Cell-penetrating peptides (CPPs) as a vector for the delivery of siRNAs into cells
Ikuhiko Nakase, Gen Tanaka and Shiroh Futaki*
Institute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: futaki@scl.kyoto-u.ac.jp; Fax: +81-774-32-3038; Tel: +81-774-38-3210
First published on 12th December 2012
Abstract
“Cell-penetrating peptides (CPPs)” is an inclusive term describing relatively small peptides (6−30 amino acid residues) having membrane translocation activity. Due to their efficacy in cellular internalisation and the accompanying low cytotoxicity, CPPs are regarded as promising vectors for intracellular delivery of various membrane-impermeable bioactive molecules. This review provides an overview of the current approaches and describes the potential of CPP-based siRNA delivery systems, specifically those using arginine-rich CPPs.
 Ikuhiko Nakase | Ikuhiko Nakase obtained his PhD in 2005 from Kyoto University. He completed his postdoctoral training at the Department of Chemistry, University of Washington (Seattle, WA, USA). He has been an Assistant Professor of Biochemistry at the Institute for Chemical Research, Kyoto University, since 2006. His research interests include peptide–membrane interactions in cells. |
 Gen Tanaka | Gen Tanaka obtained his PhD in 2007 from the Tokyo Institute of Technology under the supervision of Professor Eiry Kobatake. After obtaining his PhD, he joined the research group of Professor Shiroh Futaki, Kyoto University as a postdoctoral fellow. His main research focus is in the field of chemical biology concerning arginine-rich cell-penetrating peptides. |
 Shiroh Futaki | Shiroh Futaki obtained his PhD in 1989 from Kyoto University. Following his appointment as a Research Associate and Associate Professor at the University of Tokushima, he moved to Kyoto University in 1997. Meanwhile, he spent 16 months (1989–1991) in the USA as a Postdoctoral Associate in the Department of Biochemistry, Rockefeller University (New York City, NY). He has been a full Professor of Biochemistry at the Institute for Chemical Research, Kyoto University, since 2005. |
Introduction
Small interfering RNAs (siRNAs) lead to specific knockdown or suppression of target genes, and thus have great therapeutic potential.1–3 Due to the impermeability of nucleic acid pharmaceuticals, including siRNAs, effort has been pursued to establish safe and efficient methodologies for delivery of these molecules into target cells or tissues. These nucleic acid pharmaceuticals are often taken up by cells via endocytosis and delivered within endosomes to lysosomes, where they are degraded by lysosomal enzymes. Efficient methods must be established to release nucleic acid pharmaceuticals from endosomes to the cytosol before they are transported to lysosomes.Various vectors have been developed for these purposes, including those based on polymers, liposomes and viral vectors. Non-viral vectors often suffer low intracellular delivery efficiency in comparison with viral vectors, while concerns persist regarding the immunogenicity and pathogenicity of viral vectors.1–3 Therefore, opportunities remain for the development of vectors with improved properties.
A novel approach for the intracellular delivery of various bioactive molecules has recently been proposed using peptides with membrane permeation activity collectively named cell-penetrating peptides (CPPs).4–6 CPPs are generally composed of 6–30 amino acid residues. The conjugation or stable complex formation of CPPs with molecules of interest facilitates the internalization by target cells, which yields improved bioactivity. CPPs have been regarded as promising vectors for siRNA delivery, leading to a considerable amount of research focused on the intracellular delivery of siRNA using CPPs. In this review, we present recent approaches to siRNA delivery by CPPs for the suppression of gene expression and also describe the potential of CPP-based methods for siRNA delivery.
Arginine-rich CPPs and their use in intracellular delivery
Peptides with different physicochemical properties have been reported to have activities consistent with CPPs (Table 1). Arginine-rich CPPs, such as human immunodeficiency virus (HIV)-1 Tat (48−60)7 and oligoarginines,8,9 are the most frequently employed classes of CPPs because of their efficacy in bringing exogenous molecules into cells. The efficient internalisation of arginine-rich CPPs has been suggested to depend on the highly basic guanidino moiety of arginine (pKa = ∼12). Indeed, guanidino functional groups can form divalent hydrogen bonds with various functional groups associated with cellular molecules (e.g. phosphates, carboxylates and sulphates), which serves to enhance the affinity of arginine-rich CPPs for cell surfaces (Fig. 1).10,11 Neutralisation of the positive charges on arginine residues by these counter ions is expected to be beneficial to the translocation of arginine-rich CPPs through membranes.12,13Another benefit of arginine-rich CPPs as carriers for intracellular delivery is their low cytotoxicity. It is well known that cationic polymers, such as polyethyleneimine, have high cytotoxicity resulting from the strong interaction of these polymers with cell-surface and -membrane molecules, leading to perturbation of membranes and cellular damage. Polyarginines composed of more than 30−50 residues have high cytotoxicity for the same reason. Reducing the number of arginine residues in peptides to 6−12, which is the range used in CPPs, lowers the membrane perturbation ability of these peptides while retaining sufficient cell surface adsorption affinities.14 The affinity of arginine-rich CPPs for cell membranes is also important to their translocation into the cytoplasm. When their affinity for cell membranes is too high, CPPs cannot be readily released into cells following membrane translocation. The low immunogenicity of arginine-rich CPPs is also suggested to result from their relatively small molecular weights and the fact that they are readily degraded by cellular trypsin-like proteases.15 Some cationic liposomes and polymers also display high transfection efficacy, however, their use is often accompanied by high cytotoxicity. The high efficacy and low toxicity of CPPs are expected to be advantageous in terms of cellular delivery of bioactive molecules.
The methods by which arginine-rich CPPs are internalised depend on the physicochemical properties of both the CPPs and the cargo molecules.16 CPPs alone and those in complex with cargo molecules having a molecular weight within a certain range can directly pass through plasma membranes.14 Intracellular delivery of relatively large cargo molecules, including macromolecules and liposomes, often involves the uptake of CPP complexes by endocytosis.4 This also holds true for highly negatively charged cargo molecules, such as nucleic acids. In addition to clathrin endocytosis and caveolae endocytosis, involvement of an actin-dependent fluid-phase endocytosis pathway (macropinocytosis) has been suggested.17,18 Arginine-rich CPPs has been proposed to induce macropinocytosis, a process that leads to accelerated internalisation of cell–surface adsorbed CPPs and CPP–cargo complexes.19–21 This process likely results from interactions between arginine-rich CPPs and the cell surface, and yields high biological activities. Since macropinocytosis is considered a non-specific fluid-phase endocytosis pathway, its induction should facilitate the engulfment of cell–surface adsorbed molecules and materials into cells. The diameter of macropinosomes has been reported to frequently exceed 1 μm. This is significantly larger than that of clathrin endosomes (∼120 nm) and caveolae endosomes (∼80 nm).22 Large endosomes may preferentially take up macromolecules and liposomes used for siRNA delivery, since their diameter often exceeds 100 nm.
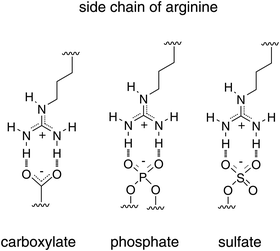 | ||
| Fig. 1 Potential ability of arginine to form hydrogen bonds with various membrane-associated molecules. | ||
Another advantage of using these peptide-based carriers is the ease of molecular design. Various approaches using CPPs have been employed to attain stable complex or conjugate formation with siRNAs and efficient delivery. In the following sections, we discuss approaches that use simple complexes of CPPs and siRNA as well as provide an introduction to approaches that utilise macromolecules and liposomes modified with CPPs.
Intracellular delivery of siRNA using CPPs
The discovery of the membrane translocation activity of HIV-1 Tat (48−60),23Drosophila antennapedia (43−58) (referred to as penetratin),24 transportan,25 various RNA/DNA binding peptides,26 oligoarginines26,27 and other CPPs (Table 1) was soon followed by reports of their ability to transport exogenous peptides and proteins into cells. We examined whether arginine-rich CPPs also have the ability to deliver plasmid DNA into cells.28 Simple complexes formed by luciferase-encoded plasmids and oligoarginines, however, yielded only a slight improvement in transfection efficiency in comparison with plasmid-only control experiments. In addition, the resulting luciferase activities were much lower than those of commercially available cationic liposomes (i.e. Lipofectamine).28 A hydrophobic moiety was introduced to arginine-rich CPPs to enhance compaction of plasmid DNA and to improve the transfection efficiency. Stearic acid was selected as a hydrophobic moiety in order to yield the best transfection efficiency and was incorporated by the N-terminal modification of octa-arginine, referred to as stearyl-R8.28 A significant improvement in transfection efficiency was obtained by the simple mixing of stearyl-R8 with the plasmid DNA. The resulting luciferase activity was comparable to that of Lipofectamine, however, it did not display any significant cytotoxicity. Results of atomic force microscopy revealed that stearyl-R8, but not R8, effectively condensed the plasmids into complexes containing core–shell structures that can be adsorbed onto the cell surface and efficiently internalised by cells.29Stearylation was also found to be useful for improving the efficiency of gene and siRNA delivery into cells using other CPPs. For example, Langel and co-workers reported that stearylated transportan (TP)-10 (stearyl-TP10; TP-10 is a shorter version of transportan) efficiently delivered a splice-correcting phosphorothioate 2′-O-methyl RNA (2′-OMe ON) into cells.30,31 The same research group also developed the novel carrier peptide PepFect 6 (PF6), which is a stearyl-TP10 analogue modified with the chloroquine derivative trifluoromethylquinoline to improve endosomal escape (Fig. 2).32 The PF6 peptide was effective at transfecting human umbilical vein endothelial cells (HUVEC) and Jurkat cells with siRNA, while commercially available transfection reagents were inefficient.32 In addition, the high RNAi response produced by PF6/siRNA was retained even in the presence of serum containing proteins that often reduce transfection efficacy.32 Intravenous administration of the PF6/siRNA complex resulted in a significant reduction of HPRT1 mRNA levels (∼50−70% knockdown efficacy) in kidneys, lungs and liver.32 In addition, liver transaminase (ALT/AST) and creatinine levels in mouse blood were not affected by the treatment, suggesting this siRNA delivery method to be less cytotoxic.32 These results show the effectiveness of siRNA delivery using PF6 in vitro and in vivo. This group also recently reported a new delivery peptide, PepFect 14 (PF14), in which stearly-TP10 lysines were substituted with ornithines.33 PF14/siRNA nanocomplexes were formulated by a solid dispersion technique and the resulting solid formulation displayed comparable RNAi activity with that of the freshly prepared nanocomplexes in solution.
Dowdy and co-workers employed an alternative approach using a fusion protein with three repeats of a Tat peptide containing a double-stranded RNA binding domain (PTD-DRBD).34 As stated above, the positive charges on arginine-rich CPPs are important to ensure their efficient cellular uptake. Complex formation with negatively charged molecules, such as nucleic acids, often reduces the internalisation efficiency. DRBD accommodates the siRNA in a binding pocket with high affinity, which retains the cationic features of arginine-rich CPPs.34 PTD-DRBD-delivered siRNA successfully induced rapid and efficient RNAi responses in various primary and transformed cells, including T cells, HUVEC and human embryonic stem cells, which were difficult to transfect using commercially available lipofection reagents.34 The cytotoxicity observed with PTD-DRBD treatment was also much lower than that of other lipofection reagents.34In vivo assays were employed using transgenic ROSA26 mice expressing luciferase in the nasal and tracheal passages. ROSA26 mice were treated intranasally with PTD-DRBD/siRNA complexes and monitored for 15 days. While the complex of control siRNA and PTD-DRBD had no significant effect on luciferase expression levels in mice, treatment with PTD-DRBD/siRNA complexes yielded a significant reduction in luciferase expression throughout the nasal and tracheal passages even after 1 day.34 Additionally, combinatorial RNAi knockdown of the epidermal growth factor receptor (EGFR) and Akt2 using PTD-DRBD was shown to induce tumour-specific apoptosis and significantly increases survival in intracerebral glioblastoma mouse models.35
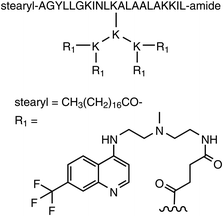 | ||
| Fig. 2 Structure of the novel carrier peptide PepFect 6 (PF6). | ||
Ohtsuki and co-workers reported the use of the Tat-U1A fusion protein for the specific delivery of a short hairpin RNA (shRNA) containing a U1A binding sequence in its loop region (Fig. 3).36,37 Tat-U1A is composed of a Tat peptide fused to a small nuclear ribonucleoprotein A (U1A)-derived RNA-binding protein (RBP) that contains the N-terminus 98 amino acids of U1A. The Tat-U1A/shRNA complexes were internalised by cells through endocytosis (Fig. 3).36,37 Light irradiation induced the efficient release of Tat-U1A and shRNA complexes from endosomes into the cytosol through the excitation of Alexa546 moieties attached to the C-terminus of Tat-U1A, presumably due to local membrane perturbation by short-lived reactive oxygen species generated by light.38 Photo-controlled gene silencing was achieved with specific cells by controlling the irradiation area in cell culture, suggesting this technique has potential in photodynamic therapy.36,37
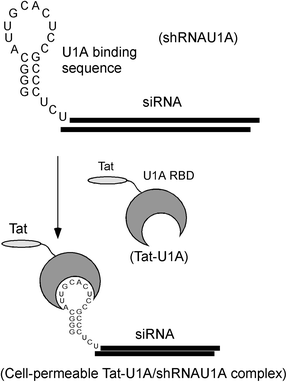 | ||
| Fig. 3 Outline of the specific delivery of a short hairpin RNA (shRNA) using Tat-U1A fusion protein. | ||
Approaches that employ hybrid peptides of oligoarginines with targeting peptides for in vivo siRNA delivery have also been reported.39,40 Kumar and co-workers employed a short peptide derived from rabies virus glycoprotein (RVG), which binds to the acetylcholine receptor expressed by neuronal cells, for the transvascular siRNA delivery to the brain. Addition of nonaarginine (R9) segment at the C-terminus of the RVG peptide allowed the binding of siRNA to the RVG-9R peptide and the eventual transduction of siRNA into neuronal cells in vitro.39 Intravenous injection of the RVG-9R/siRNA complex into mice resulted in the delivery of siRNA into the neuronal cells, yielding specific gene silencing within the brain.39 Using the same strategy, T cell-specific siRNA delivery was accomplished in mice reconstituted with human lymphocytes (Hu-PBL) or CD34+ hematopoietic stem cells (Hu-HSC) with the help of a CD7-specific single-chain antibody-R9 conjugate (scFvCD7-9R). In HIV-infected Hu-PBL mice, treatment with anti-viral coreceptor (C–C chemokine receptor type 5, CCR5) and antiviral siRNAs complexed to scFvCD7-9R controlled viral replication and prevented the CD4 T cell loss.40 Suppression of endogenous virus and restoration of CD4 T cell counts in mice reconstituted with HIV+ peripheral blood mononuclear cells were also attained by this treatment,40 suggesting the potential of this strategy for siRNA therapy against HIV infection.
siRNA delivery using macromolecules and liposomes modified with CPPs
Approaches employing macromolecules and liposomes have been reported for the delivery of siRNA, which is readily biodegraded and difficult to modify and chemically conjugate. Encapsulation of siRNA by these macromolecules and liposomes can be beneficial in terms of siRNA compaction, which facilitates the endosomal siRNA uptake and protects the siRNA from degradation enzymes. The modification of these delivery macromolecules and liposomes with functional moieties has the potential to increase the target efficiency, as well as other properties. CPPs may also be employed to increase the internalisation efficacy of the siRNA complexes.41–44Harashima and co-workers have developed octa-arginine (R8)-modified liposomes that encapsulate plasmid DNA (pDNA) into condensed pDNA cores that are coated with lipid membranes. These functionalised liposomes are produced by “Programmed Packaging” and act as multifunctional envelope-type nanodevices (MEND) that serve as vehicles for non-viral gene-delivery.44 Stearyl-R8 was employed to decorate the liposomal surfaces with R8, with the stearyl moiety serving as the anchoring moiety. High-density decoration of liposomal surfaces with R8 produced a transfection activity as high as that of adenovirus, but produced little cytotoxicity.45,46
This MEND system was also employed for the delivery of siRNA into cells, where it was found that compaction of siRNA into MEND complexes was a critical factor for RNAi responses.47 Poly-L-lysine (PLL), stearyl-R8 and protamine were compared as siRNA condensing agents, however, only stearyl-R8 yielded a condensed core with a diameter less than 100 nm. The MEND having a stearyl-R8/siRNA condensed core showed the highest RNAi response using a luciferase assay.47 The RNAi response induced by R8-MEND containing a stearyl-R8/siRNA core was also higher than that produced by the commercially available transfection reagent TransIT-TKO, and showed no notable cytotoxicity.45 Combining a pH-dependent fusogenic peptide (GALA) surface decoration with a lipid mixture that had been optimised for endosomal fusion significantly improved the endosomal escape of R8-MEND. The R8/GALA-modified MEND was successfully applied to ex vivo siRNA delivery using dendritic cells for cancer vaccination.48
Studies of the internalisation mechanisms of R8-MEND elucidated a mechanism by which R8-MEND binds electrostatically to negatively charged proteoglycans on cell surfaces. The binding between MEND and proteoglycans results in induction of macropinocytosis, which causes R8-MEND to be taken up by cells (Fig. 4). The charging ratio of R8 on MEND was also reported to be important for determining cellular uptake pathways. When 0.5 mol% R8 was charged on liposomal surfaces, the R8-MEND was taken up by the cells mainly through clathrin-mediated endocytosis and subjected to lysosomal degradation.45 A charging value of 5 mol% R8 on MEND resulted in the uptake of R8-MEND primarily through macropinocytosis, which reduced its susceptibility to lysosomal degradation.45,49 The mechanism of R8-MEND cytoplasmic transport was also investigated and compared to that of adenovirus.50 Treatment of plasmid DNA-encapsulated R8-MEND with the microtubule-disruption reagent nocodazole significantly diminished the transfection efficiency in comparison with that of adenovirus, suggesting that MEND is highly dependent on microtubules for the delivery of DNA to the nucleus.48 Microtubule-dependent transport plays an important role in gene expression by MEND; however, the internalisation pathway is expected to differ from that of adenovirus.50
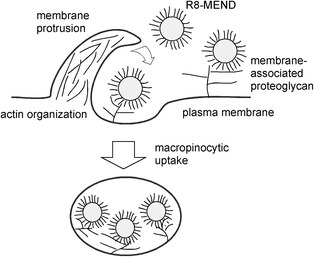 | ||
| Fig. 4 Outline of the internalisation mechanism of R8-MEND. | ||
A biodegradable core–shell polyplex carrier molecule bearing several different functional peptides was also reported. Zhang et al. described a multifunctional peptide-PEG-tris-acridine conjugate modified with Tat, a nuclear localisation signal (NLS) and a brain-specific brain-homing peptide (BH) that was designed for the delivery of genes past the blood-brain barrier.51 Nam et al. also reported the use of CPPs for siRNA delivery. The primary cardiomyocyte-specific peptide (PCM) and HIV-Tat (49−57) peptide were conjugated to the bioreducible polymer cystamine bisacrylamide-diaminohexane (CBA-DAH) to increase delivery of RNAi to target cardiomyocyte cells. After incubation in a hypoxic environment, the cells were treated with either a Src homology domain 2 (SH2)-containing tyrosine phosphatase-1 (SHP-1) siRNA complex or a Fas siRNA complex, which resulted in an increase in cell viability and a reduction in cytotoxicity.52
Cationic dendrimers are often used for gene and siRNA delivery. Han et al. employed a unique combination of dendrimers, magnetic nanoparticles and CPPs to increase siRNA delivery efficiency. Polyamidoamine (PAMAM) dendrimers and Tat peptides were conjugated to bacterial magnetic nanoparticles (BMPs).53 An expression vector for siRNA corresponding to the open reading frame of the human epidermal growth factor receptor gene (psiRNA-EGFR) was packaged into Tat-BMPs-PAMAM by means of electrostatic interactions and used successfully to reduce the growth rate of tumours in the U251 subcutaneous nude mouse model.53
Anticancer drug treatment employing siRNA for P-glycoprotein silencing was also reported. Xiong et al. developed a siRNA carrier based on poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL) micelles. These carrier molecules were decorated with integrin αvβ3 targeting peptide (RGD4C) and CPP (Tat) on the PEO shell and contained a polycation (spermine)-modified PCL core for siRNA binding.54 The authors observed enhanced cellular uptake and effective endosomal escape of siRNA delivered by peptide-functionalised micelles to MDA435/LCC6-resistant cells. The activity was most pronounced when the micelles were modified with both the RGD and Tat peptides. P-glycoprotein silencing by the RGD/Tat-micellar mdr1 siRNA complexes led to a significant increase in the accumulation of doxorubicin (a typical P-glycoprotein substrate) in MDA435/LCC6 cells, leading to eventual cell death.
Conclusion
The growing use of CPPs indicates their potential as effective delivery vectors. Due to the demand for development of safe and efficient vectors for siRNA delivery, an increasing number of CPP-based applications have been reported recently. In this review, the current trends in siRNA delivery by CPPs are summarised. Conjugation with CPP increases cell–surface affinity and eventual cellular uptake of bioactive molecules. In the case of arginine-rich CPPs, the interaction of the peptides with the cell surface leads to induction of macropinocytosis. The high efficiency of intracellular delivery obtained using arginine-rich CPPs may result from the large capacity engulfment of extracellular fluids and materials produced by macropinocytosis. Understanding the uptake mechanisms of CPPs will facilitate the design of more efficient and sophisticated delivery systems in the future. Studying the pathways of arginine-rich CPP internalisation, as well as their conjugates and complexes, will also contribute to the improvement of other delivery systems. With regard to applications involving siRNA delivery, simple mixing of CPPs with siRNA may not yield a marked effect. However, use of stearylated-CPPs and a core–shell structure facilitates the cellular uptake of complexes containing siRNAs, leading to a greater RNAi effect. Approaches have also been employed to facilitate the cellular uptake of macromolecule- or liposome-based siRNA delivery systems by modifying CPPs. Indeed, CPP-modified MEND offers great flexibility for the design of delivery systems since MEND components can be modified with various functional moieties to enhance key delivery pathways, including cellular targeting and endosomal escape. The examples described in this review will be beneficial for selection of the most suitable delivery systems depending on the needs of the in vivo system of interest. Further studies will clarify the true potential of these CPP-based delivery systems.Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. This study was also supported by Strategic Japanese-Swedish Cooperative Programme on “Multidisciplinary BIO” from Japan Science and Technology Agency (JST) and VINNOVA.References
- C. Scholz and E. Wagner, J. Controlled Release, 2012, 161, 554–565 CrossRef CAS.
- H. Margus, K. Padari and M. Pooga, Mol. Ther., 2012, 20, 525–533 Search PubMed.
- I. Nakase, H. Akita, K. Kogure, A. Gräslund, U. Langel, H. Harashima and S. Futaki, Acc. Chem. Res., 2012, 45, 1132–1139 Search PubMed.
- A. El-Sayed, S. Futaki and H. Harashima, AAPS J., 2009, 11, 13–22 Search PubMed.
- I. Nakase, T. Takeuchi, G. Tanaka and S. Futaki, Adv. Drug Delivery Rev., 2008, 60, 598–607 CrossRef CAS.
- Cell-Penetrating Peptides. Methods and Protocols. In Methods in Molecular Biology, ed. Ü. Langel, 2011, vol. 683 Search PubMed.
- H. Brooks, B. Lebleu and E. Vivès, Adv. Drug Delivery Rev., 2005, 57, 559–577 CrossRef CAS.
- J. B. Rothbard, T. C. Jessop and P. A. Wender, Adv. Drug Delivery Rev., 2005, 57, 495–504 CrossRef CAS.
- S. Futaki, Adv. Drug Delivery Rev., 2005, 57, 547–558 CrossRef CAS.
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS.
- N. Sakai, T. Takeuchi, S. Futaki and S. Matile, ChemBioChem, 2005, 6, 114–122 CrossRef CAS.
- T. Takeuchi, M. Kosuge, A. Tadokoro, Y. Sugiura, M. Nishi, M. Kawata, N. Sakai, S. Matile and S. Futaki, ACS Chem. Biol., 2006, 1, 299–303 CrossRef CAS.
- F. Perret, M. Nishihara, T. Takeuchi, S. Futaki, A. N. Lazar, A. W. Coleman, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 1114–1115 CrossRef CAS.
- M. Kosuge, T. Takeuchi, I. Nakase, A. T. Jones and S. Futaki, Bioconjugate Chem., 2008, 19, 656–664 CrossRef CAS.
- J. M. Gump and S. F. Dowdy, Trends Mol. Med., 2007, 13, 443–448 CrossRef CAS.
- G. Tünnemann, R. M. Martin, S. Haupt, C. Patsch, F. Edenhofer and M. C. Cardoso, FASEB J., 2006, 20, 1775–1784 CrossRef.
- J. S. Wadia, R. V. Stan and S. F. Dowdy, Nat. Med., 2004, 10, 310–315 CrossRef CAS.
- I. Nakase, M. Niwa, T. Takeuchi, K. Sonomura, N. Kawabata, Y. Koike, M. Takehashi, S. Tanaka, K. Ueda, J. C. Simpson, A. T. Jones, Y. Sugiura and S. Futaki, Mol. Ther., 2004, 10, 1011–1022 CrossRef CAS.
- I. Nakase, A. Tadokoro, N. Kawabata, T. Takeuchi, H. Katoh, K. Hiramoto, M. Negishi, M. Nomizu, Y. Sugiura and S. Futaki, Biochemistry, 2007, 46, 492–501 CrossRef CAS.
- S. Futaki, I. Nakase, A. Tadokoro, T. Takeuchi and A. T. Jones, Biochem. Soc. Trans., 2007, 35, 784–787 CrossRef CAS.
- I. Nakase, H. Hirose, G. Tanaka, A. Tadokoro, S. Kobayashi, T. Takeuchi and S. Futaki, Mol. Ther., 2009, 17, 1868–1876 Search PubMed.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS.
- E. Vives, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CAS.
- M. Pooga, M. Hällbrink, M. Zorko and U. Langel, FASEB J., 1998, 12, 67–77 CAS.
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS.
- J. B. Rothbard, S. Garlington, Q. Lin, T. Kirschberg, E. Kreider, P. L. McGrane, P. A. Wender and P. A. Khavari, Nat. Med., 2000, 6, 1253–1257 CrossRef CAS.
- S. Futaki, W. Ohashi, T. Suzuki, M. Niwa, S. Tanaka, K. Ueda, H. Harashima and Y. Sugiura, Bioconjugate Chem., 2001, 12, 1005–1011 CrossRef CAS.
- I. A. Khalil, S. Futaki, M. Niwa, Y. Baba, N. Kaji, H. Kamiya and H. Harashima, Gene Ther., 2004, 11, 636–644 CrossRef CAS.
- U. Soomets, M. Lindgren, X. Gallet, M. Hällbrink, A. Elmquist, L. Balaspiri, M. Zorko, M. Pooga, R. Brasseur and U. Langel, Biochim. Biophys. Acta, 2000, 1467, 165–176 Search PubMed.
- M. Mäe, S. El Andaloussi, P. Lundin, N. Oskolkov, H. J. Johansson, P. Guterstam and U. Langel, J. Controlled Release, 2009, 134, 221–227 CrossRef.
- S. E. L. Andaloussi, T. Lehto, I. Mäger, K. Rosenthal-Aizman, I. I. Oprea, O. E. Simonson, H. Sork, K. Ezzat, D. M. Copolovici, K. Kurrikoff, J. R. Viola, E. M. Zaghloul, R. Sillard, H. J. Johansson, F. Said Hassane, P. Guterstam, J. Suhorutšenko, P. M. D. Moreno, N. Oskolkov, J. Hälldin, U. Tedebark, A. Metspalu, B. Lebleu, J. Lehtiö, C. I. E. Smith and U. Langel, Nucleic Acids Res., 2011, 39, 3972–3987 CrossRef.
- K. Ezzat, S. E. L. Andaloussi, E. M. Zaghloul, T. Lehto, S. Lindberg, P. M. D. Moreno, J. R. Viola, T. Magdy, R. Abdo, P. Guterstam, R. Sillard, S. M. Hammond, M. J. A. Wood, A. A. Arzumanov, M. J. Gait, C. I. E. Smith, M. Hällbrink and Ü. Langel, Nucleic Acids Res., 2011, 39, 5284–5298 Search PubMed.
- A. Eguchi, B. R. Meade, Y. C. Chang, C. T. Fredrickson, K. Willert, N. Puri and S. F. Dowdy, Nat. Biotechnol., 2009, 27, 567–571 CrossRef CAS.
- H. Michiue, A. Eguchi, M. Scadeng and S. F. Dowdy, Cancer Biol. Ther., 2009, 8, 2306–2313 Search PubMed.
- T. Endoh, M. Sisido and T. Ohtsuki, J. Controlled Release, 2009, 137, 241–245 Search PubMed.
- T. Endoh, M. Sisido and T. Ohtsuki, Bioconjugate Chem., 2008, 19, 1017–1024 Search PubMed.
- J. R. Maiolo 3rd., E. A. Ottinger and M. Ferrer, J. Am. Chem. Soc., 2004, 126, 15376–15377 Search PubMed.
- P. Kumar, H. Wu, J. L. McBride, K. E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39–43 CrossRef CAS.
- P. Kumar, H. S. Ban, S. S. Kim, H. Wu, T. Pearson, D. L. Greiner, A. Laouar, J. Yao, V. Haridas, K. Habiro, Y. G. Yang, J. H. Jeong, K. Y. Lee, Y. H. Kim, S. W. Kim, M. Peipp, G. H. Fey, N. Manjunath, L. D. Shultz, S. K. Lee and P. Shankar, Cell, 2008, 134, 577–586 CrossRef CAS.
- V. P. Torchilin, Biopolymers, 2008, 90, 604–610 CrossRef CAS.
- V. P. Torchilin, T. S. Levchenko, R. Rammohan, N. Volodina, B. Papahadjopoulos-Sternberg and G. G. M. D'Souza, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1972–1977 CrossRef CAS.
- C. Zhang, N. Tang, X. Liu, W. Liang, W. Xu and V. P. Torchilin, J. Controlled Release, 2006, 112, 229–239 CrossRef CAS.
- H. Hatakeyama, H. Akita, K. Kogure and H. Harashima, Adv. Biochem. Eng./Biotechnol., 2010, 119, 197–230 Search PubMed.
- I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, J. Biol. Chem., 2006, 281, 3544–3551 Search PubMed.
- I. A. Khalil, K. Kogure, S. Futaki, S. Hama, H. Akita, M. Ueno, H. Kishida, M. Kudoh, Y. Mishina, K. Kataoka, M. Yamada and H. Harashima, Gene Ther., 2007, 14, 682–689 CrossRef CAS.
- Y. Nakamura, K. Kogure, S. Futaki and H. Harashima, J. Controlled Release, 2007, 119, 360–367 CrossRef CAS.
- H. Akita, K. Kogure, R. Moriguchi, Y. Nakamura, T. Higashi, T. Nakamura, S. Serada, M. Fujimoto, T. Naka, S. Futaki and H. Harashima, J. Controlled Release, 2010, 143, 311–317 Search PubMed.
- A. El-Sayed, I. A. Khalil, K. Kogure, S. Futaki and H. Harashima, J. Biol. Chem., 2008, 283, 23450–23461 Search PubMed.
- H. Akita, K. Enoto, T. Masuda, H. Mizuguchi, T. Tani and H. Harashima, Mol. Ther., 2010, 18, 955–964 Search PubMed.
- H. Zhang, T. Gerson, M. L. Varney, R. K. Singh and S. V. Vinogradov, Pharm. Res., 2010, 27, 2528–2543 Search PubMed.
- H. Y. Nam, J. Kim, S. Kim, J. W. Yockman, S. W. Kim and D. A. Bull, Biomaterials, 2011, 32, 5213–5222 CrossRef CAS.
- L. Han, A. Zhang, H. Wang, P. Pu, X. Jiang, C. Kang and J. Chang, Hum. Gene Ther., 2010, 21, 417–426 CrossRef CAS.
- X. B. Xiong, H. Uludağ and A. Lavasanifar, Biomaterials, 2010, 31, 5886–5893 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |