Navigations of chemical space to further the understanding of polypharmacology in human nuclear receptors†
Antonio
Macchiarulo
*,
Andrea
Carotti
,
Marco
Cellanetti
,
Roccaldo
Sardella
and
Antimo
Gioiello
Dipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, via del Liceo 1, 06123 Perugia, Italy. E-mail: antonio@chimfarm.unipg.it; Fax: +39 075 585 5161; Tel: +39 075 585 5160
First published on 13th September 2012
Abstract
Recent years have seen an increasing awareness that drugs often bind to more than one molecular target, exhibiting polypharmacology. Although this aspect has commonly been considered as undesirable, promiscuity being responsible for unwanted side effects, in many cases it is a key component of the therapeutic efficacy of drugs. Nuclear receptors (NRs) are ligand-dependent transcription factors that offer important druggable targets for therapeutic interventions in multiple disease areas. Many NRs are promiscuous with respect to the wide range of ligands that act as modulators, and many NR modulators are not specific with respect to the number of NRs they bind. In this article, we aim to investigate aspects of ligand polypharmacology in the superfamily of human NRs. To this end, the construction of a target-centric and a ligand-centric chemical space is first discussed as instrumental in charting geometrical and physicochemical properties of NR binding sites and cognate ligands that, being characteristic of specific groups of receptors and/or modulators, could underlie their promiscuity. In the second part of the work, generating a graph network, we depict relationships among geometrical and physicochemical properties of binding sites that are used to infer aspects of polypharmacology in human NRs. Working hypotheses of NR ligand polypharmacology are thus generated and discussed in the light of case studies found in the literature.
Introduction
The human genome encodes 48 ligand-dependent transcription factors that belong to the superfamily of nuclear receptors (NRs).1 Besides members of NR0 subfamily, namely SHP and DAX-1,2–4 NRs are endowed with a common structural organization composed of three domains: a N-terminal ligand independent transactivation domain, a central DNA binding domain (DBD), and a C-terminal ligand binding domain (LBD). Crystallographic studies have shown that, in response to ligand binding, the LBD can adopt several conformational states, with two major extreme conformations corresponding to agonist and antagonist binding. Although this observation implies the possibility to design ligands endowed with different degrees of agonism and antagonism, it suggests the allosteric nature of NR modulation that may respond, beyond ligand binding, to a great variety of regulatory signals, including phosphorylation or interaction with additional transcription factors.5,6 The notion of NRs as allosteric switches is also supported by the existence of allosteric sites in the LBD endowed with regulatory functions.7On the basis of the signaling pathways involved in NR activation, two major groups of NRs have been suggested: endocrine hormone NRs and metabolic NRs.8 Members of the first group include the glucocorticoid (GR), mineralocorticoid (MR), estrogen (ER), androgen (AR), and progesterone (PR) receptors. These receptors bind to DNA as homodimers and show high affinity for their ligands in the nanomolar range of concentration. Notably, the biosynthesis of endogenous ligands for endocrine hormone receptors is regulated by negative-feedback control of the hypothalamic-pituitary axis. The signaling pathways of the first group of NRs regulate a plethora of metabolic and developmental functions, including carbohydrate metabolism, reproduction and sexual differentiation.
Metabolic NRs comprise early orphan receptors that have been adopted by disclosing their endogenous or synthetic ligands. Members of this group comprise receptors for fatty acids (PPARs), oxysterols (LXRs), bile acids (FXR), and xenobiotics (pregnane X receptor, PXR, and constitutive androstane receptor, CAR).
At odds with endocrine receptors, they generally bind to DNA as heterodimers, with the retinoid X receptor (RXR) being the common dimerization partner. Furthermore, ligands of metabolic receptors mostly include dietary products, intermediate metabolites and xenobiotics that bind to the LBD with affinities in the micromolar range of concentration.
According to the notion that metabolic NRs act as sensors of dietary metabolites and toxic compounds, they are primarily involved in the homeostatic control of metabolism and detoxification, favoring the adaptation of the organism to different environmental changes.
In addition to these two major groups of receptors, thyroid hormone receptors (TRs), retinoic acid receptors (RARs) and vitamin D receptor (VDR) form a unique cluster that does not fit into the above groups adopting features of both endocrine and metabolic NRs.
Collectively, NRs offer important druggable targets for therapeutic interventions in multiple disease areas, including cancer, metabolic and liver disorders to say a few.9–11 As a consequence, many projects are in the pipeline to develop small molecules that selectively modulate NR functions. In this context, addressing the inherent promiscuity of NRs represents a great challenge in the dissection of therapeutic and adverse polypharmacology of NR ligands.12–15
Accordingly, different computational studies have been reported in the literature investigating NR polypharmacology with either ligand-based or structure-based approaches.16–19
Recently, we have reported that exploring a target-centric chemical space of binding site properties may represent an effective strategy for studying bioisosteric relationships among ligands.20,21 The analysis of binding sites has also been previously exploited for assessing the specificity and/or promiscuity of ligand recognition in protein families.22–26
By charting a target-centric and a ligand-centric chemical space of NRs, in the first part of this work we aim to shed light on specific properties that could be characteristic of groups of NR binding sites and/or cognate ligands, accounting for their promiscuity. In the second part of the work, generating graph networks, we attempt to depict relationships among geometrical and physicochemical properties of binding sites as a way to infer ligand polypharmacology in human NRs.
