Approaches to discover non-ATP site kinase inhibitors†
Lori Krim
Gavrin
* and
Eddine
Saiah
Pfizer Research, Rare Disease Chemistry and Chemical Biology, BioTherapeutics Chemistry, 200 Cambridgepark Drive, Cambridge, MA 02140, USA. E-mail: lori.gavrin@pfizer.com; Tel: +1 617-665-5616
First published on 28th August 2012
Abstract
The catalytic domain of kinases shows a high degree of sequence homology, especially for kinases that belong to the same family. They share a common ATP binding site with a conserved activation loop and similar three-dimensional structure. Consequently, a major challenge in kinase research exists in achieving selectivity among the >500 family members, since they all process the same substrate. In addition to requiring selectivity against other kinases, ATP site inhibitors must also bind tightly to overcome the high physiological concentration of ATP in the cell. Furthermore, the development of novel ATP site inhibitors is becoming increasingly challenging, as many ATP competitive scaffolds have previously been disclosed. In order to develop compounds with better selectivity among kinases, inhibitors that bind outside the ATP site show great promise and are currently being explored by many groups. This review will highlight the most commonly used methods to discover small molecule Type III and IV kinase inhibitors.
 Lori Krim Gavrin | Lori earned her Ph.D. from the University of Pennsylvania in 2001 in Professor Jeffrey Winkler's laboratory. She started her professional career as a medicinal chemist at Wyeth Research (now Pfizer). Over the past 10+ years, her work has spanned various stages of research from early exploratory to late discovery. Lori has led several teams from HTS to lead discovery. She also advanced several late stage programs and as a member of the norepinephrine reuptake inhibitor (NRI) program at Wyeth, Lori designed and synthesized a compound that entered Phase I clinical trials for the treatment of vasomotor symptoms (VMS). She has worked in multiple therapeutic areas, including inflammation, cardiovascular, metabolic diseases, and women's health. At present, Lori is leading the chemistry design efforts as a Principal Research Scientist within Pfizer's Orphan and Genetic Disease Research Unit. |
 Eddine Saiah | Eddine received a Ph.D. from Pierre & Marie Curie University in Paris in 1992 followed by a postdoc at the Mayo Clinic Florida. He has worked at several companies, including DuPont Pharmaceuticals and Wyeth. He joined Pfizer in 2009 through the Wyeth acquisition where he was leading the Med Chem group in Cambridge, MA. He has worked on a range of programs including oncology, cardiovascular, metabolic diseases and inflammation with multiple compounds advancing to the clinic. He has championed the use of fragments on multiple programs. He is currently working in the Orphan and Genetic Disease Research Unit with a focus on understanding and tackling protein misfolding with small molecules. He is co-chair of the Pfizer Medicinal Chemistry Design Network Group. |
1. Introduction
Protein phosphorylation is pivotal in a diverse range of cellular processes including cell division, proliferation, differentiation and apoptosis. Protein phosphorylation is regulated by kinases, as they catalyze the transfer of a phosphoryl group of ATP to the hydroxyl group of a serine, threonine or tyrosine amino acid in the substrate. The protein kinase family encompasses 518 kinases1 and due to their critical role in protein phosphorylation, they are believed to play vital roles in all aspects of cellular physiology. Therefore, the inhibition of kinases has been pursued by the pharmaceutical industry for over 20 years. There are currently 14 FDA approved small molecule kinase drugs on the market, in addition to many more kinase inhibitors currently in the clinic. Furthermore, it is estimated that over a quarter of all pharmaceuticaldrug targets are protein kinases.2 The competitive search for modulators of these essential protein targets underscores the importance of being able to discover novel chemical scaffolds that could become kinase inhibitordrugs.Historically, the main challenge of developing kinase inhibitors has been to obtain sufficient selectivity within the kinase family to achieve an acceptable therapeutic index. The vast majority of the kinases in the human body have highly conserved ATP binding pockets making selectivity with an ATP mimetic an enormous challenge. Most of the reported kinase inhibitors bind in this ATP pocket. However, it has been recognized that pockets outside the ATP binding site are highly divergent among the different kinases, and therefore offer opportunities for building in target selectivity. Fig. 1 portrays the typical structure of a protein kinase. Irrespective of the kinase subfamily, the structural features of the protein are well defined and conserved.1 The kinase diagram in Fig. 1 depicts the structure of a typical kinase domain. Specifically highlighted are the binding pockets of Type III and IV inhibitors, the hinge region, the activation loop and the α-C-helix.
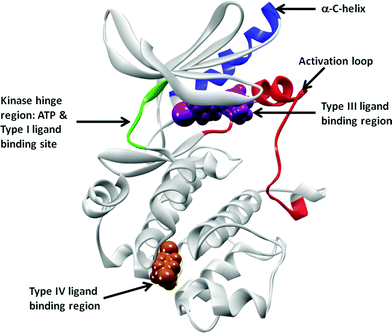 | ||
| Fig. 1 Schematic representation of Type III and Type IV binding pockets. The cMET crystal structure in DFG-in, α-C-helix-out conformation (pdb id: 3EQG) is to used highlight these pockets. The protein kinase is shown in grey, with the hinge region connecting the N- and the C-loops in green. ATP, Type I, II and kinase inhibitors have one to three hydrogen bond interactions with the hinge residues and extend into various pockets nearby (not highlighted here). Type III inhibitor (PD-325089) binding at the allosteric pocket, adjacent to the ATP pocket, is represented by magenta van der Waals surface. The region where the Type IV inhibitor GNF-2 binds, the myristoyl pocket in cAbl, is shown in this cartoon representation using a russet surface. The activation loop between the DFG and PE sequences are colored in red, and the α-C-helix is represented in blue. | ||
The pharmaceutical industry has developed a nomenclature of kinase inhibitor “type” based on mechanism of action. These classifications are briefly explained below.
Type I kinase inhibitors: these compounds interact directly with the ATP binding pocket and bind to the active conformation of the kinase in which the activation loop is phosphorylated and is in the “DFG-in” conformation. Type I inhibitors typically form one, two or three hydrogen bond interactions in a syn-periplanar motif with the hinge region of the kinase, thus mimicking the hydrogen bonds formed by the adenine ring of ATP. The fact that many heterocyclic rings mimic the hydrogen binding motif of adenine could explain why a large number of kinase inhibitors bind in the ATP pocket. Even though the adenine region is occupied by all Type I inhibitors, selectivity can still be obtained. In fact, the FDA recently voted in favor of the approval of Pfizer's Tofacitnib, a highly selective Type I JAK family kinase inhibitor. Tofacitinib, a drug in Ph III for rheumatoid arthritis, signals a new era for kinase inhibitors – one that moves beyond the traditional oncology applications.
Type II kinase inhibitors: these compounds bind in a pocket made possible by the DFG residues of the activation loop being folded away from the kinase conformation required for ATP to bind.3 This “DFG-out” conformation is due to the compound binding to the inactive form of the kinase or results from the compound inducing a large movement of the DFG motif. In this conformation, the aspartic acid of the DFG motif points away from the ATP binding site and the phenylalanine flips inside to partially occupy the ATP binding site. The movement of the DFG motif exposes a newly accessible hydrophobic binding pocket that is adjacent to the ATP site. The first reported example of a kinase inhibitor that binds to an inactive form of a kinase was Gleevec (also known as Imatinib).4 In this inactive conformation, the activation loop of ABL kinase is not phosphorylated and the conformation of this loop is distinct from the conformation of active protein kinases. This seminal finding suggested that compounds that exploit the distinctive inactivation mechanisms of individual protein kinases could achieve both high affinity and high specificity. Since the serendipitous discovery of Imatinib, there have been several additional Type II inhibitors identified with excellent potency and selectivity.5 The amino acid residues surrounding this “allosteric pocket” are less conserved as compared to the ATP binding pocket residues. As a result, Type II inhibitors are able to achieve a higher level of selectivity compared to Type I inhibitors.6
Type III kinase inhibitors: these are small molecule inhibitors that exclusively bind in a pocket adjacent to the ATP site, without making any interactions with the hinge region of the ATP binding pocket. These compounds bind in a site known as the “back pocket” of the kinase. A prototype and extremely well characterized Type III inhibitor is the MEK inhibitor, CI-1040.7 This compound class induces a change in the activation loop that forces the α-C-helix to adopt an inactive conformation, preventing a glutamate essential for catalysis from properly aligning in the ATP binding site.
Type IV kinase inhibitors: Rauh et al.8 defined these compounds as those that bind to a site remote from the ATP binding pocket. These inhibitors bind to a distinct allosteric site on the kinase and induce conformational changes that make the protein inactive. The exact location of the allosteric site of Type IV kinase inhibitors is not specifically defined and may be anywhere on the kinase except for the pocket adjacent to the ATP site (as those are considered Type III inhibitors).
Type V kinase inhibitors: this class refers to bi-substrate inhibitors. For instance, a peptide analog of the kinase's protein substrate is attached covalently to an ATP competitive small molecule.9 The preparation of bivalent inhibitors is viewed as a strategy to increase potency and selectivity and has been applied to several kinases.10 The challenge for such inhibitors is maintaining a balance between potency and selectivity on one hand and reasonable physico-chemical properties (molecular weight, solubility, permeability) and cellular activity on the other hand.
Another class of kinase inhibitors consists of those that can form an irreversible, covalent bond to the kinase active site. In most cases, these inhibitors react with a nucleophilic cysteine residue in the active site.11 Irreversible kinase inhibitors can overcome competition with high endogenous concentrations of ATP and have the potential to prevent resistance conferred by protein kinase mutations. The most advanced irreversible kinase inhibitor is Neratinib (also known as HKI-272).12 This compound irreversibly binds to a cysteine residue in the ATP binding site of EGFR kinase and is being investigated for the treatment of breast cancer and other solid tumors.
An extensive selectivity analysis of 72 kinase inhibitors tested against 442 kinases covering >80% of the human catalytic protein kinome was published recently.6 The data showed that as a class, Type II inhibitors are more selective than Type I inhibitors. Type III and IV kinase inhibitors bind to less conserved allosteric sites outside the ATP pocket and would be expected to have an even greater selectivity profile. An allosteric site also provides an opportunity to obtain novelty with unique chemical scaffolds. Furthermore, active site inhibitors must compete with millimolar concentrations of ATP in the cell. On the other hand, allosteric inhibitors are non-competitive with ATP and could offer improved biochemical efficiency and thus open the door to tackle kinase targets where achieving the acceptable therapeutic index had been challenging in the past.13 In addition, Type I/II kinase inhibitors are very sensitive to mutations affecting the direct contact residues in the ATP pocket but also mutations located in regions that affect the kinase conformation.14 In contrast, an allosteric BCR-ABL kinase inhibitor, GNF-2, showed activity against Imatinib-resistant mutants.14 These resistant mutants, such as E255V, are frequently the cause of relapse during Imatinib treatment in the clinic and are associated with a poor prognosis. Even though allosteric inhibitors are also subject to resistance through point mutations, a recent report has shown that the combined use of compounds with different mechanisms of action, namely both ATP and non-ATP competitive inhibitors, decreases the number of resistant clones that emerge as a response to continued exposure to a single agent.15 Combined treatment with GNF-5 (a GNF-2 analog that binds to the allosteric myristate binding site) and nilotinib (an ATP competitive inhibitor) led to in vivo efficacy, resulting in disease remission in a murine bone-marrow transplantation model using the T315I BCR-ABL mutant. These findings open the door to combining allosteric and ATP-competitive inhibitors in order to overcome resistance to either agent alone.
The future of novel and selective kinase inhibitors rests on the discovery of compounds that bind outside of the ATP site while retaining a high level of potency and efficacy. Recent kinase reviews summarize the challenges of and advancements to kinase inhibition.16 The majority of non-ATP site binders have been discovered through serendipity, illustrating the urgent need for new methods to assist in the discovery of these important compounds. The focus of this review is to exemplify the primary methods used to discover small molecule Type III or IV kinase inhibitors that bind outside of the ATP pocket. There are additional discovery approaches employed to find Type II (DFG-out) kinase inhibitors,17 but those will not be discussed herein.
2. Screens to identify non-ATP site kinase binders
The value of kinase inhibitors that do not bind in the ATP site has been widely recognized by the pharmaceutical industry. Yet there is a distinct lack of direct screening methods to discover such compounds. In many cases, compounds initially found via high throughput screening (HTS) are identified as non-ATP site binders by secondary experiments to rigorously determine their mechanism of kinase inhibition. Typically, the hypothesized mechanism of binding is confirmed by the use of X-ray crystallography. This screening paradigm is depicted in Fig. 2.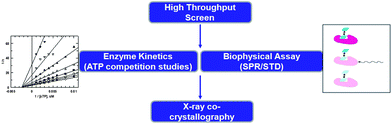 | ||
| Fig. 2 Screening tree used to serendipitously discover non-ATP site binders. | ||
The disadvantages of such a screening strategy are two-fold. First, since most biochemical kinase activity assays preferentially detect Type I compounds, this is not a very direct mode of identifying allosteric inhibitors.18 It is not surprising that Type I inhibitors are more inclined to be discovered using biochemical screens since historically these screens use highly active recombinant kinase catalytic domains at very low concentrations of ATP, conditions in which hydrophobic screening compounds are likely to bind in the ATP cleft.19 Second, this screening sequence requires thorough and time consuming enzyme kinetic assays and/or biophysical characterization of the binding mechanism in order to determine that the compound is indeed a non-ATP site binder. If the original screen, secondary analysis or crystallography was performed using the kinase domain alone, it should be confirmed that the binders are truly functional kinase inhibitors using a full length kinase20 or cell-based assay. Nevertheless, the use of this circuitous screening strategy has resulted in the successful discovery of non-ATP site binders that have provided useful leads for drug discovery programs.21
Though certainly not extensive, some select examples where screening campaigns have generated non-ATP site hits are described below in more detail. These illustrations differ in the type of primary screen (biochemical HTS, ASMS, cell based HTS), yet are alike in their meticulous characterization of the mechanism of binding and ultimate substantiation with a co-crystal structure. In some cases, further assay development has led to new screening tools for the identification of non-ATP site binders.
At Pfizer, an enzymatic HTS of the Tec family tyrosine kinase, IL2-inducible T-cell kinase (ITK) identified a range of inhibitors. The group used a surface plasmon resonance (SPR)22 assay to confirm the hits. The SPR analysis highlighted compounds that showed super-stoichiometry and complexity in the dissociation phase in the binding sensograms.23 Subsequent co-crystallization of one of the super-stoichiometric compounds with ITK uncovered that this compound bound to two distinct pockets on the kinase. The crystal structure showed simultaneous binding of the compound in the ATP site and in a second binding site, adjacent to, but distinct from the ATP site, Fig. 3.
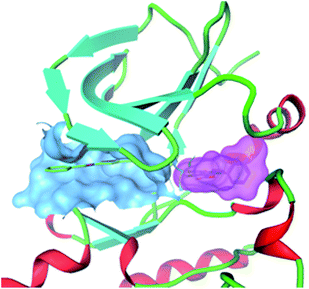 | ||
| Fig. 3 Co-crystal structure of an ITK inhibitor binding simultaneously to two distinct sites on the kinase. Blue surface: compound in ATP binding pocket; Pink surface: compound bound simultaneously to adjacent pocket. | ||
In order to determine the binding kinetics of the compound to the non-ATP site, the team developed an SPR competition assay, measuring the binding of test compounds in the presence of a high affinity ITK ATP site inhibitor. This SPR competition assay was utilized to determine if a series of compounds had mono or dual site binding. Co-crystallography of this same set of compounds with ITK validated the SPR competition assay, as there was 100% agreement between the two methods in the identification of dual-site binders.23 This work underscores the use of SPR as a tool to identify whether a compound binds to one or more sites on the target and suggests that it can be used to find compounds that bind to a second, non-ATP site, albeit in addition to an ATP site, on a target kinase. Compounds that bind to two distinct sites on a kinase could potentially provide a novel strategy for developing kinase drugs, especially against kinases that have shown drug resistance.15
In an effort to discover non-ATP site binders, researchers at Abbott Labs performed an affinity-based screen against un-phosphorylated (and therefore inactive) c-Jun N-terminal kinase 1 (JNK1).24 Affinity selection followed by mass spectroscopy (ASMS)25 is a method where mixtures of compounds are equilibrated with protein in solution and then filtered through a 10 kDa molecular weight cutoff membrane in order to separate binders to the protein from non-binders. In the end, mass spectrometry is used to directly identify the bound ligands, after a terminal chemical extraction step. Abbott screened ∼500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 small molecules in the JNK1 ASMS screen and from that effort, 68 were found to bind. In order to confirm binding and provide further information on where these compounds were binding to the protein, these 68 molecules were examined in 13C-JNK1 NMR binding studies. Of the 68 compounds studied, two were found to bind to a different site, distinct from the ATP binding pocket on the protein. NMR binding competition studies between these two compounds and an ATP competitive JNK1 inhibitor confirmed that these compounds were non-competitive with ATP. Importantly, they were found to inhibit the activity of JNK1 in a cellular assay. Co-crystallography of one of these analogs, 1, with JNK1 elucidated that its binding site is quite remote from the ATP site, and is located in the MAP insert region. Using this compound as a starting point, the group optimized the potency and cellular permeability of the original hit, 1, to yield compound 2 that exhibited robust cellular activity, Fig. 4. Compound 2 was also confirmed to bind at the MAP insert region on JNK1.
000 small molecules in the JNK1 ASMS screen and from that effort, 68 were found to bind. In order to confirm binding and provide further information on where these compounds were binding to the protein, these 68 molecules were examined in 13C-JNK1 NMR binding studies. Of the 68 compounds studied, two were found to bind to a different site, distinct from the ATP binding pocket on the protein. NMR binding competition studies between these two compounds and an ATP competitive JNK1 inhibitor confirmed that these compounds were non-competitive with ATP. Importantly, they were found to inhibit the activity of JNK1 in a cellular assay. Co-crystallography of one of these analogs, 1, with JNK1 elucidated that its binding site is quite remote from the ATP site, and is located in the MAP insert region. Using this compound as a starting point, the group optimized the potency and cellular permeability of the original hit, 1, to yield compound 2 that exhibited robust cellular activity, Fig. 4. Compound 2 was also confirmed to bind at the MAP insert region on JNK1.
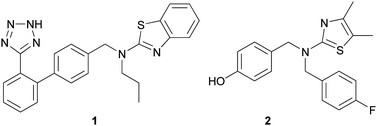 | ||
| Fig. 4 Allosteric JNK1 kinase inhibitors, 1 and 2. | ||
Recent work published from ArQule used a similar approach to identify non-ATP site binders of AKT (protein kinase B or PKB).26 Analogous to the JNK1 example above, this group used an initial ASMS screen to search for AKT binders. In order to gain insights on the mode of binding, the hits were characterized with both a thermal shift assay (TSA)27 and an SPR assay using full length AKT protein kinase. From this work, an imidazopyridine hit was recognized as binding in a non-ATP site. Optimization for potency and cellular activity provided compound 3, Fig. 5. Eventual co-crystallization of 3 with AKT unambiguously confirmed that it was indeed binding in a non-ATP binding pocket. In fact, compound 3 occupies a deep cavity at the interface of the kinase and pleckstrin homology (PH) domains, demonstrating the significance of using full length kinase in the initial screen. Inhibitor3 was determined to be selective for AKT over ∼300 other kinases, in all likelihood due to its unique mode of binding.
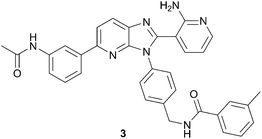 | ||
| Fig. 5 Structure of allosteric AKT inhibitor, 3. | ||
The success of the BCR-ABL inhibitor, Imatinib,4 is being challenged clinically by the emergence of acquired resistance. Therefore, Novartis, and others, are looking to develop BCR-ABL kinase inhibitors with new modes of action. Towards this end, Novartis used a high throughput cellular screen in order to find inhibitors working through an alternate mechanism.14 The cellular screen yielded GNF-1 (4), a compound that stood out due to its excellent differential cytotoxic activity (IC50 = 400 nM). Subsequent optimization of this compound's cellular activity led to GNF-2 (5), Fig. 6.28 A significant amount of work was undertaken in order to elucidate the target and mechanism of action of GNF-2. Recently, co-crystallization of GNF-2 with ABL has unambiguously confirmed that GNF-2 is bound to the myristoyl binding pocket of ABL kinase.15 This is a nice demonstration of the application of a cellular screen used to identify a non-ATP site kinase inhibitor. Due to the unique mechanism of action, GNF-2 could provide a new class of chronic myelogenous leukemia (CML) agents with no off-target activity or it could at least serve as a new research tool to assist in the investigation of ABL-dependent biology.
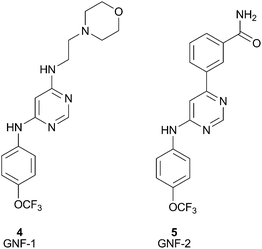 | ||
| Fig. 6 Structures of GNF-1 (4), GNF-2 (5). | ||
Direct screening to find non-ATP site binders has been hindered by the fact that the most potent molecules resulting from traditional biochemical kinase HTS campaigns are often ATP competitive inhibitors. Therefore, efforts have been initiated to see if screens can be re-purposed to generate non-ATP competitive hits. Towards this end, researchers at Life Technologies Corporation have examined the use of a time-resolved fluorescence resonance energy transfer (TR-FRET) kinase binding assay to detect known non-ATP site binders.29 This assay detects compounds that displace an ATP competitive active site fluorescent probe, called a tracer, Fig. 7.30 The theory was that since there are structural perturbations in the active site even in the presence of non-ATP site binders, perhaps the perturbations could be detected by the displacement of active site probes.31 Towards this end, 15 commercially available non-ATP competitive kinase inhibitors were screened in this assay for activity against their respective kinase targets. The TR-FRET screening method successfully detected the binding of 14 of these structurally diverse inhibitors to their particular kinase target. To date this method has only been used to detect previously known non-ATP site binders. However, their work suggests that such an assay could be effective at detecting novel non-ATP competitive kinase inhibitors for a broad spectrum of kinase targets.
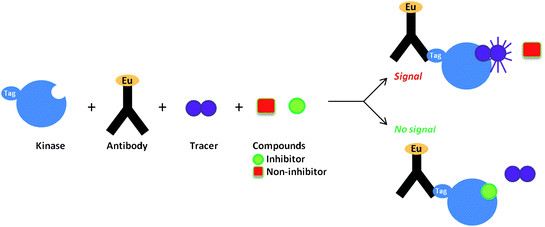 | ||
| Fig. 7 TR-FRET kinase binding assay. | ||
The use of NMR screens to identify binders to protein targets in lead discovery has been applied broadly in the pharmaceutical industry.32NMR screening is most appreciated for its robustness in eliminating false positive hits and for its sensitivity to weak interactions. Since NMR screening can detect weak binders (i.e. IC50 in millimolar range), it is a particularly useful tool for the detection of ligands for a novel pocket or target. There are a few NMR screening techniques that can be employed to specifically discover second site binding, namely SAR by NMR,33 SAR by NOE34 and SAR by ILOE.35 SAR by NMR requires large amounts of isotopically labeled protein, limiting its use to highly expressed and soluble proteins. To date, there are several examples of the use of NMR to identify second site binders to a variety of protein targets.36 However, even though these NMR screens seem poised to identify novel non-ATP site kinase binders, they have not yet been exploited in this fashion. An obvious extension would be to use the SAR by NMR/NOE/ILOE methods to uncover compounds that bind in the pocket adjacent to, but not overlapping with, the ATP site of kinases.
Screens to ascertain allosteric inhibitors will provide leads to address the most defined challenges in kinase drug discovery, limited selectivity and drug resistance. Yet the current bottleneck in allosteric inhibitor discovery is the lack of direct screening methods to identify such compounds. The potential value of library screening for binders to the allosteric pocket will only be unlocked if HTS methods are available which can discriminate between Type I and the more desirable Type III/Type IV inhibitors. There is a conspicuous lack of screening methods to directly identify these types of kinase inhibitors and therefore many research groups are still relying on chance to acquire such compounds.
3. Computational methods to identify non-ATP site kinase binders
Rational design of highly selective kinase inhibitors is remarkably challenging.16 Traditionally, developing molecular models for non-ATP binding pockets in kinases has been difficult due to the protein docking sites being largely solvent exposed and relatively shallow.37 Nonetheless, there are a few cases of computational chemistry guiding the design of non-ATP site binders and some of these examples are reported below.Very recently, researchers at the University of Texas have described a three-dimensional molecular model of the MAPK-ERK2 complex, which has provided a tool to help in the design of non-ATP site ERK2 inhibitors.38 This model was built using computational insights from both X-ray crystallography and molecular dynamics data. Although this was the first molecular model developed specifically for the MAPK substrate on ERK2, it was not the first time modeling was used in the design of non-ATP competitive ERK2 inhibitors. The first non-ATP competitive small molecule inhibitor of ERK2 was identified using computer aided drug design in 2005.39 The Hancock group performed a virtual screen on ∼800000 compounds using the program SPHGEN to detect possible binding sites on the surface of inactive ERK2. From their in silico screen, 80 compounds were identified for in vitro assay determination. Several of the compounds originally identified through the virtual screen were determined to bind to a pocket distinct from the ERK2 ATP site. One of the compounds identified from the virtual screen was shown to have a Kd of 5 μM to ERK2, 6, Fig. 8.
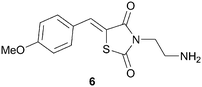 | ||
| Fig. 8 Structure of ERK2 inhibitor, 6. | ||
Although there are many known inhibitors of p38α kinase, there are very few p38α inhibitors that do not bind exclusively at the ATP site. The most notable compound is the clinical candidate, BIRB-796, which binds in a pocket adjacent to the active site and locks the kinase in an inactive confirmation.40 In 2009, Perry and co-workers41 described a novel C-terminal domain pocket on p38α that his group fully characterized using both X-ray crystallography and computational analyses such as AutoLigand.42 Perry examined both the optimal ligand size and the flexibility of this pocket and deemed it a promising allosteric site for the design of inhibitors. Although a virtual screen was not carried out on the C-terminal domain pocket, their analysis paved the way for such an investigation. In fact, in 2011, Comess and coworkers reported the discovery of several inhibitors that do bind to this allosteric site on p38α, including compound 7, Fig. 9.24 Detailed NMR studies and co-crystallization of 7 with p38α confirmed that it is binding in the C-terminal domain pocket described by Perry, and is not binding in the ATP site. The identification of 7 is the result of computational analyses that elucidated the novel binding pocket.
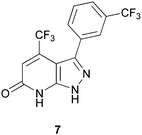 | ||
| Fig. 9 Allosteric inhibitor of p38α, 7. | ||
Grant et al. just published work that illustrates the coupling of several different computational techniques to identify small molecule allosteric pockets on Ras kinase.43 In order to account for receptor flexibility in their ligand design,44 Grant's team combined the analysis of multiple crystallographic structures with a molecular dynamic (MD) trajectory analysis.45 This integrated bioinformatics effort successfully identified novel binding pockets on Ras kinase. A virtual screen of these novel pockets using commercially available drug sets resulted in 56 compounds scoring high in their model. From those compounds, 19 ligands were selected for in vitro testing. Several of the compounds tested had micromolar inhibitor activity against Ras kinase and were found to be active in cellular assays. Further investigations are required to determine if the ligands actually do bind at the predicted sites, however, it is encouraging that these compounds show inhibitory activity against Ras kinase.
D. E. Shaw Research (“DESRES”) is an independent research laboratory that conducts basic scientific research in the field of computational biochemistry.46 DESRES uses very high level computational approaches to try to solve complex problems. Recently, researchers at DESRES published a manuscript describing the use of molecular dynamic simulations to capture the process of a small molecule finding its binding site within a protein.47 For instance, in a simulation, the well known kinase inhibitor PP1 bound to SRC kinase in an identical location as captured by X-ray crystallography.48 In this model, PP1 found its binding site on SRC kinase without any programmed prior knowledge of the binding location. Interestingly, the ligand circled the protein target extensively before eventually locating its optimal binding site. In fact, in their simulation of PP1 binding to SRC kinase, PP1 frequently inhabited a previously unreported binding pocket. Therefore, DESRES began a drug discovery effort which targets the Src kinase putative allosteric binding site. The authors underscore the great potential of this tool for discovering non-ATP site kinase binders.
As evidence strengthens that sites outside of the ATP pocket in kinases are druggable, there is increasing importance to develop new computational methods that will uncover novel inhibitors. Using these tools to help design potent non-ATP competitive kinase inhibitors has been realized in some cases. Yet with all the structural information on kinases, it is notable that few virtual screening approaches have been reported to successfully discover non-ATP site leads. Also of note, similar to biochemical screening, it is important to consider the dynamics of the catalytic domain and the full length kinase to target non-ATP site leads. There is great opportunity for advancing computational modeling methods to aid in the discovery of this important class of compounds.
4. Small molecule spin-labeled probes to identify non-ATP site kinase binders
The use of NMR to detect Type III kinase inhibitors has been achieved by screening in the presence of a paramagnetic spin-label on a first site ligand. The use of spin-labeled probes in NMR is a technique that was pioneered by Jahnke et al. in 2000.49 The theory for a spin-labeled NMR screen was based on the premise that a true second site binder will bind to the target at the same time as the first site binder, since they will be binding at separate, non-overlapping sites. Spin-labels are organic radicals, such as the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) moiety, that lead to dramatically increased relaxation rates of neighboring protons on both proteins and ligands in NMR. TEMPO radicals are widely used as spin probes due to their electron paramagnetic resonance, slow electronic relaxation time and prolonged radical stability.50 In some cases, the resonances of neighboring protons are completely quenched and can even be invisible in the NMR spectrum.51 Therefore, compounds that bind within 25 angstroms of a bound spin-labeled compound will be detected in a screen, simply by measuring if there is paramagnetic relaxation enhancement (PRE) in the NMR spectrum. To amplify the line broadening in the NMR experiment, a technique called T1ρ NMR measurement is utilized.52There are several assumptions made when using a spin-label probe including (1) there is no cooperative binding effect of the two ligands and (2) the compounds have similar binding kinetics. In terms of kinases, the quenching effect of the spin-labeled ATP site binder on the test compound will be observed if, and only if, both compounds bind at the same time and in the same vicinity (within 25 angstroms) of the paramagnetic center. If the screening compound binds to the ATP site, it will never bind at the same time as the spin-labeled ligand, and therefore, cannot produce a false positive result. Similarly, if it does not bind to the target at all, it will not be in close enough vicinity to the spin-labeled ligand and therefore will not experience any quenching effects in the NMR spectrum. As depicted in Fig. 10, only simultaneous binding of the two compounds in close proximity on the protein will result in PRE of the second site binder by the spin-labeled compound.
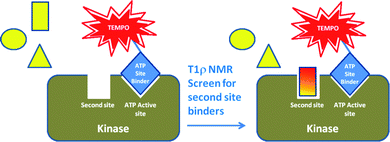 | ||
| Fig. 10 Schematic representation of the second-site screening approach using PRE of a non-ATP site binder by the spin-labeled ATP site binder. | ||
This method can be used to identify small molecule binders to serve as starting points for designing Type III kinase inhibitors. In order to have a functional kinase inhibitor, ATP should not be able to bind concomitantly with the inhibitor, so additional drug discovery efforts will most likely be needed to develop a functional Type III kinase inhibitor from the binder hit identified in such a screen. In some cases, however, the non-ATP site binder detected may bind significantly more tightly than ATP and therefore could already have functional inhibitory activity. Alternatively, groups have used spin labels to identify non-ATP site binders that may then be linked to the ATP site binder (vide supra). Due to the additive binding energies and favorable entropic effects, the resulting linked compound may have nanomolar affinity, even if the 2 ligands alone had micromolar or even millimolar affinity.53 The use of several spin-labeled compounds that have been designed as tools to identify non-ATP site kinase binders will be described below.
In order to detect compounds binding near, but not overlapping with the ATP site, the Jahnke group prepared a spin-labeled ATP mimetic, 8, for use in PRE NMR screening, Fig. 11.54 Due to the paramagnetic relaxation effects of the TEMPO moiety, 8 can be used to unambiguously identify ligands binding outside of the ATP binding site. However, there should be care in the selection of kinases to be screened for allosteric inhibitors with the use of this tool compound, since the spin-labeled adenine analog was found to bind non-specifically to several kinases (targets not explicitly identified by the authors). In order to reduce the non-specific binding of the spin probe, the authors are working toward designing more hydrophilic spin-labeled adenine analogs.54
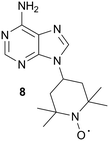 | ||
| Fig. 11 Adenine spin-labeled compound, 8. | ||
McCoy55 describes the use of Mn–ATP as a probe to detect novel kinase inhibitors where the paramagnetic atom manganese (Mn+2) was chelated to ATP in place of the typical magnesium ion (Mg+2). When Mn–ATP binds to a kinase, the protons near the ATP site show significant paramagnetic relaxation. This spin-labeled probe was used in a proof of principle experiment to detect a potent, non-ATP competitive inhibitor of the serine/threonine kinase, MEK1, namely the Type III inhibitor PD318088 (9), Fig. 12.7 Upon binding of PD318088 to MEK1, in the presence of Mn–ATP, its 1H proton resonances were broadened due to the relaxation imparted by the Mn–ATP probe.
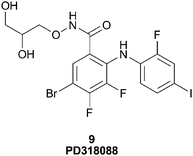 | ||
| Fig. 12 Structure of PD318088, 9. | ||
The advantages of the use of Mn–ATP as a tool to detect non-ATP site kinase binders include the fact that it can be used with any kinase that binds ATP, therefore providing a versatile tool for the detection of non-ATP site binders in kinases. However, Mn–ATP binds weakly to most proteins and therefore requires a large excess to ensure full binding occupancy. Another potential disadvantage is the non-specific binding of Mn2+ within the protein. However, the authors suggest that keeping the concentration of MnCl2 lower than 100 μM will minimize this unintended consequence.55
More recently, an indazole analog containing TEMPO was synthesized as a probe for second site screening against JNK1 kinase.56 Previous work demonstrated that binding of JNK interacting protein-1 (JIP1) or a JIP1 peptide (pepJIP1) inhibits its activity in vitro.57 Additionally, the X-ray co-crystal structure of JNK1 in complex with pepJIP1 and an ATP-mimetic, SP-600125 (10),58 revealed a close proximity between the ATP and JIP1 binding sites.59 This data provided Pellechia and co-workers with the inspiration to design a bi-dentate molecule (i.e. Type V inhibitor), concomitantly inhibiting both the ATP and JIP1 binding sites. Therefore, the group initiated an effort based on PRE screening to identify small molecule binders in the JIP1 binding pocket on JNK1. The design of the indazole probe, 11, was based on the structure of the known JNK1 ATP site binder, SP-600125 (10), as shown in Fig. 13.
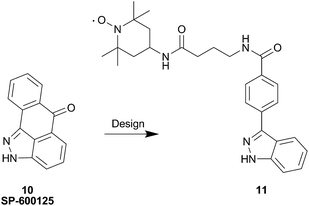 | ||
| Fig. 13 Design of the JNK1 spin labeled probe 11 from SP-600125, 10. | ||
The JNK1 probe 11 was used to determine that BI-78D3 (12), Fig. 14, binds in the site adjacent to the ATP site, namely the JIP1 pocket.60 The 1H NMR signals of BI-78D3 were strongly attenuated in the presence of the indazole-TEMPO spin probe. Moreover, the signals of the aliphatic protons on the benzodioxane moiety were the most affected and the proton on the thiazole ring was the least affected. Therefore the probe not only informed of the simultaneous binding of BI-78D3 with the ATP mimetic, but it also provided useful information of the orientation of molecular binding. The NMR data was corroborated with in silico docking of BI-78D3 in the JIP1 pocket of JNK1. The Pellechia group is continuing to explore the identification of non-ATP site JNK1 inhibitors by the use of their indazole probe. Recently, they described linking the inhibitors of both of these JNK1 pockets (ATP and JIP1) in order to prepare more potent and selective Type V JNK1 inhibitors.53
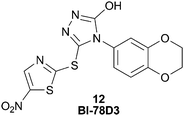 | ||
| Fig. 14 BI-78D3, 12, a JNK1 inhibitor that binds in the JIP1 pocket. | ||
Recent work in our laboratory61 focused on developing a spin-labeled ATP analog that may overcome some of the limitations associated with the previously described probes. For instance, some of the probes discussed above have low kinase affinity or show non-specific binding to certain kinases. Other literature probes were designed for a specific kinase (i.e. the JNK1 indazole probe) and therefore may not have broad applicability for screening a broad range of kinases. Therefore, our goal was to develop a spin-labeled compound that binds exclusively at the ATP binding site and has promiscuity across multiple kinases. In this way, we hoped to derive a screening tool which would have applicability for the use of detecting non-ATP site binders across a wide range of targets. The culmination of these efforts resulted in the design and synthesis of the TEMPO kinase probe, 13, Fig. 15. As desired, 13 has high affinity to several kinases, with an IC50 < 0.040 μM in 8 of the 19 kinases assayed. Additionally, compound 13 was found to bind exclusively in the ATP site by the X-ray co-crystallographic structure of 13 with LCK, KD (Lymphocyte-specific protein-tyrosine kinase, kinase domain).61
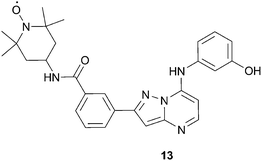 | ||
| Fig. 15 Structure of pyrazolopyrimidine kinase probe, 13. | ||
In order to validate 13 as a potential screening tool, we investigated if it could be used to detect already known non-ATP site binders. Kinex Pharmaceuticals had reported on 14 (Fig. 16), an inhibitor of SRC that is not competitive with ATP and was postulated to bind to the substrate binding domain.62 The unique binding mode and weak affinity for the SRC KD made 14 an ideal test compound to demonstrate the potential of 13 to identify second site binders via NMR relaxation experiments. Gratifyingly, we observed PRE in the 1H NMR spectrum of 14, indicating that 13 and 14 bind simultaneously to SRC. Similarly, the utility of 13 was demonstrated by characterizing compound 15, Fig. 16, binding to LCK kinase. Compound 15 was chosen as a test analog since it binds to LCK KD as evidenced in a saturation transfer difference (STD) NMR experiment, yet did not compete with staurosporine.61 We verified that 15 binds in a site adjacent to the ATP site since proton relaxation was observed in the T1ρ experiments performed. The demonstration of this spin-labeled NMR probe perceiving non-ATP site kinase binders of two different kinases suggests that 13 will have broad applicability to probe for adjacent, but not ATP site, kinase binders.
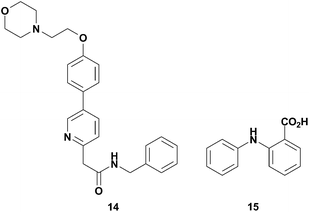 | ||
| Fig. 16 Structures of non-ATP site binders, 14 and 15. | ||
5. Conclusions
Protein kinases have been pursued by the pharmaceutical industry for over 20 years and have been proven to be exceptionally important targets for drug discovery. There are over 14 FDA approved small molecule kinase inhibitors currently on the market, and an enormous number of kinase inhibitors in the clinic. Kinase inhibitors have revolutionized treatments for oncology patients and are on the cusp of breaking into other indications as well. For instance, the recent FDA vote in favor to approve Tofacitnib, a highly selective JAK family kinase inhibitor for rheumatoid arthritis, highlights the advancements that the field has made in efforts to develop a kinase inhibitor for a chronic indication. Historically, the main challenges faced in developing kinase inhibitors as drug candidates have included, selectivity, robust efficacy (due to pathway redundancies), acceptable physico-chemical and pharmacokinetic properties, the ability to overcome endogenous ATP levels, and novel chemical equity. These challenges have triggered the need to develop alternative modalities for inhibiting kinases and will enable us to advance beyond Type I paradigms.Type III and IV kinase inhibitors bind to less conserved allosteric sites outside of the ATP pocket and will provide compounds with a greater selectivity profile. Additionally, allosteric inhibitors are non-competitive with ATP and therefore can offer improved biochemical efficiency, opening the door to tackle kinase targets where achieving acceptable therapeutic index had been challenging in the past. Small molecule spin-labeled probes are an excellent tool to have in one's arsenal of methods to discover Type III binders. The probes have the advantage of being able to simply and directly identify non-ATP site binders, without having to go through the rigorous de-convolution studies required when such compounds are detected in a typical HTS. Moreover, the Pellechia group has successfully used a probe to identify Type III JNK1 inhibitors, that when linked with ATP competitive inhibitors provide exquisitely potent and selective compounds. The use of a spin-labeled tool for the discovery of non-ATP competitive compounds for multiple kinases could be useful for kinase inhibitor programs targeting novel mechanisms of action.
In addition to exemplifying the use of spin-labeled probes to discover non-ATP site binders, this review has summarized other methods used to identify such compounds, namely Type III or IV kinase inhibitors. In the past, the majority of these compounds have been accidentally discovered, illustrating the critical need for new methods to assist in the discovery of these important bioactive molecules. The improvement of biochemical and virtual screens to discover kinase inhibitors in Type III or IV is essential in order to provide leads that will address kinase selectivity and drug-resistance issues. Selectivity and resistance are two of the most prominent issues that plague today's kinase programs. Another major issue, mentioned briefly above, is the dire need for kinase inhibitors with novel chemical scaffolds. More than 10000 patents or patent applications for protein kinase inhibitors were filed from 2001 to 2009.63 The majority of these patented scaffolds are for compounds that are competitive with ATP, making it a challenge for kinase drug researchers to find novel chemical space.
The screening bottleneck to discover allosteric kinase inhibitors must be alleviated, as hits from such a screen have an improved likelihood to achieve all the desired qualities of an ideal kinase inhibitor, including kinase selectivity, chemical novelty, reduced sensitivity to kinase resistance, and the ability to overcome high ATP concentration in cells. As seen in the examples herein, screening on full length kinase or cells, as opposed to just the kinase domain, is fundamental to delivering quality non-ATP site hits for discovery research. We believe that the future of kinase inhibitors rests upon new and innovative approaches, such as those mentioned above, in order to identify inhibitors working by a new mode of action.
Acknowledgements
The authors wish to thank R. Aldrin Denny for the preparation of Fig. 1 and for a critical reading of this manuscript.References
- G. Manning, D. Whyte, R. Martinez, T. Hunterand and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS.
- Y. Liu and N. S. Gray, Nat. Chem. Biol., 2006, 2, 358–364 CrossRef CAS.
- T. Schindler, W. Bornmann, P. Pellicen, W. T. Miller, B. Clarkson and J. Kuriyan, Science, 2000, 289, 1938–1942 CrossRef CAS.
- C. Pargellis, L. Tong, L. Churchill, P. F. Cirillo, T. Gilmore, A. G.Graham, P. M. Grob, E. R. Hickey, N. Moss, S. Pav and J. Regan, Nat. Struct. Biol., 2002, 9, 268–272 CrossRef CAS.
- M. I. Davis, J. P. Hunt, S. Herrgard, P. Ciceri, L. M. Wodicka, G. Pallares, M. Hocker, D. K. Treiber and P. P. Zarrinkar, Nat. Biotechnol., 2011, 29, 1046–1051 CrossRef CAS.
- J. F. Ohren, H. Chen, A. Pavlovsky, C. Whitehead, E. Zhang, P. Kuffa, C. Yan, P. McConnell, C. Spessard, C. Banotai, W. T. Mueller, A. Delaney, C. Omer, J. Sebolt-Leopold, D. T. Dudley, I. K. Leung, C. Flamme, J. Warmus, M. Kaufman, S. Barrett, H. Tecle and C. A. Hasemann, Nat. Struct. Mol. Biol., 2004, 11, 1192–1197 CAS.
- J. Simard, S. Klüter, C. Grütter, M. Getlik, M. Rabiller, H. B. Rode and D. Rauh, Nat. Chem. Biol., 2009, 5, 394–396 CrossRef CAS.
- K. J. Cox, C. D. Shomin and I. Ghosh, Future Med. Chem., 2011, 3, 29–43 CrossRef CAS.
- S. C. Meyer, C. D. Shomin, T. Gaj and I. Ghosh, J. Am. Chem. Soc., 2007, 129, 13812–13813 CrossRef CAS; A. J. Poot, J. van Ameijde, M. Slijper, A. van den Berg, R. Hilhorst, R. Ruijtenbeek, D. T. Rijkers and R. M. Liskamp, ChemBioChem, 2009, 10, 2042–2051 CrossRef; K. Parang, J. H. Till, A. J. Ablooglu, R. A. Kohanski, S. R. Hubbard and P. A. Cole, Nat. Struct. Biol., 2001, 1, 37–41 Search PubMed.
- M. S. Cohen, C. Zhang, K. M. Shokat and J. Taunton, Science, 2005, 308, 1318–1321 CrossRef CAS; E. L. Kwak, R. Sordella, D. W. Bell, N. Godin-Heymann, R. A. Okimoto, B. W. Brannigan, P. L. Harris, D. R. Driscoll, P. Fidias, T. J. Lynch, S. K. Rabindran, J. P. McGinnis, A. Wissner, S. V. Sharma, K. J. Isselbacher, J. Settleman and D. A. Haber, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7665–7670 CrossRef.
- S. K. Rabindran, C. M. Discafani, E. C. Rosfjord, M. Baxter, M. Brawner-Floyd, J. Golas, W. A. Hallett, B. D. Johnson, R. Nilakantan, E. Overbeek, M. F. Reich, R. Shen, X. Shi, H.-R. Tsou, Y.-F. Wang and A. Wissner, Cancer Res., 2004, 64, 3958–3965 CrossRef CAS.
- R. I. Mourey, B. L. Burnette, S. J. Brustkern, J. S. Daniels, J. L. Hirsch, W. F. Hood, M. J. Meyers, S. J. Mnich, B. S. Pierce, M. J. Saabye, J. F. Schindler, S. A. South, E. G. Webb, J. Zhang and D. R. Anderson, J. Pharmacol. Exp. Ther., 2010, 333, 797–807 CrossRef CAS.
- J. J. Adrián, Q. Ding, T. Sim, A. Velentza, C. Sloan, Y. Liu, G. Zhang, W. Hur, S. Ding, P. Manley, J. Mestan, D. Fabbro and N. S. Gray, Nat. Chem. Biol., 2006, 2, 95–102 CrossRef.
- J. Zhang, F. J. Adrian, W. Jahnke, S. W. Cowan-Jacob, A. G. Li, R. E. Iacob, T. Sim, J. Powers, C. Dierks, F. Sun, G.-R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P. W. Manley, J. R. Engen, G. Q. Daley, M. Warmuth and N. S. Gray, Nature, 2010, 463, 501–507 CrossRef CAS.
- A. C. Dar and K. M. Shokat, Annu. Rev. Biochem., 2011, 80, 769–795 CrossRef CAS; T. Anastassiadis, S. W. Deacon, K. Devarajan, H. Ma and J. R. Peterson, Nat. Biotechnol., 2011, 29, 1039–1045 CrossRef.
- J. R. Simard, C. Grutter, V. Pawar, B. Aust, A. Wolf, M. Rabiller, S. Wulfert, A. Robubi, S. Kluter, C. Ottmann and D. Rauh, J. Am. Chem. Soc., 2009, 131, 18478–18488 CrossRef CAS; P. Bonnet, D. Mucs and R. A. Bryce, Med. Chem. Commun., 2012, 3, 434–440 RSC.
- A. Card, C. Caldwell, H. Min, B. Lokchander, X. Hualin, S. Sciabola, A. V. Kamath, S. L. Clugston, W. R. Tschantz, W. Leyu and D. J. Moshinsky, J. Biomol. Screening, 2009, 14, 31–42 CrossRef CAS.
- J. Oishi, X. Han, J. H. Kang, Y. Asami, T. Mori, T. Niidome and Y. Katayama, Anal. Biochem., 2008, 373, 161–163 CrossRef CAS.
- E. V. Bobkova, M. J. Weber, Z. Xu, Y.-L. Zhang, J. Jung, P. Blume-Jensen, A. Northrup, P. Kunapuli, J. N. Andersen and I. Kariv, J. Biol. Chem., 2010, 285, 18838–18846 CrossRef CAS.
- J. A. Hardy and J. A. Wells, Curr. Opin. Struct. Biol., 2004, 14, 1–10 CrossRef CAS; S. F. Barnett, D. Defeo-Jones, S. Fu, P. J. Hancock, K. M. Haskell, R. E. Jones, J. A. Kahana, A. M. Kral, K. Leander, L. L. Lee, J. Malinowski, E. M. McAvoy, D. D. Nahas, R. G. Robinson and H. E. Huber, Biochem. J., 2005, 385, 399–408 CrossRef; J. R. Burke, M. A. Pattoli, K. R. Gregor, P. J. Brassil, J. F. MacMaster, K. W. McIntyre, X. Yang, V. S. Iotzova, W. Clarke, J. Strand, Y. Qiu and F. C. Zusi, J. Biol. Chem., 2003, 278, 1450–1456 CrossRef; W. Davidson, L. Frego, G. W. Peet, R. R. Kroe, M. E. Labadia, S. M. Lukas, R. J. Snow, S. Jakes, C. A. Grygon, C. Pargellis and B. G. Werneburg, Biochemistry, 2004, 43, 11658–11671 CrossRef; M. A. Bogoyevitch and D. P. Fairlie, Drug Discovery Today, 2007, 12, 622–633 CrossRef.
- T. Neumann, H. D. Junker, K. Schmidt and R. Sekul, Curr. Top. Med. Chem., 2007, 7, 1630–1642 CrossRef CAS.
- I. Navratilova, G. Macdonald, C. Robinson, S. Hughes, J. Mathias, C. Phillips and A. Cook, J. Biomol. Screening, 2012, 17, 183–193 CrossRef CAS.
- K. M. Comess, C. Sun, C. Abad-Zapatero, E. R. Goedken, R. J. Gum, D. W. Borhani, M. Argiriadi, D. R. Groebe, Y. Jia, J. E. Clampit, D. L. Haasch, H. T. Smith, S. Wang, D. Song, M. L. Coen, T. E. Cloutier, H. Tang, X. Cheng, C. Quinn, B. Liu, Z. Xin, G. Liu, E. H. Fry, V. Stoll, T. I. Ng, D. Banach, D. Marcotte, D. J. Burns, D. J. Calderwood and P. J. Hajduk, ACS Chem. Biol., 2011, 6, 234–244 CrossRef CAS.
- K. M. Comess, J. D. Trumbull, C. Park, Z. Chen, R. A. Judge, M. J. Voorbach, M. Coen, L. Gao, H. Tang, P. Kovar, X. Cheng, M. E. Schurdak, H. Zhang, T. Sowin and D. J. Burns, J. Biomol. Screening, 2011, 11, 755–764 CrossRef.
- M. A. Ashwell, J.-M. Lapierre, C. Brassard, K. Bresciano, C. Bull, S. Cornell-Kennon, S. Eathiraj, D. S. France, T. Hall, J. Hill, E. Kelleher, S. Khanapurkar, D. Kizer, S. Koerner, J. Link, Y. Liu, S. Makhija, M. Moussa, N. Namdev, K. Nguyen, R. Nicewonger, R. Palma, J. Szwaya, M. Tandon, U. Uppalapati, D. Vensel, L. P. Volak, E. Volckova, N. Westlund, H. Wu and R.-Y. Yang, J. Med. Chem., 2012, 55, 5291–5310 CrossRef CAS.
- J. Kean, R. M. Cleverley, L. O'Ryan, R. C. Ford, S. M. Prince and J. P. Derrick, J. Biomol. Screening, 2008, 25, 653–661 CAS.
- A. Q. Hassan, S. V. Sharma and M. Warmuth, Cell Cycle, 2010, 9, 3710–3714 CrossRef CAS.
- C. S. Lebakken, L. J. Reichling, J. E. Ellefson and S. M. Riddle, J. Biomol. Screening, 2012, 17, 813–821 CrossRef CAS.
- C. S. Lebakken, S. M. Riddle, U. Singh, W. J. Frazee, H. C. Eliason, Y. Gao, L. J. Reichling, B. D. Marks and K. W. Vogel, J. Biomol. Screening, 2009, 14, 924–935 CrossRef CAS.
- http://www.invitrogen.com/site/us/en/home/Products-and-Services/Applications/Drug-Discovery/Target-and-Lead-Identification-and-Validation/KinaseBiology/Kinase-Activity-Assays/lanthascreentm-eu-kinase-binding-assay.html .
- M. Pellecchia, D. S. Sem and K. Wuthrich, Nat. Rev. Drug Discovery, 2002, 1, 211–219 CrossRef CAS; W. Jahnke, A. Florsheimer, M. J. J. Blommers, G. Paris, J. Heim, C. M. Nalin and L. B. Perez, Curr. Top. Med. Chem., 2003, 3, 69–80 CrossRef; B. J. Sockman and C. Dalvit, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 41, 187–231 CrossRef.
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Science, 1996, 274, 1531–1534 CrossRef CAS.
- A. Kline, G. Bravi and J. Wikel, The NMR Newsletter, 1997, 472, 13 Search PubMed.
- M. F. Rega, B. Wu, J. Wei, Z. Zhang, J. F. Cellitti and M. Pellecchia, J. Med. Chem., 2011, 54, 6000–6013 CrossRef CAS.
- M. Pellechia, I. Bertini, D. Cowburn, C. Dalvit, E. Giralt, W. Jahnke, T. L. James, S. W. Homans, H. Kessler, C. Luchinat, B. Meyer, H. Oschkinat, J. Peng, H. Schwalbe and G. Siegal, Nat. Rev. Drug Discovery, 2008, 7, 738–745 CrossRef.
- L. N. Johnson, Q. Rev. Biophys., 2009, 42, 1–40 CrossRef CAS.
- M. J. Schnieders, T. S. Kaoud, C. Yan, K. N. Dalby and P. Ren, Curr. Pharm. Des., 2012, 18, 1173–1185 CrossRef CAS.
- C. N. Hancock, A. Macias, E. K. Lee, S. Y. Yu, A. D. MacKerell, Jr and P. Shapiro, J. Med. Chem., 2005, 48, 4586–4595 CrossRef CAS.
- M. R. Lee and C. Dominguez, Curr. Med. Chem., 2005, 12, 2979–2994 CrossRef CAS.
- J. J. Perry, R. M. Harris, D. Moiani, A. J. Olson and J. A. Tainer, J. Mol. Biol., 2009, 391, 1–11 CrossRef CAS.
- R. Harris, A. J. Olson and D. S. Goodsell, Proteins: Struct., Funct., Bioinf., 2008, 70, 1506–1517 CrossRef CAS.
- B. J. Grant, S. Lukman, H. J. Hocker, J. Sayyah, J. H. Brown, J. A. McCammon and A. A. Gorfe, PLoS One, 2011, 6, e25711 CAS.
- E. Laine, C. Goncalves, J. C. Karst, A. Lesnard, S. Rault, W.-J. Tang, T. E. Malliavin, D. Ladant and A. Blondel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11277–11282 CrossRef CAS.
- L. Chang, Y. Miyata, P. M. Ung, E. B. Bertelsen, T. J. McQuade, H. A. Carlson, E. R. P. Zuiderweg and J. E. Gestwicki, Chem. Biol., 2011, 18, 210–221 CrossRef CAS.
- www.deshawresearch.com/index.html .
- Y. Shan, E. T. Kim, M. P. Eastwood, R. O. Dror, M. A. Seeliger and D. E. Shaw, J. Am. Chem. Soc., 2011, 133, 9181–9183 CrossRef CAS.
- M. Getlik, C. Gruter, J. R. Simard, S. Kluter, M. Rabiller, H. B. Rode, A. Robubi and D. J. Rauh, J. Med. Chem., 2009, 52, 3915–3926 CrossRef CAS.
- W. Jahnke, L. B. Perez, G. Paris, A. Strauss, G. Fendrich and C. M. Nalin, J. Am. Chem. Soc., 2000, 122, 7394–7395 CrossRef CAS.
- M. L. Deschamps, E. S. Pilka, J. R. Potts and I. D. Campbell, J. Biomol. NMR, 2005, 31, 155–160 CrossRef CAS.
- P. A. Kosen, Methods Enzymol., 1989, 177, 86–121 CrossRef CAS.
- P. J. Hajduk, E. T. Olejniczak and S. W. Fesik, J. Am. Chem. Soc., 1997, 119, 12257–12261 CrossRef CAS.
- J. L. Stebbins, S. K. De, P. Pavlickova, V. Chen, T. Machleidt, L.-H. Chen, C. Kuntzen, S. Kitada, M. Karin and M. Pellecchia, J. Med. Chem., 2011, 54, 6206–6214 CrossRef CAS.
- W. Jahnke, M. J. J. Blommers, C. Fernandez, C. Zwingelstein and R. Amstutz, ChemBioChem, 2005, 6, 1607–1610 CrossRef CAS.
- M. A. McCoy, M. M. Senior and D. F. Wyss, J. Am. Chem. Soc., 2005, 127, 7978–7979 CrossRef CAS.
- J. Vazquez, S. K. De, L.-H. Chen, M. Riel-Mehan, A. Emdadi, J. Cellitti, J. L. Stebbins, M. F. Rega and M. Pellecchia, J. Med. Chem., 2008, 51, 3460–3465 CrossRef CAS.
- R. K. Barr, T. S. Kendrick and M. A. Bogoyevitch, J. Biol. Chem., 2002, 277, 10987–10997 CrossRef CAS.
- B. L. Bennett, D. T. Sasaki, B. W. Murray, E. C. O'Leary, S. T. Sakata, W. Xu, J. C. Leisten, A. Motiwala, B. S. Pierce, Y. Satoh, S. S. Bhagwat, A. M. Manning and D. W. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13681–13686 CrossRef CAS.
- Y. S. Heo, S.-K. Kim, C. I. Seo, Y. K. Kim, B.-J. Sung, H. S. Lee, J. I. Lee, S.-Y. Park, J. H. Kim, K. Y. Hwang, Y.-L. Hyun, Y. H. Jeon, S. Ros, J. M. Cho, T. G. Lee and C.-H. Yang, EMBO J., 2004, 23, 2185–2195 CrossRef CAS.
- J. L. Stebbins, S. K. De, T. Machleidt, B. Becattini, J. Vazquez, C. Kuntzen, L.-H. Chen, J. F. Cellitti, M. Riel-Mehan, A. Emdadi, G. Solinas and M. Karin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16809–16813 CrossRef CAS.
- F. J. Moy, A. Lee, L. K. Gavrin, Z. B. Xu, A. Sievers, E. Kieras, W. Stochaj, L. Mosyak, J. McKew and D. H. H. Tsao, J. Med. Chem., 2010, 53, 1238–1249 CrossRef CAS.
- T. H. Marsilje, K. L. Milkiewicz and D. G. Hangauer, Bioorg. Med. Chem. Lett., 2000, 10, 477–481 CrossRef CAS; K. L. Milkiewicz, T. H. Marsilje, R. P. Woodworth, Jr, N. Bifulco, Jr, M. J. Hangauer and D. G. Hangauer, Bioorg. Med. Chem. Lett., 2000, 10, 483–486 CrossRef.
- I. Akritopoulou-Zanze and P. J. Hajduk, Drug Discovery Today, 2009, 14, 291–297 CrossRef CAS.
Footnote |
| † This article is part of a MedChemComm ‘New Talents’ issue highlighting the work of outstanding rising scientists in medicinal chemistry research. |
| This journal is © The Royal Society of Chemistry 2013 |