 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sterol 3β-glucosyltransferase biocatalysts with a range of selectivities, including selectivity for testosterone
Vatsala Malika,
Meng Zhanga,
Lynn G. Dovera,
Julian S. Northenb,
Anthony Flinnb,
Justin J. Perrya and
Gary W. Black*a
aIndustrial Biotechnology Research Group, Department of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: gary.black@northumbria.ac.uk
bOnyx Scientific Limited, Sunderland, SR5 2TQ, UK
First published on 30th August 2013
Abstract
The main objectives of this work were to characterise a range of purified recombinant sterol 3β-glucosyltransferases and show that rational sampling of the diversity that exists within sterol 3β-glucosyltransferase sequence space can result in a range of enzyme selectivities. In our study the catalytically active domain of the Saccharomyces cerevisiae 3β-glucosyltransferase was used to mine putative sterol 3β-glucosyltransferases from the databases. Selected diverse sequences were expressed in and purified from Escherichia coli and shown to have different selectivities for the 3β-hydroxysteroids ergosterol and cholesterol. Surprisingly, three enzymes were also selective for testosterone, a 17β-hydroxysteroid. This study therefore reports for the first time sterol 3β-glucosyltransferases with selectivity for both 3β- and 17β-hydroxysteroids and is also the first report of recombinant 3β-glucosyltransferases with selectivity for steroids with a hydroxyl group at positions other than C-3. These enzymes could therefore find utility in the pharmaceutical industry for the green synthesis of a range of glycosylated compounds of medicinal interest.
Introduction
Many lead compounds under development by the pharmaceutical industry are poorly water soluble and hence have non-ideal pharmacokinetic properties. One strategy, to overcome this problem is to link the drug substance to a water soluble moiety such as a carbohydrate, as the drug can be cleaved from this adduct by normal metabolic processes, ideally at the desired site of action. For example, the oral pro-drug testosterone-glucoside is currently under investigation by ProStrakan as an alternative to testosterone for patients suffering from hypogonadism.1 Glycosidic derivatives are also of interest because they are commonly encountered in the in vivo metabolism of xenobiotic compounds as more water soluble products more suitable for excretion. Consequently, synthesis of appropriate carbohydrate derivatives might be employed to provide either more effective medicinal compounds or as a means to investigate drug pharmacokinetics through adsorption, distribution, metabolism and excretion (ADME) of a drug substance.2Chemical methods of glycosylation are challenging and usually require a laborious protection–deprotection strategy which drives down overall chemical yield especially with complex aglycones.3,4 In addition, this approach proves industrially unattractive due to the formation of intractable mixtures of side-products.3,4 In contrast, the chemo- and regioselective glycosylation of hydroxyl functionality is a common biotransformation and several glycosyltransferase (GTase) biocatalysts that perform efficient and selective conversions have been characterised.5 The majority of characterised GTases catalyze the transfer of the monosaccharide units from an activated nucleotide donor, such as a uridine diphosphate sugar, to specific acceptor molecules, during the formation of a glycosidic bond.6
Glucosylation of steroids with a hydroxyl group at the C-3 position is a common biotransformation catalyzed by GTases known as sterol 3β-glucosyltransferases (systematic name, uridine diphosphate-glucose:sterol 3-O-β-D-glucosyltransferases; abbreviation, UDPG-SGTases; EC number, 2.4.1.173). UDPG-SGTase are members of GTase family 1 (GT1) of the Carbohydrate Active Enzymes database; a database that classifies enzymes that degrade, modify, or create glycosidic bonds into distinct protein sequence homology-based families.7 Characterized examples of UDPG-SGTases include UGT51 from Saccharomyces cerevisiae and UGT51B1 from Pichia pastoris.8 UGT51 and UGT51B1 each contain a GT1 GT-like conserved domain (cd03784) and pleckstrin homology/GRAM conserved domains (cd13215 and cd13216) that are often involved in targeting proteins to the plasma membrane.9–11 Warnecke et al.8 also showed, by successively deleting the N-terminus of UGT51, followed by in vitro analysis, that it is only the GT1 GT-like conserved domain that is responsible for UDPG-SGTase activity.
There are several reports of characterised UDPG-SGTases, but many of these were performed with crude homogenate, purified membrane or partially purified enzyme preparations that may have contained mixtures of UDPG-SGTases.12 Unfortunately, there are only a few reports of characterised recombinant UDPG-SGTases.12 Our work therefore aimed to characterise a range of recombinant UDPG-SGTase catalytic domains, identified via sequence similarity to that of UGT51. Additionally, we aimed to show that rational sampling of the diversity that exists within sequence space, i.e. the number of possible amino acid sequences that can generate a particular enzyme activity, results in a range of enzyme selectivities. We show that a range of selectivities, including specificity for testosterone, a 17β-hydroxysteroid, were present among the six recombinant UDPG-SGTases that were characterised in this study.
Results and discussion
Identification of putative GT1 GT-like domains
Difficulties in producing and characterising GTases are well-documented and are associated with the presence of non-catalytic domains involved in membrane spanning13 or non-integral membrane associations, such as the pleckstrin homology/GRAM domains.9–11 Therefore the N-terminally truncated catalytic domains, comprising of only the GT1 GT-like domain of several UDPG-SGTases were expressed in E. coli. This was achieved by conducting a BLASTP search using the amino acid sequence of the shortest C-terminal domain of the S. cerevisiae UGT51 that encoded a catalytically active UDPG-SGTase.8 The BLASTP search revealed similarities with the previously characterised UDPG-SGTase, UGT51B1 from P. pastoris, and several uncharacterised protein products. Fig. 1 shows the ClustalW2 multiple sequence alignment (version 2.1) comparison of the deduced N-terminally truncated domains of the sequences selected from the BLASTP search. The catalytic UDPG-SGTase domain of the S. cerevisiae UGT51, UGT51′, is comprised of a polypeptide of 475 amino acids.8,11 This domain has 61–64% identity to the C-terminal domains of the selected sequences: UGT51B1 of P. pastoris (termed UGT51B1′), A7KAK6 of P. angusta (termed A7KAK6′), A5DNB9 of M. guilliermondii (termed A5DNB9′), Q6BN88 of D. hansenii (termed Q6BN88′) and Q6CUV2 of K. lactis (termed Q6CUV2′); with the selected proteins showing a good level of sequence space diversity, i.e. 58–66% identity amongst themselves. Gene fragments corresponding to deletions of 723, 747, 770, 1065, 1030 and 718 codons from the 5′ end of the sequences encoding UGT51, UGT51B1, A7KAK6, A5DNB9, Q6BN88 and Q6CUV2, respectively, were amplified, cloned and expressed in E. coli. The N-terminal residues of these sequences showed low levels of sequence identity (between 18–35%). | ||
| Fig. 1 Clustal W2 multiple sequence alignment of the deduced N-terminally truncated catalytic domains of the UDPG-SGTases expressed in this study. UGT51B1′, P. pastoris UDPG-SGTase; A7KAK6′, P. angusta UDPG-SGTase; A5DNB9′, M. guilliermondii UDPG-SGTase; Q6BN88′, D. hansenii UDPG-SGTase; UGT51′, S. cerevisiae UDPG-SGTase; Q6CUV2′, K. lactis UDPG-SGTase. Black boxes indicate identical amino acids in all 6 sequences. Grey boxes indicate identical amino acids in 4 or 5 sequences. The alignment was conducted with residues 724 to 1198 of UGT51′, 748 to 1211 of UGT51B1, 771 to 1241 of A7KAK6, 1066 to 1599 of A5DNB9, 1031 to 1574 of Q6BN88, and 719 to 1209 of Q6CUV2. | ||
Expression of putative UDPG-SGTases using a range of E. coli BL21(DE3) strains
The N-terminally hexa-histidine tagged protein products with a calculated molecular mass of 54.6 kDa for UGT51′, 53.5 kDa for UGT51B1′, 54.2 kDa for A7KAK6′, 60.6 kDa for A5DNB9′, 61.8 kDa for Q6BN88′ and 55.9 kDa for Q6CUV2′ were expressed in E. coli BL21(DE3) strains. In all cases the largest amount of soluble protein expression was achieved after 18 h of post-induction growth at 23 °C and no expression was observed in the absence of induction with isopropyl-1-thio-β-D-galactopyranoside. In two cases, UGT51′ and A5DNB9′, low expression levels in E. coli BL21(DE3) were seen (data not shown). To maximise expression of these genes E. coli BL21(DE3) CodonPlus strains were transformed with the LIC-constructs encoding UGT51′ and A5DNB9′. Following this, increase in recombinant protein expression levels was observed for A5DNB9′ and UGT51′ encoding plasmids transformed in E. coli BL21(DE3) CodonPlus (RP) and E. coli BL21(DE3) Codon Plus (RIPL), respectively (data not shown). All gene fragments encoded soluble protein products (with yields between 10–100 mg L−1) were purified using immobilised metal ion (Ni2+) chromatography. Multiple nonredundant peptides were identified by peptide fragmentation fingerprinting of UGT51′, UGT51B1′, A7KAK6′, A5DNB9′, Q6BN88′ and Q6CUV2′; confirming the identity of the purified proteins (data not shown).Identification of UDPG-SGTases with different selectivities
Each of the six recombinant proteins was assayed for activity against the following steroidal acceptors: ergosterol, cholesterol, estrone, estradiol, androstenedione and testosterone (Table 1). UGT51′ from S. cerevisiae was shown to be active against the 3β-hydroxysteroids, cholesterol and ergosterol (Fig. 2A and B, respectively); confirming the results seen by Warnecke et al.8 and revealing that activity is also observed with a purified N-terminally hexa-histidine tagged version of the enzyme. UGT51B1′ from P. pastoris was also active against the 3β-hydroxysteroids (Fig. 2A and B); again confirming the results seen by Warnecke et al.8 and revealing that it is only the GT1 GT-like domain of the P. pastoris enzyme that is responsible for UDPG-SGTase activity. The N-terminally truncated previously uncharacterised enzyme, A7KAK6, from P. angusta was also shown to be active against the 3β-hydroxysteroids (Fig. 2A and B); adding further support to the suggestion that it is only the GT1 GT-like domain of UDPG-SGTases that is responsible for activity. The N-terminally truncated previously uncharacterised enzyme, Q6CUV2, from K. lactis was shown to be active against cholesterol (Fig. 2A) but not against ergosterol (data not shown). Interestingly, UGT51′, UGT51B1′ and A7KAK6′ were also shown to be active against the 17β-hydroxysteroid, testosterone (Fig. 2C) and to our knowledge this is the first report of UDPG-SGTases with activity against both 3β- and 17β-hydroxyl steroids. In fact, any reports of UDPG-SGTases with specificity towards glucosylation of steroids at positions other than C-3 are limited and importantly none involving the characterisation of a recombinant enzyme have been reported. For example, Madina et al.14 have shown that a near to pure 27β-hydroxy glucosyltransferase from the cytosolic fraction of the medicinal plant, Withania somnifera, showed activity against testosterone; Poppenberger et al.15 have shown that wild-type seedlings of Arabidopsis converted the brassinosteroid brassinolide to the 23-O-glucoside, but in the transgenic plants silenced in UGT73C5 expression no 23-O-glucoside was detected; and O'Reilly et al.16 have shown that a baculovirus ecdysteroid UDP-glucosyltransferase produces ecdysone 22-O-β-D-glucopyranoside.| Acceptor substrate | Structure |
|---|---|
| Ergosterol |  |
| Cholesterol |  |
| Estrone |  |
| Estradiol |  |
| Androstenedione | 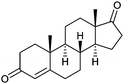 |
| Testosterone | 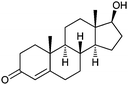 |
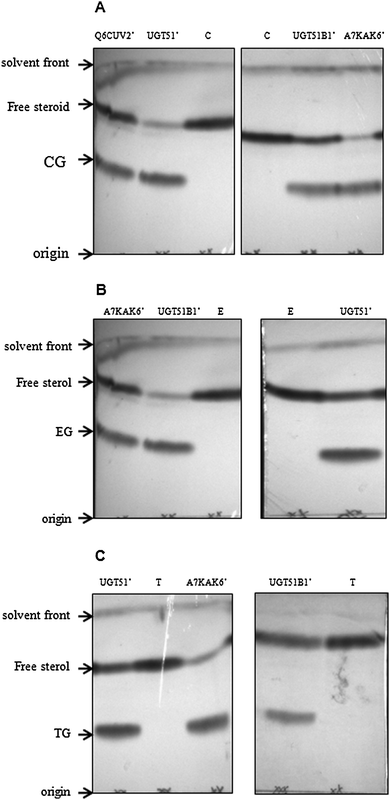 | ||
| Fig. 2 Thin layer chromatograms of extracted reaction products following 2 h incubations. Reaction products of UGT51B1′, P. pastoris UDPG-SGTase; A7KAK6′, P. angusta UDPG-SGTase; UGT51′, S. cerevisiae UDPG-SGTase; Q6CUV2′, K. lactis UDPG-SGTase using cholesterol (panel A), ergosterol (panel B) and testosterone (panel C) as acceptors. Lanes C, T and E are the cholesterol, testosterone and ergosterol controls, respectively, without enzyme. CG, TG and EG are cholesterol glucoside, testosterone glucoside and ergosterol glucoside, respectively. | ||
As expected, the UDPG-SGTases in this study exhibited no activity towards β-naphthol, a non-steroidal aromatic alcohol; estrone and estradiol, hydroxysteroids with the OH group at the C-3 position in a flat configuration; and androstenedione, a steroid with a similar structure to testosterone but with no OH group at the C-17 position (data not shown). Additionally, the other two N-terminally truncated UDPG-SGTases expressed in this study, A5DNB9′ from M. guilliermondii and Q6BN88′ from D. hansenii did not demonstrate activity towards any of the acceptors used, even though they have previously been implicated in autophagy and related processes.17,18 UDP-galactose was found not to be accepted as the sugar donor for any of the UDPG-SGTases with any of the screened acceptors. This was in accordance with previous studies where similar specificity for donor sugars has been reported for S. cerevisiae and other eukaryotic GTases.8 It is worth noting that while using testosterone as the steroidal acceptor, glucosylated products had similar Rf values to an authentic β-testosterone glucoside standard (data not shown). This was further confirmed by co-spotting of reaction products with the standard (data not shown).
LC-MS analysis was used to confirm the results from the TLC experiments. Characteristic signals for the monoprotonated testosterone (m/z = 289 with a retention time of 2.0 min) and monoprotonated testosterone glucoside (m/z = 451 with a retention time of 1.4 min) were determined using authentic standards. Both were observed in the reaction mixture after 2 h (Fig. 3) but after an overnight incubation only the peak for the testosterone glucoside could be observed. This was also supported by TLC analysis of an overnight incubation with testosterone (data not shown) and was also the case for cholesterol and ergosterol overnight incubations (data not shown). An added advantage to this analytical method was that testosterone and the corresponding glucosylated compound were separated by a chromatographic method with a run time of 5 min at room temperature, an assay which compares favourably with previous studies where separation was achieved with longer retention times at elevated column temperatures.19,20
![LC-chromatogram and MS spectrum of extracted reaction products. Extraction from P. angusta UDPG-SGTase A7KAK6′ catalysed reaction following a 2 h incubation. A characteristic peak at m/z = 451 [M + H]+ at a retention time (RT) of 1.38 min confirms the synthesis of testosterone glucoside (inset). Peak at retention time 2.06 (m/z = 289 [M + H]+) shows the residual testosterone in the reaction mixture. Selected ion monitoring mode of m/z = 288.0–290.0 and 450.0–452.0 was used.](/image/article/2013/MB/c3mb70303g/c3mb70303g-f3.gif) | ||
| Fig. 3 LC-chromatogram and MS spectrum of extracted reaction products. Extraction from P. angusta UDPG-SGTase A7KAK6′ catalysed reaction following a 2 h incubation. A characteristic peak at m/z = 451 [M + H]+ at a retention time (RT) of 1.38 min confirms the synthesis of testosterone glucoside (inset). Peak at retention time 2.06 (m/z = 289 [M + H]+) shows the residual testosterone in the reaction mixture. Selected ion monitoring mode of m/z = 288.0–290.0 and 450.0–452.0 was used. | ||
Kinetic analysis of UDPG-SGTases
Kinetic analysis was performed for all active UDPG-SGTases using cholesterol and testosterone as acceptors and UDP-[14C] glucose as the donor. All reactions demonstrated classic Michaelis–Menten kinetics with goodness-of-fit statistical analysis of the linear trend lines of the resulting Lineweaver–Burke plots producing R2 values between 0.8052 to 0.9599 (data not shown). For enzymes active against cholesterol and testosterone, the binding capacity of the enzymes for cholesterol was higher (≥4.5-fold) than that for testosterone (Table 2). Similarly, the reaction rate of the enzymes active against cholesterol was higher (≥1.8-fold) than that against testosterone (Table 2). It should be noted that the apparent Km values of the enzymes with testosterone were derived at less than saturating substrate concentrations due to the lack of solubility of testosterone at higher concentrations in the aqueous reaction mixture. UGT51B1′ from P. pastoris displayed the highest level of apparent catalytic efficiency (kcat/Km value) on both substrates (Table 2) and its catalytic efficiency against testosterone was comparable to that of the plant 27β-hydroxy glucosyltransferase against testosterone.21 The presence of Mn2+ ions was shown not to affect the activity of the enzymes in this study (data not shown) which is in agreement with that of the Madina et al.14 who have reported that the presence of Mn2+ ions did not affect the activity of the plant 3β-glucosyltransferase.| UDPG-SGTase | Acceptor | Km (μM) | Vmax (μmol min−1 mg−1) | kcat (s−1) | kcat/Km (s−1 μM−1) |
|---|---|---|---|---|---|
| UGT51′ | C | 79.9 ± 21.7 | 1447.0 ± 218.2 | 1319.2 | 16.5 |
| T | 362.5 ± 26.85 | 538.7 ± 23.75 | 491.13 | 1.35 | |
| UGT51B1′ | C | 34.90 ± 10.80 | 4337 ± 551.2 | 3897.0 | 111.6 |
| T | 211.1 ± 27.5 | 288.7 ± 20.5 | 488.67 | 2.3 | |
| A7KAK6′ | C | 67.2 ± 31.63 | 719.1 ± 176.8 | 650.6 | 9.7 |
| T | 837.9 ± 182.23 | 540.1 ± 76.24 | 351.9 | 0.42 | |
| Q6CUV2′ | C | 65.92 ± 31.09 | 509.0 ± 124.6 | 475.0 | 7.21 |
Conclusions
We have shown that the approach of rational sampling of the diversity that exists within sequence space identified via a BLAST search generated a range of UDPG-SGTases with the same activity but different selectivity. We suggest that this class of enzyme could become valuable biocatalysts in the pharmaceutical industry (and fine chemical industry) due to their ability to glycosylate a range of sterols without the need to use protection–deprotection strategies.Experimental
Yeast strains and propagation
Saccharomyces cerevisiae (ATCC 204508) and Pichia pastoris (ATCC 201178) were obtained from the LGC Standards (UK). Pichia angusta (DSM 70277), Meyerozyma guilliermondii (DSM 6381) and Kluyveromyces lactis (DSM 70799) were obtained from DSMZ (Germany). Debaryomyces hansenii (CBS 767) was obtained from the CBS Fungal Biodiversity Centre (Netherlands). S. cerevisiae and P. pastoris were grown overnight at 30 °C, 100 rpm in yeast extract peptone dextrose medium (2% [w/v] Bacto Peptone [BD, UK], 1% [w/v] yeast extract [Oxoid, UK], 2% [w/v] glucose [Sigma, UK]) whereas the other strains were grown overnight at 25 °C, 100 rpm in yeast extract–malt extract medium (0.3% [w/v] yeast extract [Oxoid, UK], 0.3% [w/v] malt extract [Oxoid, UK], 0.5% peptone [Oxoid, UK], 1% [w/v] glucose [Sigma, UK]).Identification and cloning of UDPG-SGTase encoding genes
A BLASTP (version 2.2.27)22 search of the databases was conducted using UGT51′ (residues 724 to 1198 of UGT51) as a query to identify similar sequences. Gene sequences encoding N-terminal truncated versions of the proteins identified were selected via rationally sampling the diversity that exists within sequence space. The selected genes (Table 3 for UniProtKB accession numbers) were then cloned and expressed in Escherichia coli. Genomic DNA was prepared from overnight cultures using the DNeasy Blood and Tissue Kit [Qiagen, UK] and the primer pairs [Eurofins MWG Operon, Germany], shown in Table 3, were used to amplify the gene fragments via PCR using KOD Hot Start DNA Polymerase [Merck Chemicals, UK]. PCR was performed for 5 cycles at an annealing temperature of 50 °C then for 25 cycles at an annealing temperature of 73 °C. The amplified products were inserted into pET-YSBLIC (kindly provided by Dr Mark Fogg of the York Structural Biology Laboratory, York University, UK) via ligation independent cloning, as described by Bonsor et al.23| Source organism and UniProtKB accession number | Primer pairs | E. coli strain used for expression |
|---|---|---|
| S. cerevisiae Q06321 | 5′-caccaccaccacatgATTGATGAGAATCCGCAC-3′ | BL21(DE3) CodonPlus (RP) (Agilent Technologies, UK) |
| 5′-gaggagaaggcgcgTTAAATCATCGTCCACCCTTCA-3′ | ||
| P. pastoris Q9Y751 | 5′-caccaccaccacatgAACCAGTATTCTACGTCAGA-3′ | BL21(DE3) CodonPlus (RIPL) (Agilent Technologies, UK) |
| 5′-gaggagaaggcgcgTTACACTTCAAACCATGATC-3′ | ||
| M. guilliermondii A5DNB9 | 5′-caccaccaccacatgTTAGAAGATTCACCACTCATC-3′ | BL21(DE3) CodonPlus (RP) (Agilent Technologies, UK) |
| 5′-gaggagaaggcgcgTTAGTCGTTAGTTGTCGTTG-3′ | ||
| P. angusta A7KAK6 | 5′-caccaccaccacatgATAGAAGAGCATCCATTGACC-3′ | BL21(DE3) (Merck Chemicals, UK) |
| 5′-gaggagaaggcgcgttAAACCAGTAACCACGAGCC-3′ | ||
| K. lactis Q6CUV2 | 5′-caccaccaccacatgGTTGAAAAAAACCCGTATT-3′ | BL21(DE3) (Merck Chemicals, UK) |
| 5′-gaggagaaggcgcgTTATATCAGAAGCCAGGATC-3′ | ||
| D. hansenii Q6BN88 | 5′-caccaccaccacatgTTAGAAGATTCTCCATTCTAT-3′ | BL21(DE3) (Merck Chemicals, UK) |
| 5′-gaggagaaggcgcgttaTTATTTGTTAGCATCTGTAAC-3′ | ||
Protein expression, purification and identification
Recombinant proteins were expressed from different E. coli BL21(DE3) strains as indicated in Table 3. Cells were grown at 37 °C with shaking at 200 rpm in 1 L LB medium supplemented with 100 μg mL−1 kanamycin for LIC-constructs transformed in E. coli BL21(DE3) and 100 μg mL−1 kanamycin plus 34 μg mL−1 chloramphenicol for LIC-constructs transformed in E. coli BL21(DE3) CodonPlus (RP/RIPL) to an absorbance of 0.6 at 600 nm. Induction was performed by the addition of isopropyl-1-thio-β-D-galactopyranoside, to a concentration of 240 μg mL−1, followed by further incubation for 3 h and 18 h at 23 °C, 30 °C and 37 °C, 100 rpm. Cells were harvested by centrifugation (15 min, 4000 × g, 4 °C), resuspended in 5 mL of 50 mM Na2HPO4–HCl, 0.5 M NaCl, 10 mM imidazole, pH 7.4 followed by cell disruption by sonication (6 × 10 s at 14 microns). N-terminally hexahistidine tagged protein products were purified via immobilised metal ion (Ni2+) chromatography (GE Healthcare, UK) using a linear gradient of 10 to 500 mM imidazole. Protein concentration and buffer exchange into 5 mM Tris–HCl (pH 8.0) of the purified proteins was achieved using 30 kDa cut-off concentrator units [Vivaproducts, USA]. The purity of UDPG-SGTases was judged by SDS-PAGE and Coomassie blue staining.24 Peptide fragmentation fingerprinting of purified proteins was used to confirm the identity of the protein according to the methods and criteria of Yang et al.25UDPG-SGTase activity analysis
The purified enzymes were tested for selectivity using ergosterol [Sigma-Aldrich, UK], cholesterol [Sigma-Aldrich, UK], estrone [Sigma-Aldrich, UK], estradiol [Sigma-Aldrich, UK], androstenedione [TCI, UK] and testosterone [Sigma-Aldrich, UK] as steroidal acceptor substrates (Table 1 for chemical structures of these steroids). In addition, β-naphthol, [Sigma-Aldrich, UK] was used as a non-steroidal aromatic acceptor substrate. All acceptor stock solutions were prepared in 100% (v/v) ethanol. For TLC/LC-MS analysis, the assay mixture contained (in a total volume of 100 μL) the following: 400 μM acceptor, 360 μM UDP-glucose [Sigma-Aldrich, UK] or UDP-galactose [Sigma-Aldrich, UK], 50 μM MnCl2 [Sigma-Aldrich, UK] when appropriate, 2 μg purified enzyme and 10 mM Na2HPO4–HCl, pH 7.8. The assays were performed in triplicate and terminated after 2 h incubation at 30 °C. The identity of the testosterone glucoside was confirmed by comparison with an authentic standard kindly provided by ProStrakan Group Plc, UK. Assays for TLC analysis were terminated by the addition of 0.9 mL of 0.45% (w/v) NaCl and 4 mL of chloroform–methanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), 2 μL separated on silica gel 60 TLC plates [Merck Chemicals, UK] according to the procedure described in Warnecke et al.8 and visualised by spraying with 15% (w/v) phosphotungstic acid followed by charring using a heat gun. Assays for LC-MS analysis were only performed with testosterone and were terminated by the addition of 100 μL of 0.3 M HCl, then diluted with 800 μL of 18.2 MΩ cm−1 water and 20 μL applied to a Phenomenex Luna 5 μm CN column (5.0 cm × 2 mm). Isocratic reverse phase separation was achieved in 5 min with a flow rate of 150 μL min−1 of 30% (v/v) acetonitrile, 1% (v/v) acetic acid mobile phase and the analytes detected using a ThermoFinnigan LCQ Advantage ion-trap mass-spectrometer in selected ion monitoring mode with a spray voltage of 4.5 kV, capillary voltage of 5 V and capillary temperature of 230 °C. For kinetic analysis, the assay mixture contained (in a total volume of 75 μL) the following: acceptor (cholesterol or testosterone) ranging from 12.5–200 μM, 50000 dpm UDP-[14C] glucose (final concentration 0.88 μM, specific activity 302 mCi mmol−1, Perkin Elmer, UK), 0.5 μg purified enzyme diluted in 10 mM Na2HPO4–HCl, pH 7.8. After incubation for 20 min at 30 °C, the reaction was terminated by the addition of 125 μL 0.45% (w/v) NaCl followed by extraction using 300 μL of water-saturated ethyl acetate. For each sample, 150 μL of the organic layer was counted for 3 min by liquid scintillation counting using the Tricarb 2200 CA (Perkin Elmer, UK).
:1, v/v), 2 μL separated on silica gel 60 TLC plates [Merck Chemicals, UK] according to the procedure described in Warnecke et al.8 and visualised by spraying with 15% (w/v) phosphotungstic acid followed by charring using a heat gun. Assays for LC-MS analysis were only performed with testosterone and were terminated by the addition of 100 μL of 0.3 M HCl, then diluted with 800 μL of 18.2 MΩ cm−1 water and 20 μL applied to a Phenomenex Luna 5 μm CN column (5.0 cm × 2 mm). Isocratic reverse phase separation was achieved in 5 min with a flow rate of 150 μL min−1 of 30% (v/v) acetonitrile, 1% (v/v) acetic acid mobile phase and the analytes detected using a ThermoFinnigan LCQ Advantage ion-trap mass-spectrometer in selected ion monitoring mode with a spray voltage of 4.5 kV, capillary voltage of 5 V and capillary temperature of 230 °C. For kinetic analysis, the assay mixture contained (in a total volume of 75 μL) the following: acceptor (cholesterol or testosterone) ranging from 12.5–200 μM, 50000 dpm UDP-[14C] glucose (final concentration 0.88 μM, specific activity 302 mCi mmol−1, Perkin Elmer, UK), 0.5 μg purified enzyme diluted in 10 mM Na2HPO4–HCl, pH 7.8. After incubation for 20 min at 30 °C, the reaction was terminated by the addition of 125 μL 0.45% (w/v) NaCl followed by extraction using 300 μL of water-saturated ethyl acetate. For each sample, 150 μL of the organic layer was counted for 3 min by liquid scintillation counting using the Tricarb 2200 CA (Perkin Elmer, UK).
Acknowledgements
We would like to thank Edwin Ludkin for technical help with LC-MS.References
- ProStrakan, R&D, in http://www.prostrakan.nl/rd.htm, 2013.
- E. S. Hansen, S. A. Osmani, C. Kristensen, B. L. Moller and J. Hansen, Phytochemistry, 2009, 70, 473–482 CrossRef CAS PubMed.
- D. E. Robertson and B. A. Steer, Curr. Opin. Chem. Biol., 2004, 8, 141–149 CrossRef CAS PubMed.
- M. M. Palcic, Curr. Opin. Biotechnol., 1999, 10, 616–624 CrossRef CAS.
- S. Rabbani, O. Schwardt and B. Ernst, Chimia, 2006, 60, 23–27 CrossRef CAS.
- K. Hashimoto, T. Tokimatsu, S. Kawano, A. C. Yoshizawa, S. Okuda, S. Goto and M. Kanehisa, Carbohydr. Res., 2009, 344, 881–887 CrossRef CAS PubMed.
- B. L. Cantarel, P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard and B. Henrissat, Nucleic Acids Res., 2009, 37, D233–D238 CrossRef CAS PubMed.
- D. Warnecke, R. Erdmannn, A. Fahl, B. Hube, F. Muller, T. Zank, U. Zahringer and E. Heinz, J. Biol. Chem., 1999, 274, 13048–13059 CrossRef CAS PubMed.
- M. A. Lemmon, Cell, 2005, 120, 574–576 CrossRef CAS PubMed.
- T. Doerks, M. Strauss, M. Brendel and P. Bork, Trends Biochem. Sci., 2000, 25, 483–485 CrossRef CAS.
- M. Oku, D. Warnecke, T. Noda, F. Muller, E. Heinz, H. Mukaiyama, N. Kato and Y. Sakai, EMBO J., 2003, 22, 3231–3241 CrossRef CAS PubMed.
- S. Grille, A. Zaslawski, S. Thiele, J. Plat and D. Warnecke, Prog. Lipid Res., 2010, 49, 262–288 CrossRef CAS PubMed.
- P. M. Coutinho, E. Deleury, G. D. Davies and B. Henrissat, J. Mol. Biol., 2003, 328, 307–317 CrossRef CAS.
- B. R. Madina, L. K. Sharma, P. Chaturvedi, R. S. Sangwan and R. Tuli, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 392–402 CrossRef CAS PubMed.
- B. Poppenberger, S. Fujioka, K. Soeno, G. L. George, F. E. Vaistij, S. Hiranuma, H. Seto, S. Takatsuto, G. Adam, S. Yoshida and D. Bowles, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15253–15258 CrossRef CAS PubMed.
- D. R. O'Reilly, O. W. Howarth, H. H. Rees and L. K. Miller, Insect Biochem., 1991, 21, 795–801 CrossRef CAS.
- A. R. Bellu and J. A. K. W. Kiel, Microsc. Res. Tech., 2003, 61, 161–170 CrossRef CAS PubMed.
- C. A. G. M. Weijers, M. C. R. Franssen and G. M. Visser, Biotechnol. Adv., 2008, 26, 436–456 CrossRef CAS PubMed.
- X. F. Li, M. S. Ma, A. Cheng, J. Zheng and Y. K. Tam, Anal. Chim. Acta, 2002, 457, 165–171 CrossRef CAS.
- C. J. Zea and N. L. Pohl, Anal. Biochem., 2004, 328, 196–202 CrossRef CAS PubMed.
- B. R. Madina, L. K. Sharma, P. Chaturvedi, R. S. Sangwan and R. Tuli, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 1199–1207 CrossRef CAS PubMed.
- S. F. Altschul, T. L. Madden, A. A. Schäffer, Z. Z. Zhang, W. Miller and D. Lipman, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS PubMed.
- D. Bonsor, S. F. Butz, J. Solomons, S. Grant, I. J. S. Fairlamb, M. J. Fogg and G. Grogan, Org. Biomol. Chem., 2006, 4, 1252–1260 CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- Q. Yang, M. Zhang, D. J. Harrington, G. W. Black and I. C. Sutcliffe, Microbes Infect., 2011, 13, 757–760 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2013 |