 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective inhibition of the unfolded protein response: targeting catalytic sites for Schiff base modification
Susana M.
Tomasio
a,
Heather P.
Harding
b,
David
Ron
b,
Benedict C. S.
Cross
*bc and
Peter J.
Bond
*a
aUnilever Centre for Molecular Science Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: pjb91@cam.ac.uk; Tel: +44 (0)1223 763981
bUniversity of Cambridge Metabolic Research Laboratories and NIHR Cambridge Biomedical Research Centre, Cambridge CB2 0QQ, UK. E-mail: bcc33@cam.ac.uk; Tel: +44 (0)1223 588040
cDepartment of Haematology, University of Cambridge, Cambridge, CB2 2PT, UK
First published on 19th July 2013
Abstract
Constitutive protein misfolding in the endoplasmic reticulum (ER) can lead to cellular toxicity and disease. Consequently, the protein folding environment within the ER is highly optimised and tightly regulated by the unfolded protein response (UPR). The apparent convergence of myriad diseases upon proteostasis in the ER has triggered a broad effort to identify selective inhibitors of the UPR. In particular, the most ancient component of this cellular stress pathway, the transmembrane protein IRE1, represents an appealing target for pharmacological intervention. Several inhibitors of IRE1 have recently been reported, each containing an aldehyde moiety that forms an unusual, highly selective Schiff base with a single key lysine (K907) within the RNase domain. Here we review the progress made in chemical genetic manipulation of IRE1 and the unfolded protein response and discuss computational strategies to rationalise the selectivity of covalently active small molecules for their targets. As an exemplar, we provide additional evidence that K907 of IRE1 is buried within a particularly unusual environment that facilitates Schiff base formation. New free-energy calculations within a molecular dynamics (MD) simulation framework show that the pKa of K907 is reduced by ∼3.6 pKa units, relative to the model pKa of lysine in water. This significant pKa perturbation provides additional insights into the precise requirements for inhibition and for RNase catalysis by IRE1. Our computational method may represent a general approach for identifying potential covalent inhibitory lysine sites within buried protein cavities.
The unfolded protein response and IRE1
The fitness and survival of eukaryotic organisms is contingent upon the fidelity of protein biosynthesis within their cells. Loss-of-function mistakes in protein folding damage cellular efficiency, whilst proteotoxic gain-of-function misfolding is associated with a heavy burden of human disease. In healthy cells, quality control mechanisms act to preclude toxicity and maintain protein folding homeostasis. However, in a diseased cell these same pathways can also be corrupted and the rectifying intentions subverted to cause dysfunction.1 This apparent conflict and the convergence of multiple diseases on protein folding homeostasis has precipitated a need for new tools to understand the cellular response to perturbations in proteostasis, and garnered a broad effort for discovery.Whilst protein biosynthesis begins in the cytosol of the cell, a large proportion of the eukaryotic proteome is trafficked through the luminal space of the endoplasmic reticulum (ER) soon after the initiation of synthesis.2 Thus, the ribosome-studded rough ER is a major site of protein synthesis and this dominance is reflected in the complexity and sophistication of the systems operating to defend homeostasis in this compartment. In metazoans, accumulated unfolded or misfolded proteins are recognised by three transmembrane resident ER sensors: PERK (PKR-like endoplasmic reticulum eIF2α kinase), IRE1 (inositol-requiring enzyme 1) and ATF6 (activating transcription factor 6). These components adapt the ER to the folding client load using both translational and transcriptional interventions in a pathway collectively known as the unfolded protein response (UPR). Of these components, only IRE1 is conserved from fungus to man. It is the most studied element of the UPR, is linked most promiscuously to human disease, and its unusual dual enzymatic activity is a salient draw for chemical biologists.
