 Open Access Article
Open Access ArticleBuilding synthetic gene circuits from combinatorial libraries: screening and selection strategies
Yolanda
Schaerli
ab and
Mark
Isalan
ab
aEMBL/CRG Systems Biology Research Unit, Centre for Genomic Regulation (CRG), Dr Aiguader 88, 08003 Barcelona, Spain. E-mail: yolanda.schaerli@crg.eu, isalan@crg.es
bUniversitat Pompeu Fabra (UPF), 08003 Barcelona, Spain
First published on 23rd January 2013
Abstract
The promise of wide-ranging biotechnology applications inspires synthetic biologists to design novel genetic circuits. However, building such circuits rationally is still not straightforward and often involves painstaking trial-and-error. Mimicking the process of natural selection can help us to bridge the gap between our incomplete understanding of nature's design rules and our desire to build functional networks. By adopting the powerful method of directed evolution, which is usually applied to protein engineering, functional networks can be obtained through screening or selecting from randomised combinatorial libraries. This review first highlights the practical options to introduce combinatorial diversity into gene circuits and then examines strategies for identifying the potentially rare library members with desired functions, either by screening or selection.
Introduction
Synthetic biologists apply engineering approaches to build new biological networks. These synthetic circuits are composed of biological parts1 and are helping us to understand design principles in nature, as well as having numerous potential applications in therapeutics, bioremediation, biofuels, agriculture and biosensing.2–4 While the field includes designing metabolic pathways5–8 or even whole cells,9–11 this review concentrates on transcriptional circuits. We focus in particular on the prospect of applying techniques from protein engineering for building gene networks, such as the screening or selection of combinatorial libraries.Whereas early synthetic genetic circuit engineering studies began with simple devices like a negative feedback loop12 and a toggle switch,13 nowadays the community focuses on circuits displaying more complex behaviours. Recent examples include oscillators comprising thousands of synchronised bacterial colonies,14 layered logic gates in bacteria,15 or mammalian cells performing programmable half-subtractor and half-adder calculations.16 Despite this progress, and the growing interest of the scientific community, it is well-known that synthetic biology faces serious technical difficulties.17–20 For example, an article entitled “Five hard truths for synthetic biology”17 outlined several problems in the field: many parts are not well characterised, are incompatible with the host cell and do not work as predicted when assembled into circuits. Therefore, building a synthetic network is usually still a challenging, slow and difficult process, involving “tweaking” and “debugging” the initial design to obtain a working device. The devil is often in the detail of small context-dependent effects, thus limiting our ability to build more complicated networks easily.
Although rationally designing and building any given device remains the goal, we have to admit that our current knowledge and understanding of how biology works is frequently insufficient. Therefore, synthetic biologists have started to apply the powerful method of directed evolution to synthetic networks.21–25 Combinatorial libraries of network variants are produced and then the variants with the desired properties are found by screening or competitive selection. Typically, the identified variants are subjected to one or more rounds of diversification and selection or screening.
In this review, after briefly touching upon the origins of directed evolution, we describe how combinatorial diversity can be introduced into synthetic transcriptional networks. We then focus on selection and screening systems to identify the functional devices in a library. As most of the reviewed work has been carried out in prokaryotes, the emphasis is on bacterial systems but analogous techniques could be applied to eukaryotic cells.
Protein engineering – the precursor of synthetic biology
The idea of synthetic biology has its roots in molecular cloning and recombinant DNA technologies, where genetic components such as transcription promoters and coding regions are now routinely combined to make protein expression constructs or other new plasmids.26 Making synthetic gene networks is seemingly just one level of complexity higher, simply harnessing the appropriate recombinant constructs to make networks.One field of biological engineering which is now relatively mature, and where new functional constructs are routinely made, is protein engineering. New proteins are constructed, often using structural information and an element of rational design,27,28 but also through screening or selecting from large randomised combinatorial libraries. In a screening assay, a specific output of the individual library members (e.g. their fluorescence) is measured and the best variants are taken to the next round of randomisation or screening. By contrast, in a selection, the desired behaviour of a variant is linked to a competitive survival advantage, so that only positive clones should ultimately survive the selection procedure. Many highly efficient selection systems (e.g. phage display29) as well as screening systems (e.g. based on flow cytometry30) have been successfully applied to protein engineering.31,32 It is therefore manifest that generating diversity and selecting or screening for the desired variants could also be a powerful tool for the engineering of synthetic networks.
Where to introduce the combinatorial diversity?
One important question is where to introduce diversity in the network. The target sites of mutations should be chosen in order to maximise the success rate of obtaining a functional network (mutational robustness33). By analogy to protein engineering, scaffolds such as zinc fingers tolerate mutation while retaining rich functional diversity. It is therefore necessary to find equivalent scaffolds for network engineering.34Another consideration should be the size of the library that can be screened or selected. For instance, when engineering synthetic transcription networks, it would be wasteful to randomise each transcription factor residue; most mutants would be non-functional or similarly functional when compared with the original network, and the library size would quickly become too big to screen. Rather, it would be smarter to mutate around the transcription factor DNA-binding interface, either mutating the key amino acid residues making DNA contacts, or the corresponding DNA bases in the promoter region. Thus, targeted mutations can provide functional diversity in relatively small, easy-to-handle libraries. The options for introducing diversity are numerous (Fig. 1) and include: network connectivities, promoters, ribosomal binding sites, codon variations, intergenic regions, protein parts, degradation tags and others. Here we discuss the different possibilities:
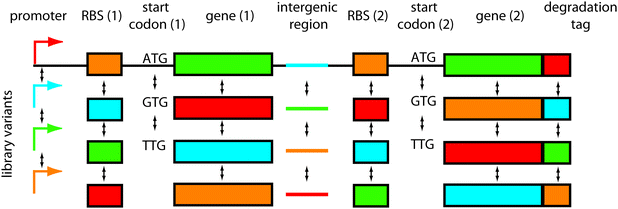 | ||
| Fig. 1 Where to introduce combinatorial diversity in bacterial gene network libraries. An operon containing two genes is shown as an example. The options of where to introduce variations are numerous: in the network connectivities (not shown), promoters, ribosomal binding sites, (start) codons, genes, intergenic regions, degradation tags, and combinations thereof. | ||
Network connectivities
In the first combinatorial network engineering study, Guet et al. built genetic circuits containing three transcriptional regulators (lacI, TetR and cI) and a green fluorescent protein (GFP) reporter.35 Five different promoters, including binding sites for the transcription factors, were randomly cloned in front of the three genes, thus connecting them in 125 (53) different ways. By analysing the behaviour of the resulting networks in the presence or absence of the chemical inducers of lacI (IPTG) and TetR (aTc), the logic gates NAND, NOR and NOT IF could be identified.While it is conceivable that this approach could be used to identify more new circuits, a different procedure predominates in the literature: for a given desired output a possible network topology is rationally designed and suitable parts are chosen and assembled. This process is potentially guided by a model.36–41 Directed evolution is then used only if the built circuit is non-functional or needs improvement.15,42,43
Promoters
Closely related to changing the network connectivity is altering promoter regions and thereby changing input–output relationships at network nodes. This is achieved by mutating the promoters in one or several of their functional units: RNA polymerase binding site, transcription start site and transcription factor binding sites and enhancers. The tuning of promoters has been used to reduce leakiness and to increase the dynamic range of expression15 or to fine-tune expression levels of gene parts.38The group of Elowitz built a combinatorial library of random promoter architectures containing up to three transcription factor binding sites, which could be placed in the distal, core or proximal regions of the promoter.44 A subset of the library was characterised and promoter strengths were observed that varied over five orders of magnitude. From this analysis, empirical rules were derived for bacterial promoter design, for example repression is strongest when the repressor binding site is located in the core part of the promoter and is weakest in the distal part.
Alternatively, the architecture of the promoter is kept constant and diversity is introduced by randomising all bases,45 or only a subset of the promoter while leaving key motifs unchanged.15,38,46–49 The latter strategy has the advantage that the promoter function is retained in most library members and that the library size can be kept small.
After the generation of the promoter library two different approaches have been pursued. First, the diversity is directly incorporated into the synthetic circuit and a screening or selection is performed to obtain the final working device.15 Alternatively, members of the library are characterised in the context of a basic device, to obtain a collection of promoters covering a wide range of strengths. The promoter matching the required strength is then used to build the intended network.38,45,48,49 The second approach requires a good idea of what promoter characteristics will render the device functional, i.e. from detailed in silico modelling. The advantage is that the collection of well-characterised promoters can be re-used for building different devices. While promoter engineering alters transcription expression levels, it is also possible to tune post-transcriptional processes, as described in the following sections.
Ribosomal binding sites
The translation levels of genes in synthetic circuits are most commonly adjusted by changing the ribosomal binding sites (RBSs) in mRNAs. An algorithm for the forward engineering of synthetic RBSs has recently been developed, for achieving specific expression levels.50 However, when the optimal expression level of one or more components in a circuit is not known, screening a RBS library is a powerful approach. For example, Anderson et al.51 wanted to engineer bacteria that can invade cancerous tumors upon a signal. For this purpose they put the gene of an invasin, that can initiate bacterial adhesion and invasion of mammalian cells, under the control of a signal-responsive promoter. However, with two of the three tested promoters, the device showed high leaky expression even in the absence of a signal. Functional circuits were ultimately obtained after introducing diversity at the RBS, with a positive selection in the presence of the signal and a negative screen in the absence of the signal.In another example, mutations were simultaneously targeted to the RBSs of two transcription factors, thus allowing a search for the right balance of their expression levels.52 Similarly, RBS libraries were applied in the construction of logic gates,15,43,53,54 orthogonal transcription–translation networks,47,55,56 a rewritable digital data storage device42,57 and bistable switches.57,58
Codon variations
Moving downstream of the RBS on an mRNA, start codons are where the ribosome begins translation and changing these codons offers another way of modulating translation levels. As prokaryotes mainly use three start codons, these can be rapidly tested. If more start codons are varied simultaneously – or if combined with other library diversity – a more elaborate screening or selection might become necessary. In one study, for example, a library targeted the RBS as well as the start codon of a gene.54Not only can the start codon be exchanged, but also the other codons. While it has long been known that codon usage alters gene expression levels, a recent study quantified this effect and found a 250-fold variation in GFP expression levels in E. coli, using different synonymous codons.59 Design parameters can be obtained to control synthetic gene expression in E. coli,60 and this could provide a source of variation for combinatorial network libraries. Various mechanisms may allow variation, including the use of rare codons or altering RNA secondary structure, via the presence or absence of hairpins.
Intergenic regions
The RNA secondary structure of untranslated regions between genes in an operon has been shown to affect transcription termination, mRNA stability and translation initiation.61 As a result of these combined effects, libraries of intergenic regions can vary the relative expression levels of two proteins in the same operon by up to 100-fold.61 Such regions therefore provide an alternative site to introduce diversity in a circuit.Protein parts
Of course it is not only possible to modify regulatory parts, but also the protein parts themselves. Often protein parts are obtained by directed evolution using a suitable screening or selection system that is independent of the rest of the device. Only once a functional protein with the desired characteristics has been found and tested is it incorporated into the synthetic network. This simplifies the screening or selection process and therefore allows the use of bigger libraries. The latter can be necessary because protein sequence space (21n, where n is a randomised position) rapidly overtakes promoter sequence space (4n). However, the protein mutagenesis can often be targeted to a subregion more likely to result in useful mutations, by using structural or biochemical information.31,32 Examples of such part engineering include altering the affinity of a protein to a signalling molecule62 or to a promoter.43A frequent goal of the directed evolution of protein parts for synthetic biology is to achieve orthogonality, i.e. the parts should only interact with their defined molecular partner, but not with other cell components.63 For example Zhan et al. evolved new variants of the lacI repressor to recognise different DNA sequences than the wild-type protein, and which no longer bind to the natural lacI operator (lacO).64 Collins et al. engineered a version of the LuxR transcription factor that activates transcription after binding to a different signalling molecule than its parent protein and which no longer recognises the natural quorum sensing signal.65 Similarly, orthogonal transcription factors,66–69 ribosomes,70 polymerases,49,71 receptor–ligand pairs72 and chaperones15 have been created to expand the repertoire of available protein parts for synthetic biology. In contrast to standard protein engineering (e.g. the improvement in the catalytic efficiency for a new substrate), directed evolution of an orthogonal part not only requires a positive selection or screen for improved binding or activity, but also a negative one in order to exclude undesired cross-talk.
Degradation tags
The half-life of proteins is an important parameter in synthetic networks displaying dynamic behaviour such as switching between different states upon an external signal. Degradation allows an output to be reversible and thus the system can fine-tune responses to varying inputs. The group of Endy recently constructed a circuit that can switch between a red and a green state in response to distinct inputs.42 In the absence of any input signal the last state is maintained. This was achieved by using two proteins involved in site-specific DNA integration – an integrase and an excisionase. Crucially, the device was only functional after the half-lives of these two enzymes were fine-tuned. To achieve this, the final residues of the ssrA degradation tags73 were randomised and the library was screened for functional circuits.Although the randomisation categories described above cover many possibilities, in principle, many other positions and processes could encode useful functional diversity. The main aim of any library design is to cover a useful range of variations (e.g. ‘low’, ‘middle’ or ‘high’ activity) with as few variants as possible, so as not to increase library size beyond practical limits. Having designed libraries, the next stage is to search for required outputs under particular conditions.
Screening and selection systems for synthetic networks
Having decided where to introduce combinatorial diversity in the synthetic circuit, the second important issue is how to identify the (rare) variants with desired properties. While for protein engineering many selections and screening systems are well established, for synthetic networks they are only just emerging and the field could still benefit from innovative new approaches. The challenge is that most synthetic devices not only have one state (e.g. being ON), but several (e.g. being ON or OFF or changing dynamically). Consequently, to identify a functional circuit, all the states have to be tested. While this might still be quite straightforward in the case of a switch or a logic gate (ON/OFF), it is rather more complicated to set up selections or screening systems for networks with outputs such as spatial or temporal patterns – and these are essential for many key biological processes. For example, the spots, stripes or waves found in Turing or Gierer–Meinhardt patterns74,75 are thought to be important in developmental patterning, but are difficult to engineer with current approaches. Therefore, the chosen strategy must strongly depend on the desired network behaviour. Nevertheless, the key considerations for choosing the method are common to all synthetic devices and are also shared with protein engineering: what is the throughput of the method? How does this compare to the library size? What is the dynamic range of the screening or selection? What are the enrichment rates? What are the frequencies of false positives and negatives? Next, we give an overview of the selection and screening systems used so far in synthetic biology projects and highlight their advantages and disadvantages.Screening systems
The majority of synthetic circuits are designed to have a fluorescent output. Therefore, it is often convenient to use a screening system with fluorescence as readout, as no further parts have to be added. For example to screen for an AND gate responding to arabinose and salicylate, a positive screening was performed in the presence of both chemicals.54 Library members displaying high fluorescence under these conditions were further subjected to three negative screens (no inducer, arabinose only, salicylate only). Only mutants showing the required behaviour under all four conditions were further characterized (Fig. 2).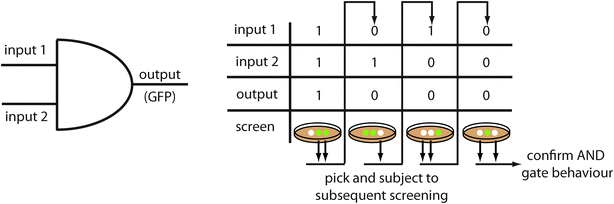 | ||
| Fig. 2 Example of a screening protocol. Screening for an AND gate, as performed by the Voigt group,54 requires a positive screen in the presence of the two inputs and three negative screens in the presence of one input or in the absence of any input. | ||
Such systems generate their own set of practical considerations. Often it might not be clear where to set the fluorescence threshold between positive and negative clones. Cells in the OFF state might already have some fluorescence, but an even higher signal in the ON state. Depending on the application, different thresholds can be acceptable. If so, it might be advisable to measure the ON/OFF ratios for all the members, thus avoiding discarding functional variants. Moreover, there can be a difference in responses at the levels of cell populations and individual cells. Depending on the application, it can sometimes be useful to retain even clones where only a small percentage of cells in the ‘clonal’ population display the desired behaviour when induced. To understand such systems, flow cytometry measurements can be essential.
