Discovery of a highly selective FLT3 kinase inhibitor from phenotypic cell viability profiling†
Sanghee
Lee
a,
Ala
Jo
a and
Seung Bum
Park
*ab
aDepartment of Chemistry, Seoul National University, Seoul, Korea. E-mail: sbpark@snu.ac.kr; Fax: +82 2 884 4025; Tel: +82 2 880 9090
bDepartment of Biophysics and Chemical Biology/BioMAX institute, Seoul National University, Seoul 151-747, Korea
First published on 11th September 2012
Abstract
We discovered a novel molecular framework 4 containing a heterobiaryl pyrazolopyridine moiety as a selective FLT3 kinase inhibitor from phenotype-based viability profiling. Compound 4g showed outstanding selectivity in cellular cytotoxicity against MV-4-11 leukemic cells via the induction of apoptosis. The hypothesis-driven deconvolution elucidated that compound 4g selectively blocked the phosphorylation of FLT3 and its downstream effectors, such as ERK and STAT5, only in MV-4-11 cells. The inhibitory effect of 4g on in vitro enzyme function and FLT3 phosphorylation in cells proved that FLT3 kinase is a direct molecular target of 4g. Finally, the kinase activity profiling of 4g verified its excellent selectivity toward FLT3 over 40 representative kinases, including the receptor tyrosine kinase (RTK) family.
Introduction
The current focus in drug discovery research has been shifted from the multi-targeting promiscuous drug to the highly selective drug. Target-specific small-molecule modulators can lead to the acquisition of mechanistic clues to understand the function of biopolymers in a complex biological system. In fact, a series of studies reported that specific small-molecule modulators were extremely important as chemical probes, which can interrogate the function of target proteins, suggest a new paradigm for complex biological functions,1 and eventually expand the therapeutic window of those drug candidates.2 In addition, their selectivity toward certain drug targets ensure therapeutic activities with minimal adverse effects for various diseases. Therefore, the discovery of highly specific small-molecule modulators remains a challenging issue, but a worthwhile strategy in the field of drug discovery and chemical biology.Under the name of rational drug discovery, the major drug companies have used the target-based enzymatic assay as a primary screening platform. High throughput screening with conventional drug targets has been a major workhorse in the drug discovery industry and allowed the launching of various new drugs in the market, however, some of those drugs were withdrawn from the market, not because of their insufficient efficacy, but because of their unexpected side effects.3 This limitation of the target-based approach calls forth more interest in phenotypic screening, which allows the elimination of candidate molecules with undesirable side effects in cells or model animals at the early stage of drug discovery. Therefore, phenotype-based screening systems recently received huge attention as one of the major initial screening platforms for biomedical researchers in academia as well as in the pharmaceutical industry. In addition to the reduction in the late-stage attrition, the phenotypic screening shows a great contribution to the discovery of first-in-class drugs with new molecular entities, which can surpass the conventional target-based approach.4 For this reason, a large number of drug discovery centres in the academic environment are much more focused on the development and application of phenotype-based screening.5 However, even though the phenotype-based screening approach might increase the potential to identify novel molecular frameworks with unique biological activity and superb selectivity, these bioactive small molecules discovered by this strategy might be limited in their usage as potential therapeutics or as chemical biology research tools for the study of mysterious biological process until the identification or deconvolution of molecular targets of small-molecule modulators.6 Therefore, the phenotype-based approach might be a great strategy for the discovery of novel molecular frameworks with a desired efficacy toward certain disease areas, when it is tightly combined with the subsequent identification of the target proteins, which can elucidate the molecular mechanism of the efficacy and selectivity.
Herein, we report the discovery of a novel molecular framework with highly selective cellular cytotoxicity toward an acute myeloid leukemia (AML) cell line using phenotype-based viability profiling and the subsequent identification of FMS-like tyrosine kinase-3 (FLT3) as its target protein via hypothesis-driven deconvolution. In fact, FLT3 has been considered as a therapeutic target for the treatment of AML patients and it is activated by FLT3 ligand (FLT3L), which is known to have a crucial role in the proliferation and apoptosis of hematopoietic cells.7
Results and discussion
Recently, we introduced the concept of the privileged substructure-based diversity-oriented synthesis (pDOS) strategy for the efficient construction of skeletally diverse small-molecule collections that maximize molecular diversity with ensured biological relevance via the incorporation of privileged substructures into drug-like polyheterocycles.8 With this unique resource in hand for the discovery of novel bioactive small molecules, we pursued the identification of novel small molecules with selective cytotoxic activity toward certain cancer cells: the pDOS-derived small-molecule collection containing over 3000 members with 60 unique molecular frameworks was subjected to high-throughput cell viability screening (HTS) against 6 different cells using WST, a water-soluble tetrazolium salt assay. The extensive HTS screening lead us to the identification of the pyrazolopyridine-based molecular framework 4 as an initial hit compound with a specific cellular toxicity pattern for MV-4-11 cells (see ESI†). To enhance the potency along with the understanding of their structure–activity relationship, we then constructed a focused library of 13 compounds using a synthetic procedure developed in our group.9 In brief, we prepared the properly functionalized indole moieties 1, which were transformed into indole-3-carboxaldehyde analogues 2via the Vilsmeier–Haack reaction. The resulting dielectrophilic 3-formyl indoles 2 can undergo indole ring opening and subsequent cyclization with various aminopyrazoles 3 as dinucleophiles to yield the desired heterobiaryl pyrazolopyridines in a single step (Scheme 1). The substrate scope of this transformation is quite broad, which allows the formation of various analogues via the combination of R2-substituted 3-aminopyrazoles and R1,R3-substituted 3-formyl indole derivatives with various substituents, including halogens, nitro groups, and esters, at the different positions on the indole rings. This synthetic route allows the formation of heterobiaryl pyrazolopyridine derivatives 4a–4m in moderate to excellent yields (29–74%). The detailed synthetic procedure and reaction yields are reported in the ESI.†![The synthetic procedure for heterobiaryl pyrazolo[3,4-b]pyridine derivatives. Reagents and condition: (i) R1,R3-substituted indole, POCl3, DMF, 0 °C, 0.5–2 h; (ii) R2-substituted 3-aminopyrazole, AlCl3, MeOH, 70 °C, 4 h.](/image/article/2013/MD/c2md20169k/c2md20169k-s1.gif) | ||
| Scheme 1 The synthetic procedure for heterobiaryl pyrazolo[3,4-b]pyridine derivatives. Reagents and condition: (i) R1,R3-substituted indole, POCl3, DMF, 0 °C, 0.5–2 h; (ii) R2-substituted 3-aminopyrazole, AlCl3, MeOH, 70 °C, 4 h. | ||
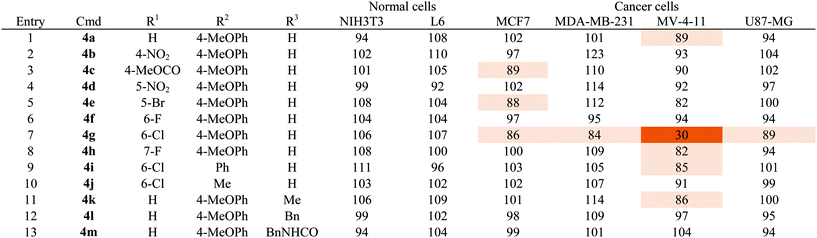
As shown in Table 1, a series of heterobiaryl pyrazolopyridine derivatives 4a–4m was subjected to the cell viability profiling assay against 6 representative cell lines, including two normal cell lines (NIH3T3 and L6) and four cancer cell lines from different tissue origins, such as MCF7 and MDA-MB-231 breast cancer cell lines, MV-4-11 acute myeloid leukemia, and U87-MG brain glioblastoma cell lines. First of all, we compared the cell viability pattern upon substituent changes at the R1 position while keeping 4-methoxyphenyl moiety as the fixed R2 substituent (entries 1–8, Table 1). Even though we tested a broad range of positioning and substitution effects at the R1 position, only a chlorine substituent at C-6 position 4g showed selective cellular cytotoxicity toward MV-4-11 AML cells. To determine whether the 4-methoxyphenyl moiety at the R2 position has an essential role in its activity, we changed the R2 substituent to either phenyl 4i or methyl 4j with a 6-chloro substituent at the R1 position, but we did not observe any cellular cytotoxic effects for these compounds under the identical condition used for compound 4g (entries 9 and 10, Table 1). The further N-modification of aniline with methyl 4k, benzyl 4l, and benzyl urea 4m did abolish the cytotoxic effects toward all tested cell lines (entries 11–13, Table 1). On the basis of our SAR study, we observed a quite tight SAR pattern of 4g toward selective cellular cytotoxicity and achieved no improvement of potency and selectivity from our initial hit compound 4g, even though we introduced diverse structural modifications on the heterobiaryl pyrazolopyridine skeleton 4.
| a Cell viability was measured by WST assay. The series of compounds were treated for 24 h with 1 μM as final concentration. Each compound was tested in triplicate. Results are mean value and designated by % which is normalized by DMSO as a control. Data is illustrated by color coding from dark color for low values to white for high values. |
|---|
|
Before probing into the mechanistic study of 4g to understand the mode of action, we performed an in vitro evaluation to confirm the selective cytotoxicity pattern induced by 4g. At first, we evaluated the inhibitory effect of 4g on the cell proliferation in a dose-dependent manner. As shown in Fig. 1b, 4g inhibited the cell growth in MV-4-11 cells and resulted in an IC50 value of 592 nM. We also examined the cell viability profiling upon treatment with 4g at 1 μM in a panel of twelve different cell lines, including the original 6 cell lines along with 5 additional cancer cells from different tissue origins (A549 lung cancer, HCT116 colon cancer, SW480 colon cancer, DU145 prostate cancer, PC3 prostate cancer) and Raw264.7 macrophage cells. Surprisingly, 4g showed excellent selectivity only toward MV-4-11 AML cells in cellular cytotoxicity (see Fig. 1c).
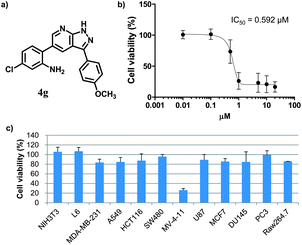 | ||
| Fig. 1 Characterization of the cell-specific cytotoxic agent, 4g. (a) The chemical structure of compound 4g. (b) Dose-dependent inhibitory activity on cell proliferation and its IC50 toward MV-4-11 AML cells. (c) Cell viability profiling data against 12 different cell lines with the treatment of 4g at the final concentration of 1 μM. Individual cell viability data was normalized to DMSO control as 100%. Each experiment was performed in triplicate; data was figured as mean and SD. | ||
With a highly selective cytotoxic agent 4g in hand, we pursued the in vitro confirmation of 4g for its selective cytotoxic effect toward MV-4-11 AML cells to explore its mechanism of action. As shown in Fig. 2, our selective cytotoxic agent 4g induced apoptosis, which was confirmed by western blot analysis of cleaved PARP and caspase-3 in MV-4-11 AML cells and the protein level of cleaved PARP and caspase-3 was also increased in a dose-dependent manner. These results indicated that our selective cytotoxic agent 4g inhibited cell proliferation via the induction of apoptotic cell death in MV-4-11 AML cells. We then moved forward to study the related protein signalling pathways in the apoptotic process to access important clues for the identification of its plausible target proteins. On the basis of the outstanding selectivity of 4g toward MV-4-11 AML cells over other cancer cells, we searched for different phosphorylation patterns among key players in the apoptotic process upon treatment of 4g. After extensive western blotting analyses on those key factors, we observed the dose-dependent reduction in the phosphorylation of ERK and STAT5 on MV-4-11 cells (see Fig. 3a). In fact, AML is a type of hematopoietic malignancy, characterized by the rapid growth of abnormal white blood cells, resulting in the interruption of the production of normal blood cells. On the basis of a literature survey, we investigated what factors might induce the specific cytotoxicity of 4g, observed in MV-4-11 AML cells, and hypothesized that the FLT3 signalling pathway might be a suitable target because MV-4-11 cells are known to have a high expression level of FLT3 with internal tandem duplication (ITD) mutation, which is commonly found in AML patients and results in constitutive activation of FLT3.7 FLT3 is one of the class III receptor tyrosine kinases (RTK) that is activated by FLT3L and has important roles in the proliferation and apoptosis of hematopoietic cells. Even though the expression level of FLT3L varies in a wide range of tissues, the expression pattern of FLT3 is quite limited in early hematopoietic progenitor cells, which suggested the tissue-specific activation of FLT3.10 Therefore, FLT3 has been recognized as a therapeutic target for the treatment of AML patients. In addition, ERK and STAT5 are the representative downstream proteins of FLT3 as well as other RTK families.10
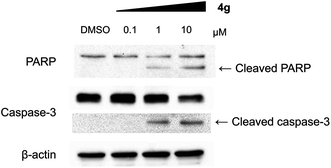 | ||
| Fig. 2 Compound 4g induced apoptosis in MV-4-11 cells. Western blot analysis for cleaved PARP and caspase-3. | ||
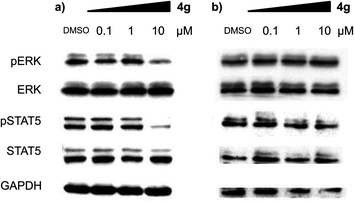 | ||
| Fig. 3 Compound 4g inhibits the phosphorylation of key signalling factors in the apoptotic process in a dose-dependent manner. Western blot analysis of phosphorylated and total ERK and STAT5 upon treatment of 4g at the different concentrations. Immunoblotting was performed with (a) MV-4-11 cells and (b) HL60 cells. | ||
We were particularly intrigued by the observation that there was no significant difference in the phosphorylation level of ERK and STAT5 in HL60 AML cells, which is known to have a quite low or no expression of FLT3,11 upon treatment of 4g under identical conditions with MV-4-11 AML cells (see Fig. 3b). In addition, there was no meaningful cellular toxicity in HL60 cells, unlike the high cytotoxicity in MV-4-11 cells (see Fig. S2 in ESI†). This observation, supported by our hypothesis, led us to examine whether our selective cytotoxic agent 4g can directly modulate FLT3 activity by inhibiting its phosphorylation. As shown in Fig. 4a, 4g inhibited the phosphorylation of FLT3, confirmed by the immunoblotting of phosphorylated and total FLT3 in MV-4-11 AML cells, in a dose-dependent manner. We further validated the inhibition of 4g on FLT3 in vitro activity by the treatment of 4g using a Kinase Hot Spot assay. Consistent with our western blotting data, 4g directly inhibited FLT3 activity with an IC50 of 1.3 μM. These results suggested that 4g blocked the phosphorylation of FLT3 and its kinase activity, which led to the subsequent blockage of the downstream signalling pathway, and inhibited the proliferation of MV-4-11 AML cells by triggering apoptosis. Therefore, we concluded that FLT3 is the direct molecular target of our selective cytotoxic agent 4g.
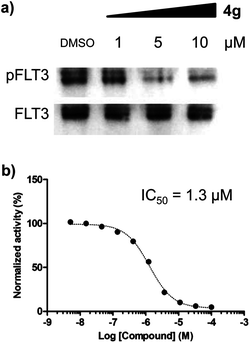 | ||
| Fig. 4 Compound 4g directly inhibited FLT3 activity. (a) Immunoprecipitation and western blot analysis for phosphorylated and total FLT3 in MV-4-11 cells. (b) A dose-dependent inhibitory effect of 4g on the in vitro enzymatic assay of FLT3. | ||
Selective inhibitory activity is an essential element for the development of kinase inhibitors as therapeutic agents or research tools.2 Therefore, after confirming the inhibitory activity of 4g against FLT3 along with its selective cytotoxicity to MV-4-11 AML cells, we tested the selectivity in its inhibitory activity against a panel of representative kinases, including the class III receptor tyrosine kinase (RTK) family, which has a high structural similarity with FLT3.12 As shown in Table 2, 4g showed excellent selectivity toward FLT3 over 40 different representative kinases. It is worth mentioning that 4g showed no inhibitory activities toward other class III RTKs, such as FMS, c-KIT, PDGFRα and PDGFRβ, at 10 μM, which is 10-fold higher than the IC50 value of 4g for FLT3. Therefore, we are confident that the selective cellular cytotoxicity of 4g to MV-4-11 AML cells is based on its selective inhibition of the kinase activity of FLT3 over other kinases.
| Kinase | Activity (%) | Kinase | Activity (%) |
|---|---|---|---|
| a Each experiment was performed in duplicate at 10 μM of 4g. The result was normalized by a DMSO control. | |||
| Class III receptor tyrosine kinase family | |||
| FMS | 101.5 | PDGFRα | 93.8 |
| FLT3 | 9.5 | PDGFRβ | 96.3 |
| c-KIT | 99.2 | ||
| Representative protein kinases | |||
| ABL1 | 88.9 | Lck | 95.2 |
| Akt1 | 93.6 | LYN | 86.6 |
| ALK | 96.5 | LKB1 | 101.1 |
| ASK1/MAP3K5 | 97.8 | MEK1 | 102.4 |
| ARAF | 77.0 | P38a/MAPK14 | 110.3 |
| Aurora A | 87.7 | P38b/MAPK11 | 97.8 |
| CDK1/cyclin A | 97.4 | p70S6K | 94.1 |
| CDK5/p35 | 87.2 | PAK1 | 98.9 |
| c-MET | 118.2 | PKA | 93.0 |
| c-Src | 78.7 | PKCa | 84.5 |
| EGFR | 91.2 | PLK1 | 107.5 |
| Erk1 | 95.4 | Raf1 | 94.9 |
| Erk2/MAPK1 | 94.0 | RET | 83.3 |
| FAK/PTK2 | 96.8 | STK2 | 85.8 |
| FGFR1 | 90.1 | TYK/LTK | 92.9 |
| IR | 95.0 | VEGFR1/FLT1 | 90.0 |
| JAK1 | 102.7 | VEGFR2/KDR | 98.6 |
| JNK1 | 101.4 | VEGFR3/FLT4 | 101.2 |
Conclusions
In summary, we identified a novel cell-specific cytotoxic agent containing the heterobiaryl pyrazolopyridine core skeleton 4 from phenotype-based cell viability profiling with over 3000 compounds, synthesized in-house using a pDOS strategy, against various cell lines. Our hit compound 4g induces apoptosis with an excellent selectivity in cellular cytotoxicity with an IC50 of 0.6 μM toward MV-4-11 AML cells over 12 different cell lines. To elucidate the mode of action of 4g, we pursued the hypothesis-driven deconvolution of protein targets and identified FLT3 kinase as a plausible target due to its specific expression in hematopoietic cells. Systematic western blot analysis revealed that the phosphorylation of ERK and STAT5, downstream signalling proteins of the FLT3 cascade, was blocked upon treatment of 4g only in FLT3-positive AML cells, but not in FLT3-negative HL60 AML cells. The western blotting and in vitro enzyme assay confirmed that compound 4g directly inhibits the phosphorylation of FLT3 in a dose-dependent manner with an IC50 of 1.3 μM on FLT3 kinase activity. The key feature of 4g is an excellent selectivity in the inhibition of FLT3 activity over 40 representative kinases, including class III receptor tyrosine kinase (RTK) family. Moreover, these results clearly proved the value of a cell-based phenotype assay as initial platform for the discovery of highly selective small molecules when it was combined with hypothesis-driven deconvolution or target identification. In fact, a continuous community effort has been devoted to the discovery of FLT3 inhibitors due to the importance of FLT3 as a potential drug target for the treatment of AML patients.13 Among the reported FLT3 inhibitors, AC220, developed by Bhagwat and co-workers,13a is one of the advanced FLT3 inhibitors because of its high selectivity over a panel of kinase and is currently in phase II clinical trials. However, the build-up resistance in AML relapsed patients toward AC220 was also reported in a recent paper.14 Therefore, it might be quite important to identify the new molecular framework 4g as a FLT3 inhibitor with high selectivity to address this resistance issue, which suggested beneficial information about selective anti-cancer therapy for AML.Acknowledgements
This study was supported by (1) a Chemical Genomics Research grant (NRF-2011-0000058); (2) a Global Frontier Project grant (NRF-M1AXA002-0032150); (3) the World Class University program (R31-2010-000-10032-0) of the National Research Foundation, funded by the Korean Ministry of Education, Science, and Technology. S.L and A.J are grateful for fellowships awarded by the BK21 Program. S.L received a Seoul Science Fellowship.References
- E. Gavathiotis, D. E. Reyna, J. A. Bellairs, E. S. Leshchiner and L. D. Walensky, Nat. Chem. Biol., 2012, 8, 639–645 Search PubMed.
- (a) H. Sakamoto, T. Tsukaguchi, S. Hiroshima, T. Kodama, T. Kobayashi, T. A. Fukami, N. Oikawa, T. Tsukuda, N. Ishii and Y. Aoki, Cancer Cell, 2011, 19, 679–690 Search PubMed; (b) J. W. Chang, M. J. Niphakis, K. M. Lum, A. B. Cognetta, C. Wang, M. L. Matthews, S. Niessen, M. W. Buczynski, L. H. Parsons and B. F. Cravatt, Chem. Biol., 2012, 19, 579–588 Search PubMed; (c) K. R. Brandvold, M. E. Steffey, C. C. Fox and M. B. Soellner, ACS Chem. Biol., 2012, 7, 1393–1398 Search PubMed.
- F. Sams-Dodd, Drug Discovery Today, 2005, 10, 139–147 CrossRef CAS.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS.
- S. Frye, M. Crosby, T. Edwards and R. Juliano, Nat. Rev. Drug Discovery, 2011, 10, 409–410 Search PubMed.
- G. C. Terstappen, C. Schlupen, R. Raggiaschi and G. Gaviraghi, Nat. Rev. Drug Discovery, 2007, 6, 891–903 CrossRef CAS.
- (a) C. L. Sawvers, Cancer Cell, 2002, 1, 413–415 Search PubMed; (b) T. Kindler, D. B. Lipka and T. Fischer, Blood, 2010, 116, 5089 Search PubMed.
- S. Oh and S. B. Park, Chem. Commun., 2011, 47, 12754–12761 RSC.
- S. Lee and S. B. Park, Org. Lett., 2009, 11, 5214–5217 CrossRef CAS.
- D. L. Stirewalt and J. P. Radich, Nat. Rev. Cancer, 2003, 3, 650–665 Search PubMed.
- K. Kojima, M. Konopleva, T. Tsao, M. Andreeff, H. Ishida, Y. Shiotsu, L. Jin, Y. Tabe and H. Nakakuma, Leukemia, 2010, 24, 33–43 Search PubMed.
- P. Blume-Jensen and T. Hunter, Nature, 2001, 411, 355–365 CrossRef CAS.
- (a) Q. Chao, K. G. Sprankle, R. M. Grotzfeld, A. G. Lai, T. A. Carter, A. M. Velasco, R. N. Gunawardane, M. D. Cramer, M. F. Gardner, J. James, P. P. Zarrinkar, H. K. Patel and S. S. Bhagwat, J. Med. Chem., 2009, 52, 7808–7816 CrossRef CAS; (b) B. D. Smith, M. Levis, M. Beran, F. Giles, H. Kantarjian, K. Berg, K. M. Murphy, T. Dauses, J. Allebach and D. Small, Blood, 2004, 103, 3669–3676 CrossRef CAS; (c) A.-M. O. 'Farrell, T. J. Abrams, H. A. Yuen, T. J. Ngai, S. G. Louie, K. W. H. Yee, L. M. Wong, W. Hong, L. B. Lee, A. Town, B. D. Smolich, W. C. Manning, L. J. Murray, M. C. Heinrich and J. M. Cherrington, Blood, 2003, 101, 3597–3605 Search PubMed; (d) W.-W. Li, X.-Y. Wang, R.-L. Zheng, H.-X. Yan, Z.-X. Cao, L. Zhong, Z.-R. Wang, P. Ji, L.-L. Yang, L.-J. Wang, Y. Xu, J.-J. Liu, J. Yang, C.-H. Zhang, S. Ma, S. Feng, Q.-Z. Sun, Y.-Q. Wei and S.-Y. Yang, J. Med. Chem., 2012, 55, 3852–3866 Search PubMed.
- C. C. Smith, Q. Wing, C.-S. Chin, S. Salemo, L. E. Damon, M. J. Levis, A. E. Perl, K. J. Travers, S. Wang, J. P. Hunt, P. P. Zarrinkar, E. E. Schadt, A. Kasarskis, J. Kuriyan and N. P. Shah, Nature, 2012, 485, 260–263 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cell viability data, detailed procedures for synthetic and biological experiments, and spectroscopic data of all new compounds. See DOI: 10.1039/c2md20169k |
| This journal is © The Royal Society of Chemistry 2013 |