Methods
A dataset of 339 structures of the ligand binding domain (LBD) of human NRs in complex with small molecules was collected from the RCSB protein database (www.pdb.org).27 Although structures with poorly solved regions in the binding pocket (namely residues with alternate positions and/or residues with missing atoms lying within 5 Å from the co-crystallized ligand) were not included in the dataset, human NRs with point mutations in the LBD were also included in the dataset, representing 15% of the total entries. The composition of the dataset is reported in Table 1 and Fig. 1; it spans 27 human NRs with an average resolution factor of 2.26 ± 0.41 for the crystal structures.| Name | Class | Group | Entries | Agonists | Partial agonists | Antagonists |
|---|---|---|---|---|---|---|
| TRα | NR1A1 | Endocrine/metabolic | 1 | 1 | 0 | 0 |
| TRβ | NR1A2 | Endocrine/metabolic | 17 | 16 | 0 | 1 |
| RARα | NR1B1 | Endocrine/metabolic | 1 | 0 | 0 | 1 |
| RARβ | NR1B2 | Endocrine/metabolic | 1 | 1 | 0 | 0 |
| RARγ | NR1B3 | Endocrine/metabolic | 9 | 9 | 0 | 0 |
| PPARα | NR1C1 | Metabolic | 9 | 8 | 0 | 1 |
| PPARβ,δ | NR1C2 | Metabolic | 12 | 11 | 1 | 0 |
| PPARγ | NR1C3 | Metabolic | 59 | 43 | 13 | 3 |
| RORα | NR1F1 | Metabolic | 2 | 2 | 0 | 0 |
| LXRα | NR1H3 | Metabolic | 4 | 4 | 0 | 0 |
| LXRβ | NR1H2 | Metabolic | 7 | 7 | 0 | 0 |
| FXRα | NR1H4 | Metabolic | 13 | 13 | 0 | 0 |
| VDR | NR1I1 | Endocrine/metabolic | 13 | 13 | 0 | 0 |
| PXR | NR1I2 | Metabolic | 7 | 6 | 0 | 1 |
| CAR | NR1I3 | Metabolic | 2 | 2 | 0 | 0 |
| HNF4α | NR2A1 | Metabolic | 2 | 2 | 0 | 0 |
| HNF4γ | NR2A2 | Metabolic | 1 | 1 | 0 | 0 |
| RXRα | NR2B1 | Metabolic | 24 | 21 | 3 | 0 |
| RXRβ | NR2B2 | Metabolic | 2 | 2 | 0 | 0 |
| ERα | NR3A1 | Endocrine | 55 | 28 | 9 | 18 |
| ERβ | NR3A2 | Endocrine | 20 | 15 | 1 | 4 |
| ERRα | NR3B1 | Metabolic | 1 | 0 | 0 | 1 |
| ERRγ | NR3B3 | Metabolic | 9 | 5 | 0 | 4 |
| GR | NR3C1 | Endocrine | 9 | 7 | 0 | 2 |
| MR | NR3C2 | Endocrine | 11 | 9 | 0 | 2 |
| PR | NR3C3 | Endocrine | 7 | 5 | 1 | 1 |
| AR | NR3C4 | Endocrine | 41 | 41 | 0 | 0 |
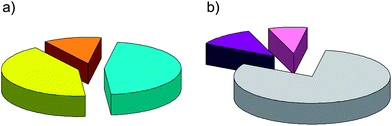 | ||
| Fig. 1 (a) Pie chart showing the composition of the dataset according to the signaling group of NRs: endocrine receptors (42%, yellow), metabolic/endocrine receptors (12%, orange), and metabolic receptors (46%, cyan). (b) Pie chart showing the composition of the subset of cognate ligands according to the pharmacological profile: agonists (80%, gray), partial agonists (8%, pink), and antagonists (12%, violet). | ||
The Protein Preparation tool implemented in Maestro28 was used to delete water molecules, detergent molecules and protein chains not containing co-crystallized ligand in the binding site. In particular, ligand unbound RXR chains were removed from heterodimeric NRs (metabolic and metabolic/endocrine NRs), while ligand bound RXRs were kept in the dataset (Table s1,†e.g.: 1FM6_A, 1FM9_A, 1XV9_A, 1XVP_A, and 2ACL_A). In the case of endocrine NRs, only one chain containing the co-crystallized ligand was included in the dataset.
All hydrogen atoms were added to the residues and bond orders were fixed using the OPLS-2005 force field. During this step, the protonation state of charged residues and cognate ligands was set at the physiological pH of 7.0. The most stable tautomeric form of histidines was established to maximize the hydrogen bond network. The complete list of pdb codes, chains, and cognate ligands of human NRs used in this study is provided in the ESI (Table s1†). The SiteMap program was instrumental to define the binding site of each receptor, taking the co-crystallized ligand as a reference template. Briefly, SiteMap calculation is composed of three steps: initially, grids in rectilinear boxes are computed around the crystallized structures with a grid-point separation of 0.7 Å; site points are then placed and subsequently grouped into sets in order to define the binding cleft; finally, fifteen molecular descriptors are calculated, including the number of site points (size), exposure (exp), enclosure (encl), number of van der Waals contacts with ligand (contact), hydrophobic character (phobic), hydrophilic character (philic), hydrophobic/hydrophilic balance (balance), ratio between hydrogen bond donors and hydrogen bond acceptors (donor/acceptors), site score (S-score), druggability score (D-score), volume, and four ligand positioning descriptors (refdist, refmin, refavg, and sitemin values). The description of each of these descriptors can be found in the original publication.29
Ligands co-crystallized in the LBD of the dataset were extracted in their relative bioactive conformations and used to define a second subset for this study (cognate ligand subset).
A set of descriptors was then calculated using Quikprop and the cognate ligand subset. This set of descriptors included molecular weight (MW), volume (Vol), number of hydrogen bond donor groups (H-don), number of hydrogen bond acceptor groups (H-Acc), octanol/water partition coefficient (QPLogPo/w), solvent accessible surface area (SASA), solvent accessible polar surface area (PSA), hydrophobic component of the solvent accessible surface area (FOSA), hydrophilic component of the solvent accessible surface area (FISA), aromatic component of the solvent accessible surface area (PISA), and weakly polar component of the solvent accessible surface area (WPSA).
Statistical analysis of these descriptors was carried out using principal component analysis (PCA). In particular, two PCAs were carried out in this study. PCA-1 was performed on binding site descriptors (SiteMap) as calculated from the LBD dataset of human NRs in complex with small-molecule ligands and PCA-2 was run using molecular descriptors (Quikprop) as calculated on the subset of co-crystallized ligands. The skewness value measures the asymmetry of the distribution and was calculated using the following formula:
| Skewness = [(Average) − (Median)]/(Standard deviation) | (1) |
Principal components from PCA-1 were also used to calculate Pearson correlation coefficients between pairs of binding sites, thereby generating a similarity matrix of 339 × 339. Binding sites endowed with a coefficient higher than 0.9 were selected as nodes connected with edges to draw a network of property relationships. The network graph was constructed using Cytoscape and the edge-weighted spring embedded layout method, as implemented in the program.30 The average clustering coefficient (〈C〉) of the network was calculated using the following formula:
| 〈C〉 = (n − 1)/k | (2) |
Results
The datasets
A binding site dataset and a cognate ligand subset were defined by collecting the crystal structures of 339 ligand binding domains (LBD) of 27 human NRs. The composition of the datasets is shown in Fig. 1 according to the pharmacological profile of the ligand co-crystallized in the LBD and to the paradigm of three signaling groups of NRs (endocrine, metabolic, and endocrine/metabolic), respectively. Table 1 reports the number of structures and cognate ligands for each of the 27 human NRs sampled in the datasets. In particular, we deemed it of interest to keep all the structures and cognate ligands for a given human NR in the datasets, since they may represent different experimental conformations of the relative LBD in response to ligand binding. In this regard, it should be mentioned that ligand affinity was not taken into account as a cutoff to restrict the number of structures in the datasets, given the following considerations: (i) the emphasis of the study was placed on binding site properties of NRs rather than properties of cognate ligands, and affinity is mostly a ligand's property rather than a target's property; and (ii) only 18% of NR-ligand complexes in our dataset have ligand affinity data annotated in the PDBbind database.31,32A target-based chemical space of NR binding sites
The binding site dataset was instrumental to calculate a pool of molecular descriptors of NR binding sites which were used to perform a principal component analysis (PCA-1), thereby generating a target-centric chemical space of NR ligand binding pockets (Fig. 2). The first four components showed eigenvalues higher than 1, covering 81% of the variance of the dataset. Table 2 reports the loadings of the original variables into these components, revealing the contribution of the original descriptors to the relative component.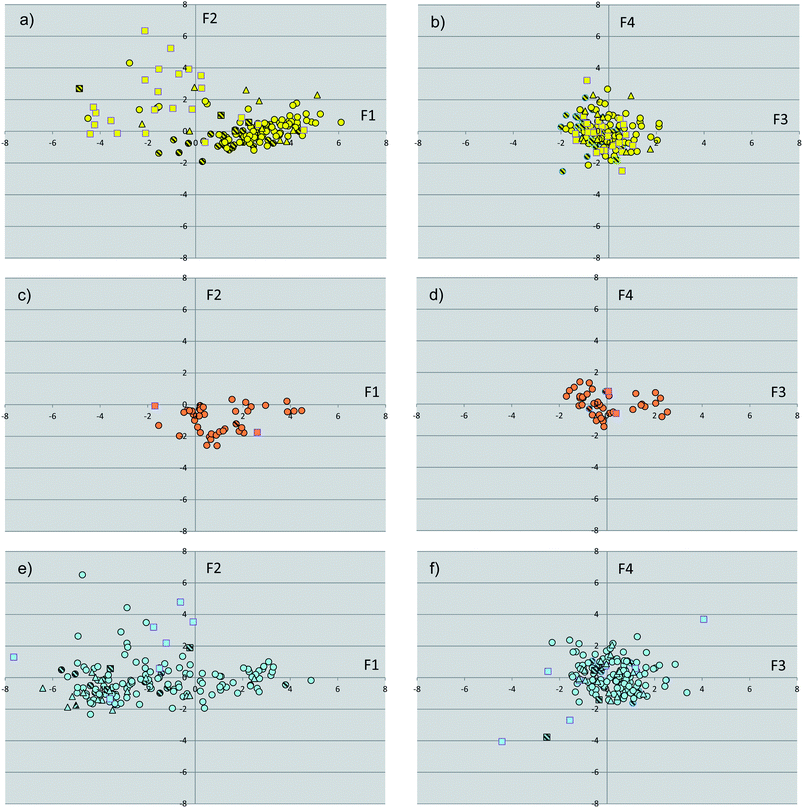 | ||
| Fig. 2 Plots of the scores of first two components (F1, F2, PCA-1) for binding sites of endocrine receptors (a), endocrine/metabolic receptors (c) and metabolic receptors (e). Plots of the scores of the third and fourth component (F3, F4, PCA-1) for binding sites of endocrine receptors (b), endocrine/metabolic receptors (d) and metabolic receptors (f). Binding sites in complex with agonists are shown as circles, with partial agonists as triangles, with antagonists as squares. Binding sites with point mutations are filled with line patterns. | ||
| Descriptors | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| Eigenvalue = 8.26 (55.11%) | Eigenvalue = 1.79 (11.92%) | Eigenvalue = 1.11 (7.42%) | Eigenvalue = 1.01 (6.73%) | |
| SiteScore | 0.337 | 0.033 | 0.135 | 0.042 |
| Size | −0.282 | −0.227 | 0.282 | 0.063 |
| D-score | 0.328 | 0.143 | 0.204 | 0.023 |
| Volume | −0.305 | −0.057 | 0.275 | 0.069 |
| Exposure | −0.295 | 0.176 | 0.112 | −0.038 |
| Enclosure | 0.297 | −0.293 | −0.100 | 0.082 |
| Contact | 0.223 | −0.473 | −0.075 | 0.067 |
| Phobic | 0.324 | 0.031 | 0.222 | 0.073 |
| Philic | −0.283 | −0.329 | −0.310 | 0.012 |
| Balance | 0.287 | 0.156 | 0.399 | 0.062 |
| Don/acc | 0.041 | 0.343 | −0.367 | −0.422 |
| Refdist | −0.270 | 0.037 | 0.270 | 0.000 |
| Refmin | −0.027 | 0.145 | −0.284 | 0.868 |
| Refavg | −0.118 | 0.541 | 0.003 | 0.176 |
| Sitemin | −0.156 | −0.134 | 0.406 | 0.060 |
The inspection of Fig. 2 reveals that binding sites with point mutations in the LBD (15% of the whole dataset) are not outliers in chemical space, occupying similar regions of the property space wherein wild-type NRs are located. These structures were thus kept in the dataset, not influencing the overall analysis.
Although Fig. 2 shows a discrete clustering of NRs along the first component (F1), they mostly overlap at values of the remaining components (F2–F4). Accordingly, we deemed it of interest to deepen the analysis of the molecular descriptors contributing to the first component. The component F1 mainly encodes hydrophobic effects (phobic), size (volume) and druggability (SiteScore, DrugScore) of the binding sites. More in detail, negative values of F1 are characteristic of large and hydrophilic binding sites with poor druggability score. Conversely, positive values of F1 feature smaller and hydrophobic binding sites with high druggability scores. Fig. 3 reports distribution histograms of F1 values according to the paradigm of signaling groups of NRs. The inspection of these histograms shows that metabolic NRs mostly occupy negative values of the first component (F1 mean = −1.81 ± 2.56), while skewing to positive values of F1 (skewness = 0.155). In contrast, endocrine NRs are preferentially located at positive values of F1 (F1 mean = 1.64 ± 2.34) with a negative skewness (skewness = −0.267). Metabolic/endocrine NRs are grouped in a narrow region at low values of F1 (F1 mean = 1.04 ± 1.44), showing a skewness at positive F1 values (skewness = 0.263).
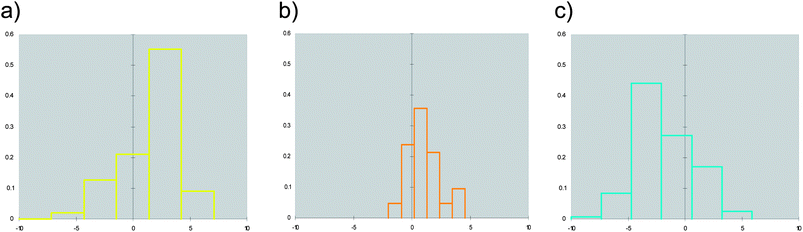 | ||
| Fig. 3 Distributions of F1 values for binding sites of endocrine receptors (a), endocrine/metabolic receptors (b) and metabolic receptors (c). | ||
A ligand-based chemical space of NR ligands
The subset of cognate ligands was instrumental to calculate a pool of molecular shape descriptors and run an additional principal component analysis (PCA-2). The results of this analysis are shown in Fig. 4 and Table 3. Collectively, the first four components (F1′–F4′) explain approximately 87% of the variance of the dataset of ligands, with F1′–F3′ showing eigenvalues higher than 1. Inspection of the loadings (Table 3) of original variables into these components reveals that the first component (F1′, variance 37%) is correlated to the size of ligands (SASA, volume, and MW), with positive and negative values encoding large and small molecules, respectively. On the other side, positive values of F2′ (F2′, variance 25%) mainly encode hydrophilic ligands (FISA, PSA), whereas negative values of the second component are mostly affected by the octanol/water partition coefficient (cLogP) of ligands. Positive values of F3′ encode the hydrophobic component of the solvent accessible surface area (FOSA), whereas negative F3′ values are related to the aromatic and weakly polar components of the solvent accessible surface area (PISA and WPSA). Finally, the fourth component (F4′) discriminated at positive values the contribution of WPSA and at negative values the contribution of PISA.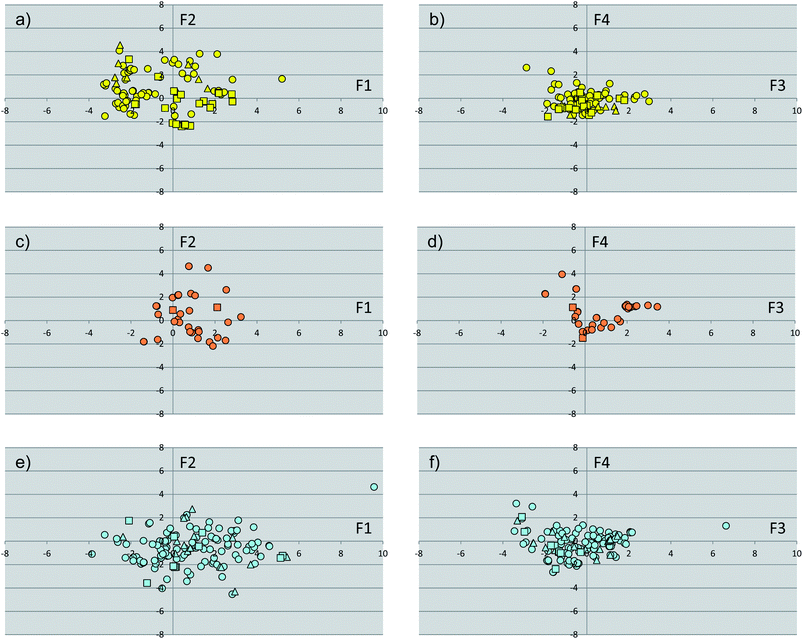 | ||
| Fig. 4 Plots of the scores of first two components (F1, F2, PCA-2) for the subset of cognate ligands of endocrine receptors (a), endocrine/metabolic receptors (c) and metabolic receptors (e). Plots of the scores of the third and fourth component (F3, F4, PCA-1) for the subset of cognate ligands of endocrine receptors (b), endocrine/metabolic receptors (d) and metabolic receptors (f). Agonists are shown as circles, partial agonists as triangles, and antagonists as squares. | ||
| Descriptors | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| Eigenvalue = 4.03 (36.66%) | Eigenvalue = 2.81 (25.59%) | Eigenvalue = 1.77 (16.09%) | Eigenvalue = 0.95 (8.63%) | |
| Volume | 0.461 | −0.203 | 0.081 | −0.034 |
| DonorHB | 0.070 | 0.345 | 0.202 | 0.265 |
| AccptHB | 0.360 | 0.182 | 0.225 | −0.090 |
| QPlogPo/w | 0.287 | −0.409 | −0.198 | 0.020 |
| MW | 0.438 | 0.065 | −0.152 | 0.314 |
| SASA | 0.458 | −0.194 | 0.049 | −0.080 |
| FOSA | 0.087 | −0.394 | 0.533 | 0.170 |
| FISA | 0.096 | 0.478 | 0.254 | −0.109 |
| PISA | 0.210 | 0.110 | −0.482 | −0.592 |
| WPSA | 0.129 | 0.199 | −0.467 | 0.638 |
| PSA | 0.301 | 0.402 | 0.196 | −0.129 |
As far as the paradigm of NR signaling groups is concerned, the analysis of Fig. 4 shows the absence of specific regions of the space occupied by discrete classes of compounds, making the identification of specific properties that characterize NR ligands difficult. Nevertheless, in agreement with the high chemical diversity of metabolic NR modulators, a wider spreading of ligands for this group of receptors is observed with respect to ligands for endocrine and metabolic/endocrine NRs.
Networks of NR binding sites
Networks are graphs representing relationships. They are composed of nodes and edges, namely the objects and their relationships, respectively. In a biological context, the latter may be physical interactions (e.g. ligand–protein interactions and/or protein–protein interactions), regulatory interactions (e.g. activation or inhibition events), genetic interactions (e.g. gene expression correlations), metabolic reactions, or relationships in a broad sense.33 In particular, relationships among NRs may be assessed by calculating the Pearson correlation coefficients on the geometrical and physicochemical properties of the binding sites. In this context, the presence of high correlated properties between a couple of NR binding sites may hint at the presence of conserved patterns of interactions, providing clues to the promiscuous cross-recognition of the relative ligands. Furthermore, the presence of strong relationships between binding site properties of NRs may be envisaged as the existence of an evolutionary linkage between their genes.In this part of the study, a network graph was generated for NR binding sites on the basis of the Pearson correlation coefficient as calculated on the principal components of the target-based chemical space. In particular, all of the principal components were used in order to keep the full variance of the original set of descriptors. The advantage of using principal components instead of the original descriptors grounds on the fact that these new variables are uncorrelated, arising from linear combinations of the original variables. Uncorrelated variables are fully orthogonal and, as such, more useful than correlated variables to highlight relationships among objects.
Thus, all pairs of binding sites showing Pearson correlation coefficients higher than 0.9 were selected as nodes and connected with edges to draw a network of property relationships. As a result, the network (Fig. 5) is composed of 246 nodes and 1060 edges with an average clustering coefficient (〈C〉) of 0.021.
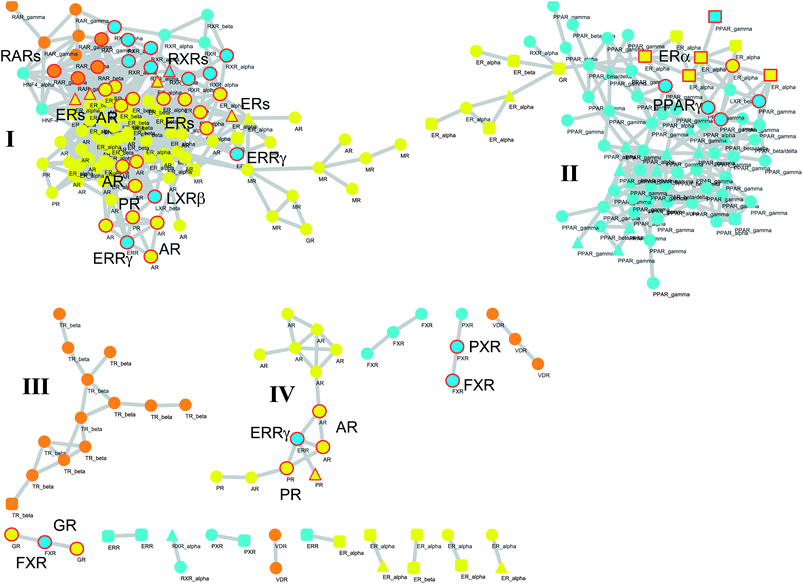 | ||
| Fig. 5 Network graph of binding sites. Each node represents a binding site bound to agonist (circle), partial agonist (triangle), or antagonist (square). Colour codes are referred to endocrine receptors (yellow), endocrine/metabolic receptors (orange) and metabolic receptors (cyan). Edges are placed between couples of nodes if the Pearson correlation coefficient between the relative properties of the binding sites is higher than 0.9. Large and medium clusters are labelled with roman numbers (I, II, III, and IV). NRs selected according to the guilt by association principle and discussed in the manuscript are labeled and highlighted with red circles. | ||
Discussion
Human NRs comprise 48 members of ligand-dependent transcription factors that offer important druggable targets for therapeutic interventions in multiple disease areas. Many NRs are promiscuous with respect to the wide range of ligands that act as modulators, and many NR ligands are not specific with respect to the number of NRs they bind.12,13 Furthermore, promiscuity of NRs has also been reported with respect to their involvement in biological pathways.17 Ligand, protein and pathway promiscuity are thus at the basis of polypharmacology in human NRs.In this work, we have first investigated geometrical and physicochemical properties of binding sites and cognate ligands that, being characteristic of specific signaling groups of NRs, could underlie their promiscuity. Second, generating graph networks, we have attempted to depict relationships among properties of binding sites to further the understanding of ligand polypharmacology in human NRs.
To address the first issue, we have investigated the distribution of specific groups of NR binding sites and ligands in a target-centric and a ligand-centric chemical space of geometrical and physicochemical properties, respectively.
Although our results revealed the absence of specific regions of the ligand-centric chemical space occupied by discrete classes of NR ligands, a nice clustering of NR binding sites could be observed along the first component (F1) of the target-centric chemical space, suggesting the presence of specific properties that characterize the ligand binding cleft of NRs according to the paradigm of different signaling groups.
Another observation concerns the regions occupied by NR binding sites and cognate ligands according to the pharmacological profile of agonism (circles in Fig. 2 and 4), partial agonism (triangles in Fig. 2 and 4) and antagonism (squares in Fig. 2 and 4). Again, no difference is observed as far as the ligand-centric chemical space is considered. Conversely, in the target-centric chemical space binding sites of endocrine NRs in complex with antagonist ligands (yellow squares) mainly populate a region located at negative F1 values and positive F2 values, suggesting the engagement of large and more hydrophilic conformations.
At first glance, these findings may not be surprising for medicinal chemists working in the field of NRs, being in agreement with the general properties of small lipophilic agonists and large hydrophilic antagonists of estrogen receptors, as well as the general knowledge of metabolic NRs having large and rather hydrophilic binding sites and endocrine NRs showing narrow and hydrophobic binding clefts. However, our explorations in chemical space suggest that these agreements are clear as far as a target-centric chemical space is considered and that NR binding sites are able to cover continuous distributions of physicochemical and geometrical properties, by engaging diverse conformations, with relatively overlapping parts at the extreme of the distributions. In particular, the inspection of the distribution histogram of endocrine/metabolic NRs on F1 values (Fig. 3) reveals that these receptors have mixed properties with respect to metabolic and endocrine NRs.
Interestingly, while this latter observation provides a suggestive link to functional aspects of TRs, RARs and VDR that adopt features of both endocrine and metabolic NRs, the rather opposite (albeit continuous) properties of the other two NR signaling groups may account for the low and high affinity binding of their respective endogenous ligands, as well as provide clues on the molecular basis of NR promiscuity in ligand recognition.
Number and types of factors affecting protein promiscuity have been recently reviewed by Nobeli and coworkers.34 At the protein level, these factors include the alternative or combined presence of multiple binding sites, single larger site with diverse interacting residues, extensive hydrophobic interactions, and protein flexibility.35–37
In agreement with the presence of a large site with diverse interacting residues (a sort of volume-dependent site promiscuity), the larger binding site featuring the LBD of metabolic NRs may account for promiscuous ligand recognition at this group of receptors. For instance, the metabolic nuclear receptor FXR shows a binding site volume of 536 Å3 (Fig. 6). Obeticholic acid (OCA, 1, Chart 1), a bile acid derivative now advancing in the phase III clinical study for primary biliary cirrhosis (PBC),38,39 occupies only 47% of the cavity (pdb code: 1OSV).40,41 The large binding site of FXR may thus provide an explanation to why it is not a difficult task finding potent and efficacious non-steroidal FXR ligands using structure-based drug design approaches, as evidenced by the many successful studies reported in the literature.42–49 Furthermore, supporting our results, several crystal structures of FXR have been disclosed in complex with different classes of ligands (2–4), showing diverse binding modes in the large LBD cleft that involve interactions with a conserved motif of residues (Fig. 7).
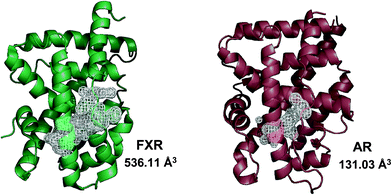 | ||
| Fig. 6 Metabolic NRs feature a larger binding site than endocrine NRs. The cartoon structure of FXR (green cartoon, pdb code 3DCT) and its binding site volume with respect to the binding site volume observed in the crystal structure of AR (brown cartoon, pdb code 1AX6) is shown here. | ||
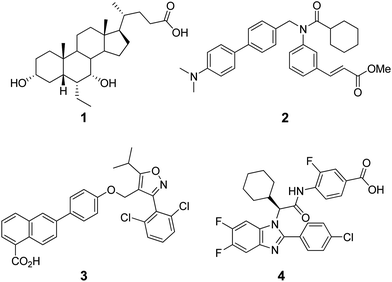 | ||
| Chart 1 Structures of steroidal (1) and non-steroidal (2–4) FXR agonists. | ||
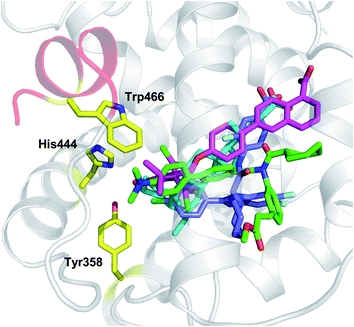 | ||
| Fig. 7 Examples of crystal structures of FXR (pdb codes 1OSV, 1OSH, 3DCU, 3OMM) in complex with steroidal (OCA, 1 cyan) and non-steroidal ligands (Fexaramine 2, green; 3, magenta; 4, blue) show diverse binding modes and interactions with a conserved motif of residues (Tyr358, His444, Trp466). | ||
The calculated low druggability of metabolic NRs may in part account for their poor exploitation as drug targets by the pharmaceutical industry. According to the recent analysis of Rask-Andersen and coworkers,50 metabolic NRs cover only 0.8% of 2242 annotated drug–target interactions, akin to 0.8% of metabolic/endocrine NRs and opposed to 3.0% of endocrine NRs.
In contrast to metabolic NRs, endocrine NRs are endowed with narrow and pretty hydrophobic binding clefts. In line with the aforementioned factors contributing to protein promiscuity, it is the presence of extensive hydrophobic interactions that may account for promiscuous molecular recognition in this signaling group of NRs. Supporting this observation, very well-known examples in the literature are glucocorticoid hormones, such as cortisol (5, Chart 2) and corticosterone (6), which bind to MR with a similar affinity to aldosterone (7), the main endogenous hormone of MR. It is worth noting that the specificity of aldosterone binding to MR is ascribed to factors beyond direct hydrophobic and/or hydrogen bonding interactions with the receptor, and including indirect interactions with a set of 25 residues (820–844) located outside the binding cleft, as well as to the cellular co-expression of 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) which metabolizes cortisol and corticosterone into inactive metabolites at MR.51
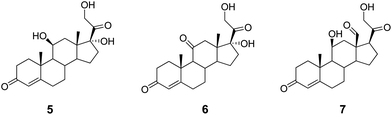 | ||
| Chart 2 Structures of glucocorticoid (5, 6) and mineralcorticoid (7) hormones. | ||
In the second part of the study, given the nice agreement between binding site properties and specific signaling groups of NRs, we attempted to relate geometrical and physicochemical properties of NR binding sites with the aim of identifying clefts with high correlated properties as a means of inferring ligand polypharmacology in human NRs. Accordingly, a network graph was generated using a correlation matrix among NR binding sites on the basis of their geometrical and physicochemical properties. The inspection of the network reveals the presence of a specific pattern composed of two large clusters (I and II, Fig. 5), two medium clusters (III and IV) and several small clusters comprising three or two binding sites.
The two large clusters are in agreement with the paradigm of signaling groups of NRs, with the first cluster (I) being mainly composed of endocrine NR binding sites and the second cluster (II) comprising mostly metabolic NR binding sites. Accordingly, relationships exist between geometrical and physicochemical properties of NR binding sites within groups of signaling NRs that might be envisaged as arising from a common evolutionary process of an early ligand bound ancestor.52
Another observation concerns the change of geometrical and physicochemical properties of the binding site occurring in different ligand bound conformations of the same receptor. For instance, this is evidenced by the diverse geometrical and physicochemical properties of the ERα binding site conformations that may alternatively be either related to properties occurring in NR binding sites of cluster-I and binding sites of cluster-II or even form unique small cluster of binding sites with specific properties. These data evidence the remarkable plasticity of the binding site of some NRs, such as ERα, that may adopt diverse geometrical and physicochemical properties to favor the binding of different classes of compounds.53
Assuming the guilt by association principle, cross-binding can thus be expected in ligands targeting NRs of linked nodes in the network. More in detail, the core of cluster-I is composed of endocrine NR binding sites that are linked to two small groups of interconnected endocrine/metabolic NRs and metabolic NRs. These latter comprise RARs, RXRs and HNF4α, while a few other metabolic NRs form additional nodes of this cluster, including ERRγ and LXRβ receptors (Fig. 5).
From a thorough search in the literature, compounds containing a diphenylamine scaffold (8, Chart 3) were found to preferentially bind a number of NRs including RARs, RXRs, AR and ERs.54-56 Likewise, diethylstilbestrol (9), tamoxifen (10), and 4-hydroxytamoxifen (11) were described in the literature as high-affinity ligands for both ERs and ERRγ.57 The regulation of ERα, AR, PR and ERRγ activities was reported as the molecular basis by which some phenols (e.g.12–14) modulate the endocrine systems of wildlife and humans.58 Recently, the most used agonist (T0901317, 15) to investigate the physiological roles of LXRs has been demonstrated to compete with androgen hormones for AR binding, showing antiandrogen activity.59
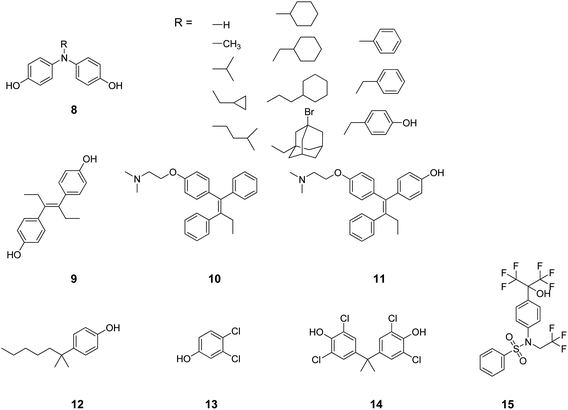 | ||
| Chart 3 Structures of ligands showing cross-binding at NRs of cluster I. | ||
The second cluster (II) is mainly composed of metabolic NRs connected to a small number of endocrine NRs, including ERα (Fig. 5). Again, in the literature it is reported that phytoestrogens (e.g.16 and 17, Chart 4), plant-derived non-steroidal compounds with estrogen-like activity, can dose-dependently activate PPARγ, thereby inducing divergent effects on osteogenesis and adipogenesis.60
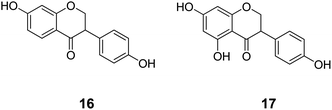 | ||
| Chart 4 Structures of ligands showing cross-binding at NRs of cluster II. | ||
Although these observations collectively support the results of our study, it should be mentioned that the network approach we propose is not a predictive tool, but rather a way to generate working hypotheses by relating binding site properties. This is exemplified by the case of FXR, which is linked to PXR and GR in the network, forming small clusters of binding sites with correlated properties.
On the basis of the guilt by association principle, as a first hypothesis, PXR cross-activity can be expected in ligands targeting FXR (Fig. 5). Sustaining this hypothesis, cafestol (18, Chart 5), a diterpene compound found in unfiltered coffee brews, has been shown to act as an agonist of FXR and PXR, regulating cholesterol homeostasis.61 Some polyhydroxylated steroids (e.g.19–21) from the marine sponge Theonella swinhoei have recently been reported as PXR and FXR ligands.62–64 Litocholic acid (LCA, 22), a toxic secondary bile acid formed by the bacterial biotransformation of chenodeoxycholic acid (CDCA, 23), regulates bile acid homeostasis by binding and modulating the activity of FXR and PXR.65,66 As a second hypothesis, GR cross-activity can alternatively be expected in ligands binding FXR (Fig. 5). Interestingly, UDCA (24), another natural bile acid structurally related to the endogenous FXR ligand (20), has been reported in the literature to be able to activate and induce GR translocation in the low micromolar range of potency by binding to the LBD of the receptor.67,68 Conversely to LCA and CDCA, however, UDCA does not bind FXR,69–71 suggesting how the stereochemistry at position C7 of the bile acid scaffold is crucial for bestowing FXR or GR activity. The fallacy of the second hypothesis may thus be ascribed to the molecular descriptors of the binding site used in this study, which do not capture the 3D information related to the stereospecificity of the molecular recognition.
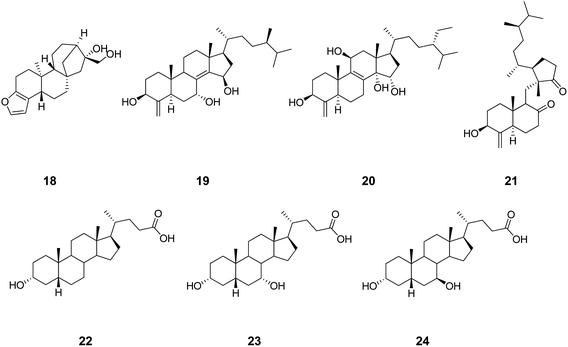 | ||
| Chart 5 Structures of ligands showing cross-binding at FXR, PXR and GR. | ||
Although the results of this study may be used retrospectively to get insights into the mechanism of action of already identified NR ligands, another use of the network graph of NR binding sites can be envisaged in aiding the design and/or optimization of NR lead compounds, taking into proper consideration issues of polypharmacology at the superfamily level in an early stage of the drug discovery process. In this context, the inspection of the network may provide clues to medicinal chemists on binding cleft conformations of additional NRs (targets or anti-targets) that, being closely related to the target NR binding site, can be incorporated into the drug discovery pipeline.
It is also important to stress that, having neglected ligand affinity data in the analysis, the hypotheses generated from the inspection of the network can only have a qualitative basis, being not informative on the potency of ligands that are expected to show cross-activity at linked target nodes.
Conclusions
The objective of this work has been the investigation of aspects of polypharmacology in the superfamily of human NRs. Pursuing this aim, we have first explored a target-centric and a ligand-centric chemical space of NRs to identify specific properties of binding sites and/or cognate ligands that could provide clues to the molecular basis of NR promiscuity.Reflecting the paradigm of NR signaling groups, three regions of binding sites could thus be observed along the first component of the target-centric chemical space of NRs, while no specific regions could be identified in the property space of cognate ligands. The properties encoded by these regions are in agreement with the general knowledge of NRs, with binding sites of metabolic NRs being large and rather hydrophilic, endocrine NRs showing narrow and hydrophobic binding clefts, and endocrine/metabolic NRs having mixed properties with respect to the former groups. Hence, the presence of a large binding site with diverse interacting residues explains protein promiscuity in metabolic NRs, whereas extensive hydrophobic interactions account for promiscuous molecular recognition in endocrine NRs.
In the second part of the work, generating a graph network and using a guilt by association principle, we have investigated whether the presence of cross-correlations among geometrical and physicochemical properties of NR binding sites could be instrumental to infer ligand polypharmacology in NRs. Case studies were found in the literature that support the results of this approach, sustaining the hypothesis that binding sites with correlated properties may share cross-binding of ligands. However, it should be stressed that our approach is not a predictive tool but a method to generate hypotheses and drive future experiments of ligand profiling at NRs, with the aim of investigating therapeutic and adverse polypharmacology.
References
- H. Gronemeyer, J. A. Gustafsson and V. Laudet, Nat. Rev. Drug Discovery, 2004, 3, 950–964 CrossRef CAS.
- A. Ehrlund and E. Treuter, J. Steroid Biochem. Mol. Biol., 2012, 130, 169–179 Search PubMed.
- A. Macchiarulo, G. Rizzo, G. Costantino, S. Fiorucci and R. Pellicciari, J. Mol. Graphics Modell., 2006, 24, 362–372 Search PubMed.
- E. P. Sablin, A. Woods, I. N. Krylova, P. Hwang, H. A. Ingraham and R. J. Fletterick, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18390–18395 Search PubMed.
- H. Faus and B. Haendler, Biomed. Pharmacother., 2006, 60, 520–528 CrossRef CAS.
- J. Osz, Y. Brelivet, C. Peluso-Iltis, V. Cura, S. Eiler, M. Ruff, W. Bourguet, N. Rochel and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E588–E594 Search PubMed.
- T. W. Moore, C. G. Mayne and J. A. Katzenellenbogen, Mol. Endocrinol., 2010, 24, 683–695 CrossRef CAS.
- A. Chawla, J. J. Repa, R. M. Evans and D. J. Mangelsdorf, Science, 2001, 294, 1866–1870 Search PubMed.
- C. J. Ryan and D. J. Tindall, J. Clin. Oncol., 2011, 29, 3651–3658 Search PubMed.
- T. P. Burris, S. A. Busby and P. R. Griffin, Chem. Biol., 2012, 19, 51–59 Search PubMed.
- M. Trauner, A. Baghdasaryan, T. Claudel, P. Fickert, E. Halilbasic, T. Moustafa and G. Zollner, Dig. Dis., 2011, 29, 98–102 Search PubMed.
- N. Noy, Biochemistry, 2007, 46, 13461–13467 CrossRef CAS.
- C. Pilgrim, Eur. J. Histochem., 1999, 43, 261–264 Search PubMed.
- A. D. Boran and R. Iyengar, Curr. Opin. Drug Discovery Dev., 2010, 13, 297–309 Search PubMed.
- G. V. Paolini, R. H. Shapland, W. P. van Hoorn, J. S. Mason and A. L. Hopkins, Nat. Biotechnol., 2006, 24, 805–815 CrossRef CAS.
- G. Wohlfahrt, J. Sipila and L. O. Pietila, Biopolymers, 2009, 91, 884–894 Search PubMed.
- K. Hettne, M. Cases, S. Boyer and J. Mestres, Curr. Top. Med. Chem., 2007, 7, 1530–1536 Search PubMed.
- J. Mestres, L. Martin-Couce, E. Gregori-Puigjane, M. Cases and S. Boyer, J. Chem. Inf. Model., 2006, 46, 2725–2736 CrossRef CAS.
- M. Cases, R. Garcia-Serna, K. Hettne, M. Weeber, J. van der Lei, S. Boyer and J. Mestres, Curr. Top. Med. Chem., 2005, 5, 763–772 CrossRef CAS.
- A. Macchiarulo, R. Nuti, G. Eren and R. Pellicciari, J. Chem. Inf. Model., 2009, 49, 900–912 Search PubMed.
- A. Macchiarulo and R. Pellicciari, J. Mol. Graphics Modell., 2007, 26, 728–739 Search PubMed.
- R. J. Najmanovich, A. Allali-Hassani, R. J. Morris, L. Dombrovsky, P. W. Pan, M. Vedadi, A. N. Plotnikov, A. Edwards, C. Arrowsmith and J. M. Thornton, Bioinformatics, 2007, 23, e104–e109 Search PubMed.
- M. Bashton, I. Nobeli and J. M. Thornton, J. Mol. Biol., 2006, 364, 836–852 CrossRef CAS.
- F. Milletti and A. Vulpetti, J. Chem. Inf. Model., 2010, 50, 1418–1431 CrossRef CAS.
- S. L. Kinnings and R. M. Jackson, J. Chem. Inf. Model., 2009, 49, 318–329 Search PubMed.
- G. Ausiello, P. F. Gherardini, E. Gatti, O. Incani and M. Helmer-Citterich, BMC Bioinf., 2009, 10, 182 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- L. C. C. Schrodinger, Maestro, v. 9.2, New York, NY, 2009 Search PubMed.
- T. A. Halgren, J. Chem. Inf. Model., 2009, 49, 377–389 Search PubMed.
- M. E. Smoot, K. Ono, J. Ruscheinski, P. L. Wang and T. Ideker, Bioinformatics, 2011, 27, 431–432 CrossRef CAS.
- R. Wang, X. Fang, Y. Lu, C.-Y. Yang and S. Wang, J. Med. Chem., 2005, 48, 4111–4119 CrossRef CAS.
- R. Wang, X. Fang, Y. Lu and S. Wang, J. Med. Chem., 2004, 47, 2977–2980 CrossRef CAS.
- D. Merico, D. Gfeller and G. D. Bader, Nat. Biotechnol., 2009, 27, 921–924 Search PubMed.
- I. Nobeli, A. D. Favia and J. M. Thornton, Nat. Biotechnol., 2009, 27, 157–167 CrossRef CAS.
- M. R. Redinbo, Drug Discovery Today, 2004, 9, 431–432 Search PubMed.
- N. Tokuriki and D. S. Tawfik, Science, 2009, 324, 203–207 CrossRef CAS.
- A. Babtie, N. Tokuriki and F. Hollfelder, Curr. Opin. Chem. Biol., 2010, 14, 200–207 CrossRef CAS.
- R. Pellicciari, S. Fiorucci, E. Camaioni, C. Clerici, G. Costantino, P. R. Maloney, A. Morelli, D. J. Parks and T. M. Willson, J. Med. Chem., 2002, 45, 3569–3572 CrossRef CAS.
- K. D. Lindor, Curr. Opin. Gastroenterol., 2011, 27, 285–288 Search PubMed.
- L. Z. Mi, S. Devarakonda, J. M. Harp, Q. Han, R. Pellicciari, T. M. Willson, S. Khorasanizadeh and F. Rastinejad, Mol. Cell, 2003, 11, 1093–1100 CrossRef CAS.
- G. Costantino, A. Macchiarulo, A. Entrena-Guadix, E. Camaioni and R. Pellicciari, Bioorg. Med. Chem. Lett., 2003, 13, 1865–1868 Search PubMed.
- M. Marinozzi, A. Carotti, E. Sansone, A. Macchiarulo, E. Rosatelli, R. Sardella, B. Natalini, G. Rizzo, L. Adorini, D. Passeri, F. De Franco, M. Pruzanski and R. Pellicciari, Bioorg. Med. Chem., 2012, 20, 3429–3445 Search PubMed.
- R. Steri, J. Achenbach, D. Steinhilber, M. Schubert-Zsilavecz and E. Proschak, Biochem. Pharmacol., 2012, 83, 1674–1681 Search PubMed.
- U. Grienke, J. Mihaly-Bison, D. Schuster, T. Afonyushkin, M. Binder, S. H. Guan, C. R. Cheng, G. Wolber, H. Stuppner, D. A. Guo, V. N. Bochkov and J. M. Rollinger, Bioorg. Med. Chem., 2011, 19, 6779–6791 Search PubMed.
- G. Deng, W. Li, J. Shen, H. Jiang, K. Chen and H. Liu, Bioorg. Med. Chem. Lett., 2008, 18, 5497–5502 Search PubMed.
- H. G. Richter, G. M. Benson, D. Blum, E. Chaput, S. Feng, C. Gardes, U. Grether, P. Hartman, B. Kuhn, R. E. Martin, J. M. Plancher, M. G. Rudolph, F. Schuler, S. Taylor and K. H. Bleicher, Bioorg. Med. Chem. Lett., 2011, 21, 191–194 Search PubMed.
- K. C. Han, J. H. Kim, K. H. Kim, E. E. Kim, J. H. Seo and E. G. Yang, Anal. Biochem., 2010, 398, 185–190 Search PubMed.
- B. Flatt, R. Martin, T. L. Wang, P. Mahaney, B. Murphy, X. H. Gu, P. Foster, J. Li, P. Pircher, M. Petrowski, I. Schulman, S. Westin, J. Wrobel, G. Yan, E. Bischoff, C. Daige and R. Mohan, J. Med. Chem., 2009, 52, 904–907 CrossRef CAS.
- K. C. Nicolaou, R. M. Evans, A. J. Roecker, R. Hughes, M. Downes and J. A. Pfefferkorn, Org. Biomol. Chem., 2003, 1, 908–920 RSC.
- M. Rask-Andersen, M. S. Almen and H. B. Schioth, Nat. Rev. Drug Discovery, 2011, 10, 579–590 CrossRef CAS.
- J. B. Pippal and P. J. Fuller, J. Mol. Endocrinol., 2008, 41, 405–413 Search PubMed.
- J. T. Bridgham, G. N. Eick, C. Larroux, K. Deshpande, M. J. Harms, M. E. Gauthier, E. A. Ortlund, B. M. Degnan and J. W. Thornton, PLoS Biol., 2010, 8, e1000497 Search PubMed.
- V. I. Perez-Nueno and D. W. Ritchie, Expert Opin. Drug Discovery, 2012, 7, 1–17 Search PubMed.
- Y. Endo, T. Iijima, H. Kagechika, K. Ohta, E. Kawachi and K. Shudo, Chem. Pharm. Bull., 1999, 47, 585–587 Search PubMed.
- B. Takahashi, K. Ohta, E. Kawachi, H. Fukasawa, Y. Hashimoto and H. Kagechika, J. Med. Chem., 2002, 45, 3327–3330 Search PubMed.
- K. Ohta, Y. Chiba, T. Ogawa and Y. Endo, Bioorg. Med. Chem. Lett., 2008, 18, 5050–5053 Search PubMed.
- P. Coward, D. Lee, M. V. Hull and J. M. Lehmann, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8880–8884 Search PubMed.
- J. Li, M. Ma and Z. Wang, Toxicol. In Vitro, 2010, 24, 201–207 Search PubMed.
- C. P. Chuu, R. Y. Chen, R. A. Hiipakka, J. M. Kokontis, K. V. Warner, J. Xiang and S. Liao, Biochem. Biophys. Res. Commun., 2007, 357, 341–346 Search PubMed.
- Z. C. Dang, V. Audinot, S. E. Papapoulos, J. A. Boutin and C. W. Lowik, J. Biol. Chem., 2003, 278, 962–967 Search PubMed.
- M. L. Ricketts, M. V. Boekschoten, A. J. Kreeft, G. J. Hooiveld, C. J. Moen, M. Muller, R. R. Frants, S. Kasanmoentalib, S. M. Post, H. M. Princen, J. G. Porter, M. B. Katan, M. H. Hofker and D. D. Moore, Mol. Endocrinol., 2007, 21, 1603–1616 Search PubMed.
- S. De Marino, R. Ummarino, M. V. D'Auria, M. G. Chini, G. Bifulco, C. D'Amore, B. Renga, A. Mencarelli, S. Petek, S. Fiorucci and A. Zampella, Steroids, 2012, 77, 484–495 CrossRef CAS.
- V. Sepe, R. Ummarino, M. V. D'Auria, M. G. Chini, G. Bifulco, B. Renga, C. D'Amore, C. Debitus, S. Fiorucci and A. Zampella, J. Med. Chem., 2012, 55, 84–93 CrossRef CAS.
- S. De Marino, R. Ummarino, M. V. D'Auria, M. G. Chini, G. Bifulco, B. Renga, C. D'Amore, S. Fiorucci, C. Debitus and A. Zampella, J. Med. Chem., 2011, 54, 3065–3075 CrossRef CAS.
- W. Xie, A. Radominska-Pandya, Y. Shi, C. M. Simon, M. C. Nelson, E. S. Ong, D. J. Waxman and R. M. Evans, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3375–3380 CrossRef.
- J. L. Staudinger, B. Goodwin, S. A. Jones, D. Hawkins-Brown, K. I. MacKenzie, A. LaTour, Y. Liu, C. D. Klaassen, K. K. Brown, J. Reinhard, T. M. Willson, B. H. Koller and S. A. Kliewer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3369–3374 Search PubMed.
- R. Sharma, D. Prichard, F. Majer, A. M. Byrne, D. Kelleher, A. Long and J. F. Gilmer, J. Med. Chem., 2011, 54, 122–130 Search PubMed.
- H. Tanaka and I. Makino, Biochem. Biophys. Res. Commun., 1992, 188, 942–948 Search PubMed.
- M. Makishima, A. Y. Okamoto, J. J. Repa, H. Tu, R. M. Learned, A. Luk, M. V. Hull, K. D. Lustig, D. J. Mangelsdorf and B. Shan, Science, 1999, 284, 1362–1365 CrossRef CAS.
- D. J. Parks, S. G. Blanchard, R. K. Bledsoe, G. Chandra, T. G. Consler, S. A. Kliewer, J. B. Stimmel, T. M. Willson, A. M. Zavacki, D. D. Moore and J. M. Lehmann, Science, 1999, 284, 1365–1368 CrossRef CAS.
- H. Wang, J. Chen, K. Hollister, L. C. Sowers and B. M. Forman, Mol. Cell, 1999, 3, 543–553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table s1 contains the list of nuclear receptors, pdb codes, cognate ligands and molecular descriptors used in this study. See DOI: 10.1039/c2md20157g |
| This journal is © The Royal Society of Chemistry 2013 |