As the functions of synthetic circuits become more complicated, the screening systems must also become more sophisticated. Lou and colleagues built a device in bacteria that switches from green to red after a first UV exposure and back to green after a second UV exposure (push-on push-off switch).52 To screen for this behaviour many steps were necessary: after transformation green colonies were picked and transferred to two agar plates, only one of which was irradiated with UV. Colonies that switched to red after exposure to UV, but did not in the absence of the signal were chosen. Subsequently the same procedure was repeated, but this time red cells switching to green after UV irradiation were selected.
If cells are grown in liquid culture the fluorescence can be measured with a fluorescence spectrometer or a flow cytometer. For cells growing on plates, a fluorescence microscope or an UV transilluminator can be used, the latter only if the excitation range of the fluorescent protein falls in the UV range (e.g. GFPuv78). Liquid cultures are usually handled in a 96-well format and standard agar plates fit about 200 colonies, meaning that the throughput of plate-based fluorescence screens is commonly 100–2000 library members.
Flow cytometry can be applied in two ways: as a fluorescence measuring tool or as a fluorescence activated cell sorting (FACS) tool. The advantage of using a flow cytometer instead of a fluorescence spectrometer for measurement is that the fluorescence of the individual cells is recorded. This allows one to determine the distribution of the cells within a population, rather than only measuring an average. For example a population might display an unexpected bimodal distribution due to the variable metabolic burden that is imposed on the host by the synthetic device.79 In other networks, it is only a part of the population that actually responds to the signal.16,52
A FACS machine can screen about 107 cells per hour80 and thus enables access to much bigger libraries than other screening methods based on fluorescence. Rather surprisingly, with the exception of few examples,46,47 this technique has hardly been applied so far to synthetic genetic circuits. The common occurrence of heterogeneous cell populations for one synthetic network, as mentioned above, might be one reason, as these will cause many false positives or negatives in a screening based on single cells. Nevertheless, the advantage of the superior throughput of this method probably outweighs the effort of eliminating errors post-sorting. We therefore expect that FACS will be used more often for the screening of synthetic transcriptional circuits in the future.
Although fluorescent proteins are by far the most commonly used markers for screenings, they are not the only option. Enzymes producing a colorimetric or a luminescent readout are also feasible. For example, in the above mentioned evolution of orthogonal lacI repressors, blue/white colony screening employed β-galactosidase expression and X-gal staining.64
Selection systems
By applying a Darwinian selection pressure to the randomised network, so that only the desired variants survive, much bigger libraries can be processed than when having to screen all library members one-by-one. In fact, when using a selection system, the transformation efficiency commonly becomes the limiting factor at around 1010 bacterial transformants per μg of DNA (although 106 is more achievable in practice26). The challenge is how to link the correct readout of a synthetic circuit, in each of its states, to the survival of it hosts; the output of synthetic networks can be coupled to a rescue gene in positive selections and a killer gene in negative selections. This means that under ON conditions only the networks expressing a resistance or a metabolic mutant complementation gene will survive. Conversely, under OFF conditions only the circuits that do not express the (conditionally) toxic gene will grow.Initial selections of this type used two independent genes on different plasmids,65 on the same plasmid81 or as a genetic fusion70 for the two selection conditions. The disadvantage of this strategy is that any mutation disabling the function of the killer gene results in false positives. Since false positives can quickly outgrow the rare positive library members, especially if the selections are performed in liquid culture, it is important to plate out cells on Petri dishes during selections. Moreover, it is necessary to remove and replace the selection marker plasmid after each round of selection because otherwise this will accrue false positive mutations under the strong selection pressure.70,81
To make selections more robust and to simplify processes, systems have been developed where one gene can function as a selection marker for both positive and negative selection. To achieve this, the selection marker gene can either enable cell survival or induce cell death under defined conditions.53,55,56,76,77 Because any mutation that eliminates the function of the protein will most likely affect both the ON and OFF selections, the chances of the emergence of false positives are much lower. This enables one to perform selections in liquid cultures, further increasing throughput and speeding up the process.
TetA, a tetracycline/H+ antiporter is just such a dual selector.55,56,76,77 Its expression confers resistance to tetracycline and can therefore be used for positive selections in the ON state. As a membrane-bound protein, its overexpression also makes the host bacteria more susceptible to toxic metal salts, including NiCl2. Therefore, cells with TetA expression will not grow well in the presence of NiCl2 allowing cells in the OFF state to be selected (negative selection) (Fig. 3A). This strategy was successfully applied in synthetic circuits based on riboswitches.55,56,77 Additionally, fusing TetA to GFPuv allows a quantitative readout without further subcloning.55,56
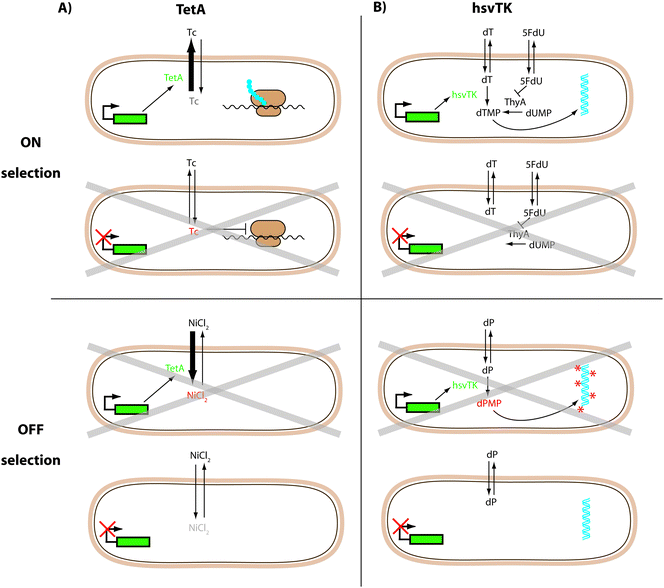 | ||
| Fig. 3 Dual selection protocols. (A) TetA confers resistance to tetracycline (ON selection) and makes the host bacteria more susceptible to toxic NiCl2 (OFF selection).55,56,76,77 (B) HsvTK can rescue the thymidine deficiency of a thymidine kinase deficient strain in the presence of a thymidylate synthase (ThyA) inhibitor (5FdU) (ON selection). HsvTK can also make cells sensitive to synthetic dP nucleosides; these become toxic upon phosphorylation by hsvTK (OFF selection).53 Grey crosses indicate cells where the conditions reduce viability. | ||
The Herpes simplex virus thymidine kinase (hsvTK) is an alternative dual selector with demonstrated use in E. coli.53 In the presence of a thymidylate synthase inhibitor (5FdU), a thymidine kinase-deficient strain does not grow, due to the lack of thymidine. In ON selections hsvTK expression can rescue this deficiency. The OFF selection is performed in the presence of synthetic nucleosides (e.g. deoxyribosyl-dihydropyrimido[4,5-c][1,2]oxazin-7-one: dP) that only become toxic upon phosphorylation by hsvTK (Fig. 3B). As with TetA, hsvTK can be fused C-terminally to GFP without losing its function (unpublished data).
Although impressive enrichment factors (1300–33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times per ON/OFF cycle) have been demonstrated for these two dual selectors in model selections,53,56 both systems are still waiting to be applied more widely. One reason might be that it is difficult to match the rather limited dynamic range of these selection systems to the functional range of the synthetic circuits. For example, while it is possible to select circuits that are ON among those that are completely OFF, it is more difficult to select against slightly leaky devices: low expression levels of TetA are already enough to confer tetracycline resistance. The use of less active TetA mutants82 might alleviate this problem. The upper expression limit of the dual selectors is also constrained as they contribute to the metabolic load imposed on the host cell, even in the absence of the negative selection conditions. Moreover, TetA overexpression is known to be detrimental to cell growth.84 Another concern is whether the use of mutagenic nucleosides in the hsvTK OFF selection will introduce undesired mutations. Therefore, while the throughput of selection systems is generally higher than that of their screening counterparts, they are often less flexible. However, it is still early days and time will show whether the dual selectors discussed here are robust enough to be adopted by the community.
000 times per ON/OFF cycle) have been demonstrated for these two dual selectors in model selections,53,56 both systems are still waiting to be applied more widely. One reason might be that it is difficult to match the rather limited dynamic range of these selection systems to the functional range of the synthetic circuits. For example, while it is possible to select circuits that are ON among those that are completely OFF, it is more difficult to select against slightly leaky devices: low expression levels of TetA are already enough to confer tetracycline resistance. The use of less active TetA mutants82 might alleviate this problem. The upper expression limit of the dual selectors is also constrained as they contribute to the metabolic load imposed on the host cell, even in the absence of the negative selection conditions. Moreover, TetA overexpression is known to be detrimental to cell growth.84 Another concern is whether the use of mutagenic nucleosides in the hsvTK OFF selection will introduce undesired mutations. Therefore, while the throughput of selection systems is generally higher than that of their screening counterparts, they are often less flexible. However, it is still early days and time will show whether the dual selectors discussed here are robust enough to be adopted by the community.
For devices intended for targeted applications, the selection can be tailored to be more specific for the purpose of the device. An elegant example was demonstrated for the above-mentioned bacteria that can invade cancer cells upon a signal:51 the bacteria carrying the device library were incubated with the cultured cancer cells, followed by the addition of an antibiotic. Bacteria unable to invade the mammalian cells were killed, but bacteria inside the cancer cells were protected from the antibiotic effect. Internalised bacteria were then released by mammalian cell lysis and grown on plates. Positive clones were subsequently screened for loss of invasiveness in the absence of the signal.
After any process requiring several rounds of selection or screening, the resulting circuits always have to be analysed carefully. Controls should ensure a good understanding of the function of the individual circuit components. This analysis should also uncover the cases where the observed behaviour is caused by (unexpected) interactions of the synthetic network and the host cells.79,85
The work cited in this section has been mostly carried out in bacterial cells. However, synthetic biology in eukaryotes is fast catching up86 and we expect to see the development of analogous screening systems in the near future.
Challenges and future prospects
Synthetic biology has made significant advances in recent years and is developing a set of community-based resources.2 However, the dream that characterised parts can simply be put together to yield new genetic circuits still remains a dream, at least for now. In the meantime, directed evolution is a powerful method to help us bridge the gap between our incomplete understanding of nature's design rules and our desire to build synthetic networks. Techniques to manipulate DNA are already mature87 and, as we have discussed in this review, many practical options are available to diversify the devices combinatorially. In contrast to its precursor – the field of protein engineering – rather few screening and selection systems have been described so far. This is especially true if the desired circuits are intended to have behaviour that is more complicated than switches or logic gates as, for example, in oscillatory networks or spatial patterns. We expect that the integration of cutting edge technologies within synthetic biology labs will give rise to new ways of performing the screening of genetic circuit libraries. Among these promising techniques are screenings based on microfluidics88,89 and/or automated high-content imaging.90–92 For example, the replacement of 96-well plates with a microfluidic device producing water-in-oil emulsion droplets could increase the throughput of screenings by several orders of magnitude.93 Automated systems, able to take and analyse pictures or movies, could be imagined for screening complicated patterns, such as Turing or Gierer–Meinhardt patterns74,75 (Fig. 4). However, this remains a truly challenging goal for the synthetic biology community. A combination of rational design, guided by a model, combined with screenings of combinatorial libraries is in our opinion a promising approach to work towards this goal.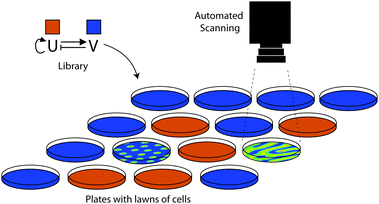 | ||
| Fig. 4 Automated screening of spatial patterning. The schematic shows a thought-experiment on how one might screen a combinatorial library scaffold for a Gierer–Meinhardt system.83 Thousands of randomised candidates might have to be tested to find the correct behaviour. The library would comprise variants of an activator (U; red) and an inhibitor (V; blue) which would communicate local, non-linear self-activation and long range inhibition signals to other cells. By plating library members on dishes or multiwell plates (here, one per plate) thousands of randomised parameter sets might be screened for potential patterning behaviour. Reproduced from ref. 34. | ||
Another emerging trend is the engineering of multicellular traits in cell consortia where single cells carry out different simple tasks.94 This makes particular sense because our ability to increase the size and complexity of synthetic networks in one host cell has begun to stagnate.17 Two recent studies95,96 applied this approach in a rather elegant way: cells, each carrying out a simple function, were combined in multiple ways so that the whole consortia carried out far more complicated distributed computational tasks than the individual cells. Perhaps this concept of distributed networks truly represents the future of synthetic biology and individual robust functions could be made relatively easily using the screening or selection of combinatorial libraries. Ultimately, the power of directed evolution has yet to be fully harnessed and should provide us with an efficient new generation of engineering tools in the years to come.
Acknowledgements
We thank Javier Macía and Diego Bárcena for critical reading. The group of MI is funded by the European Research Council, FP7-ERC-201249-ZINC-HUBS, the MEC-EMBL agreement and Ministerio de Ciencia e Innovación Grant MICINN BFU2010-17953. YS receives funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007–2013) under REA grant agreement PIEF-GA-2011-298348.References
- B. Canton, A. Labno and D. Endy, Nat. Biotechnol., 2008, 26, 787–793 CrossRef CAS.
- J. Peccoud and M. Isalan, PLoS One, 2012, 7, e43231 CAS.
- M. Folcher and M. Fussenegger, Curr. Opin. Chem. Biol., 2012, 16, 345–354 CrossRef CAS.
- A. A. Cheng and T. K. Lu, Annu. Rev. Biomed. Eng., 2012, 14, 155–178 CrossRef CAS.
- H. H. Wang, F. J. Isaacs, P. A. Carr, Z. Z. Sun, G. Xu, C. R. Forest and G. M. Church, Nature, 2009, 460, 894–898 CrossRef CAS.
- H. Tsuruta, C. J. Paddon, D. Eng, J. R. Lenihan, T. Horning, L. C. Anthony, R. Regentin, J. D. Keasling, N. S. Renninger and J. D. Newman, PLoS One, 2009, 4, e4489 Search PubMed.
- S. K. Lee, H. Chou, T. S. Ham, T. S. Lee and J. D. Keasling, Curr. Opin. Biotechnol., 2008, 19, 556–563 CrossRef CAS.
- H. Alper and G. Stephanopoulos, Metab. Eng., 2007, 9, 258–267 CrossRef CAS.
- D. G. Gibson, J. I. Glass, C. Lartigue, V. N. Noskov, R. Y. Chuang, M. A. Algire, G. A. Benders, M. G. Montague, L. Ma, M. M. Moodie, C. Merryman, S. Vashee, R. Krishnakumar, N. Assad-Garcia, C. Andrews-Pfannkoch, E. A. Denisova, L. Young, Z. Q. Qi, T. H. Segall-Shapiro, C. H. Calvey, P. P. Parmar, C. A. Hutchison, 3rd, H. O. Smith and J. C. Venter, Science, 2010, 329, 52–56 CrossRef CAS.
- T. Oberholzer, R. Wick, P. L. Luisi and C. K. Biebricher, Biochem. Biophys. Res. Commun., 1995, 207, 250–257 CrossRef CAS.
- K. Kurihara, M. Tamura, K. Shohda, T. Toyota, K. Suzuki and T. Sugawara, Nat. Chem., 2011, 3, 775–781 CrossRef CAS.
- A. Becskei and L. Serrano, Nature, 2000, 405, 590–593 CrossRef CAS.
- T. S. Gardner, C. R. Cantor and J. J. Collins, Nature, 2000, 403, 339–342 CrossRef CAS.
- A. Prindle, P. Samayoa, I. Razinkov, T. Danino, L. S. Tsimring and J. Hasty, Nature, 2012, 481, 39–44 CrossRef CAS.
- T. S. Moon, C. Lou, A. Tamsir, B. C. Stanton and C. A. Voigt, Nature, 2012, 491, 249–253 CrossRef CAS.
- S. Auslander, D. Auslander, M. Muller, M. Wieland and M. Fussenegger, Nature, 2012, 487, 123–127 Search PubMed.
- R. Kwok, Nature, 2010, 463, 288–290 CrossRef CAS.
- J. T. Kittleson, G. C. Wu and J. C. Anderson, Curr. Opin. Chem. Biol., 2012, 16, 329–336 CrossRef CAS.
- S. Cardinale and A. P. Arkin, Biotechnol. J., 2012, 7, 856–866 CrossRef CAS.
- A. L. Slusarczyk and R. Weiss, Sci. Signaling, 2012, 5, pe16 CrossRef.
- E. L. Haseltine and F. H. Arnold, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 1–19 CrossRef CAS.
- M. J. Dougherty and F. H. Arnold, Curr. Opin. Biotechnol., 2009, 20, 486–491 CrossRef CAS.
- M. Porcar, Syst. Synth. Biol., 2010, 4, 1–6 CrossRef.
- R. E. Cobb, T. Si and H. Zhao, Curr. Opin. Chem. Biol., 2012, 16, 285–291 CrossRef CAS.
- R. E. Cobb, N. Sun and H. Zhao, Methods, 2012 DOI:10.1016/j.ymeth.2012.03.009.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 1989 Search PubMed.
- B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, B. L. Stoddard and D. Baker, Science, 2003, 302, 1364–1368 CrossRef CAS.
- S. J. Fleishman, T. A. Whitehead, D. C. Ekiert, C. Dreyfus, J. E. Corn, E. M. Strauch, I. A. Wilson and D. Baker, Science, 2011, 332, 816–821 CrossRef CAS.
- G. P. Smith, Science, 1985, 228, 1315–1317 CAS.
- A. D. Griffiths and D. S. Tawfik, EMBO J., 2003, 22, 24–35 CrossRef CAS.
- M. Isalan, A. Klug and Y. Choo, Nat. Biotechnol., 2001, 19, 656–660 CrossRef CAS.
- G. Winter, A. D. Griffiths, R. E. Hawkins and H. R. Hoogenboom, Annu. Rev. Immunol., 1994, 12, 433–455 CrossRef CAS.
- A. Wagner, FEBS Lett., 2005, 579, 1772–1778 CrossRef CAS.
- M. Isalan and Y. Schaerli, SEBBM, 2012, 171, 16–20 Search PubMed.
- C. C. Guet, M. B. Elowitz, W. Hsing and S. Leibler, Science, 2002, 296, 1466–1470 CrossRef CAS.
- K. Wu and C. V. Rao, Curr. Opin. Chem. Biol., 2012, 16, 318–322 CrossRef CAS.
- J. M. Carothers, J. A. Goler, D. Juminaga and J. D. Keasling, Science, 2011, 334, 1716–1719 CrossRef CAS.
- T. Ellis, X. Wang and J. J. Collins, Nat. Biotechnol., 2009, 27, 465–471 CrossRef CAS.
- G. Rodrigo, J. Carrera and A. Jaramillo, Nucleic Acids Res., 2011, 39, e138 CrossRef CAS.
- G. Rodrigo, J. Carrera, T. E. Landrain and A. Jaramillo, FEBS Lett., 2012, 586, 2037–2042 CrossRef CAS.
- A. L. Slusarczyk, A. Lin and R. Weiss, Nat. Rev. Genet., 2012, 13, 406–420 CrossRef CAS.
- J. Bonnet, P. Subsoontorn and D. Endy, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8884–8889 CrossRef CAS.
- Y. Yokobayashi, R. Weiss and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16587–16591 CrossRef CAS.
- R. S. Cox, 3rd, M. G. Surette and M. B. Elowitz, Mol. Syst. Biol., 2007, 3, 145 Search PubMed.
- H. Alper, C. Fischer, E. Nevoigt and G. Stephanopoulos, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12678–12683 CrossRef CAS.
- M. R. Schlabach, J. K. Hu, M. Li and S. J. Elledge, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2538–2543 CrossRef CAS.
- W. An and J. W. Chin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8477–8482 CrossRef CAS.
- B. A. Blount, T. Weenink, S. Vasylechko and T. Ellis, PLoS One, 2012, 7, e33279 CAS.
- K. Temme, R. Hill, T. H. Segall-Shapiro, F. Moser and C. A. Voigt, Nucleic Acids Res., 2012, 40, 8773–8781 CrossRef CAS.
- H. M. Salis, E. A. Mirsky and C. A. Voigt, Nat. Biotechnol., 2009, 27, 946–950 CrossRef CAS.
- J. C. Anderson, E. J. Clarke, A. P. Arkin and C. A. Voigt, J. Mol. Biol., 2006, 355, 619–627 CrossRef CAS.
- C. Lou, X. Liu, M. Ni, Y. Huang, Q. Huang, L. Huang, L. Jiang, D. Lu, M. Wang, C. Liu, D. Chen, C. Chen, X. Chen, L. Yang, H. Ma, J. Chen and Q. Ouyang, Mol. Syst. Biol., 2010, 6, 350 CrossRef.
- Y. Tashiro, H. Fukutomi, K. Terakubo, K. Saito and D. Umeno, Nucleic Acids Res., 2011, 39, e12 CrossRef.
- J. C. Anderson, C. A. Voigt and A. P. Arkin, Mol. Syst. Biol., 2007, 3, 133 CrossRef.
- N. Muranaka, K. Abe and Y. Yokobayashi, ChemBioChem, 2009, 10, 2375–2381 CrossRef CAS.
- N. Muranaka, V. Sharma, Y. Nomura and Y. Yokobayashi, Nucleic Acids Res., 2009, 37, e39 CrossRef.
- R. G. Egbert and E. Klavins, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16817–16822 CrossRef CAS.
- K. D. Litcofsky, R. B. Afeyan, R. J. Krom, A. S. Khalil and J. J. Collins, Nat. Methods, 2012, 9, 1077–1080 CrossRef CAS.
- G. Kudla, A. W. Murray, D. Tollervey and J. B. Plotkin, Science, 2009, 324, 255–258 CrossRef CAS.
- M. Welch, S. Govindarajan, J. E. Ness, A. Villalobos, A. Gurney, J. Minshull and C. Gustafsson, PLoS One, 2009, 4, e7002 Search PubMed.
- B. F. Pfleger, D. J. Pitera, C. D. Smolke and J. D. Keasling, Nat. Biotechnol., 2006, 24, 1027–1032 CrossRef CAS.
- D. J. Sayut, Y. Niu and L. Sun, ACS Chem. Biol., 2006, 1, 692–696 CrossRef CAS.
- C. V. Rao, Curr. Opin. Biotechnol., 2012, 23, 689–694 CrossRef CAS.
- J. Zhan, B. Ding, R. Ma, X. Ma, X. Su, Y. Zhao, Z. Liu, J. Wu and H. Liu, Mol. Syst. Biol., 2010, 6, 388 CrossRef.
- C. H. Collins, J. R. Leadbetter and F. H. Arnold, Nat. Biotechnol., 2006, 24, 708–712 CrossRef CAS.
- S. K. Lee, H. H. Chou, B. F. Pfleger, J. D. Newman, Y. Yoshikuni and J. D. Keasling, Appl. Environ. Microbiol., 2007, 73, 5711–5715 CrossRef CAS.
- M. Krueger, O. Scholz, S. Wisshak and W. Hillen, Gene, 2007, 404, 93–100 CrossRef CAS.
- R. Daber and M. Lewis, J. Mol. Biol., 2009, 391, 661–670 CrossRef CAS.
- A. S. Khalil, T. K. Lu, C. J. Bashor, C. L. Ramirez, N. C. Pyenson, J. K. Joung and J. J. Collins, Cell, 2012, 150, 647–658 CrossRef CAS.
- O. Rackham and J. W. Chin, Nat. Chem. Biol., 2005, 1, 159–166 CrossRef CAS.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S. Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS.
- K. Chockalingam, Z. Chen, J. A. Katzenellenbogen and H. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5691–5696 CrossRef CAS.
- J. B. Andersen, C. Sternberg, L. K. Poulsen, S. P. Bjorn, M. Givskov and S. Molin, Appl. Environ. Microbiol., 1998, 64, 2240–2246 CAS.
- A. M. Turing, Philos. Trans. R. Soc. London, Ser. B, 1952, 237, 37–72 CrossRef.
- A. Gierer and H. Meinhardt, Kybernetik, 1972, 12, 30–39 CAS.
- Y. Nomura and Y. Yokobayashi, BioSystems, 2007, 90, 115–120 CrossRef CAS.
- Y. Nomura and Y. Yokobayashi, J. Am. Chem. Soc., 2007, 129, 13814–13815 CrossRef CAS.
- A. Crameri, E. A. Whitehorn, E. Tate and W. P. Stemmer, Nat. Biotechnol., 1996, 14, 315–319 CrossRef CAS.
- C. Tan, P. Marguet and L. You, Nat. Chem. Biol., 2009, 5, 842–848 CrossRef CAS.
- D. Davies, Methods Mol. Biol., 2012, 878, 185–199 CAS.
- Y. Yokobayashi and F. H. Arnold, Nat. Comput., 2005, 4, 245–254 CrossRef CAS.
- P. McNicholas, I. Chopra and D. M. Rothstein, J. Bacteriol., 1992, 174, 7926–7933 CAS.
- H. Meinhardt and A. Gierer, Bioessays, 2000, 22, 753–760 CrossRef CAS.
- J. K. Griffith, D. H. Cuellar, C. A. Fordyce, K. G. Hutchings and A. A. Mondragon, Mol. Membr. Biol., 1994, 11, 271–277 CrossRef CAS.
- P. Marguet, Y. Tanouchi, E. Spitz, C. Smith and L. You, PLoS One, 2010, 5, e11909 Search PubMed.
- W. Weber and M. Fussenegger, Chem. Biol., 2009, 16, 287–297 CrossRef CAS.
- T. Ellis, T. Adie and G. S. Baldwin, Integr. Biol., 2011, 3, 109–118 RSC.
- B. Lin and A. Levchenko, Curr. Opin. Chem. Biol., 2012, 16, 307–317 CrossRef CAS.
- S. Gulati, V. Rouilly, X. Niu, J. Chappell, R. I. Kitney, J. B. Edel, P. S. Freemont and A. J. deMello, J. R. Soc., Interface, 2009, 6(Suppl 4), S493–S506 CrossRef CAS.
- F. Zanella, J. B. Lorens and W. Link, Trends Biotechnol., 2010, 28, 237–245 CrossRef CAS.
- P. Brodin and T. Christophe, Curr. Opin. Chem. Biol., 2011, 15, 534–539 CrossRef CAS.
- M. Bickle, Anal. Bioanal. Chem., 2010, 398, 219–226 CrossRef CAS.
- Y. Schaerli and F. Hollfelder, Mol. BioSyst., 2009, 5, 1392–1404 RSC.
- J. S. Chuang, Curr. Opin. Chem. Biol., 2012, 16, 370–378 CrossRef CAS.
- S. Regot, J. Macia, N. Conde, K. Furukawa, J. Kjellen, T. Peeters, S. Hohmann, E. de Nadal, F. Posas and R. Sole, Nature, 2011, 469, 207–211 CrossRef CAS.
- A. Tamsir, J. J. Tabor and C. A. Voigt, Nature, 2011, 469, 212–215 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |