The status of type I polyketide synthase ketoreductases†
Jianting
Zheng
and
Adrian T.
Keatinge-Clay
*
Department of Chemistry and Biochemistry, The University of Texas at Austin, USA. E-mail: adriankc@mail.utexas.edu
First published on 13th August 2012
Abstract
Complex polyketides are a large group of structurally diverse and pharmaceutically important natural products known for their multiple chiral centers harboring hydroxyl and alkyl substituents. Numerous studies to decipher how stereochemistry is controlled during their biosyntheses have determined that ketoreductases usually set most of their chiral centers. While the reliable engineering of ketoreductases within modular polyketide synthases to set new stereocenters in complex polyketides has yet to be realized, this review summarizes the contributions of biochemical studies, engineering efforts, and structural biology to understanding these stereocenter-molding enzymes.
Introduction
Complex polyketides are a large class of natural products most commonly synthesized by bacteria and fungi. They are distinguished from aromatic polyketides by their more reduced, stereochemically-enriched scaffolds.1 The diverse chemical structures and biological activities of complex polyketides has made them attractive in drug development (Fig. 1A). Azithromycin, a semisynthetic derivative of erythromycin A, is one of the most commonly prescribed antibacterials. Simvastatin, a derivative of lovastatin, is a top-selling anticholesterol agent. Ixabepilone, an anticancer drug derived from epothilone B, represents yet another human medicine developed from a complex polyketide. Thus, determining how such stereochemically complex compounds are biosynthesized is not only of fundamental interest but also of importance in medicinal chemistry.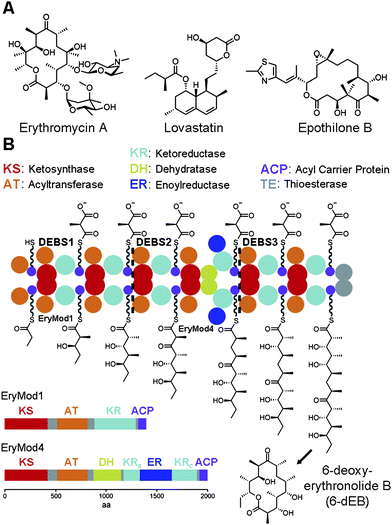 | ||
| Fig. 1 The stereocenters of complex polyketides. (A) The complex polyketides erythromycin A, lovastatin, and epothilone B are the precursors of the antibacterial azithromycin, the anticholesterol agent simvastatin, and the anticancer drug ixabepilone. (B) The three proteins of the 6-dEB synthase (DEBS1-3) form an assembly line composed of many domain types. Each module contains a ketoreductase (KR) that helps set the orientations of the α- and β-substituents after each chain elongation reaction. In the fourth module (EryMod4), ER is inserted between the structural subdomain (KRs) and the catalytic subdomain (KRc) of KR. | ||
The carbon backbones of complex polyketides are assembled by type I polyketide synthases (PKSs) through sequential decarboxylative condensation of acyl-coenzyme A (CoA) building blocks similar to fatty acid biosynthesis (Fig. 1B).1–5 Polyketides like the erythromycin precursor 6-deoxyerythronolide B (6-dEB) are biosynthesized by modular type I PKSs that contain sets of enzymes called modules organized in an assembly line. Each module catalyzes one round of chain elongation and optional processing during the construction of a polyketide. Thus, the smallest module only contains the domains necessary for chain extension: a ketosythase (KS), an acyltransferase (AT), and an acyl carrier protein (ACP). Within the module AT transfers an extender unit, such as the methylmalonyl group of (2S)-methylmalonyl-CoA, to the 18 Å phosphopantetheinyl arm of ACP, which presents this extender unit to KS for condensation with a growing polyketide chain. Modules may also contain a ketoreductase (KR), a dehydratase (DH), and an enoylreductase (ER) that modify the ACP-bound β-ketoacyl intermediate. Combinations of these processing enzymes can thus transform a β-keto group into a β-hydroxyl group, an α,β-double bond, or a β-methylene.
This review focuses on how KRs set stereocenters within complex polyketides through the formation of β-hydroxyacyl intermediates. The chemistry and shape of KR active sites are known to control the production of D- or L-oriented hydroxyl substituents. Now it is also known that KR active sites can control the orientations of alkyl substituents through a combination of epimerase and epimer-specific reductase activities. The recent structural elucidation of each KR-type has provided new insights into the mechanisms employed by these chirality-generating enzymes. Strategies used to engineer KRs that enable PKSs to synthesize new complex polyketides are also discussed.
KR enzymology
When the relationship between the biosynthesis of fatty acids and complex polyketides was revealed it was surprising that the 6-dEB synthase (DEBS) could produce intermediates with L-β-hydroxyl groups since the metazoan fatty acid synthase was known to produce intermediates with D-β-hydroxyl groups.6–8 The KRs from the second, fifth, and sixth modules of DEBS (EryKR2, EryKR5, and EryKR6; PKS fragments are specified by their synthase and module of origin) were suspected of performing the reductions that resulted in the oppositely oriented hydroxyl groups. A domain-swapping experiment later proved that KR domains were responsible for setting the orientation of hydroxyl groups within complex polyketides. EryKR2 of the engineered triketide synthase DEBS1+TE (the first polypeptide of DEBS fused to its thioesterase) was replaced by RapKR2 (and an inactive RapDH2) (Fig. 2).9,10 RapKR2 is expected from the stereochemistry of rapamycin to generate an intermediate with a D-β-hydroxyl group; indeed, the RapKR2-swapped DEBS1+TE produced a D-β-hydroxyl group instead of the L-β-hydroxyl group produced by DEBS1+TE.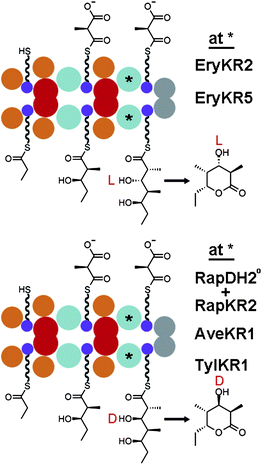 | ||
| Fig. 2 KR-swapping. Experiments with the engineered triketide lactone synthase DEBS1+TE helped determine that KRs control the stereochemistry of hydroxyl substituents and revealed that some KRs can reduce substrates not naturally encountered in a stereocontrolled manner. | ||
The activities of KRs were further explored with α-substituted, β-ketoacyl N-acetylcysteamine (NAC) thioester substrate analogs. Incubation of (±)-2-methyl-3-oxopentanoyl-S-NAC, which spontaneously epimerizes in water, with DEBS1+TE and NADPH yielded L-α-methyl-D-β-hydroxyl and D-α-methyl-L-β-hydroxyl products in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.11 When EryKR2 of DEBS1+TE was inactivated, only the L-α-methyl-D-β-hydroxyl isomer was produced. These results suggested that EryKR1 is specific for the L-α-methyl isomer of the racemic substrate mixture. Thus, through substrate selection and stereoselective hydride addition, EryKR1 controls the orientations of both the α-methyl and β-hydroxy substituents as anticipated from the stereochemistry of 6-dEB.
:1 ratio.11 When EryKR2 of DEBS1+TE was inactivated, only the L-α-methyl-D-β-hydroxyl isomer was produced. These results suggested that EryKR1 is specific for the L-α-methyl isomer of the racemic substrate mixture. Thus, through substrate selection and stereoselective hydride addition, EryKR1 controls the orientations of both the α-methyl and β-hydroxy substituents as anticipated from the stereochemistry of 6-dEB.
For many years it was challenging to assay individual KR domains for stereocontrol as their boundaries had not been established. A combination of limited proteolysis and successful KR domain-swaps eventually revealed their locations and enabled the stereocontrol of isolated KR domains towards α-substituted, β-ketoacyl NAC thioester substrates to be studied (Fig. 3A).12–16 KRs that reduce shorter polyketide intermediates within their native PKSs (e.g. EryKR1, TylKR1, AmpKR2, and AmpKR1) were found to be more active and stereocontrolled towards diketide-S-NAC substrates compared to KRs that naturally reduce longer polyketide intermediates. While isolated EryKR2, EryKR5, and EryKR6 showed poor stereocontrol towards (±)-2-methyl-3-oxopentanoyl-S-NAC, they showed excellent stereocontrol towards triketides tethered to ACP.15,17 This observation suggests that some KRs process intermediates in a more stereocontrolled manner when the intermediates are tethered to the ACP phosphopantetheinyl arm compared to the shorter NAC phosphopantetheine mimic.
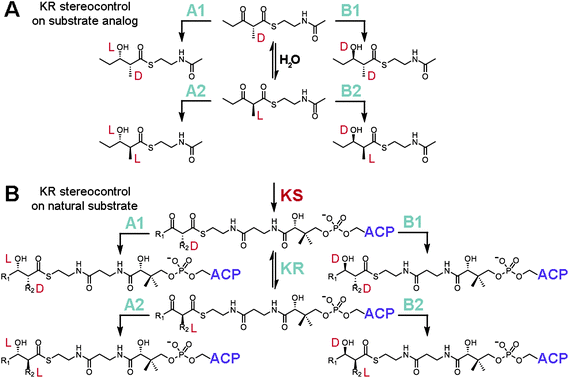 | ||
| Fig. 3 KR stereocontrol. (A) Reduction assays with the substrate analog (±)-2-methyl-3-oxopentanoyl-S-NAC have been employed to study how stereocontrol is mediated by KRs. Many isolated A1-, A2-, B1-, and B2-type KRs retain a high degree of stereocontrol reducing this small molecule. Epimerization is catalyzed by water. (B) Within an intact PKS, an α-substituted intermediate is presented to a KR as D-α-alkyl-β-ketoacyl-S-ACP. A1- and B1-type KRs will reduce this substrate, while A2- and B2-type KRs must await a KR-catalyzed epimerization reaction (this activity is necessary as the D-α-substituent is otherwise configurationally stable) before reduction can proceed. | ||
How epimerization is mediated during the synthesis of complex polyketides has frequently been the subject of debate. Many KRs selectively reduce intermediates containing α-substituents of L-orientation, yet α-substituents have been shown through sodium borohydride-trapping experiments to exit KS in a D-orientation.17 There were hints from isotope incorporation studies of erythromycin biosynthesis by Saccharopolyspora erythraea that differences existed between the generation of D-methyl and L-methyl groups.18 The hydrogens geminal to D-methyl groups were derived from propionate precursors, while the hydrogens geminal to L-methyl groups were derived from water. Subsequent experiments confirmed this through the incubation of DEBS1+TE with deuterium-labeled (2S)-methylmalonyl-CoA.19 That the α-deuterium of the methylmalonyl extender unit is retained by the second module but not the first indicated that an epimerization step occurs in the first module but not in the second module to produce an L-oriented methyl group. The epimerase activity was ultimately found to be associated with KR through the process of elimination.17,20 Reconstitution experiments did not link KSs to the epimerizing activity, and water-catalyzed epimerization was determined to be too slow. Perhaps it is not surprising that KRs possess epimerase activity since EryKR3 is likely to help install the epimerized L-methyl group at C-8 of 6-dEB.21 That epimerization is catalyzed by KRs also agrees with two additional observations: (1) modules lacking processing enzymes have not been observed to catalyze epimerization,22 and (2) the D-α-substituent of α-substituted β-ketoacyl-S-ACPs is resistant, or configurationally stable, to water-catalyzed epimerization.17
From the above biochemical studies a picture has emerged for how KR enforces stereocontrolled reduction within a module (Fig. 3B). According to the aforementioned isotope-labelling experiments, KRs can catalyze reduction faster than epimerization. Thus, if a KR is specific for an α-substituent in the D-orientation, then reduction ensues when the D-α-methyl-β-ketoacyl-S-ACP product from KS binds to the KR active site. If a KR is specific for an α-substituent in the L-orientation, then reduction occurs only after the epimerization reaction is catalyzed by KR. From the variety of substrates assayed with many isolated KRs, substrate binding energy seems usually to be obtained through interactions with the polyketide and the portion of the phosphopantetheinyl arm closest to the thioester linkage, although some KRs must also obtain binding energy from other portions of the natural substrate, such as more distal regions of the phosphopantetheinyl arm and the surface of ACP.
KRs are classified into different types, often distinguishable through fingerprints in their amino acid sequences.23 A-type KRs generate L-β-hydroxyl groups, B-type KRs generate D-β-hydroxyl groups, and C-type KRs are reductase-incompetent. KRs that reduce α-unsubstituted intermediates are termed A0-type or B0-type; however, KRs that reduce α-substituted intermediates are subdivided such that those producing a D-α-substituent are denoted with a “1” while those producing an L-α-substituent are denoted with a “2”. Thus A1-, A2-, B1-, and B2-type KRs generate D-α-alkyl-L-β-hydroxyl, L-α-alkyl-L-β-hydroxyl, D-α-alkyl-D-β-hydroxyl, and L-α-alkyl-D-β-hydroxyl products, respectively. Reductase-incompetent KRs are classified as C1-type KRs, which are catalytically nonfunctional, and C2-type KRs, which function as epimerases to help convert an α-substituent from a D- to an L-orientation. Residues unique to each type of KR were identified through multiple sequence alignments.23–25 A leucine-aspartate-aspartate motif is consistently present in the B-type KRs, whereas a conserved tryptophan is present in A-type KRs. A2-type KRs can often be distinguished from A1-type KRs through the presence of a histidine three residues N-terminal to the catalytic tyrosine, and B2-type KRs can often be distinguished from B1-type KRs through the presence of a proline two residues C-terminal to the catalytic tyrosine. C2-type KRs possess the catalytic tyrosine but lack the NADPH-binding motif (TGGTGxLG), while C1-type KRs lack both elements.21 These fingerprints have proven valuable in predicting the alkyl and hydroxyl orientations of complex polyketides prior to experimental assignment.26–28
KR structures
Six crystal structures of KRs from modular PKSs have now been reported – the A1-type AmpKR2, the A2-type AmpKR11, the B0-type SpnKR2, the B1-type TylKR1, the B2-type EryKR1, and the C2-type PikKR3 (PDB codes: 3MJS, 4DI7, 3SLK, 2Z5L, 2FR0, 3QP9).21–23,29–31 Each of these KRs are monomeric and comprised of both an N-terminal structural subdomain (KRs) and a slightly larger, ∼250-residue C-terminal catalytic subdomain (KRc) (Fig. 4A). The β-sheets of the Rossmann-like fold of each of the subdomains are bridged by the first and last β-strands of KRs to create an essentially continuous β-sheet through the entire KR domain. In the absence of an ER domain a ∼20-residue loop joins the C-terminal end of KRs to KRc. When an ER domain is present, it is inserted into this loop. Without any obvious catalytic residues of its own, KRs has no known role other than stabilizing KRc. The position of the first β-strand of KRs, β1, and thus the N-terminal boundary of the KR are indicated by the sequence motif R(H/L/M/F/Y)xxxW in modules that do not contain other processing domains and L(L/F/Y)x(L/V)xW in modules that contain other processing domains.22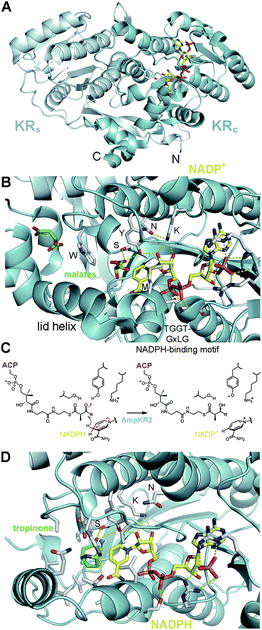 | ||
| Fig. 4 Reductase architecture. (A) In each KR a structural subdomain (KRs) stabilizes a catalytic subdomain (KRc). (B) NADP+ and two malate molecules are bound in the active site of the A1-type AmpKR2. The active site residues tyrosine, serine, lysine, and asparagine are indicated in one-letter code. The characteristic A-type tryptophan may help guide a phosphopantetheinyl-bound polyketide into the active site groove. An essential methionine folds over the nicotinamide coenzyme. (C) AmpKR2 binds its D-α-methyl-β-ketoacyl-S-ACP substrate and catalyzes the stereoselective transfer of the NADPH pro-4S hydride to the substrate β-keto group. (D) KRs may bind phosphopantetheinyl-bound polyketides similar to how tropinone reductase II stereospecifically binds tropinone (PDB code: 1IPF). | ||
The KR crystal structures of have enabled a physical connection between enzymology of KRs and the residues within their active sites (Fig. 4B). They are classified in the ‘complex’ subfamily within the short-chain dehydrogenase/reductase (SDR) superfamily and likely operate similar to related SDR enzymes.32 By analogy, the KR tyrosine and serine are thought to bind the β-keto group of the polyketide substrate and the tyrosine is anticipated to donate its proton to the carbonyl oxygen as the NADPH pro-4S hydride is transferred to the β-carbon (Fig. 4C). The lysine is expected to aid proton transfer by decreasing the pKa of the tyrosine side chain, and a conserved asparagine is anticipated to have an organizing role. Curiously, relative to other SDR enzymes, the positions of lysine and asparagine are swapped in type I PKS KRs (Fig. 4D). The crystal structures of each of the reductase-competent KRs reveal that the tyrosine, serine, lysine, and asparagine are essentially in the same orientations. The tyrosine, serine, and lysine of the C2-type PikKR3 are in equivalent orientations to those of the reductase-competent KRs; however, the asparagine is substituted by a smaller threonine residue.
In addition to the orientations of the catalytic residues changing little between KR-types, the nicotinamide coenzyme binds to both A- and B-type KRs in the same location and conformation (Fig. 4B).22,29–31 NADPH occupies a characteristic groove in the Rossmann fold of KRc and presents its pro-4S hydride to the active site, consistent with the observation that both A- and B-type KRs transfer the NADPH pro-4S hydride to substrates.33,34 Several contacts are formed with the nicotinamide coenzyme including hydrogen bonds from the NADPH-binding motif (TGGTGxLG) to the pyrophosphate moiety and from the tyrosine and lysine to the nicotinamide ribose. Upon the nicotinamide coenzyme binding to AmpKR2, a loop becomes ordered and the side chain of a semi-conserved methionine folds over the coenzyme to make several contacts with it. Intriguingly, the mutation of this methionine into alanine renders the mutant AmpKR2 nearly inactive.29
An under-appreciated feature of KRs is the lid helix, αFG. In most crystal structures, this helix is invisible or weakly visible in the electron density maps due to high B-factors. However, the lid helix of TylKR1 was observed in a closed conformation with several helix residues making interactions with the substrate-binding groove (Fig. 5A).23 If KRs operate similar to tropinone reductases, the lid helix may clamp over a bound substrate.35,36 The conservation of a mysterious surface-exposed arginine at the C-terminal end of the helix suggests a role in binding ACP.
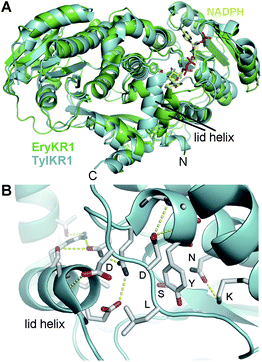 | ||
| Fig. 5 The lid helix. (A) TylKR1 crystallized with its lid helix in a closed conformation. NADPH is from the EryKR1 structure. (B) The TylKR1 lid helix makes several interactions with the well-folded portion of KR. The aspartates of the B-type Leu-Asp-Asp motif cap both the catalytic tyrosine-containing helix and the lid helix. The leucine may be positioned to help guide a phosphopantetheinyl-bound polyketide in the active site groove. | ||
The crystal structures reveal that the leucine-aspartate-aspartate motif of B-type KRs is on a loop adjacent to the active site. As with the lid helix, this loop is not usually visible due to high temperature factors; however, the closed TylKR1 structure shows the first aspartate capping helix αF and the second aspartate capping the lid helix (Fig. 5B). Interestingly, the leucine does not form any contacts. In the AmpKR2 and AmpKR11 crystal structures, the characteristic A-type tryptophan is positioned on the opposite side of the substrate-binding groove from where the leucine-aspartate-aspartate motif is positioned in B-type KRs. For A- and B-type KRs to generate different hydroxyl group orientations, polyketide intermediates must be bound differently in the substrate-binding groove.15,24,25 The leucine of the leucine-aspartate-aspartate motif of a B-type KR may contact the terminal portion of the phosphopantetheinyl arm to position the si face of a β-keto group for attack by the NADPH pro-4S hydride. Likewise, the tryptophan of an A-type KR may contact the terminal portion of the phosphopantetheinyl arm to position the re face of a β-keto group for attack by the NADPH pro-4S hydride. Other fingerprint residues such as the residue three before the catalytic tyrosine (often histidine in A2-type KRs, leucine in B2-type KRs, and glutamine in A1- and B1-type KRs) and two after the catalytic tyrosine (often a proline in B2-type KRs) are also observable in the crystal structures; how they aid in regulating stereocontrol is not yet known.
KRs may enforce stereocontrol similar to the related tropinone reductases.35,36 Tropinone reductase I and tropinone reductase II possess the same fold and bind the same tropinone substrate but in orientations that present different faces of the tropinone keto group to the NADPH pro-4S hydride. The opposite tropinone-binding modes of these enzymes thus yields products with hydroxyl groups of opposite stereochemical orientation. The ternary complex of tropinone reductase II with NADPH and tropinone (PDB code: 1IPF) shows the equivalent of a lid helix clamping down such that all faces of the tropinone substrate are contacted by the enzyme and nicotinamide coenzyme (Fig. 4D). The lid helix may likewise surround the phosphopantetheinyl-bound polyketide in the Michaelis complex of KRs. As in the ternary complex of tropinone reductase II and the closed form of TylKR1, many helix residues may not directly contact the substrate but instead contact the body of the enzyme to facilitate the reaction between the reactive hydride and the polyketide β-keto group. Induced fit may indeed be necessary for KR-mediated catalysis. If true, a ternary complex of a KR with a nicotinamide coenzyme and a phosphopantetheinyl-bound polyketide would elucidate which residues contact polyketide intermediates and how they exert stereocontrol. The epimerase activity of KRs is mysterious; however, if C2-type KRs catalyze epimerization the mechanism likely involves the tyrosine, serine, and lysine conserved in those KRs. These enzymes may only need to align the thioester and β-keto groups to decrease the pKa of the α-proton so that it can be abstracted by a water molecule. The enolized polyketide may then tautomerize back to the original substrate or its epimer.21
Interactions between a polyketide intermediate and a KR determine the stereochemical fate of the β-hydroxy product. While most isolated KRs reduce β-ketoacyl-S-NAC substrate analogs as expected, some exceptions have been observed.16 For example, TylKR1 reduces (±)-2-methyl-3-oxopentanoyl-S-NAC as anticipated for a B1-type KR, but when the α-unsubstituted substrate 3-oxopentanoyl-S-NAC is encountered it primarily behaves like an A-type KR. In some iterative type I PKSs the KR operates as an A-type KR towards some intermediates and as a B-type KR towards other intermediates.37 Such observations imply that the lid helix must be able to close over a complementary substrate in order for the reduction reaction to proceed.
KR engineering
One strategy to generate derivatives of complex polyketides is to replace domains within the modular PKS assembly line. KR-swapping experiments in which the stereochemistry of a resulting polyketide product was thoroughly analyzed include replacements of the A1-type EryKR2 of DEBS1+TE, such as by the A1-type EryKR5, the B0-type RapKR2, the B1-type AveKR1, and the B1-type TylKR1 (Fig. 2).10,38 These results demonstrate that the same polyketide-bound ACP can be reduced by different KRs and suggest that the interface between KRs and ACPs is minimal. Attempts to swap the A2-type RifKR7 or the B2-type EryKR1 for the A1-type EryKR2 of DEBS1+TE were unsuccessful. However, even the swap with A1-type EryKR6 was unsuccessful.38 Apparently some β-ketoacyl intermediates and KRs are incompatible.Another strategy to generate derivatives of complex polyketides is to knock out KRs within a modular PKS. By mutating the catalytic tyrosine to a phenylalanine in EryKR6 within DEBS, the 3-hydroxy group of 6-dEB was substituted by a 3-keto group.25 Several derivatives of amphotericin were generated through the same strategy;39 however, inactivation of AmpKR10 resulted in the production of a pyrone shunt product, presumably due to incompatibility of the unnatural intermediate with a downstream enzyme.40
Through the mutagenesis of key residues, isolated KRs have been converted from one KR-type to another, as assayed through the reduction of β-ketoacyl-S-NAC substrate analogs. By removing the B-type leucine-aspartate-aspartate motif and introducing the A-type tryptophan, the B2-type EryKR1 was converted into an A2-type KR.41 The A1-type AmpKR2 converts (±)-2-methyl-3-oxopentanoyl-S-NAC primarily to the expected A1-type product, L-3-hydroxyl-D-2-methylpentanoyl-S-NAC (92% of products).29 Introduction of the A2 histidine resulted in AmpKR2(Q364H) principally generating the A2-type product, L-3-hydroxyl-L-2-methylpentanoyl-S-NAC (55% of products). The additional introduction of an A2-type residue empowered AmpKR2(Q364H,G335T) to almost exclusively produce the A2-type product (94% of products).30
Reintroducing KRs optimized as isolated domains through in vitro assays on β-ketoacyl-S-NAC substrate analogs back into intact PKSs has not met with success thus far.30,42 When mutants of the B2-type EryKR1 that behaved like A2-type KRs were reintroduced into DEBS1+TE only the triketide lactone normally produced by DEBS1+TE was detected. When mutations that enabled the isolated A1-type EryKR2 to generate mixtures of reduced products were introduced into DEBS1+TE only the unreduced, 3-keto triketide lactone was detected. These results do not seem to auger well for the site-directed mutagenesis of KRs within intact synthases; however, an observation from the mycolactone PKS provides inspiration: the B1-type KR of the fourth module of MlsA1 differs from the B2-type KR of the second module of MlsA1 by only three residues, only one of which is in the active site.43 A glutamine is present three residues N-terminal to the catalytic tyrosine in the B1-type KR, while the same position is occupied by a leucine in the B2-type KR. Mutagenesis of a single residue within a PKS may thus be sufficient to switch the substituent orientations set by certain KRs.
Summary
Our understanding of the stereochemistry of polyketide biosynthesis has significantly advanced over the last two decades. The functional dissection of modular PKSs has established that KRs most commonly set the orientations of the hydroxyl and alkyl substituents of complex polyketides. Crystal structures of several reductase-competent KRs have revealed that the catalytic residues and bound nicotinamide coenzyme are essentially in the same locations. Thus, the orientations of polyketide substituents primarily depend on how KRs bind β-ketoacyl intermediates for reduction reactions during the growth of the polyketide chain. An A-type KR positions the re face of a β-keto group to be attacked by the NADPH pro-4S hydride to generate an L-hydroxyl group, while a B-type KR positions the si face of the β-keto group to be attacked by the NADPH pro-4S hydride to generate a D-hydroxyl group. Stereocenter formation is more complicated when a β-ketoacyl intermediate also possesses an α-substituent. α-Substituents are positioned in the D-orientation by KS through the condensation reaction. If a KR is specific for this D-orientation, reduction quickly follows to fix the orientation of the D-alkyl substituent; however, if a KR is specific for the L-orientation then a KR-catalyzed epimerization occurs before the reduction that fixes the orientation of the L-alkyl substituent.KRs likely catalyze reduction similar to related SDR enzymes. The ternary complex of tropinone reductase II reveals the equivalent of a lid helix clamping over tropinone and NADPH through induced fit. The AmpKR2 crystal structures shows that a KR loop folds over a bound nicotinamide coenzyme, and the TylKR1 crystal structure suggests that the lid helix also clamps down when a polyketide substrate enters the substrate-binding groove. KRs primarily bind substrates through interactions close to the thioester bond; however, some KRs rely on interactions with more distal portions of the phosphopantetheinyl arm and potentially the surface of ACP.
Progress in engineering KRs to produce new complex polyketides has been made, yet to achieve the reliable biosynthesis of desired complex polyketides more mechanistic details need to be revealed. Structural biology will undoubtedly play a major role in elucidating the precise contacts between polyketide intermediates and KR active sites and determining which interactions govern stereocontrol. Ultimately, however, a combination of enzymology, biosynthetic engineering, and structural biology will be necessary to enable the rational design of complex polyketides of medicinal importance.
Acknowledgements
A.T.K is funded by the Robert A. Welch Foundation (F-1712).References
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS.
- C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane, Structure and mechanism of the 6-deoxyerythronolide B synthase, Annu. Rev. Biochem., 2007, 76, 195–221 CrossRef CAS.
- S. Smith and S. C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 RSC.
- D. H. Kwan and F. Schulz, Molecules, 2011, 16, 6092–6115 CrossRef CAS.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012 10.1039/c2np20019h.
- J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt and P. F. Leadlay, Nature, 1990, 348, 176–178 CrossRef CAS.
- S. Donadio, M. J. Staver, J. B. McAlpine, S. J. Swanson and L. Katz, Science, 1991, 252, 675–679 CAS.
- V. E. Anderson and G. G. Hammes, Biochemistry, 1984, 23, 2088–2094 CrossRef CAS.
- J. Cortes, K. E. Wiesmann, G. A. Roberts, M. J. Brown, J. Staunton and P. F. Leadlay, Science, 1995, 268, 1487–1489 CAS.
- C. M. Kao, M. McPherson, R. N. McDaniel, H. Fu, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 1998, 120, 2478–2479 CrossRef CAS.
- I. E. Holzbaur, R. C. Harris, M. Bycroft, J. Cortes, C. Bisang, J. Staunton, B. A. Rudd and P. F. Leadlay, Chem. Biol., 1999, 6, 189–195 CrossRef CAS.
- J. F. Aparicio, P. Caffrey, A. F. Marsden, J. Staunton and P. F. Leadlay, J. Biol. Chem., 1994, 269, 8524–8528 CAS.
- J. Staunton, P. Caffrey, J. F. Aparicio, G. A. Roberts, S. S. Bethell and P. F. Leadlay, Nat. Struct. Biol., 1996, 3, 188–192 CrossRef CAS.
- D. Bedford, J. R. Jacobsen, G. Luo, D. E. Cane and C. Khosla, Chem. Biol., 1996, 3, 827–831 CrossRef CAS.
- A. P. Siskos, A. Baerga-Ortiz, S. Bali, V. Stein, H. Mamdani, D. Spiteller, B. Popovic, J. B. Spencer, J. Staunton, K. J. Weissman and P. F. Leadlay, Chem. Biol., 2005, 12, 1145–1153 CrossRef CAS.
- S. K. Piasecki, C. A. Taylor, J. F. Detelich, J. Liu, J. Zheng, A. Komsoukaniants, D. R. Siegel and A. T. Keatinge-Clay, Chem. Biol., 2011, 18, 1331–1340 CrossRef CAS.
- R. Castonguay, W. He, A. Y. Chen, C. Khosla and D. E. Cane, J. Am. Chem. Soc., 2007, 129, 13758–13769 CrossRef CAS.
- D. E. Cane, T. C. Liang, P. B. Taylor, C. Chang and C. C. Yang, J. Am. Chem. Soc., 1986, 108, 4957–4964 CrossRef CAS.
- K. J. Weissman, M. Timoney, M. Bycroft, P. Grice, U. Hanefeld, J. Staunton and P. F. Leadlay, Biochemistry, 1997, 36, 13849–13855 CrossRef CAS.
- C. R. Valenzano, R. J. Lawson, A. Y. Chen, C. Khosla and D. E. Cane, J. Am. Chem. Soc., 2009, 131, 18501–18511 CrossRef CAS.
- J. Zheng and A. T. Keatinge-Clay, J. Mol. Biol., 2011, 410, 105–117 CrossRef CAS.
- A. T. Keatinge-Clay and R. M. Stroud, Structure, 2006, 14, 737–748 CrossRef CAS.
- A. T. Keatinge-Clay, Chem. Biol., 2007, 14, 898–908 CrossRef CAS.
- P. Caffrey, ChemBioChem, 2003, 4, 654–657 CrossRef CAS.
- R. Reid, M. Piagentini, E. Rodriguez, G. Ashley, N. Viswanathan, J. Carney, D. V. Santi, C. R. Hutchinson and R. McDaniel, Biochemistry, 2003, 42, 72–79 CrossRef CAS.
- D. Janssen, D. Albert, R. Jansen, R. Muller and M. Kalesse, Angew. Chem., Int. Ed., 2007, 46, 4898–4901 CrossRef CAS.
- D. Menche, F. Arikan, O. Perlova, N. Horstmann, W. Ahlbrecht, S. C. Wenzel, R. Jansen, H. Irschik and R. Muller, J. Am. Chem. Soc., 2008, 130, 14234–14243 CrossRef CAS.
- S. Takahashi, A. Toyoda, Y. Sekiyama, H. Takagi, T. Nogawa, M. Uramoto, R. Suzuki, H. Koshino, T. Kumano, S. Panthee, T. Dairi, J. Ishikawa, H. Ikeda, Y. Sakaki and H. Osada, Nat. Chem. Biol., 2011, 7, 461–468 CrossRef CAS.
- J. Zheng, C. A. Taylor, S. K. Piasecki and A. T. Keatinge-Clay, Structure, 2010, 18, 913–922 CrossRef CAS.
- J. Zheng and A. T. Keatinge-Clay, 2012, Manuscript in preparation.
- J. Zheng, D. C. Gay, B. Demeler, M. A. White and A. T. Keatinge-Clay, Nat. Chem. Biol., 2012, 8, 615–621 CrossRef CAS.
- K. L. Kavanagh, H. Jornvall, B. Persson and U. Oppermann, Cell. Mol. Life Sci., 2008, 65, 3895–3906 CrossRef CAS.
- M. McPherson, C. Khosla and D. E. Cane, J. Am. Chem. Soc., 1998, 120, 3267–3268 CrossRef CAS.
- Y. Yin, R. Gokhale, C. Khosla and D. E. Cane, Bioorg. Med. Chem. Lett., 2001, 11, 1477–1479 CrossRef CAS.
- K. Nakajima, A. Yamashita, H. Akama, T. Nakatsu, H. Kato, T. Hashimoto, J. Oda and Y. Yamada, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4876–4881 CrossRef CAS.
- A. Yamashita, M. Endo, T. Higashi, T. Nakatsu, Y. Yamada, J. Oda and H. Kato, Biochemistry, 2003, 42, 5566–5573 CrossRef CAS.
- H. Zhou, Z. Gao, K. Qiao, J. Wang, J. C. Vederas and Y. Tang, Nat. Chem. Biol., 2012, 8, 331–333 CrossRef CAS.
- L. Kellenberger, I. S. Galloway, G. Sauter, G. Bohm, U. Hanefeld, J. Cortes, J. Staunton and P. F. Leadlay, ChemBioChem, 2008, 9, 2740–2749 CrossRef CAS.
- P. Power, T. Dunne, B. Murphy, L. Nic Lochlainn, D. Rai, C. Borissow, B. Rawlings and P. Caffrey, Chem. Biol., 2008, 15, 78–86 CrossRef CAS.
- N. Khan, B. Rawlings and P. Caffrey, Biotechnol. Lett., 2011, 33, 1121–1126 CrossRef CAS.
- A. Baerga-Ortiz, B. Popovic, A. P. Siskos, H. M. O'Hare, D. Spiteller, M. G. Williams, N. Campillo, J. B. Spencer and P. F. Leadlay, Chem. Biol., 2006, 13, 277–285 CrossRef CAS.
- D. H. Kwan, M. Tosin, N. Schlager, F. Schulz and P. F. Leadlay, Org. Biomol. Chem., 2011, 9, 2053–2056 CAS.
- T. P. Stinear, A. Mve-Obiang, P. L. Small, W. Frigui, M. J. Pryor, R. Brosch, G. A. Jenkin, P. D. Johnson, J. K. Davies, R. E. Lee, S. Adusumilli, T. Garnier, S. F. Haydock, P. F. Leadlay and S. T. Cole, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1345–1349 CrossRef CAS.
Footnote |
| † This article is part of a MedChemComm ‘New Talents’ issue highlighting the work of outstanding rising scientists in medicinal chemistry research. |
| This journal is © The Royal Society of Chemistry 2013 |