IRE1 exists as two isoforms in mammals: IRE1α is ubiquitously expressed, whilst IRE1β, exhibiting only slightly differing enzymology,3 is restricted to the epithelial cells of the intestinal tract. IRE1 comprises a tripartite structure with a luminal domain linked via a single transmembrane segment to the cytosolic kinase and endoribonuclease domains. The luminal domain of IRE1 is closely related to that of PERK and detects the accumulation of unfolded proteins by one of several proposed mechanisms. Both the direct binding of unfolded clients to IRE14 and manipulation of the membrane composition5 can impact on IRE1 activity. Additionally, the constitutively IRE1-bound molecular chaperone BiP (binding immunoglobulin protein) is dislocated away from IRE1 as unfolded clients accumulate.6,7 These conditions shift the monomeric pool of IRE1 towards oligomerization, triggering the kinase activity of IRE1 and trans-autophosphorylation of adjacent IRE1 protomers.8 Phosphorylated IRE1 is activated, and is arranged in order to promote the endoribonuclease function that defines the role of IRE1 in the UPR. ER stress-activated IRE1 cleaves the latent mRNA for the XBP1 (X-box binding protein 1) transcription factor, liberating a 26 bp fragment and generating a frame-shifted transcript for XBP1 upon re-ligation.9,10 Processed XBP1 then upregulates the expression of genes encoding ER molecular chaperones, ER biosynthetic enzymes, translocation components, ER-associated degradation factors and ER membrane biogenesis enzymes.11,12 Thus, IRE1 responds to the saturation of ER client load with a broad regulatory programme that resolves ER stress by generating improved folding conditions, enhancing degradation and swelling the ER volume to thermodynamically favour folding over aggregation.13 In addition to these functions, IRE1 can also cleave a subset of other ER-bound mRNAs,14 in a process that may have links to cellular physiology,15,16 but which at present is only partially understood. Regulated IRE1-dependent mRNA decay (RIDD) attenuates protein load on the ER by pre-emptively degrading ER client mRNA,17 and has also recently been linked to a surprising innate immune response to infection by cholera toxin.18 In this study, RIDD products were shown to act as part of a signalling cascade to initiate a cytokine response to the toxin, where IRE1 was able to directly bind the cholera toxin to trigger the effect. IRE1 can also provide a node for pro-apoptotic signals emanating from the ER via the ASK1 (apoptosis signalling kinase) and JNK (c-Jun N-terminal kinase) pathways.19,20 This bipartisanship of IRE1 in the adaptive and apoptotic response of the cell to unfolded protein stress hints at a complex relationship with disease and places the enzyme at a compelling nexus for pharmacological intervention. Metabolic diseases including type II diabetes mellitus, neurodegeneration, cancer and inflammatory diseases have all been linked to the IRE1/XBP1 pathway,1 and in most cases pathogenesis is linked with a hyperactive UPR.
Pharmacological inhibition of the IRE1 endonuclease
The enzymology and structure of IRE1 make it an ideal candidate for reverse chemical genetics. The kinase domain of IRE1 has been targeted by nucleotide analogues, allosterically inactivating the RNase of IRE1 and blocking high order assembly of the IRE1 multimers,21 but direct inhibitors of the RNase have only been discovered following large-scale screening efforts. Remarkably, each of these studies converged upon molecules with very similar chemistry. The mammalian protein was adapted to a FRET-derepression assay for high-throughput screening and libraries of >220![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00022 and >24000017 compounds were screened for kinetic disruption of the fluorescence signal. In a third programme, a cell-based XBP1-lufierase assay was used to screen >60000 compounds for their effect on in vivo IRE1 activity.23 The lead compounds selected following each independent effort yielded small hydrophobic molecules containing an ortho-hydroxyl aryl aldehyde moiety: the salicylaldehydes,22 the coumarin 4μ8C17 and STF08301023 (Fig. 1). Although the aldehyde moiety of STF083010 went undetected during the screening process, it is clear that the imine bond of the library compound rapidly hydrolyses in aqueous solution to liberate an active carbaldehyde-containing napthalene.17,24 Indeed, for each of these inhibitors of IRE1, the aldehyde moiety was initially masked in the library compound by an imine linkage (Fig. 1), an important consideration when trying to deconvolute the data returned from high-throughput screening efforts and to characterise lead compounds.
00022 and >24000017 compounds were screened for kinetic disruption of the fluorescence signal. In a third programme, a cell-based XBP1-lufierase assay was used to screen >60000 compounds for their effect on in vivo IRE1 activity.23 The lead compounds selected following each independent effort yielded small hydrophobic molecules containing an ortho-hydroxyl aryl aldehyde moiety: the salicylaldehydes,22 the coumarin 4μ8C17 and STF08301023 (Fig. 1). Although the aldehyde moiety of STF083010 went undetected during the screening process, it is clear that the imine bond of the library compound rapidly hydrolyses in aqueous solution to liberate an active carbaldehyde-containing napthalene.17,24 Indeed, for each of these inhibitors of IRE1, the aldehyde moiety was initially masked in the library compound by an imine linkage (Fig. 1), an important consideration when trying to deconvolute the data returned from high-throughput screening efforts and to characterise lead compounds.
 | ||
| Fig. 1 Inhibitors of the IRE1 endonuclease identified by high-throughput screening. (A) 4μ8C identified by Cross et al.17 (B) Lead salicylaldehyde inhibitor identified by Volkman et al.22 (C) STF-083010 identified by Papandreou et al.23 In each of the screening programmes, the hit compounds were initially selected from imine linked scaffolds (upper panels). These were subsequently found to hydrolyse during the aqueous in vitro assays to yield the carbaldehyde-containing active species (lower panels). | ||
The aldehyde moiety is key to inhibition and each of these compounds operates by formation of a Schiff base with at least one key residue in the IRE1 molecule. However, despite the covalent mode of these inhibitors, they appear to retain specificity for IRE1 both in vitro and in cellular studies.17,22,23 The explanation for the surprising selectivity of the drugs was greatly aided by biochemical analysis of 4μ8C and the application of in silico docking and molecular dynamics simulations to rationalise the structural implications for the drug-inhibited IRE1 molecule.
Molecular dynamics simulations & insights into IRE1 inhibition
The molecular dynamics (MD) approach provides a Newtonian physics-based framework in which to simulate motions of biomolecules at unparalleled temporal and spatial resolution. Typically, the structure of a protein, obtained experimentally via nuclear magnetic resonance (NMR) spectroscopy or X-ray crystallography, is embedded in a simulation unit cell containing a physiologically modelled salt solution, resulting in a system containing tens of thousands of atoms.25,26 The forces acting on each atom are calculated via a classical potential function or “force field” composed of simple equations containing terms for intra- and inter-molecular interactions, which may simplistically be likened to a collection of “balls and springs”. Under the assumption that these forces are constant over a small time interval, they may be used to estimate accelerations, and hence new positions and velocities, of the atoms in the system, according to Newton's laws of motion.27,28 Repeating this cycle many millions of times enables propagation of a simulation trajectory, from which information concerning the dynamics and energetics of biomolecules may be gathered, complementing available experimental data.Continuing improvements in algorithms, force field parameters, and high-performance computing technologies have made it routinely possible to carry out accurate simulations of biomolecular systems over timescales of tens or hundreds of nanoseconds, or even beyond.29 MD also provides an exciting opportunity to supplement the drug design process. Proteins are dynamic and flexible; they breathe, change shape, and respond to the presence of other molecules.30 Simulations can identify conformational changes in biomolecules in response to ligand binding/release,31,32 and can help to discover and characterize novel druggable sites.33–35 Simulations also make it possible to accurately predict thermodynamic properties such as ligand-binding free energies, via so-called “alchemical” transformation. In such approaches, the interaction between a drug and its environment are slowly “annihilated” during a series of MD simulations, in both the solvated, protein-bound and protein-free states.25,36,37 Measuring the respective energetic changes ΔGprotein,bind and ΔGwater,bind during these processes enables “completion” of the thermodynamic cycle, yielding the relative free energy change for extracting a drug molecule from solvent and positioning it within the protein binding site – in other words, the binding free energy ΔGbind (Fig. 2A). Such calculations can yield extremely accurate results, but are computationally demanding, particularly as a result of their sensitivity to inadequate conformational sampling. Thus, they are generally of most use in the drug optimization stage, following lead compound discovery.
 | ||
| Fig. 2 Thermodynamic cycles and free-energy calculations. (A) Cycle for estimating free-energy of binding of a non-covalent drug to a protein. (B) Cycle for estimating pKa shifts in a lysine residue on a protein site. (C) Simulation workflow for obtaining free-energy estimates to complete the thermodynamic cycle illustrated in (B). In (A) and (B), ΔGwater is the free-energy change associated with the unbound drug being annihilated (ΔGwater,bind) or with an unbound model lysine sidechain becoming deprotonated (ΔGwater,deprot). ΔGprotein is the equivalent free-energy change in the protein-bound state for annihilation (ΔGprotein,bind) or deprotonation (ΔGprotein,deprot), respectively. In (A), measurement of these quantities enables calculation of the free-energy ΔGbind for drug binding along the * arm. In (B), the * arm represents ΔΔGdeprot, the relative free-energy difference for deprotonation between the protein-resident and solvated forms of lysine, which enables calculation of the pKa shift within the protein relative to the model compound, as illustrated in (C). | ||
A combination of molecular modelling, simulation, and thermodynamics calculations has recently been utilized to gain insights into the mechanism of inhibition of IRE1 by 4μ8C, complementing and extending experimental observations.17 The chemical properties of 4μ8C allowed the covalent interaction of the drug with the enzyme to be directly observed by spectroscopy. This modification was further confirmed and mapped by high-performance liquid chromatography (HPLC)-linked mass spectrometry, revealing an unusually stable Schiff base at two mechanistically critical lysines. One of these (K907) is present in the endonuclease domain at the active site of IRE1 and is required for IRE1 RNase catalysis.17,38 The other lysine, K599, located within the kinase domain, plays a role in phosphate coordination and is common to all kinase domains. Curiously, only K907 was modifiable in vivo. Thus, two key questions concerning the specificity of 4μ8C was posed by these observations: first, why does measurable Schiff-base formation by 4μ8C only occur at two out of a total of twenty-five lysine residues in its cytosolic domains? Second, why is the modifiability of K599 context-dependent?
To begin to answer these questions, Cross et al. performed explicitly solvated, all-atom MD simulations of the “apo” uninhibited state of the IRE1 dimer.17 The complete system amounted to a size on the order of a quarter of a million atoms, making these calculations computationally demanding. The first hint to the peculiar specificity of 4μ8C came from an analysis of the exposed surface area accessible to solvent of the lysine residues present in IRE1. Most lysines exhibited an average exposed surface area of ∼90 Å2 during simulation on the nanosecond timescale. This agrees reasonably well with a previous analysis of protein structures from the protein databank (PDB), showing that lysine is the most solvent-accessible of all amino acids, with a mean exposed area of ∼100 Å2.39 With the exception of two residues buried at the dimer interface, the only lysines in IRE1 that did not follow this trend were K599 and K907, each with exposed areas of just ∼50 Å2. Only ∼10% of lysines in folded proteins tend to exhibit this degree of burial, and for comparison, fully exposed lysine possesses a surface area of over 200 Å2.39
Thus, whilst most lysines in the cytosolic domain of IRE1 are surface exposed and flexible, K599 and K907 remain somewhat shielded from bulk solvent. Following Schiff base formation by 4μ8C at these sites, reduced ease of hydrolysis of the imine bond compared to other sites was hypothesized to lead to a low off-rate, helping to explain the selectivity. On the other hand, this did not provide an explanation for the lack of modifiability of K599 in vivo. To answer this, in silico docking of 4μ8C, along with extensive geometry optimization, of the inhibitor-bound state in the presence and absence of ADP·Mg2+ was performed. Strikingly, favourable, unstrained binding of 4μ8C was only possible in the absence of nucleotide; in other words, the two were predicted to sterically compete for the same site. This mutually exclusive binding is consistent with experimental evidence where co-incubation of the inhibitor with nucleotide prevents the binding of 4μ8C to IRE1.17 Likewise, in the context of the whole cell, binding of 4μ8C to IRE1 was prevented by endogenous competing nucleotide.17
Predicted mode of IRE1 inhibition
To gain further insights into the structural basis for IRE1 inhibition, possible 4μ8C-inhibition modes were modelled, using a tailored protocol designed for the covalently-bound inhibitor involving extensive sampling and optimization in torsional-angle space around each rotatable bond of the Schiff base at K907.17 A number of possible orientations for 4μ8C were identified via this approach, the most favourable (large and negative enthalpy of interaction) of which satisfied all possible interactions within the binding site. As shown in Fig. 3A, this involves a stacked conformation of one of the 4μ8C coumarin rings against the sidechain of F889, along with two hydrogen-bonds to the sidechain of D885 and/or H910. The predicted location of 4μ8C would be expected to interfere with catalysis and impede substrate binding, by forming a wedge positioned between the catalytic residues Y892 and H910. | ||
| Fig. 3 IRE1 inhibition modes predicted for (A) 4μ8C,17 (B) napthalene analogue of STF-083010,23 and (C) 3-methoxy-6-bromo-sialicylaldehyde.22 The RNase domain is shown in cartoons format, with key residues highlighted in wireframe format and labelled. The binding conformation for each inhibitor was predicted via sampling and optimization in torsional-angle space around each Schiff base rotatable bond, using the CHARMM package55 with the CHARMM22/CMAP41 and CGenFF56 forcefields for protein and inhibitor, respectively. Molecular graphics were generated using VMD.57 | ||
We now extend these observations to the other families of small, potent inhibitory molecules of IRE1: a hydrolysed carbaldehyde-containing napthalene analogue of STF-08301023 and one of the most potent salicylaldehydes, 3-methoxy-6-bromo-sialicylaldehyde.22 The same exhaustive conformational sampling and optimization protocol was used to identify favourably bound states of each to K907, as described previously.17 In each case, a similar interaction mode was predicted for the most favourable orientation as for 4μ8C (Fig. 3B and C). For the STF-083010 analogue, an almost identical conformation was observed, with comparable stacking of one of the rings against F889, and two hydrogen-bonds to D885. For the salicylaldehyde, a hydrogen-bond was formed with H910 to facilitate stacking of the inhibitor's single ring against F889, again leading to a similar inhibitory mode within the catalytic site. Thus, these independently identified aldehyde-containing small molecules appear to have converged upon a similar mechanism of inhibition.
To test the stability of this predicted binding mode, the modelled conformation of 4μ8C bound to K907 was used to initiate multiple MD simulations of the inhibited IRE1 in solvent.17 In each subsequent simulation replica, the 4μ8C rings remained stacked against the sidechain of F889, and the inhibitor relaxed into a hydrophobic pocket formed by sidechains of nearby, nonpolar residues. Strikingly, this immersion of 4μ8C within the hydrophobic pocket appeared to constrain the RNase active site, which by comparison, was relatively dynamic over tens of nanoseconds in the apo, unbound state. This would be expected to further slow the off-rate by reducing bulk water exchange and hence hydrolysis at the imine bond site.
Computational determination of IRE1 K907 pKa
The reduced solvent exposure of the K907 sidechain and the stable burial of bound 4μ8C within a hydrophobic microenvironment are of great interest, since conditions of low polarity or polarizability can shift pKa values of internal ionizable residues including lysines40 towards their unbound form. The ε-amino group of K907 must be in its uncharged form in the first step of Schiff base formation, to enable nucleophilic attack on the 4μ8C carbaldehyde carbonyl. We therefore questioned whether the hydrophobic microenvironment accessible to K907, buried within the RNase active site, might down-shift its pKa, and thus increase the rate of Schiff base formation by 4μ8C. To investigate this further, we note that the dissociation constant Ka of a titratable group is related to the free energy change of deprotonation ΔGdeprot in the following way:where k is the Boltzmann constant. The pKa may therefore be related to ΔGdeprot:
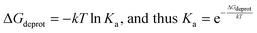

As discussed above, alchemical transformation within an MD simulation framework may be used to estimate free-energy changes associated with, for example, repositioning a solvated drug molecule into a protein binding site, as part of a thermodynamic cycle (Fig. 2A). In the same way, the free-energy associated with deprotonation may be calculated, via annihilation of the proton associated with an ionizable site. Thus, according to Fig. 2B, calculation of the free energy for deprotonation of an ionisable residue in the protein (ΔGprot,deprot) and for the model amino acid analogue in solvent (ΔGwater,deprot) enables estimation of a pKa shift, ΔpKa:

Two sets of free-energy calculations were therefore performed, of the complete IRE1 dimer in water (to estimate ΔGprotein,deprot), and of a lysine amino acid analogue in water (to estimate ΔGwater,deprot). In each case, 20–30 MD simulations of 3–4 ns each were performed, in which the system was gradually changed via a coupling parameter λ, from state λ = 0 (K907 protonated) to state λ = 1 (K907 deprotonated). The respective free energy changes (averaged over both IRE1 monomers) could then be calculated as a sum of energies over the consecutive, intermediate states. Each simulation was carried out using the same conditions as described previously.17 Briefly, the system contained dimeric IRE1 (PDB ID code 3P23) in complex with ADP·Mg2+, surrounded by a 0.1 M NaCl solution in a truncated octahedral box (∼250000 atoms total), and was described using the CHARMM22/CMAP41 forcefield within the GROMACS 4.5 simulation package.42,43
As shown in Fig. 4, the cumulative free energy change for deprotonation of isolated lysine in water (ΔGwater,deprot = 73.1 ± 0.6 kcal mol−1) and of K907 in IRE1 (ΔGprotein,deprot = 68.2 ± 0.6 kcal mol−1) yields ΔΔGdeprot = −4.9 ± 0.8 kcal mol−1. We therefore estimate a pKa down-shift for K907 within IRE1 of 3.6 ± 0.6 units. Thus, relative to the model pKa of lysine in water (∼10.5), the pKa of the K907 sidechain is likely to lie at around 7, and macroscopically, it will exist in IRE1 molecules as an equal population of charged and uncharged states at neutral pH. At the structural level (Fig. 4), this pKa shift results from the ease with which K907 becomes buried within the deep hydrophobic pocket upon deprotonation, combined with the dynamic nature of the RNase active site entrance, as observed previously.17 For example, upon complete deprotonation, the ε-amino group of K907 moves further into the binding site, and its mean distance from D885 and H910 at the active site entrance increases, respectively, from 0.5 ± 0.2 Å and 0.2 ± 0.1 Å during simulation at λ = 0, to 0.7 ± 0.2 Å and 0.5 ± 0.1 Å during simulation at λ = 1.
 | ||
| Fig. 4 Calculated pKa shift for K907 in IRE1. Above, free-energy curves are shown for K907 in IRE1 (ΔGprotein) and for a lysine sidechain in water (ΔGmodel), with corresponding snapshots shown below of the simulated structures of the RNase domain in its protonated (λ = 0) and deprotonated (λ = 1) forms. The RNase domain is shown in cartoons format. Hydrophobic residues within the binding pocket, along with key ionizable sidechains, are shown in wireframe format. Molecular graphics were generated using VMD.57 The free-energy curves were generated using the Bennett acceptance ratio (BAR) method with soft-core potentials. ΔGprotein was calculated using λ windows 0.05 or 0.025 apart, between 0 ≤ λ < 0.7 and 0.7 ≤ λ ≤ 1.0 respectively. ΔGmodel used λ windows 0.05 apart between 0 ≤ λ ≤ 1.0. Extensive energy minimisation followed by 0.2 ns equilibration was performed, prior to the production run for each window. Each simulation was run under conditions of constant temperature (298 K)58 and pressure (1 atm),59 using a 2 fs time step with LINCS constraints applied to bond lengths.60 Electrostatics were calculated using the Particle-Mesh Ewald algorithm, and Van der Waals interactions were smoothly switched off from 10 to 12 Å. | ||
Experimental support for this shift in pKa at lysine 907 is found when the efficiency of 4μ8C modification of IRE1 is measured in vitro. Binding of the fluorescent compound to IRE1 can be visualised in-gel following UV irradiation,17 and we find that whilst this modification is strictly pH-dependent, reactivity is observed even in conditions where lysines on IRE1 are expected to be deprotonated (Fig. 5). Substantially increased binding of 4μ8C is found when the buffered conditions match the pKa for lysine in solution where the compound is able to react with the other nucleophiles on IRE1 (>pH 10, Fig. 5, lane 11). Hence, the pKa perturbed K907 is able to participate in Schiff base formation even at pH 7.
 | ||
| Fig. 5 pH-dependent binding of 4μ8C to IRE1. Compound was incubated with the human IRE1 cytosolic domain17 at the indicated pH before addition of sodium borohydride and analysis by electrophoresis. Bound 4μ8C was visualised by UV excitation and the protein was then stained with coomassie brilliant blue (CBB). Quantification lane shows the calculated relative fraction of the protein that was found modified under each condition. | ||
Overall, our results suggest that K907 is more easily deprotonated than usual under normal cell conditions, consistent with it being buried in a hydrophobic microenvironment that is partially dehydrated. This would lead to increased nucleophilicity of the K907 ε-amino group, an increased rate of Schiff base formation with 4μ8C, and a slow off-rate.
RNase catalysis in IRE1
In addition to the contribution to the Schiff base formation, a shifted pKa at K907 may have consequences for the mechanism of RNase catalysis by IRE1. Structural analysis of the yeast apoenzyme provided the first detailed clues into the mechanism of endoribonucleolytic scission in IRE1.44 Catalysis by IRE1 is likely to proceed via a general acid–base mechanism, similar to that found for Archaeglobus fulgidus splicing endonuclease and yielding a 2′,3′-cyclic phosphate product.44,45 His1061/Tyr1043 in yeast IRE1 would therefore form a proton transfer acid–base pair for scission, homologous to His257/Tyr246 of the splicing endonuclease and analogous to His pairs in both RNase A and T1. R1056 in yeast IRE1 stabilises the transition state by hydrogen bond formation with the non-bridging oxygen during proton transfer to enact RNA cleavage.46,47 The contribution to catalysis for the two core catalytic residues in ScIre1p has been quantitatively demonstrated,46 and elements of the active site are also well conserved to mammalian IRE1 and RNase L. This core mechanism therefore likely largely persists through evolution, although some notable divergence in the active site from the yeast to the human enzyme is apparent. In the structure for the human enzyme for example,48 the conserved HsIRE1α R902 is found displaced from the core catalytic pair whilst the sidechain of K907 is oriented towards the His/Tyr intersection. Thus, in mammalian IRE1, K907 appears to be better placed to support transition state stabilisation than R902, and given the pronounced reduction of the pKa it is likely to perform this role better, forming a stronger hydrogen bond to the cleavage intermediate. A similar arrangement is found in RNase A where K41 is pKa perturbed, lowering the free energy of the transition state to promote catalysis.49 Our computationally derived model may therefore suggest a mechanistic rationale for excelled catalysis by human IRE1 as compared to the budding yeast enzyme – a more potent mammalian enzyme provides the necessary depth to cleavage for the emergence of RIDD and the physiological consequences of lapsed specificity. Interestingly, although the S. cerevisiae IRE1 does not appear to conduct a RIDD-like process, the fission yeast enzyme does.50 Solution of the S. Pombe IRE1 structure might therefore provide important new clues to the evolution of IRE1 cellular function.Outlook
Large-scale screening efforts have led to the independent discovery of several hydrophobic inhibitors that target the IRE1 RNase domain. Each molecule contains a common aldehyde moiety, whose presence enables selective Schiff base formation with K907.17,22,23 A combination of biochemical analysis, molecular modelling, and simulation has provided a rationale for this mode of inhibition, and in particular, immersion of each molecule within the hydrophobic pocket of the RNase domain appears to contribute to specificity. As well as slowing the off-rate by protecting the imine bond against hydrolysis, we now believe that this binding mode also directly influences the chemistry of inhibition. The K907 sidechain must be in its uncharged form in the first step of Schiff base formation, to facilitate nucleophilic attack on the inhibitor. The surrounding, nonpolar microenvironment has indeed been shown to perturb the pKa of K907 to around 7. This would favour deprotonation and increase its intrinsic nucleophilicity, making it significantly reactive even at neutral pH, as supported by our experimental data. This is not unprecedented. A lysine within an aldolase catalytic antibody was previously demonstrated to proceed via a Schiff base intermediate; its deeply buried location within a hydrophobic pocket perturbed its pKa below 7.51 Moreover, such shifts can be functionally important in catalysis, and indeed, measurements based upon engineered staphylococcal nuclease constructs have revealed down-shifts in internal lysine residues by as much as 5 pKa units.52 In this context, our results also hint at the source of differing catalytic efficiencies in human and yeast IRE1. Uncovering the divergence in enzymology in the active site of IRE1 by both computational and biochemical means may therefore provide important new understanding of these cellular pathways.As well as providing a useful new tool for manipulating IRE1 in vivo and in vitro, these newly discovered classes of inhibitors provide a possible route to treatments for diseases associated with proteostasis. For many years, covalent drugs have stimulated anxiety in the pharmaceutical industry due to their potential for off-target reactivity. Nevertheless, several of the top-selling drugs are covalent inhibitors of their targets (e.g. proton pump inhibitors), leading to suggestions that we are likely to see a resurgence of interest in covalent drugs.53 Whilst existing covalent drugs have tended to be discovered through biological screening assays, it seems possible that a more targeted approach may now be possible. As suggested by Singh et al., design could involve identifying non-reversible inhibitors for a specific target site, followed by adaptation of the structure for covalent binding.53 Given the emerging perspective that pKa-shifted lysines in buried protein cavities may be more common than expected,54 we could therefore envisage a general drug design pipeline which incorporates the identification of nearby lysine residues, combined with simulation-based pKa shift calculations, towards selective Schiff-base inhibition.
Acknowledgements
We wish to thank the Darwin Supercomputer of the University of Cambridge and the Swiss National Supercomputing Center via DECI/PRACE-2IP for computational resources. PJB and SMT thank Unilever and EPSRC for funding. HPH, DR and BCSC were supported by a Wellcome Trust Principal Research Fellowship to DR.References
- S. Wang and R. J. Kaufman, J. Cell Biol., 2012, 197, 857–867 CrossRef CAS.
- B. C. S. Cross, I. Sinning, J. Luirink and S. High, Nat. Rev. Mol. Cell Biol., 2009, 10, 255–264 CrossRef CAS.
- T. Iwawaki, A. Hosoda, T. Okuda, Y. Kamigori, C. Nomura-Furuwatari, Y. Kimata, A. Tsuru and K. Kohno, Nat. Cell Biol., 2001, 3, 158–164 CrossRef CAS.
- B. M. Gardner, D. Pincus, K. Gotthardt, C. M. Gallagher and P. Walter, Cold Spring Harbor Perspect. Biol., 2013, 5, a013169 CrossRef.
- R. Volmer, K. van der Ploeg and D. Ron, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4628–4633 CrossRef CAS.
- A. Bertolotti, Y. Zhang, L. M. Hendershot, H. P. Harding and D. Ron, Nat. Cell Biol., 2000, 2, 326–332 CrossRef CAS.
- D. Pincus, M. W. Chevalier, T. Aragon, E. van Anken, S. E. Vidal, H. El-Samad and P. Walter, PLoS Biol., 2011, 8, e1000415 Search PubMed.
- C. E. Shamu and P. Walter, EMBO J., 1996, 15, 3028–3039 CAS.
- M. Calfon, H. Zeng, F. Urano, J. H. Till, S. R. Hubbard, H. P. Harding, S. G. Clark and D. Ron, Nature, 2002, 415, 92–96 CrossRef CAS.
- H. Yoshida, T. Matsui, A. Yamamoto, T. Okada and K. Mori, Cell, 2001, 107, 881–891 CrossRef CAS.
- A. H. Lee, N. N. Iwakoshi and L. H. Glimcher, Mol. Cell. Biol., 2003, 23, 7448–7459 CrossRef CAS.
- K. J. Travers, C. K. Patil, L. Wodicka, D. J. Lockhart, J. S. Weissman and P. Walter, Cell, 2000, 101, 249–258 CrossRef CAS.
- J. E. Chambers, K. Petrova, G. Tomba, M. Vendruscolo and D. Ron, J. Cell Biol., 2012, 198, 371–385 CrossRef CAS.
- J. Hollien, J. H. Lin, H. Li, N. Stevens, P. Walter and J. S. Weissman, J. Cell Biol., 2009, 186, 323–331 CrossRef CAS.
- D. Han, A. G. Lerner, L. Vande Walle, J. P. Upton, W. Xu, A. Hagen, B. J. Backes, S. A. Oakes and F. R. Papa, Cell, 2009, 138, 562–575 CrossRef CAS.
- K. Y. Hur, J. S. So, V. Ruda, M. Frank-Kamenetsky, K. Fitzgerald, V. Koteliansky, T. Iwawaki, L. H. Glimcher and A. H. Lee, J. Exp. Med., 2012, 209, 307–318 CrossRef CAS.
- B. C. S. Cross, P. J. Bond, P. G. Sadowski, B. K. Jha, J. Zak, J. M. Goodman, R. H. Silverman, T. A. Neubert, I. R. Baxendale, D. Ron and H. P. Harding, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E869–E878 CrossRef CAS.
- J. A. Cho, A. H. Lee, B. Platzer, B. C. Cross, B. M. Gardner, H. De Luca, P. Luong, H. P. Harding, L. H. Glimcher, P. Walter, E. Fiebiger, D. Ron, J. C. Kagan and W. I. Lencer, Cell Host Microbe, 2013, 13, 558–569 CAS.
- H. Nishitoh, A. Matsuzawa, K. Tobiume, K. Saegusa, K. Takeda, K. Inoue, S. Hori, A. Kakizuka and H. Ichijo, Genes Dev., 2002, 16, 1345–1355 CrossRef CAS.
- F. Urano, X. Wang, A. Bertolotti, Y. Zhang, P. Chung, H. P. Harding and D. Ron, Science, 2000, 287, 664–666 CrossRef CAS.
- L. Wang, B. G. Perera, S. B. Hari, B. Bhhatarai, B. J. Backes, M. A. Seeliger, S. C. Schurer, S. A. Oakes, F. R. Papa and D. J. Maly, Nat. Chem. Biol., 2012, 8, 982–989 CrossRef CAS.
- K. Volkmann, J. L. Lucas, D. Vuga, X. Wang, D. Brumm, C. Stiles, D. Kriebel, A. Der-Sarkissian, K. Krishnan, C. Schweitzer, Z. Liu, U. M. Malyankar, D. Chiovitti, M. Canny, D. Durocher, F. Sicheri and J. B. Patterson, J. Biol. Chem., 2011, 286, 12743–12755 CrossRef CAS.
- I. Papandreou, N. C. Denko, M. Olson, H. Van Melckebeke, S. Lust, A. Tam, D. E. Solow-Cordero, D. M. Bouley, F. Offner, M. Niwa and A. C. Koong, Blood, 2011, 117, 1311–1314 CrossRef CAS.
- C. L. Kriss, J. A. Pinilla-Ibarz, A. W. Mailloux, J. J. Powers, C. H. Tang, C. W. Kang, N. Zanesi, P. K. Epling-Burnette, E. M. Sotomayor, C. M. Croce, J. R. Del Valle and C. C. Hu, Blood, 2012, 120, 1027–1038 CrossRef CAS.
- J. D. Durrant and J. A. McCammon, BMC Biol., 2011, 9, 71 CrossRef CAS.
- R. O. Dror, R. M. Dirks, J. P. Grossman, H. Xu and D. E. Shaw, Annu. Rev. Biophys., 2012, 41, 429–452 CrossRef CAS.
- S. Khalid and P. J. Bond, Methods Mol. Biol., 2013, 924, 635–657 CAS.
- T. Paramo, D. Garzon, D. A. Holdbrook, S. Khalid and P. J. Bond, Methods Mol. Biol., 2013, 974, 435–455 CAS.
- D. W. Borhani and D. E. Shaw, J. Comput.-Aided Mol. Des., 2012, 26, 15–26 CrossRef CAS.
- D. L. Mobley and K. A. Dill, Structure, 2009, 17, 489–498 CrossRef CAS.
- D. Garzon, C. Anselmi, P. J. Bond and J. D. Faraldo-Gomez, J. Biol. Chem., 2013, 288, 19528–19536 CrossRef CAS.
- P. J. Bond and J. D. Faraldo-Gomez, J. Biol. Chem., 2011, 286, 25872–25881 CrossRef CAS.
- R. Baron and J. A. McCammon, Annu. Rev. Phys. Chem., 2013, 64, 151–175 CrossRef CAS.
- C. J. Woods, M. Malaisree, N. Pattarapongdilok, P. Sompornpisut, S. Hannongbua and A. J. Mulholland, Biochemistry, 2012, 51, 4364–4375 CrossRef CAS.
- M. J. Harvey and G. De Fabritiis, Drug Discovery Today, 2012, 17, 1059–1062 CrossRef CAS.
- M. R. Shirts, Methods Mol. Biol., 2012, 819, 425–467 CAS.
- A. Pohorille, C. Jarzynski and C. Chipot, J. Phys. Chem. B, 2010, 114, 10235–10253 CrossRef CAS.
- W. Tirasophon, K. Lee, B. Callaghan, A. Welihinda and R. J. Kaufman, Genes Dev., 2000, 14, 2725–2736 CrossRef CAS.
- L. Lins, A. Thomas and R. Brasseur, Protein Sci., 2003, 12, 1406–1417 CrossRef CAS.
- T. K. Harris and G. J. Turner, IUBMB Life, 2002, 53, 85–98 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS.
- B. Hess, C. Kutzner, D. Van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- P. Bjelkmar, P. Larsson, M. A. Cuendet, B. Hess and E. Lindahl, J. Chem. Theory Comput., 2010, 6, 459–466 CrossRef CAS.
- K. P. Lee, M. Dey, D. Neculai, C. Cao, T. E. Dever and F. Sicheri, Cell, 2008, 132, 89–100 CrossRef CAS.
- T. N. Gonzalez, C. Sidrauski, S. Dorfler and P. Walter, EMBO J., 1999, 18, 3119–3132 CrossRef CAS.
- A. V. Korennykh, A. A. Korostelev, P. F. Egea, J. Finer-Moore, R. M. Stroud, C. Zhang, K. M. Shokat and P. Walter, BMC Biol., 2011, 9, 47 CrossRef CAS.
- S. Xue, K. Calvin and H. Li, Science, 2006, 312, 906–910 CrossRef CAS.
- M. M. Ali, T. Bagratuni, E. L. Davenport, P. R. Nowak, M. C. Silva-Santisteban, A. Hardcastle, C. McAndrews, M. G. Rowlands, G. J. Morgan, W. Aherne, I. Collins, F. E. Davies and L. H. Pearl, EMBO J., 2011, 30, 894–905 CrossRef CAS.
- J. M. Messmore, D. N. Fuchs and R. T. Raines, J. Am. Chem. Soc., 1995, 117, 8057–8060 CrossRef CAS.
- P. Kimmig, M. Diaz, J. Zheng, C. C. Williams, A. Lang, T. Aragon, H. Li and P. Walter, eLife, 2012, 1, e00048 Search PubMed.
- C. F. Barbas, 3rd, A. Heine, G. Zhong, T. Hoffmann, S. Gramatikova, R. Bjornestedt, B. List, J. Anderson, E. A. Stura, I. A. Wilson and R. A. Lerner, Science, 1997, 278, 2085–2092 CrossRef.
- W. E. Stites, A. G. Gittis, E. E. Lattman and D. Shortle, J. Mol. Biol., 1991, 221, 7–14 CrossRef CAS.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS.
- D. G. Isom, C. A. Castaneda, B. R. Cannon and B. Garcia-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS.
- B. R. Brooks, C. L. Brooks, 3rd, A. D. Mackerell, Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, Jr., J. Comput. Chem., 2010, 31, 671–690 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS , 27–38.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |