaffected, effectors accumulate intracellularly.31 Another study performed RT-PCR of virulence genes during infection of HeLa cells where compound was added at the time of infection.33 They also observed that only some virulence genes are down-regulated when incubated with active compound, and importantly, they used an inactive compound as a negative control and saw no differences between that and the DMSO sample. In E. coli, four compounds with varying potency all down-regulated genes in the locus for enterocyte effacement, but there was large variation between strains.27 When the transcriptome in Salmonella incubated with various inhibitors was examined, the needle complex ATPase InvC and the HilD regulator were down-regulated, but no major effects on HilA or the effectors themselves were observed.37 Taken together, it is obvious the salicylidene acylhydrazides have some transcriptional effects; however, it is not clear if this is a direct effect and the cause of secretion inhibition or the result of a feedback loop.
In Chlamydia33 and Salmonella,37 it has been reported that adding exogenous iron to the cultures can prevent the inhibitory effects of the salicylidene acylhydrazides, suggesting iron restriction as a potential mechanism of action. Iron transport and acquisition genes are up-regulated in Salmonella upon treatment with several inhibitors, however, neither the up-regulation of iron-related genes nor an effect from adding exogenous iron is observed in Yersinia. Also, an inactive compound was shown to bind iron to the same extent as an active compound,33 thus casting doubt on an iron-restriction mechanism.
Roe and co-workers have attempted to biochemically identify the targets of the salicylidene acylhydrazides.28 They synthesized two analogs of ME0052 (Table 1, entry 2) and ME0055 (Table 1, entry 3) for affinity purification of binding partners from E. coli lysate. After trypsinization and LC-MS/MS analysis, 16 proteins were identified, and three were suggested binding partners by Far Western analysis using a biotinylated version of a much less active compound: Tpx (thiol peroxidase), WrbA (NAD(P)H quinine oxidoreductase), and FolX (dihydroneopterin-tri-P-epimerase). Knockouts of each gene results in down-regulation of flagellar components and up-regulation of T3SS genes, the opposite of what is observed with compound treatment. Deletion mutants of Tpx and WrbA secrete more effectors, and they are as virulent in infecting macrophages, but adding ME0052 can still inhibit both processes. The authors suggest the inhibition of T3SS activity is due to a polypharmacological effect on proteins involved in metabolism, which then results in a dis-regulation of bacterial virulence. Given the diverse pharmacological effects of salicylidene acylhydrazides that have been reported, the precise mechanism of action for these compounds remains unclear.
2-Imino-5-arylidene thiazolidinones
A whole-cell HTS in Salmonella identified the 2-imino-5-arylidene thiazolidinones as broad inhibitors of T3S.41 The screen was performed by engineering a Salmonella strain where the secreted effector SipA was fused to a portion of a Yersiniaprotein YplA, that contained phospholipase A2 activity (Fig. 2B). The phospholipase A2 activity could then be measured with a fluorogenic substrate. Using this strain, they screened 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 small molecules from natural and synthetic libraries. After ruling out compounds that affected growth or were unsuitable for drug development, they followed upon 25 compounds. Excluding compounds that had an effect on general transcription, translation, or Sec-dependent secretion left seven molecules for follow-up studies. Six of these were found to affect T3S gene expression, leaving one that might specifically target the T3S process or assembly directly.
000 small molecules from natural and synthetic libraries. After ruling out compounds that affected growth or were unsuitable for drug development, they followed upon 25 compounds. Excluding compounds that had an effect on general transcription, translation, or Sec-dependent secretion left seven molecules for follow-up studies. Six of these were found to affect T3S gene expression, leaving one that might specifically target the T3S process or assembly directly.
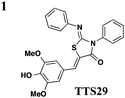
The one compound Felise et al. pursued was a 2-imino-5-arylidene thiazolidinone (TTS29) (Table 2, entry 1), and it was shown to have T3S inhibitory effects in Yersinia, Pseudomonas aeruginosa, and Francisella novicida, demonstrating broad-spectrum inhibitory activity. By purifying the needle complexes of Salmonella, they found that compound-treated samples had lower levels of needle components, but whole-cell levels were unchanged, suggesting that the compound disrupts needle assembly. No effect was observed on motility or the levels of flagellar components, suggesting the effect was specific to the T3SS. They also discovered that TTS29 affects type II secretion (T2S) and the related type IV pili assembly. Examining what is common to all affected systems and not present in the flagellar system, it was proposed that TTS29 acts by disrupting secretin interactions, thus affecting needle complex assembly or stability but not the flagellar system. These effects on the T3SS carried over into an infection setting, as the compound was able to prevent Salmonella-induced lysis of macrophages and the hypersensitivity response in tobacco plants from the plant pathogen Pseudomonas syringae.
One major drawback is the low potency of TTS29, as several hundred micromolar is required to have an inhibitory effect. Subsequent SAR analysis demonstrated the importance of the imino nitrogen, its aryl group, and the substitution pattern on the arylidene ring while the amido nitrogen tolerated modification. A TTS29-analog in this study displayed higher potency in every assay tested without affecting bacterial growth (Table 2, entry 2). This led to further modification of the amido nitrogen, with several analogs containing a dipeptide chain displaying low micromolar IC50 values for inhibition of SipA.42 Tethered thiazolidinone dimers (Table 2, entry 3), some of which are linked with a peptide, displayed similar potencies;43 however, no follow-up studies have been reported with these compounds.
Other chemotypes from HTS
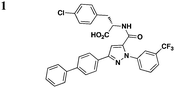
A few other HTSs have revealed several other classes of T3SS inhibitors. Janssen Pharmaceuticals employed an assay which measures levels of the secreted effectorprotein ExoU from P. aeruginosa and SipA and SopE from S. typhimurium as the readouts of T3SS inhibition.44 The majority of the inhibitors that exhibited low micromolar potencies were comprised of a common N-acyl p-Cl phenylalanine moiety (Table 3, entry 1). However, this class of compounds lacks the broad-spectrum anti-T3SS activity across different Gram-negative pathogens, and no further development has yet been reported.| Representative compounds | Regulation | Secretion/translocation | T3SS IC50 | Infection | Other pathways | Ref. |
|---|---|---|---|---|---|---|
|
S. typhimurium (SipA, SopE), P. aeruginosa (ExoU) | 44 | ||||
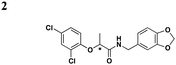
|
exoT transcription | Y. pestis (YopE-βla), P. aeruginosa (ExoS) | 12.5 μM for ExoS, 22 μM for YopE | Inhibits P. aeruginosa ExoU-dependent CHO cytotoxicity, inhibits C. trachomatis growth in Hep-2 cells | No effect on type II-mediated elastase secretion | 45 |
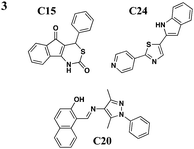
|
No effect on needle assembly | Y. pseudotuberculosis, YopE-βla translocation, P. aeruginosa | Reduces Hep-2 cell-rounding by P. aeruginosa | Increases Yop leakage, C20 reduces adherence of Hep-2 cells | 46 | |
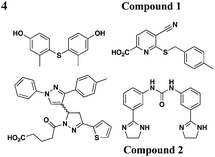
|
YopE transcription, no effect on OmpA level in EPEC | Y. pestis (effectors), YopE-βla translocation, EPEC (Tir) | 10–15 μM for Yops | Reduces HeLa cells toxicity by Y. pestis | 47 | |
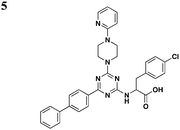
|
S. typhimurium (SipB), P. aeruginosa (ExoU) | 20–80 μM for SipB, >100 μM for ExoU | 48 | |||
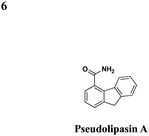
|
No effect on P. aeruginosa secretion | Protects CHO cells cytotoxicity from the P. aeruginosaeffector ExoU, prevents P. aeruginosa-mediated killing of Dictyostelium discoideum, inhibits ExoU-mediated killing of Saccharomyces cerevisiae | 49 | |||
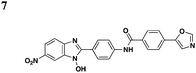
|
Y. pseudotuberculosis | Y. pseudotuberculosis cytotoxicity | 50 | |||
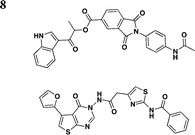
|
Y. pestis (YopE) | 20 μM | Protects Y. pestis cytotoxicity | ATPase activity of BsaS protein from Burkholderia mallei | 51 | |

|
No effect on type-V effector EspC | EPEC (EspB) | 20 μM | 52 and 53 | ||

|
EPEC | 20 nM in hemolysis assay | Prevents hemolysis of erythrocytes | 54–56 | ||
|
No effect on expression of GroEL | EPEC (EspA, EspB, EspD) | 1.5 μg ml−1 in hemolysis assay | Prevents hemolysis of erythrocytes, protects Citrobactor rodentium infection | 57 |
Microbiotix Pharmaceuticals has identified five inhibitors from a library of 80000 compounds through two distinct functional bioassays.45 First, compounds were screened against a transcriptional fusion of the Photorhabdus luminescens luxCDABE operon to the P. aeruginosa exoTeffector gene. Then the compounds were selected based on the inhibition of the secretion of a P. aeruginosa ExoS effector-β-lactamase fusion protein (Fig. 2C) and also of native ExoS by the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of culture supernatants. To demonstrate the broad-spectrum efficacy of the compounds, they were found to also antagonize Yersinia T3S and one compound, MBX1641 also targeted Chlamydia T3S (Table 3, entry 2). Interestingly, contrary to typical HTS hits, Microbiotix compounds contained chiral centers and through a series of synthetic efforts, it was shown that the R enantiomer was the preferred stereochemistry. With a stringent structure–activity relationship, these compounds might see many obstacles moving forward; however, other scaffolds identified in the screen could be explored in the future.
Harmon et al. devised a HTS to identify compounds that inhibit the T3S-dependent translocation of Yersiniaeffectorproteins, Yops, into host cells.46 This translocation-monitoring FRET-based system employed a chimeric protein, E-TEM that encodes the first 100 amino acids of YopE, containing essential secretion and translocation signals to direct it into mammalian cells, fused to a fragment of β-lactamase (TEM) (Fig. 2C). TEM can cleave the lactam ring between the substrate CCF2-AM, leading to the loss of FRET and resulting in blue fluorescence. If Y. pseudotuberculosis is unable to translocate E-TEM into host cells, the substrate will remain uncleaved and the cells will fluoresce green. The green-to-blue ratio was measured to determine whether the compounds could block the translocation of E-TEM. Notably, they have identified six compounds that specifically inhibit translocation of Yops into mammalian cells but not Yop synthesis or secretion (Table 3, entry 3). These six diverse compounds lacked a consensus pharmacophore, yet they inhibited translocation of effectors into the host cell while not affecting the synthesis of T3SS components and secreted effectors, assembly of the T3SS, nor secretion of effectors. Interestingly, C20 reduced adherence of Y. pseudotuberculosis to target cells. Additionally, the compounds were shown to cause leakage of Yops into the supernatant during infection and reduced polarized translocation. Furthermore, several molecules also inhibited P. aeruginosa ExoS-mediated cell rounding, suggesting that the compounds target factors that are conserved between these two pathogens.
Similar to the reported yopE:luxAB strain,21 Pan et al. screened a collection of 70966 compounds using a Y. pestis luciferase reporter strain.47 Four new compounds were identified to inhibit secretion of Y. pestis T3SS effectorproteins YopD, YopH, and YopM at micromolar concentrations without affecting bacterial growth. Moreover, two of the four compounds (compounds 1 and 2, Table 3, entry 4) also attenuated T3SS-mediated secretion of EPEC effectorproteins. In all, compounds discovered from this library are likely to have different mechanisms of action and targets from each other.
Li and co-workers adopted a cell-based assay that measures the T3S-dependent secretion of a chimeric SopE-β lactamase fusion and identified a class of triazine-based compounds (Table 3, entry 5) as inhibitors of T3S.48 Active compounds can selectively reduce the level of SipB in the supernatant fraction of Salmonella cultures without affecting the production of SipB intracellularly. Moreover, these compounds also diminished the level of ExoU in the supernatant of P. aeruginosa cultures. No further development has been reported since the patent.
In an effort to identify compounds that protect Chinese hampster ovary (CHO) cells against the T3S-depedent cytotoxic effects of the P. aeruginosaeffector ExoU, Lee et al. reported pseudolipasin A as a specific inhibitor for the phospholipase A2 activity of ExoU with an IC50 of 7 μM (Table 3, entry 6).49 Pseudolipasin A did not inhibit protein secretion and a collection of structural analogs was used to demonstrate a conserved pharmacophore for phospholipase A2 inhibitory activity. Starting with an in silico screen, a number of N-hydroxybenzimidazole based scaffolds has been synthesized to block the binding of the Yersinia transcription factor LcrF to the DNA (Table 3, entry 7).50 With more detailed SAR studies, a number of novel compounds potently inhibit the secretion of Yops. However, the anti-virulence activity among different Gram-negative pathogens by these compounds is still undetermined.
Swientnicki et al. has recently reported in silico studies to identify inhibitors to target the Yersinia pestis T3SS ATPase YscN (Table 3, entry 8).51 The authors validated YscN as a therapeutic target by deleting the catalytic domain of the yscN gene in Y. pestis CO92 and showed a reduction of over three million-fold of bubonic plague in the Swiss-Webster mouse model. The validated but diverse inhibitors had IC50 values below 20 μM in an in vitro ATPase assay and were also found to inhibit the homologous BsaS protein from the Burkholderia mallei animal-like T3SS at similar potency. Moreover, these compounds inhibited YopE secretion and attenuated Y. pestis toxicity in mammalian cells. These data demonstrate the possibilities of targeting and inhibiting ATPase of T3SSs in other pathogens.
Natural products: the caminosides and the guadinomines
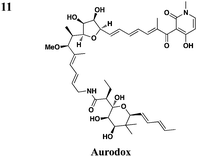
In addition to synthetic chemical libraries, natural product extracts have been surveyed for compounds with T3SS inhibitory activity. Bioassay-guided fractionation of the extracts from the marine sponge Caminus sphaeroconia led to the isolation of a series of lipidated pentasaccharides that possess potent anti-T3SS activity (Table 3, entry 9).52,53 Analysis of EPEC culture supernatants showed that Caminoside A selectively diminished the level of a T3S-dependent effectorprotein EspB while not affecting the level of EspC, a type V-dependent effector. The potency of Caminoside A is approximately 20 μM without affecting the growth of the pathogens,52 yet further development of the caminosides has not been reported. Alternatively, guadinomines isolated from Streptomyces sp. K-01-0509 were reported to inhibit EPEC T3S-dependent lysis of sheep blood erythrocytes with an IC50 value of 20 nM in a whole cell assay (Table 3, entry 10).54,55 Even though this class of natural products might be the most potent inhibitors to date, major synthetic efforts are needed to produce pure material for further studies.56From a screen of monitoring T3SS-mediated hemolysis from enteropathogenic E. coli (EPEC), Abe and co-workers identified aurodox, a linear polyketide from Streptomyces sp, as a specific T3SS inhibitor.57 Aurodox potently inhibited T3SS-mediated hemolysis with an IC50 value of 1.5 μg ml−1 without affecting bacterial growth (Table 3, entry 11). By coomassie blue-staining and Western analysis of the supernatant fraction, aurodox specifically diminished the levels of effectorproteins such as EspB, EspF, and Map without affecting the expression of the housekeeping protein GroEL. Moreover, aurodox allowed mice to survive a lethal dose of Citrobactor rodentium, a model bacterium for human pathogens such as EPEC. Beyond the antibacterial drug development, the discovery of aurodox highlights the possibility microbes may produce small molecules that can antagonize T3SSs in chemical ecology.
Additional assays for monitoring T3SSs
To facilitate the analysis of T3SSs and small molecule inhibitor discovery, new assays have been developed to more efficiently and precisely visualize protein secretion and translocation. Our laboratory recently described a carboxypeptidase G2 (CPG2)-based reporter system for fluorescence and visible detection of type III protein secretion from Salmonella (Fig. 2B).38 The orthogonal specificity of CPG2 presents an attractive enzyme–reporter system to monitor bacterial protein secretion. CPG2 is a 43 kDa metalloprotease found in rare Pseudomonas syringae strains that selectively cleaves glutamate (Glu) from small molecule metabolites.58 Importantly, the glutamyl-carboxypeptidase activity of CPG2 is not present in most species of bacteria or higher eukaryotes. The ability of CPG2 to hydrolyze urea analogues of Glu provides a unique enzymatic activity to uncage fluorogenic dyes. Based upon the tunable, photostable, and fluorescent properties of 2-dicyanomethylene-3-cyano-2,5-dihydrofuran (CyFur) fluorophores,59–61 we synthesized a Glu-modified derivative of CyFur (Glu-CyFur) as a potential fluorogenic substrate for CPG2. The time-dependent uncaging of Glu-CyFur with CPG2 can be readily observed by the naked eye with or without UV-excitation. In S. typhimurium, SopE2–CPG2–HA was cloned into the pWSK29 plasmid driven by the SopE2 promoter. Confirming that this assay measures secretion through the T3SS, S. typhimurium deficient in a structural component of the SPI-1 T3SS needle complex (ΔinvA) did not secrete SopE2–CPG2–HA and, consequently, did not exhibit CPG2 activity in the growth media.The SopE2–CPG2–HA:CyFur reporter system can differentially measure the activity of T3SS inhibitors. An INP-0007 analog lacking the dibromophenol motif showed no inhibitory activity as compared to INP-0007, which suggests that the dibromophenol motif of INP-0007 is crucial for potent inhibition of T3SSs. Moreover, INP-0007 is ∼4 times more potent in blocking SopE2–CPG2–HA secretion than 2-imino-5-arylidene-thiazolidinone with IC50 values of 5.5 and 22.6 μM, respectively. These experiments highlight the utility and sensitivity of the CPG2:Glu-CyFur reporter system in measuring differential inhibitory activity of small molecules targeted at T3SSs. The CPG2:Glu-CyFur reporter system provides a robust and sensitive method for monitoring protein expression and secretion. This system is complementary and orthogonal to other enzyme reporter systems such as luciferase and β-lactamase, making it potentially useful for dual-imaging applications in the future.
Green fluorescent protein (GFP)-based systems have been employed to monitor type III protein secretion. While direct translation fusions of GFP to bacterial effectors have not been successful, GFP-labeled effector chaperones can be used as probes to visualize translocation of bacterial effectors into the host cells (Fig. 2D). This approach allows the imaging of effector accumulation in the cytosol of the host cells by detecting clustering or accumulation of the fluorescently labeled chaperones. Schlumberger et al. used this strategy to monitor the injection of the Salmonellaeffector SipA into the host cytosol and concurrent depletion from the bacterial cytosol in real time.62 Alternatively, Palmer and co-workers recently reported a split GFP system to image T3S. They fused the small 13-amino-acid 11th strand of the GFP β-barrel to Salmonellaeffectors and expressed the complementary fragment of the split GFP in trans in the host cells (Fig. 2E).63 Upon T3SS effector translocation, the association of the two split GFP fragments leads to fluorescent tagging of the effector population in the host cells. The authors used this method to directly label and visualize the dynamics of the T3SS-dependent SPI-2 effectors PipB2, SteA, and SteC during bacterial replication in host cells. Rather than GFP-based systems, smaller protein tags such as tetracysteine sequences can also be used with fluorescein-based biarsenical dye FlAsH to image bacterial effector translocation into mammalian cells (Fig. 2F).64–66 It would be interesting to employ these assays to evaluate the precise effects of the reported T3SS inhibitors on bacterial effector translocation into mammalian cells.
Concluding remarks
The discovery of T3SSs as essential mechanisms of bacterial virulence has provided important and exciting targets for antibacterial development to address the shrinking arsenal of antibiotics. Consequently, many new classes of small molecules have been reported with inhibitory activity against T3SSs (Tables 1–3). Despite the attractiveness of these potential anti-virulence compounds, none of the currently reported T3SS inhibitors have advanced into the clinic. Indeed, major challenges still need to be addressed before these compounds can be effective therapeutics. Notably, nearly all of the T3SS inhibitors discovered from HTS cell-based assays have limited potency and lack clear targets and mechanisms of action. For example, the preliminary studies of previously described salicylidene acylhydrazides and thiazolidinones42,43 yielded structural features for blocking the activity of T3SSs, but none of ∼120 new analogs yielded more potent inhibitors. Even though tethered thiazolidinone derivatives43 and peptide-functionalized analogs42 have yielded improved inhibitory activity towards T3SS-mediated invasion in vitro, the activity of more potent second generation thiazolidinones (∼56) in cell culture or animal models of bacterial infection have not been reported. While these T3SS inhibitors present promising leads for antibacterial development, significant improvements in their potency are needed for clinical studies. The analysis of focused chemical libraries and other naturally occurring small molecules should yield new candidate T3SS inhibitors.The target identification and mechanism of action studies for candidate T3SS inhibitors are challenging. Unlike antibiotics that kill bacteria and enable selection of resistant mutants, this strategy cannot be utilized for T3SS inhibitors since this pathway is not required for growth in vitro. As T3SSs are composed of ∼20 proteins that oligomerize into the needle-like secretion apparatus, inhibition of secretion might arise from cumulative effects on many protein–protein interactions. T3SS assays typically monitor effector levels in the culture supernatant, however, researchers need to be cautious that diminished levels of effectors in the supernatant may not fully equate with the inhibition of protein secretion. For example, agglutination of the bacterial effectors by the compounds might lead to precipitation of the proteins and present a similar biochemical phenotype. Novel strategies to elucidate the genetic and biochemical targets of T3SS inhibitors are still needed. With new T3SS assays and a more detailed understanding of T3SS biogenesis, a more careful analysis and classification of the existing or new T3SS inhibitors should be possible.
Beyond in vitro screening of T3SS inhibitors, the analysis of small molecules that inhibit bacterial virulence in more complex cell culture or animals could reveal new classes of compounds and also begin to address their activity on host cells. For enteric pathogens, it will be interesting to assay compounds targeting the bacterial replication cycles in mammalian cells. These compounds may serve as better drug candidates to treat systematic infection rather than anti-virulence compounds that may only be used to prevent infection. Alternatively, analysis of compounds in small animal models such as worms67–69 and zebrafish70 are already beginning to reveal potential anti-virulence inhibitors as well as potential mechanisms of drug tolerance. While this approach may yield compounds that activate host immunity,71 this strategy may already select for compounds that can access the appropriate sites and tissues relevant for bacterial infection in vivo and provide promising leads for clinical development in humans.
Key virulence mechanisms of many bacterial pathogens like T3SSs have now been uncovered and are prime targets for chemical intervention. Potent and specific T3SS inhibitors with validated mechanisms of action are still needed to establish whether this conserved virulence pathway is indeed a druggable target and viable approach towards disarming clinically relevant bacterial pathogens. Many of the virulence factors are essential for establishment of human diseases but are dispensable for the growth of pathogens. It remains to be determined whether the use of anti-virulence compounds would exert less selective pressure on bacterial pathogens to develop drug resistance as well as preserve beneficial microbiota in vivo. A focused effort on T3SS inhibitors could address these major challenges in antibacterial drug discovery and help combat microbial infections that impact human health.
Acknowledgements
P.D.D. is a graduate student in the Weill Cornell/Memorial Sloan-Kettering/Rockefeller University Tri-Institutional Program in Chemical Biology. We thank The Rockefeller University and Lerner Trust for support.References
- D. A. Rasko and V. Sperandio, Nat. Rev. Drug Discovery, 2010, 9, 117–128 CrossRef CAS.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS.
- L. V. Hooper, D. R. Littman and A. J. Macpherson, Science, 2012, 336, 1268–1273 Search PubMed.
- B. P. Willing, S. L. Russell and B. B. Finlay, Nat. Rev. Microbiol., 2011, 9, 233–243 Search PubMed.
- P. Keyser, M. Elofsson, S. Rosell and H. Wolf-Watz, J. Intern. Med., 2008, 264, 17–29 CrossRef CAS.
- T. Kline, H. B. Felise, S. Sanowar and S. I. Miller, Curr. Drug Targets, 2012, 13, 338–351 Search PubMed.
- V. T. Lee and J. L. Kessler, IDrugs, 2009, 12, 636–641 Search PubMed.
- T. Izore, V. Job and A. Dessen, Structure, 2011, 19, 603–612 Search PubMed.
- J. E. Galan and H. Wolf-Watz, Nature, 2006, 444, 567–573 CrossRef CAS.
- G. R. Cornelis, Nat. Rev. Microbiol., 2006, 4, 811–825 Search PubMed.
- T. C. Marlovits, T. Kubori, A. Sukhan, D. R. Thomas, J. E. Galan and V. M. Unger, Science, 2004, 306, 1040–1042 CrossRef CAS.
- T. C. Marlovits, T. Kubori, M. Lara-Tejero, D. Thomas, V. M. Unger and J. E. Galan, Nature, 2006, 441, 637–640 CrossRef CAS.
- O. Schraidt and T. C. Marlovits, Science, 2011, 331, 1192–1195 Search PubMed.
- O. Schraidt, M. D. Lefebre, M. J. Brunner, W. H. Schmied, A. Schmidt, J. Radics, K. Mechtler, J. E. Galan and T. C. Marlovits, PLoS Pathog., 2010, 6, e1000824 Search PubMed.
- T. C. Marlovits and C. E. Stebbins, Curr. Opin. Microbiol., 2010, 13, 47–52 Search PubMed.
- J. E. Galan, Annu. Rev. Cell Dev. Biol., 2001, 17, 53–86 Search PubMed.
- M. Lara-Tejero, J. Kato, S. Wagner, X. Liu and J. E. Galan, Science, 2011, 331, 1188–1191 Search PubMed.
- B. Coburn, I. Sekirov and B. B. Finlay, Clin. Microbiol. Rev., 2007, 20, 535–549 Search PubMed.
- A. R. Hauser, Nat. Rev. Microbiol., 2009, 7, 654–665 Search PubMed.
- J. E. Galan, Cell Host Microbe, 2009, 5, 571–579 Search PubMed.
- A. M. Kauppi, R. Nordfelth, H. Uvell, H. Wolf-Watz and M. Elofsson, Chem. Biol., 2003, 10, 241–249 CrossRef CAS.
- J. Pettersson, R. Nordfelth, E. Dubinina, T. Bergman, M. Gustafsson, K. E. Magnusson and H. Wolf-Watz, Science, 1996, 273, 1231–1233 Search PubMed.
- A. M. Kauppi, C. D. Andersson, H. A. Norberg, C. Sundin, A. Linusson and M. Elofsson, Bioorg. Med. Chem., 2007, 15, 6994–7011 Search PubMed.
- A. Gauthier, M. L. Robertson, M. Lowden, J. A. Ibarra, J. L. Puente and B. B. Finlay, Antimicrob. Agents Chemother., 2005, 49, 4101–4109 CrossRef CAS.
- M. J. Macielag, J. P. Demers, S. A. Fraga-Spano, D. J. Hlasta, S. G. Johnson, R. M. Kanojia, R. K. Russell, Z. Sui, M. A. Weidner-Wells, H. Werblood, B. D. Foleno, R. M. Goldschmidt, M. J. Loeloff, G. C. Webb and J. F. Barrett, J. Med. Chem., 1998, 41, 2939–2945 CrossRef CAS.
- R. Nordfelth, A. M. Kauppi, H. A. Norberg, H. Wolf-Watz and M. Elofsson, Infect. Immun., 2005, 73, 3104–3114 CrossRef CAS.
- J. J. Tree, D. Wang, C. McInally, A. Mahajan, A. Layton, I. Houghton, M. Elofsson, M. P. Stevens, D. L. Gally and A. J. Roe, Infect. Immun., 2009, 77, 4209–4220 Search PubMed.
- D. Wang, C. E. Zetterstrom, M. Gabrielsen, K. S. Beckham, J. J. Tree, S. E. Macdonald, O. Byron, T. J. Mitchell, D. L. Gally, P. Herzyk, A. Mahajan, H. Uvell, R. Burchmore, B. O. Smith, M. Elofsson and A. J. Roe, J. Biol. Chem., 2011, 286, 29922–29931 Search PubMed.
- S. Gong, L. Lei, X. Chang, R. Belland and G. Zhong, Microbiology, 2011, 157, 1134–1144 Search PubMed.
- S. Muschiol, L. Bailey, A. Gylfe, C. Sundin, K. Hultenby, S. Bergstrom, M. Elofsson, H. Wolf-Watz, S. Normark and B. Henriques-Normark, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14566–14571 CrossRef CAS.
- K. Wolf, H. J. Betts, B. Chellas-Gery, S. Hower, C. N. Linton and K. A. Fields, Mol. Microbiol., 2006, 61, 1543–1555 CrossRef CAS.
- L. Bailey, A. Gylfe, C. Sundin, S. Muschiol, M. Elofsson, P. Nordstrom, B. Henriques-Normark, R. Lugert, A. Waldenstrom, H. Wolf-Watz and S. Bergstrom, FEBS Lett., 2007, 581, 587–595 CrossRef CAS.
- A. Slepenkin, P. A. Enquist, U. Hagglund, L. M. de la Maza, M. Elofsson and E. M. Peterson, Infect. Immun., 2007, 75, 3478–3489 CrossRef CAS.
- S. Muschiol, S. Normark, B. Henriques-Normark and A. Subtil, BMC Microbiol., 2009, 9, 75 Search PubMed.
- D. L. Hudson, A. N. Layton, T. R. Field, A. J. Bowen, H. Wolf-Watz, M. Elofsson, M. P. Stevens and E. E. Galyov, Antimicrob. Agents Chemother., 2007, 51, 2631–2635 CrossRef CAS.
- A. Negrea, E. Bjur, S. E. Ygberg, M. Elofsson, H. Wolf-Watz and M. Rhen, Antimicrob. Agents Chemother., 2007, 51, 2867–2876 CrossRef CAS.
- A. N. Layton, D. L. Hudson, A. Thompson, J. C. Hinton, J. M. Stevens, E. E. Galyov and M. P. Stevens, FEMS Microbiol. Lett., 2010, 302, 114–122 Search PubMed.
- J. S. Yount, L. K. Tsou, P. D. Dossa, A. L. Kullas, A. W. van der Velden and H. C. Hang, J. Am. Chem. Soc., 2010, 132, 8244–8245 Search PubMed.
- A. K. Veenendaal, C. Sundin and A. J. Blocker, J. Bacteriol., 2009, 191, 563–570 Search PubMed.
- M. K. Dahlgren, C. E. Zetterstrom, S. Gylfe, A. Linusson and M. Elofsson, Bioorg. Med. Chem., 2010, 18, 2686–2703 CrossRef CAS.
- H. B. Felise, H. V. Nguyen, R. A. Pfuetzner, K. C. Barry, S. R. Jackson, M. P. Blanc, P. A. Bronstein, T. Kline and S. I. Miller, Cell Host Microbe, 2008, 4, 325–336 Search PubMed.
- T. Kline, H. B. Felise, K. C. Barry, S. R. Jackson, H. V. Nguyen and S. I. Miller, J. Med. Chem., 2008, 51, 7065–7074 Search PubMed.
- T. Kline, K. C. Barry, S. R. Jackson, H. B. Felise, H. V. Nguyen and S. I. Miller, Bioorg. Med. Chem. Lett., 2009, 19, 1340–1343 Search PubMed.
- X. Li, US Pat. 0272784 A1, 2005.
- D. Aiello, J. D. Williams, H. Majgier-Baranowska, I. Patel, N. P. Peet, J. Huang, S. Lory, T. L. Bowlin and D. T. Moir, Antimicrob. Agents Chemother., 2010, 54, 1988–1999 Search PubMed.
- D. E. Harmon, A. J. Davis, C. Castillo and J. Mecsas, Antimicrob. Agents Chemother., 2010, 54, 3241–3254 Search PubMed.
- N. J. Pan, M. J. Brady, J. M. Leong and J. D. Goguen, Antimicrob. Agents Chemother., 2009, 53, 385–392 CrossRef CAS.
- X. Li, F.-A. Kang and M. J. Macielag, PCT/US2005/015808 WO2005/111017, 2005.
- V. T. Lee, S. Pukatzki, H. Sato, E. Kikawada, A. A. Kazimirova, J. Huang, X. Li, J. P. Arm, D. W. Frank and S. Lory, Infect. Immun., 2007, 75, 1089–1098 Search PubMed.
- O. K. Kim, L. K. Garrity-Ryan, V. J. Bartlett, M. C. Grier, A. K. Verma, G. Medjanis, J. E. Donatelli, A. B. Macone, S. K. Tanaka, S. B. Levy and M. N. Alekshun, J. Med. Chem., 2009, 52, 5626–5634 Search PubMed.
- W. Swietnicki, D. Carmany, M. Retford, M. Guelta, R. Dorsey, J. Bozue, M. S. Lee and M. A. Olson, PLoS One, 2011, 6, e19716 Search PubMed.
- R. G. Linington, M. Robertson, A. Gauthier, B. B. Finlay, R. van Soest and R. J. Andersen, Org. Lett., 2002, 4, 4089–4092 CrossRef CAS.
- R. G. Linington, M. Robertson, A. Gauthier, B. B. Finlay, J. B. MacMillan, T. F. Molinski, R. van Soest and R. J. Andersen, J. Nat. Prod., 2006, 69, 173–177 CrossRef CAS.
- M. Iwatsuki, R. Uchida, H. Yoshijima, H. Ui, K. Shiomi, A. Matsumoto, Y. Takahashi, A. Abe, H. Tomoda and S. Omura, J. Antibiot., 2008, 61, 222–229 CrossRef CAS.
- M. Iwatsuki, R. Uchida, H. Yoshijima, H. Ui, K. Shiomi, Y. P. Kim, T. Hirose, T. Sunazuka, A. Abe, H. Tomoda and S. Omura, J. Antibiot., 2008, 61, 230–236 CrossRef CAS.
- T. Hirose, T. Sunazuka, S. Tsuchiya, T. Tanaka, Y. Kojima, R. Mori, M. Iwatsuki and S. Omura, Chemistry, 2008, 14, 8220–8238 Search PubMed.
- K. Kimura, M. Iwatsuki, T. Nagai, A. Matsumoto, Y. Takahashi, K. Shiomi, S. Omura and A. Abe, J. Antibiot., 2010, 64, 197–203 Search PubMed.
- D. Hedley, L. Ogilvie and C. Springer, Nat. Rev. Cancer, 2007, 7, 870–879 Search PubMed.
- S. J. Lord, N. R. Conley, H. L. Lee, R. Samuel, N. Liu, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2008, 130, 9204–9205 CrossRef CAS.
- S. J. Lord, N. R. Conley, H. L. Lee, S. Y. Nishimura, A. K. Pomerantz, K. A. Willets, Z. Lu, H. Wang, N. Liu, R. Samuel, R. Weber, A. Semyonov, M. He, R. J. Twieg and W. E. Moerner, ChemPhysChem, 2009, 10, 55–65 CrossRef CAS.
- L. K. Tsou, M. M. Zhang and H. C. Hang, Org. Biomol. Chem., 2009, 7, 5055–5058 RSC.
- M. C. Schlumberger, A. J. Muller, K. Ehrbar, B. Winnen, I. Duss, B. Stecher and W. D. Hardt, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12548–12553 Search PubMed.
- S. B. Van Engelenburg and A. E. Palmer, Nat. Methods, 2010, 7, 325–330 Search PubMed.
- N. Simpson, L. Audry and J. Enninga, Methods Mol. Biol., 2010, 619, 241–256 Search PubMed.
- S. B. Van Engelenburg and A. E. Palmer, Chem. Biol., 2008, 15, 619–628 Search PubMed.
- J. Enninga, J. Mounier, P. Sansonetti and G. Tran Van Nhieu, Nat. Methods, 2005, 2, 959–965 CrossRef CAS.
- T. I. Moy, A. R. Ball, Z. Anklesaria, G. Casadei, K. Lewis and F. M. Ausubel, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10414–10419 Search PubMed.
- T. I. Moy, A. L. Conery, J. Larkins-Ford, G. Wu, R. Mazitschek, G. Casadei, K. Lewis, A. E. Carpenter and F. M. Ausubel, ACS Chem. Biol., 2009, 4, 527–533 CrossRef CAS.
- J. L. Tenor, B. A. McCormick, F. M. Ausubel and A. Aballay, Curr. Biol., 2004, 14, 1018–1024 CrossRef CAS.
- K. N. Adams, K. Takaki, L. E. Connolly, H. Wiedenhoft, K. Winglee, O. Humbert, P. H. Edelstein, C. L. Cosma and L. Ramakrishnan, Cell, 2011, 145, 39–53 Search PubMed.
- R. Pukkila-Worley, R. Feinbaum, N. V. Kirienko, J. Larkins-Ford, A. L. Conery and F. M. Ausubel, PLoS Genet., 2012, 8, e1002733 Search PubMed.
Footnote |
| † Current address: Institute of Biotechnology and Pharmaceutical Research, National Health Research Institutes, 35 Keyan Road, Zhunan Town, Miaoli County 35053, Taiwan, ROC. |
| This journal is © The Royal Society of Chemistry 2013 |