 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and biological evaluation of synthetic retinoids: probing length vs. stability vs. activity
Graeme Clemensa, Kevin R. Flowerb, Peter Gardnera, Andrew P. Hendersonc, Jonathan P. Knowlesc, Todd B. Mardercd, Andrew Whiting*c and Stefan Przyborski*ef
aManchester Institute of Biotechnology, Manchester University, 131 Princess Street, Manchester, M1 7DN, UK
bSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cDepartment of Chemistry, Durham University, Science Laboratories, South Road, Durham, DH1 3LE, UK. E-mail: andy.whiting@durham.ac.uk
dInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
eDepartment of Biological Sciences, Durham University, Science Laboratories, South Road, Durham, DH1 3LE, UK. E-mail: stefan.przyborski@durham.ac.uk
fReinnervate Ltd., NETPark Incubator, Thomas Wright Way, Sedgefield, TS21 3FD, UK
First published on 3rd October 2013
Abstract
All trans-retinoic acid (ATRA) is widely used to direct the differentiation of cultured stem cells. When exposed to the pluripotent human embryonal carcinoma (EC) stem cell line, TERA2.cl.SP12, ATRA induces ectoderm differentiation and the formation of neuronal cell types. We report in this study that novel polyene chain length analogues of ATRA require a specific chain length to elicit a biological responses of the EC cells TERA2.cl.SP12, with synthetic retinoid AH61 being particularly active, and indeed more so than ATRA. The impacts of both the synthetic retinoid AH61 and natural ATRA on the TERA2.cl.SP12 cells were directly compared using both RT-PCR and Fourier Transform Infrared Micro-Spectroscopy (FT-IRMS) coupled with multivariate analysis. Analytical results produced from this study also confirmed that the synthetic retinoid AH61 had biological activity comparable or greater than that of ATRA. In addition to this, AH61 has the added advantage of greater compound stability than ATRA, therefore, avoiding issues of oxidation or decomposition during use with embryonic stem cells.
Introduction
There are over 4000 retinoids, both natural and synthetic, structurally and often biochemically related to vitamin A.1 All-trans-retinoic acid 1 (ATRA), the major metabolite of vitamin A, and its two isomers, 9-cis-retinoic acid 2 (9CRA) and 13-cis-retinoic acid 3 (13CRA), have essential roles in many biological processes during both chordate embryogenesis and adult homeostasis.2,3 These include cellular differentiation, proliferation and apoptosis,4 embryonic development5 and vision.6 Due to the ability of retinoids to control differentiation and apoptosis in both normal and tumour cells, they have the potential to act as chemopreventative and chemotherapeutic agents, although toxicity has prevented widespread use.7,8 ATRA and other commercially available retinoids are, however, used to treat dermatological conditions.9The use of retinoic acids as inducers of cell differentiation has some limitations, exemplified by ATRA which readily isomerises resulting in mixtures of ATRA, 9CRA, 13CRA and other species.10 This instability derives from the five conjugated double bonds that absorb light in the 300–400 nm region, which can occur under laboratory lighting conditions.11–14 Murayama et al.11 reported that isomers of ATRA differently affect the ability of mammalian stem cells to differentiate along alternative lineages. Consequently this can lead to the differential regulation of alternative key molecular pathways involved in cellular development, which can result in increased heterogeneity within cultures of differentiating stem cells.15,16 In order to increase reproducibility in the differentiation process, stable retinoid derivatives are desirable, an approach adopted by us.10 These types of stable compounds can maintain a constant concentration in the culture system, and are readily stored and handled. This is preferable to the alternative approach of attempting to reduce the sensitivity of ATRA to isomerisation by the use of additives such as bovine serum albumin (BSA), fibrogen, lysozyme, phosphatidylcholine N-ethylmaleimide and vitamin C.17,18 None of these additives completely suppress isomerisation, and indeed, they may affect cellular processes on their own.
Retinoic acids contain three major characteristic regions: a bulky hydrophobic section; a conjugated variable linker; and a carboxylic acid end group (Fig. 1). Extensive modifications to all three regions are possible1,19 and we reported the incorporation of an aromatic ring to replace parts of the polyene chain, forming a class of retinoids sometimes termed arotinoids.10,19 Another common alteration is the inclusion of a 1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene unit as a substitute for the trimethylcyclohexenyl ring and part of the polyene chain, as this removes metabolic oxidation sites.19–22 Also, an acetylene moiety has been successfully employed to act as a non-isomerisable conjugated linker unit while retaining, or even enhancing, biological activity in systems such as EC23 4a (Fig. 1).10 Interestingly, biological characterisation of EC23 4a demonstrated its ability to induce neural tissues, which contrasts with EC19 4b which also induced cell differentiation, the latter resulting primarily in epithelial cells with only few neural phenotypes.10 These results indicated that subtle differences in molecular structure can have dramatic effects on the biological response. In this paper, we contrast these findings with recent work on the design, synthesis, stability and biological activity of a series of new synthetic retinoids designed to be closer in structure to the natural systems 1–3, but with some of the increased stability of 4a. With these types of compounds we expected to be able to probe further the effects polyene chain length and stability versus their effect on cellular development processes, particularly in stem cells or models of stem cell differentiation.
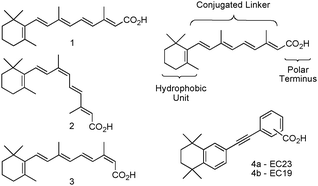 | ||
| Fig. 1 Structures for ATRA 1, 9CRA 2, 13CRA 3, generic retinoid structure, EC23 (4-position CO2H) and EC19 (3-position CO2H). | ||
Results and discussion
Preparation of retinoid analogues
It was envisaged that through a series of cross-coupling reactions, including Heck–Mizoroki and Sonogashira processes, we would be able to obtain compounds of varying size to undertake a focussed structure activity relationship (SAR) study. Alkyne 5 was prepared as a generic synthon, starting with iodination of 1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene, followed by cross-coupling to TMSA under Sonogashira conditions and finally deprotection according to optimised procedures previously reported.10 The synthesis of the different polyene chains with varying lengths began with the treatment of methyl tetrolate 6 with sodium iodide in acetic acid23,24 to provide Z-3-iodobut-2-enoic acid methyl ester 7 in 89% yield and 100% stereoselectivity. Generation of the E-isomer 8 was achieved through isomerisation of the Z-isomer with hydroiodic acid in benzene24,25 to give 3-E-iodobut-2-enoic acid methyl ester 8 in a 66% yield (Scheme 1).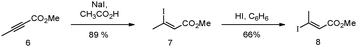 | ||
| Scheme 1 Synthesis of iodide 8 from methyl tetrolate 6. | ||
The E-isomer was subsequently used in two separate reactions: (1) direct coupling to acetylene 5 allowing for the formation of ‘short’ retinoid-like analogue 9; and (2) Heck–Mizoroki coupling to vinylboronate ester 10, followed by iododeboronation, leading to a dienyliodide 11 for further elaboration to longer derivatives as outlined in Scheme 2, which employs an iterative Heck–Mizoroki coupling16 with boronate 10 and highly stereoselective iododeboronation methodology.17 Subsequent, Sonogashira couplings of iodides 11 and 13 to acetylene 5 under standard conditions26,27 provided esters 12 (known as AH61) and 14 in 76 and 64% yields, respectively. Saponification28,29 of the methyl esters produced the corresponding carboxylic acids 12 (AH61) and 14 in 85 and 87% yields, respectively (Scheme 2).
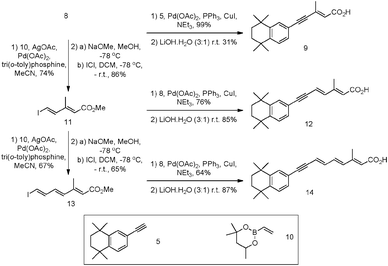 | ||
| Scheme 2 Synthesis of synthetic retinoids 9, 12 (AH61) and 14 starting with iodide 8 and acetylene 5. | ||
Stability of the synthetic retinoids versus natural ATRA
With the series of synthetic retinoids 9, 12 (AH61) and 14 in hand, their photostability was tested and compared to that of ATRA. We have previously shown, by using 1H NMR spectroscopy, that ATRA was susceptible to photoisomerisation and degradation when exposed to laboratory (fumehood) fluorescent light in the visible to near-UV range. After three days exposure, substantial isomerisation/degradation occurred with approximately 37% of ATRA remaining.10 When the synthetic retinoids 9, 12 (AH61) and 14 were exposed to the same wavelength of fluorescent light and their 1H NMR spectra compared with control (un-irradiated) samples, the severity of the isomerisation depended greatly upon the length of the conjugated linker. The short analogue 9 remained completely unaltered, with 100% of the original compound present (Fig. 2). The ‘ATRA-like’ analogue 12 (AH61) showed signs of isomerisation to one other isomer identified as (E,Z) but still retained 86% of the original compound and, more importantly, showed no signs of degradation unlike as previously reported for ATRA (Fig. 3). As one might predict, the extended analogue 14 was shown to be the least stable, with isomerisation not dissimilar to that seen for ATRA (Fig. 4). The inherent instability of ATRA lies within the conjugated linker region, as previously established, and replacement of sections of the polyene chain with both an acetylene moiety and phenyl rings as for EC23 4a provides complete stability. The stability of the systems 9, 12 (AH61) and 14 reported here varies as a function of polyene chain length, though there is a significant improvement in stability compared to similarly sized retinoic acids.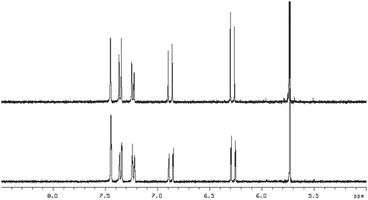 | ||
| Fig. 2 1H NMR at 400 MHz of 9 in DMSO-d6 (5.0–8.5 ppm region) in: top – in the absence of light and; bottom – after 3 days exposure to white fluorescent light at a distance of 40 cm. | ||
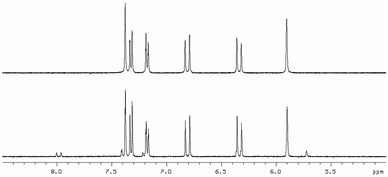 | ||
| Fig. 3 1H NMR at 400 MHz of 12 (AH61) in DMSO-d6 (5.0–8.5 ppm region) in: top – in the absence of light and; bottom – after 3 days exposure to white fluorescent light at a distance of 40 cm. | ||
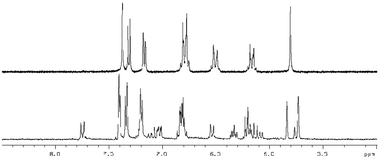 | ||
| Fig. 4 1H NMR at 400 MHz of 14 in DMSO-d6 (5.0–8.5 ppm region) in: (A) the absence of light and; (B) after 3 days exposure to white fluorescent light at a distance of 40 cm. | ||
Effect of the novel retinoids on human pluripotent stem cells
Having prepared the series of synthetic retinoids 9, 12 (AH61) and 14, they were assessed for their ability to induce differentiation carried out using a human embryonal carcinoma (EC) cell line. EC cells are the stem cells of teratocarcinomas, and therefore, retain their ability to differentiate into one or more of the embryonic germ layers. One of the earliest and most frequently used human EC cell lines is the TERA2 cell line, originally isolated from a lung metastasis originating from a testicular germ cell tumour.30 A subsequent sub-clone culture, termed TERA2.cl.SP12, was developed in 2001 by immunomagnetic isolation followed by single cell selection and culture.31 This cell line was chosen for our study, because it has been shown to differentiate in response to ATRA. After 21 days of co-culture with ATRA a heterogeneous cell population was visualised containing multiple mature functioning neurons.32–34 Consequently, this makes the TERA2.cl.SP12 cell line an effective model system for the in vitro study of neurogenesis.To compare the effect of the synthetic retinoids 9, 12 (AH61) and 14versus both known natural retinoid responses and other synthetic retinoids previously synthesised within the group, cultures of TERA2.cl.SP12 EC cells were incubated with 10 μM concentrations of each compound synthesised. Simultaneously, control cultures were assessed consisting of untreated, therefore undifferentiated, cells along with cultures exposed to the vehicle, DMSO. Induction of cellular differentiation in response to the synthetic retinoids was initially evaluated by analysing the expression profile of known markers for both stem cell and differentiated phenotypes (Fig. 5). Flow cytometry analysis was performed at 3, 7 and 14 days on cell samples, treated with 10 μM ATRA, 9, 12 (AH61) and 14. The cells were analysed for the expression of the stem cell antigens, SSEA-3 (globoseries stage specific embryonic antigen-3) and TRA-1-60 (keratin-sulfate-associated glycoprotein stem surface marker), whereby a decrease in the expression level of SSEA-3 and TRA-1-60 indicates cells committing to differentiation. Simultaneously, as cell lines of the TERA2 lineage are well characterised in their ability to form neurons in response to ATRA (33,34), flow cytometry was also used to assess the expression of A2B5 (ganglioseries antigen marking early-stage neural cells), a marker which is expressed during the early stages of neuronal development. Similar expression profiles were visualised for 12 (AH61) as were seen for ATRA over the 14 day period (Fig. 5). In cell cultures treated with the short and extended analogues 9 and 14, expression levels of SSEA-3 and TRA-1-60 remained relatively high with A2B5 staying comparatively low. Consequently, cultures treated with 9 and 14 were cultured for only 7 days and not through to 14 days due to overgrowth (Fig. 5). As both 9 and 14 did not induce any significant degree of cellular differentiation, all further studies were centred on the ATRA-sized analogue 12 (AH61).
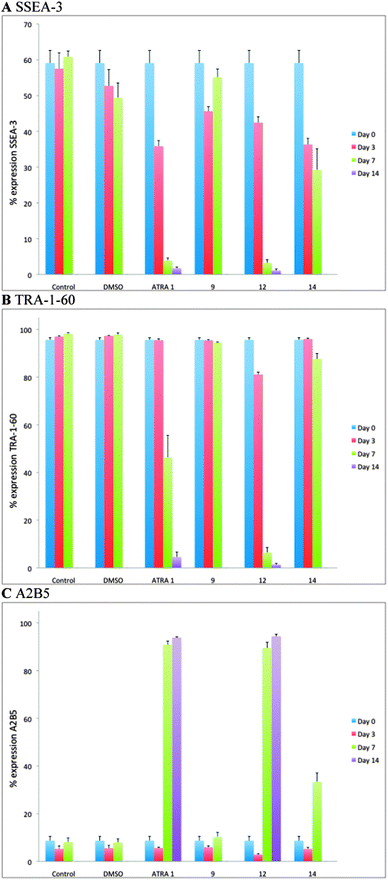 | ||
| Fig. 5 Flow cytometric analysis of the induction of TERA2.cl.SP12 cell differentiation in response to retinoids. Markers of stem cells (SSEA-3 and TRA-1-60, A and B respectively) and neural differentiation (A2B5, C) were differentially regulated during the 14 day culture period in response to 10 μM ATRA, 9, 12 and 14. Both ATRA and 12 (AH61) resulted in a rapid decrease in expression of the stem cell markers and reciprocal increase in neural marker expression after 14 days. Treatment with 9 and 14 did not induce differentiation; cells continued to proliferate and the experiment was terminated after 7 days as a results of being over-confluent. Data represent mean ± SEM, n = 3. | ||
Further assessment of cellular differentiation in response to synthetic retinoid 12 was evaluated through the expression profiles of known genes. Samples from the cells analysed by flow cytometry at 3, 7 and 14 days after treatment with 10 μM concentrations of retinoids were simultaneously analysed through RT-PCR. Three genes of interest were chosen, i.e. Nanog and Oct-4, as gatekeepers of pluripotency, and Pax-6 as a motor and ventral neural phenotype. The expression of Nanog and Oct-4 was suppressed in cultures treated with both ATRA and analogue 12 (AH61), all in a similar manner (Fig. 6). Consistent with these observations, Pax-6 expression increased to up to and just after day 7, after which it decreased slightly to day 14. Again both natural retinoid and synthetic analogue showed a similar profile (Fig. 6).
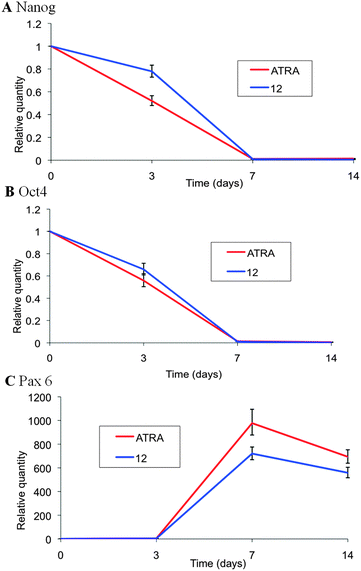 | ||
| Fig. 6 RT-PCR analysis of the induction of TERA2.cl.SP12 cell differentiation in response to retinoids. Genes associated with pluripotency (Nanog and Oct 4, A and B respectively) and motor and ventral neural phenotype (Pax 6, C) were differentially regulated during the 14 day culture period in response to 10 μM ATRA and 12 (AH61). Both ATRA and 12 (AH61) resulted in a rapid decrease in expression of the pluripotency genes and reciprocal increase in the motor and ventral neural gene after 14 days. Data represent mean ± SEM, n = 3. | ||
IR imaging of human pluripotent stem cells treated with novel retinoid derivatives
Results from the biological assays, Fig. 5 and 6, clearly show that the synthetic retinoid 12 (AH61) and ATRA induce the differentiation of the TERA2.cl.SP12 cells in a similar manner, whereas, retinoids 9 and 14 do not. Recently, Clemens et al.35 successfully used Fourier Transform Infrared Microspectroscopy (FT-IRMS) coupled with multivariate analysis to distinguish TERA2.cl.SP12 cells from their differentiating derivatives. An infrared absorption spectrum of a molecule is characteristic of the structure and bonding within the molecule and thus represents a molecular fingerprint. The infrared spectrum of a biological cell contains a superposition of spectra from all the molecules within the cell and, although these cannot be separated, the overall pattern is nevertheless characteristic of the cell. The technique can therefore be used to identify different cell types or to identify induced cellular changes such as differentiation. Accordingly, we used FT-IRMS to further assess and validate the biological activity of synthetic retinoid 12 (AH61).Fig. 7(a) shows that cells, 7 days after treated synthetic retinoid 12 (AH61), can be distinguished from the pluripotent stem cells using FT-IRMS coupled with Principle Component Linear Discriminant Analysis (PC-LDA). The biochemical changes of the differentiating cells are described in Fig. 7(b).
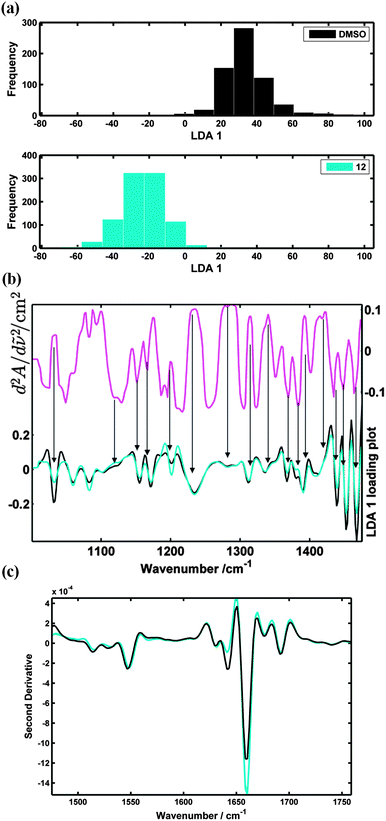 | ||
| Fig. 7 (a) PC-LDA analysis, 2 PCs extracted, spectra transformed to second derivative and then auto-scaled. Histograms display where the DMSO and synthetic retinoid 12 (AH61) treated TERA2.cl.SP12 cells lie in the LDA 1 scores plot after 7 days; (b) (upper curve) LDA 1 spectral loading, which shows the wavenumbers that contribute to the separation seen from the PC-LDA analysis (lower curves) mean spectra of the DMSO and 12 (AH61) treated cells (fingerprint spectral range); (c) mean spectral comparison of the DMSO and 12 (AH61) treated cells (protein spectral range). (Note that a positive absorption band will appear as a negative “peak” in the second derivative but at the same wavenumber position as the original.) | ||
In the LDA 1 spectral loading, Fig. 7(b), absorption increases at wavenumbers associated with proteins (1234, 1286, 1315, 1337 cm−1) can be seen from cells treated with 12 (AH61) when compared to those of the undifferentiated stem cells.36 Protein changes can also be seen from the mean spectral differences shown as second derivatives in Fig. 7(c). The negative “peaks” at 1640 and 1660 cm−1 (components of the amide I band which is very sensitive to secondary protein structure) are indicative of beta sheet and alpha helix structure, respectively. Clearly, the cells treated with 12 (AH61) have an increased level of alpha helix structure proteins compared with the control TERA2.cl.SP12 stem cells.
The LDA 1 spectral loading also shows decreases in absorption associated with RNA at 1120 (symmetric stretching of the phosphodiester band of RNA)37 and 1367–1369 cm−1 (C–N stretching vibration associated with cytosine and guanine).36 These overall changes in net RNA and protein content from the cells treated with 12 (AH61) may be a sign of mRNA stores being used up and transcription occurring to direct the synthesis of specific proteins as a result of induction and progression of cellular differentiation.
Spectra recorded from the TERA2.cl.SP12 cells treated with 12 (AH61) were also compared against cells treated with ATRA, and EC23, at 7 days using PC-LDA, Fig. 8. The LDA scores plot clearly shows that spectra recorded from the retinoid treated cells (blue, cyan, green) can be distinguished from the pluripotent stem cells (black) in LDA scores space. Fig. 8 and 9 also show that the TERA2.cl.SP12 cells treated with both ATRA and the synthetic retinoid 12 (AH61) are biochemically similar, 7 days after the introduction of the retinoids indicated by the virtual overlap of the cyan and green data. This in agreement with both the flow cytometry and RT-PCR analysis results seen in Fig. 5 and 6, respectively.
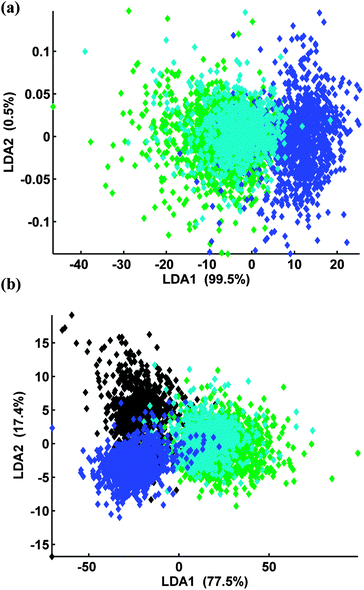 | ||
| Fig. 8 (a) PC-LDA of TERA2.cl.SP12 cells treated with ATRA (green), EC23 (blue) and 12 (AH61) (cyan) at day 7, 3 PCs extracted (b) PC-LDA of TERA2.cl.SP12 cells treated DMSO (black), ATRA (green), EC23 (blue) and 12 (AH61) (cyan) at day 7, 10 PCs extracted. | ||
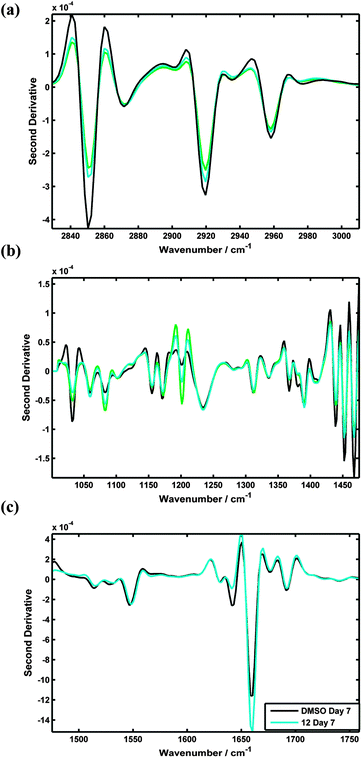 | ||
| Fig. 9 Mean spectral comparisons of ATRA, 12 and DMSO treated TERA2.cl.SP12 cells: (a) lipid spectral range (2830–3010 cm−1); (b) fingerprint spectral range (1000–1475 cm−1) (c) protein spectral range (1475–1760 cm−1). | ||
Biological assays, such as flow cytometry and RT-PCR, are seen as the industry standard and are commonly used when discriminating stem cells from their differentiating derivatives. However, these biological assays can be laborious and have been shown to have some drawbacks.38 Results seen from the FT-IRMS analysis are in agreement with those seen from the biological assays and these were achieved without intensive sample preparation, relatively cheaply, and very importantly, quickly. As an infrared absorption spectrum of a cell contains information of the macromolecular composition, protein, lipid, carbohydrate and nucleic acid changes from differentiating cells can be explained. This therefore shows the potential of FT-IRMS in screening stem cells and their differentiating derivatives. The information obtained can be seen as complementary to biological assays when investigating cell differentiation and understanding cellular development. Because of the speed of FT-IRMS, it is possible that it may be favoured when looking at the effect of new synthetic retinoids on known embryonic model systems.
Summary and conclusions
Our findings further underline the value of understanding how the structure of retinoid analogues relates to their biological activity. In this study we report on the importance of polyene chain length in relation to compound stability and their effect on cellular differentiation by human pluripotent stem cells. Increasing the polyene chain length resulted in enhanced compound instability and levels of isomerisation proportional to the degree of such structural modification. Lengthening of the polyene linker region resulted in isomerisation not dissimilar to that seen for ATRA. In terms of induction of biological activity, a short chain length did not provoke a biological response (compound 9). The longest chain length we tested (compound 14) induced only a small partial cellular response that was most likely the result of a mixture of compounds produced from the isomerised parent molecule. In relation to the molecular structures tested, the optimal structure of the polyene linker region appears to be of medium chain length [compound 12 (AH61)] in between that of retinoids 9 and 14. Using conventional molecular methods and FT-IRMS, we showed that synthetic retinoid 12 (AH61) showed biological activities comparable to ATRA. However unlike ATRA, compound 12 (AH61) remained mostly intact during the compound stability tests and showed only mild isomerisation.In conclusion, we have further demonstrated how understanding the structure activity relationship of synthetic retinoids plays an important role in compound design. Moreover, compound 12 (AH61) represents a potentially useful tool for biological investigations avoiding the issues of severe compound isomerisation experienced by ATRA whilst conserving a high level of biological activity.
Experimental
General synthetic chemistry
All cross-couplings were carried out under an argon atmosphere using standard Schlenk techniques unless otherwise stated. Other reactions were performed in the air in reagent-grade solvents. The compound 1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene acetylene 5 was prepared according to the literature.10 Trimethylsilylacetylene was obtained from Fluorochem, methyl propiolate and methyl tetrolate from Aldrich, and 4,4,6-trimethyl-2-vinyl-1,3,2-dioxaborane 10 from Frontier Scientific.Dry solvents were dried by the use of a commercial solvent purification system (SPS). Petroleum ether (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60) refers to the fraction of petroleum ether that boils between 40 and 60 °C. Where mixtures of solvents have been used for chromatography, the ratios given refer to volumes used. Thin layer chromatography (TLC) was performed on Polygram SIL G/UV254 plastic backed silica gel plates. Visualisation was achieved using a UV lamp at 254 nm, or staining with basic potassium permanganate. Column chromatography was carried out at medium pressure using silica gel, 40–60 μm, 60 Å obtained from Alfa Aesar. 1H NMR spectra were obtained in CDCl3, unless otherwise stated, at 400, 500 or 700 MHz on Varian Mercury-400, Varian Inova-500 or Varian VNMRS 700 MHz spectrometers respectively. Spectra are reported as chemical shift δ (ppm) (number of protons, multiplicity, coupling constant J (Hz), assignment). All peaks are reported using the residual protic solvent peak of CHCl3 (δH = 7.26 ppm), as an internal reference. 13C NMR spectra were obtained in CDCl3 at either 126 or 176 MHz on Varian Inova-500 or Varian VNMRS 700 MHz spectrometers, respectively. All peaks are reported using the residual protic solvent peak of CHCl3 (δC = 77.00 ppm) as an internal reference. 11B NMR spectra were recorded at 128 MHz on a Bruker Avance-400 spectrometer in ppm with respect to external BF3·OEt2. IR spectra were recorded on a Perkin-Elmer Paragon 1000 FT-IR Spectrometer. EI-MS analyses were performed on an Agilent 6890N GC equipped with a 5973N MSD Performance Turbo CI running an EI mode, and an Anatune Focus Autosampler/liquid handler, using UHP helium as the carrier gas. ES-MS analyses were performed on a Thermo-Electron Corp. Finnigan LTQ-FT mass spectrometer, using the electrospray in positive ion mode (ES+) to generate ions. Elemental analyses were obtained using an Exeter Analytical CE-440 analyser. Melting points were obtained using a Gallenkamp melting point apparatus.
:60) refers to the fraction of petroleum ether that boils between 40 and 60 °C. Where mixtures of solvents have been used for chromatography, the ratios given refer to volumes used. Thin layer chromatography (TLC) was performed on Polygram SIL G/UV254 plastic backed silica gel plates. Visualisation was achieved using a UV lamp at 254 nm, or staining with basic potassium permanganate. Column chromatography was carried out at medium pressure using silica gel, 40–60 μm, 60 Å obtained from Alfa Aesar. 1H NMR spectra were obtained in CDCl3, unless otherwise stated, at 400, 500 or 700 MHz on Varian Mercury-400, Varian Inova-500 or Varian VNMRS 700 MHz spectrometers respectively. Spectra are reported as chemical shift δ (ppm) (number of protons, multiplicity, coupling constant J (Hz), assignment). All peaks are reported using the residual protic solvent peak of CHCl3 (δH = 7.26 ppm), as an internal reference. 13C NMR spectra were obtained in CDCl3 at either 126 or 176 MHz on Varian Inova-500 or Varian VNMRS 700 MHz spectrometers, respectively. All peaks are reported using the residual protic solvent peak of CHCl3 (δC = 77.00 ppm) as an internal reference. 11B NMR spectra were recorded at 128 MHz on a Bruker Avance-400 spectrometer in ppm with respect to external BF3·OEt2. IR spectra were recorded on a Perkin-Elmer Paragon 1000 FT-IR Spectrometer. EI-MS analyses were performed on an Agilent 6890N GC equipped with a 5973N MSD Performance Turbo CI running an EI mode, and an Anatune Focus Autosampler/liquid handler, using UHP helium as the carrier gas. ES-MS analyses were performed on a Thermo-Electron Corp. Finnigan LTQ-FT mass spectrometer, using the electrospray in positive ion mode (ES+) to generate ions. Elemental analyses were obtained using an Exeter Analytical CE-440 analyser. Melting points were obtained using a Gallenkamp melting point apparatus.
:pet. ether, 5:95, as eluent) yielded (E)-3-iodobut-2-enoic acid methyl ester as a colourless oil (1.10 g, 25%) and recovered (Z)-3-iodobut-2-enoic acid methyl ester (2.90 g, 66%). All spectral and analytical properties were identical to those reported in the literature.39:pet. ether, 1:9 as eluent) gave (2E,4E)-3-methyl-5-(4,4,6-trimethyl-[1,3,2]-dioxaborinan-2-yl)-penta-2,4-dienoic acid methyl ester (780 mg, 74%) as a clear oil; νmax/cm−1 2976, 2359, 1710, 1596, 1393, 1327, 1290, 1229 and 1153; 1H NMR (700 MHz, CDCl3) δH 1.28 (3H, d, J = 6, CH3), 1.30 (3H, s, CH3), 1.31 (3H, s, CH3), 1.51 (1H, t, J = 13, CH2), 1.80 (1H, dd, J = 13 and 2.5, CH2), 2.25 (3H, d, J = 1, CH3), 3.70 (3H, s, OCH3), 4.21–4.27 (1H, m, CH), 5.87 (1H, s, CH), 5.93 (1H, d, J = 18, CH) and 6.92 (1H, d, J = 18, CH); 13C NMR (176 MHz, CDCl3) δC 13.5, 23.3, 28.3, 31.4, 46.1, 51.3, 65.2, 71.3, 121.0, 129–130, 149.2, 153.2, 167.8; 11B NMR (128 MHz, CDCl3) δB 26 (br s); m/z (ES) 252.1643 (M+, C13H21BO4, requires 252.1642). 237, 221, 196, 152, 137, 124 and 83.:pet. ether, 1:9, a eluent), gave the product as a clear oil, which slowly crystallised as an off-white solid (602 mg, 86%); all spectral and analytical properties were identical to those reported in the literature.39νmax/cm−1 3070, 3013, 2943, 1708, 1612, 1433, 1386, 1359, 1234, 1190 and 1153; 1H NMR (400 MHz, CDCl3) δH 2.24 (3H, d, J = 1.2, CH3), 3.71 (3H, s, OCH3), 5.74 (1H, q, J = 0.8, CH), 6.89 (1H, d, J = 15, CH), 7.10 (1H, d, J = 15, CH); 13C NMR (126 MHz, CDCl3) δC 13.6, 51.5, 84.8, 120.0, 148.5, 151.4, 167.2; m/z (EI) 252 (M+), 221, 193, 126 and 125.:EtOAc, 95:5, as eluent) gave (2E,4E)-2-methyl-7-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)-hepta-2,4-dien-6-ynoic acid methyl ester as a yellow oil (510 mg, 76%); νmax/cm−1 2956, 2192, 1715, 1608, 1490, 1434, 1358, 1232 and 1154; 1H NMR 500 MHz, CDCl3: δH 1.27 (6H, s, 2× CH3), 1.28 (6H, s, 2× CH3), 1.68 (4H, s, 2× CH2), 2.31 (3H, d, J = 1.2, CH3), 3.73 (3H, s, OCH3), 5.84 (1H, s, CH), 6.24 (1H, d, J = 16, CH), 6.70 (1H, d, J = 16, CH), 7.21 (1H, dd, J = 1.5 and 8, Ar), 7.26 (1H, d, J = 8, Ar), 7.41 (1H, d, J = 1.5, Ar); 13C NMR (126 MHz, CDCl3) δC 13.4, 31.9, 32.0, 34.5, 34.6, 35.1, 35.1, 51.5, 87.6, 96.0, 114.7, 120.1, 120.4, 127.0, 128.9, 130.3, 143.7, 145.4, 146.3, 151.4, 167.4; m/z (ES) 337.2164 (M + H+, C23H29O2, requires 337.2162), 337, 305 and 215.:1, 20 mL) followed by the addition of LiOH·H2O (250 mg, 5.94 mmol). The mixture was stirred at room temperature for 48 h in the absence of light after which the reaction was judged to be complete by tlc. The mixture was then acidified to pH 1 by the addition of HCl (20%), and extracted with diethyl ether (20 mL, 2×). The sample was evaporated to give the crude product as a yellow powder. This was re-crystallised from acetonitrile to yield the product as a yellow crystalline solid (394 mg, 82%); m.p. 205.1–206.7 °C; νmax/cm−1 2500–3500, 2194, 1679, 1604, 1488, 1459, 1255 and 1185; 1H NMR: (700 MHz, CDCl3) δH 1.27 (6H, s, 2× CH3), 1.28 (6H, s, 2× CH3), 1.68 (4H, s, 2× CH2), 2.32 (3H, d, J = 1, CH3), 5.87 (1H, s, CH), 6.29 (1H, d, J = 16, CH), 6.73 (1H, d, J = 16, CH), 7.21 (1H, dd, J = 1.5 and 8, Ar), 7.27 (1H, d, J = 8, Ar), 7.41 (1H, d, J = 1.5, Ar); 13C NMR: (126 MHz, CDCl3) δC 13.6, 31.9, 32.0, 34.4, 34.6, 35.1, 35.1, 87.5, 96.6, 115.6, 119.9, 120.0, 127.0, 129.0, 130.3, 143.5, 145.4, 146.4, 153.7, 172.0; m/z (ES) 321.1858 (M−, C22H26O2, requires 321.1860), 260, 186, 159 and 91; anal. calcd for C22H26O2: C, 81.95; H, 8.13. Found: C, 81.87; H, 8.10.:petroleum ether, 5:95, as eluent) gave (E)-3-methyl-5-(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)-pent-2-en-4-ynoic acid methyl ester as a pale yellow oil (530 mg, 99%). νmax/cm−1 2957, 2195, 1715, 1612, 1433, 1342, 1269, 1198, 1180 and 1135; 1H NMR (700 MHz, CDCl3) δ 1.27 (6H, s, 2× CH3), 1.28 (6H, s, 2× CH3), 1.68 (4H, s, 2× CH2), 2.40 (3H, d, J = 1.5, CH3), 3.73 (3H, s, OCH3), 6.15 (1H, q, J = 1.5, CH), 7.21 (1H, dd, J = 8 and 1.5, Ar), 7.27 (1H, d, J = 8, Ar), 7.41 (1H, d, J = 1.5, Ar); 13C NMR (176 MHz, CDCl3) δC 20.3, 31.9, 32.0, 34.5, 34.7, 35.1, 35.1, 51.5, 90.5, 94.9, 119.4, 123.4, 127.0, 129.2, 130.5, 138.8, 145.5, 146.8, 166.9; m/z (ES) 311.2011 (M + H+, C21H27O2, requires 311.2006). 279.:1, 20 mL) at room temperature. After 48 hours the reaction was judged to be complete and acidified to pH 1 to 2 by 20% HCl, extracted with ether (20 mL), dried (MgSO4) and concentrated to give an off-white solid. Recrystallisation from acetonitrile yielded the product as an off-white crystalline solid (25 mg, 31%). νmax/cm−1 2912, 2197, 1688, 1602, 1428, 1296, 1225 and 1145; 1H NMR (700 MHz, CDCl3) δ 1.27 (6H, s, 2× CH3), 1.28 (6H, s, 2× CH3), 1.68 (4H, s, 2× CH2), 2.41 (3H, d, J = 1.5, CH3), 6.18 (1H, d, J = 1.5, CH), 7.22 (1H, dd, J = 8 and 1.5, Ar), 7.28 (1H, d, J = 8, Ar), 7.42 (1H, d, J = 1.5, Ar); 13C NMR (176 MHz, CDCl3) δC 20.6, 31.9, 32.0, 34.5, 34.7, 35.0, 35.1, 90.5, 96.2, 119.3, 122.4, 127.1, 129.2, 130.6, 141.3, 145.5, 147.0, 169.0; m/z (ES) 296.1773 (M+, C20H24O2, requires 296.1771), 279, 255, 84 and 43.:pet. ether, 1:9, as eluent) to give the product (2E,4E,6E)-3-methyl-7-(4,4,6-trimethyl-[1,2,3]-dioxaborinan-2-yl)-hepta-2,4,6-trienoic acid methyl ester as a pale yellow oil (274 mg, 67%). νmax/cm−1 2973, 1711, 1614, 1391, 1303, 1237, and 1150; 1H NMR (700 MHz, CDCl3) δH 1.28 (3H, d, J = 6, CH3), 1.31 (3H, s, CH3), 1.31 (3H, s, CH3), 1.51 (1H, t, J = 14, CH2), 1.80 (1H, dd, J = 14 and 3, CH2), 2.30 (3H, s, CH3), 3.71 (3H, s, OCH3), 4.21–4.27 (1H, m, CH), 5.68 (1H, d, J = 17, CH), 5.81 (1H, s, CH), 6.36 (1H, d, J = 17, CH), 6.63 (1H, dd, J = 17 and 11, CH) and 6.99 (1H, dd, J = 17 and 11, CH); 13C NMR (176 MHz, CDCl3) δC 13.9, 23.3, 28.3, 31.4, 46.1, 51.2, 65.1, 71.1, 119.9, 130.0, 132.0, 136.6, 137.9, 146.0, 152.6, 167.5; m/z (ES) 278.1802 (M+, C15H24BO4, requires 278.1798), 190, 174, 162, 144, 136, 58, 52 and 44.:pet. ether, 1:9, cooled to 0 °C) yielded the product as a pale yellow oil (131 mg, 65%). νmax/cm−1 2946, 1710, 1609, 1557, 1433, 1390, 1357, 1238, 1189 and 1152; 1H NMR (500 MHz, CDCl3) δH 2.31 (3H, d, J = 1, CH3), 3.73 (3H, s, OCH3), 5.87 (1H, s, CH), 6.28 (1H, d, J = 15, CH), 6.51 (1H, ddd, J = 15, 11 and 5, CH), 6.62 (1H, d, J = 15, CH), and 7.16 (1H, ddd, J = 15, 11 and 5, CH); 13C NMR (126 MHz, CDCl3) δC 13.8, 51.4, 83.4, 120.7, 133.8, 136.3, 145.0, 151.8, 167.5; m/z (ES) 277.9799 (M+, C9H11IO2, requires 277.9798), 247, 219, 127, 91, 65 and 50.:EtOAc, 95:5, as eluent) gave (2E,4E,6E)-3-methyl-7-(4,4,6-trimethyl-[1,2,3]-dioxaborinan-2-yl)-hepta-2,4,6-trienoic acid methyl ester as a pale yellow oil (50 mg, 64%). νmax/cm−1 2955, 2181, 1709, 1604, 1435, 1358, 1240, and 1153; 1H NMR (700 MHz, CDCl3) δH 1.27 (6H, s, 2× CH3), 1.28 (6H, s, 2× CH3), 1.67 (4H, s, 2× CH2), 2.31 (3H, s, CH3), 3.71 (3H, s, OCH3), 5.82 (1H, s, CH), 5.99 (1H, d, J = 15, CH), 6.34 (1H, d, J = 15, CH), 6.67 (1H, ddd, J = 15, 11 and 4, CH), 6.74 (1H, ddd, J = 15, 11 and 4, CH), 7.19 (1H, dd, J = 8 and 1.5, Ar), 7.27 (1H, d, J = 8, Ar), 7.39 (1H, d, J = 1.5, Ar); 13C NMR (176 MHz, CDCl3) δC 13.9, 31.9, 31.9, 34.6, 34.6, 35.1, 35.1, 51.3, 88.2, 95.5, 98.8, 115.0, 120.2, 120.4, 127.0, 128.8, 130.1, 134.0, 137.5, 140.6, 145.3, 146.1, 167.5; m/z (ES) 363.2322 (M + H+, C25H31O2, requires 363.2319). 331, 289 and 215.:1, 10 mL) at 4 °C. After the reaction was judged to be complete by tlc, the reaction mixture was acidified to pH 1/2 by 20% HCl, extracted with Et2O (20 mL), dried (MgSO4) and evaporated to give an orange oil. Purification by silica gel chromatography (petroleum ether:EtOAc, 9:1, as eluent) gave the product as a yellow solid (100 mg, 87%) and a 2.5:1 mixture of alkene diastereoisomers. 1H NMR (700 MHz, CDCl3) δH 1.26 (6H, s, 2× CH3), 1.27 (6H, s, 2× CH3), 1.67 (4H, s, 2× CH2), 2.32 (3H, s, CH3), 5.85 (1H, s, CH), 6.01 (1H, d, J = 14, CH), 6.36 (1H, d, J = 14, CH), 6.73 (2H, ddd, J = 14, 11 and 4, 2× CH), 7.19 (1H, dd, J = 8 and 1.5, Ar), 7.28 (1H, d, J = 8, Ar), 7.39 (1H, d, J = 1.5, Ar); 13C NMR (176 MHz, CDCl3) δC 14.1, 31.9, 31.9, 32.0, 34.4, 34.6, 35.1, 35.1, 88.1, 95.8, 99.0, 115.6, 119.4, 120.2, 126.9, 128.8, 130.2, 134.8, 137.2, 140.5, 145.4, 146.1, 154.4; m/z (ES) 347.2016 (M+ − H, C24H27O2, requires 347.2017).Exposure to light
Synthetic retinoid NMR samples were prepared and left to stand in ordinary laboratory light, with additional irradiation from a standard fluorescent light ca. 30 cm above the sample.Biological analytical procedures
:20 SSEA-3 and TRA-1-60, or 1:10 A2B5) and FITC-conjugated secondary antibody (Cappell, 1:100) as previously described. Labelled cells were analysed in a Guave EasyCyte™ Plus System (Millipore) flow cytometer. Thresholds determining the numbers of positively expressing cells were set against the negative control antibody P3X.FTIR microspectroscopy
The infrared data were recorded on a Varian 670 FTIR spectrometer interfaced with a Varian-620 imaging infrared microscope. The microscope was equipped with a 128 × 128 liquid nitrogen cooled MCT focal plane array detector (FPA) with a pixel effective size on the sample of 5.5 μm. The infrared spectral images were collected in transmission mode (512 scans at 4 cm−1 resolution) and the background image was recorded from a clean CaF2 slide. Data was processed using the same methods described by Clemens et al.35Acknowledgements
We thank the BBSRC and Reinnervate Ltd. for an industrial studentship (to A. P. H.) and the National Mass Spectrometry Service, Swansea. PG acknowledges STFC for funding of beamtime. GC acknowledges support from the School of Chemical Engineering and Analytical Science at UoM for a PhD scholarship.References
- M. B. Sporn, A. B. Roberts and D. S. Goodman, The Retinoids, Academic Press, Orlando, 2nd edn, 1984 Search PubMed.
- V. B. Christie, T. B. Marder, A. Whiting and S. A. Przyborski, Mini-Rev. Med. Chem., 2008, 8, 601–608 CrossRef CAS.
- M. Maden, Nat. Rev. Neurosci., 2007, 8, 755–765 CrossRef CAS PubMed.
- J. L. Napoli, Clin. Immunol. Immunopathol., 1996, 80, S52–S62 CrossRef CAS.
- S. A. Ross, P. J. McCaffery, U. C. Drager and L. M. De Luca, Physiol. Rev., 2000, 80, 1021–1054 CAS.
- G. Wald, Nature, 1968, 219, 800–802 CrossRef CAS.
- D. R. Soprano, P. Qin and K. J. Soprano, Annu. Rev. Nutr., 2004, 24, 201–221 CrossRef CAS PubMed.
- S. J. Freemantle, K. H. Dragnev and E. Dmitrovsky, J. Natl. Cancer Inst., 2006, 98, 426–427 CrossRef CAS PubMed.
- N. Lowe and R. Marks, Retinoids: a Clinicians Guide, London, 2nd edn, 1997 Search PubMed.
- V. B. Christie, J. H. Barnard, A. S. Batsanov, C. E. Bridgens, E. B. Cartmell, J. C. Collings, D. J. Maltman, C. P. F. Redfern, T. B. Marder, S. Przybirski and A. Whiting, Org. Biomol. Chem., 2008, 6, 3497–3507 CAS.
- A. Murayama, T. Suzuki and M. Matsui, J. Nutr. Sci. Vitaminol., 1997, 43, 167–176 CrossRef CAS.
- D. K. Bempong, I. L. Honigberg and N. M. Meltzer, J. Pharm. Biomed. Anal., 1995, 13, 285–291 CrossRef CAS.
- T. Suzuki, S. R. Kunchala, M. Matsui and A. Murayama, J. Nutr. Sci. Vitaminol., 1998, 44, 729–736 CrossRef CAS.
- S. R. Kunchala, T. Suzuki and A. Murayama, Indian J. Biochem. Biophys., 2000, 37, 71–76 CAS.
- F. Mavilio, A. Simeone, E. Boncinelli and P. W. Andrews, Differentiation, 1988, 37, 73–79 CrossRef CAS.
- A. Simeone, D. Acampora, L. Arcioni, P. W. Andrews, E. Boncinelli and F. Mavilio, Nature, 1990, 346, 763–766 CrossRef CAS PubMed.
- P. Venepally, L. G. Reddy and B. P. Sani, Arch. Biochem. Biophys., 1997, 343, 234–242 CrossRef CAS PubMed.
- R. W. Curley and J. W. Fowble, Photochem. Photobiol., 1988, 47, 831–835 CrossRef CAS.
- J. H. Barnard, J. C. Collings, A. Whiting, S. A. Przyborski and T. B. Marder, Chem.–Eur. J., 2009, 15, 11430–11442 CrossRef CAS PubMed.
- A. B. Roberts, M. D. Nichols, D. L. Newton and M. B. Sporn, J. Biol. Chem., 1979, 254, 6296–6302 CAS.
- C. A. Frolik, M. B. Sporn and D. S. Goodman, Metabolism of retinoids, in The Retinoids, Academic Press, New York, 1984 Search PubMed.
- A. M. McCormick, J. L. Napoli, H. K. Schnoes and H. F. Deluca, Biochemistry, 1978, 17, 4085–4090 CrossRef CAS.
- W. E. Piers, T. Wong, P. D. Coish and C. Rogers, Can. J. Chem., 1994, 72, 1816–1819 CrossRef.
- A. S. Batsanov, J. P. Knowles and A. Whiting, J. Org. Chem., 2007, 72, 2525–2532 CrossRef CAS PubMed.
- R. Takeuchi, K. Tanabe and S. Tanaka, J. Org. Chem., 2000, 65, 1558–1561 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467–4470 CrossRef CAS.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- J. D. White, J. C. Amedio, S. Gut, S. Ohira and L. R. Jayasinghe, J. Org. Chem., 1992, 57, 2270–2284 CrossRef CAS.
- H. B. Zhai, Q. S. Chen, J. R. Zhao, S. J. Luo and X. S. Jia, Tetrahedron Lett., 2003, 44, 2893–2894 CrossRef CAS.
- J. Fogh and G. Trempe, Human tumour cells in-vitro, Plenum Press, New York, 1975 Search PubMed.
- S. Przyborski, Stem Cells, 2001, 19, 500–504 CrossRef CAS PubMed.
- R. Stewart, L. Coyne, M. Lako, R. F. Halliwell and S. A. Przyborski, Stem Cells Dev., 2004, 13, 646–657 CrossRef CAS PubMed.
- R. Stewart, V. B. Christie and S. A. Przyborski, Stem Cells, 2003, 21, 248–256 CrossRef CAS PubMed.
- S. A. Przyborski, I. E. Morton, A. Wood and P. W. Andrews, Eur. J. Neurosci., 2000, 12, 3521–3528 CrossRef CAS PubMed.
- G. Clemens, K. R. Flower, A. P. Henderson, A. Whiting, S. A. Przyborski, M. Jimenez-Hernandez, F. Ball, P. Bassan, G. Cinque and P. Gardner, Mol. BioSyst., 2013, 9, 677–692 RSC.
- Z. Movasaghi, S. Rehman and I. Rehman, Appl. Spectrosc. Rev., 2008, 43, 134–179 CrossRef CAS.
- P. G. L. Andrus and R. D. Strickland, Biospectroscopy, 1998, 4, 37–46 CrossRef CAS.
- A. Downes, R. Mouras and A. Elfick, J. Biomed. Biotechnol., 2010, 1–10 CrossRef PubMed.
- J. P. Knowles, V. E. O'Connor and A. Whiting, Org. Biomol. Chem., 2011, 9, 1876–1886 CAS.
| This journal is © The Royal Society of Chemistry 2013 |