Learning the molecular mechanisms of the reprogramming factors: let's start from microRNAs
Chao-Shun
Yang
and
Tariq M.
Rana
*
Program for RNA Biology, Sanford-Burnham Medical Research Institute, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA. E-mail: trana@sanfordburnham.org; Fax: +1 858 795 5328; Tel: +1 858 795 5325
First published on 5th October 2012
Abstract
Induced reprogramming of somatic cells has had a great impact on stem cell research, and the reprogramming technologies have evolved from four transgenic factors (Oct4, Sox2, Klf4, and c-Myc; OSKM) to just a few microRNAs (mainly miR-290/302 seed family). Despite these advances, the molecular events occurring during various stages of reprogramming remain largely unknown. Here, we concisely review current knowledge of miRNA regulation from the initiation phase of OSKM-induced reprogramming, through the transitional stage, to final maturation. At the start of reprogramming, the microRNAs miR-21, miR-29a, let-7a, and miR-34 act as guards to secure the somatic identity and genomic integrity of the cell of origin. As reprogramming proceeds, miR-155, miR-10b, miR-205, and miR-429 modulate the epithelial–mesenchymal/mesenchymal–epithelial transition (EMT/MET), which is a critical step towards transformed pluripotent status. Finally, the pluripotency regulatory network is secured in the iPSCs and fine-tuned by a group of miRNAs belonging to the miR-290/302 seed family. Among the four reprogramming factors, c-Myc plays the dominant role in regulating the miRNAs under reprogramming-specific conditions. Accumulating evidence suggests that the reprogramming efficiency can be improved by either blocking barrier miRNAs or introducing helper miRNAs. Intriguingly, induced pluripotency can be obtained by introducing a single miR-302 cluster, although the supportive molecular mechanism is still lacking. In the near future, we may be able to realize the broad potential of miRNAs in the stem cell field, such as altering cell identities with high efficiency through the transient introduction of tissue-specific miRNAs.
Chao-Shun Yang received his Master degree in Department of Biochemistry in 2000 from National Cheng Kung University, Taiwan. He is currently a PhD student at University of Massachusetts Medical School and is completing his research dissertation in Dr Tariq Rana's laboratory at Sanford-Burnham Medical Research Institute, La Jolla, California. |
Tariq M. Rana received his PhD from the University of California at Davis and was an American Cancer Society fellow at the University of California at Berkeley. He was a Professor of Biochemistry and Molecular Pharmacology and Director of Program in Chemical Biology at the University of Massachusetts Medical School before joining the Sanford-Burnham Medical Research Institute in 2008. He is currently Professor and Director of the RNA Biology Program at Sanford-Burnham Medical Research Institute, where his laboratory studies involve RNA regulation of development and disease. |
Induced reprogramming overview
The new era of reprogramming was initiated by the ectopic expression of four transcription factors in somatic cells, first demonstrated in mouse cells1 and later in human cells,2–6 which have the capacity to differentiate into different cell lineages. Using retroviral or lentiviral systems, these four factors, Oct4, Sox2, Klf4/Lin28, and c-Myc/Nanog (also referred to as OSKM or OSLN), can be easily introduced into somatic cells to induce reprogramming to an embryonic stem (ES) cell-like pluripotent state. The induced pluripotent stem cells (iPSCs) generated by this breakthrough technology have provided a valuable alternative resource to human embryonic stem cells.7 However, the low efficiency of reprogramming and concerns of genetic modification by the transgenes remain major hurdles in the therapeutic application of iPSCs.2,4,7,8 In recent years, substantial progress has been made in improving reprogramming efficiency and in substituting selected transcription factors.8–11 A few reports have also revealed the great promise of inducing reprogramming with only mRNAs or microRNAs (miRNAs).12–16 Although many windows have been opened to improve the efficiency of reprogramming and to minimize transgenic integrations into the genome, we have only just begun to understand the molecular mechanisms that control reprogramming beyond the four factors. Many studies have shown that reprogramming can be defined and achieved as a step-wise process.17–19 Furthermore, several genes and proteins have been identified that have greatly impacted reprogramming efficiency, such as PTGS2,20 Ink4a/ARF, p53/p21,21–26 TGF-β,27,28 and miRNAs.29–37MicroRNAs are ∼22 nucleotide small non-coding RNAs that are highly conserved among species.38,39 They contain short sequences in the 5′ end (“seed” regions) that direct target gene recognition of miRNA-loaded processing complexes, RISCs (RNA-induced Silencing Complexes).40 In mammals, miRNAs act as post-transcriptional regulators to reduce translation of target genes by either destabilizing mRNAs or blocking their translation. miRNAs have been shown to play critical roles in various physiological processes, including embryogenesis41–43 and tumorigenesis.44–48 In addition, numerous reports have shown that miRNAs play significant roles in somatic cell reprogramming to iPSC.29–35,49 The progress and expectations of induced reprogramming technology have been recently described in numerous review articles.8–10,50–57 Several reviews42,43,58–64 also address the improvement of reprogramming methods by introducing miRNAs upon induced reprogramming. However, intrinsic roles of miRNAs, which are regulated by OSKM at each stage of a reprogramming process, have not been addressed. In this review, we discuss the molecular mechanisms of reprogramming from this unique viewpoint, focusing on the effects of the reprogramming factors on endogenous miRNA regulation and the regulatory networks of these miRNAs during iPSC induction.
Reprogramming is a stochastic but step-wise event
Reprogramming is induced by ectopic expression in somatic cells of the four reprogramming factors that drive the cells to de-differentiate and achieve a state of pluripotency. An increasing body of evidence shows that it is a generally stochastic event65–67 but is able to achieve step-wise transition during reprogramming.17–19,65 The OSKM reprogramming factors bind their targets in a coordinated fashion19 to initiate the first step of reprogramming, the transcriptional and epigenetic changes.19,68 Furthermore, it has been suggested that OSKM may assemble an inhibitory circuit against somatic identity prior to building up the transcriptional network of pluripotency in the later stages of the transition.19,68 This observation is supported by other reports showing that a number of barriers need to be overcome to reach the next steps in the transition.9,10c-Myc plays a key role in establishing the early transition stage
The cellular phenotypes associated with the reprogramming transitions have been reported in recent studies,69,70 but a clear picture of the detailed molecular events driving the transitions is still lacking. Among the four reprogramming factors, c-Myc has been shown to play the dominant role in initiating the early transitional stage.19,68 Expression of c-Myc alone can downregulate the expression of fibroblast-specific genes and induce the molecular context of the embryonic status within 3 days of transduction.19 In addition to regulating the expression of hundreds of genes, as shown in previous reports,19,71 c-Myc regulates numerous miRNAs to promote tumorigenesis72–75 and to maintain pluripotency in ES cells.76–79 Recently, we demonstrated that c-Myc disrupts the fibroblastic network by inhibiting the mouse embryonic fibroblast (MEF)-enriched miRNAs, miR-21 and miR-29a (Fig. 1), to lower the threshold for reprogramming.29 Thus, c-Myc establishes the early molecular context of reprogramming, not only by directly interacting with promoter regions of target genes, but also by exerting inhibitory effects on somatic networks by regulating miRNAs.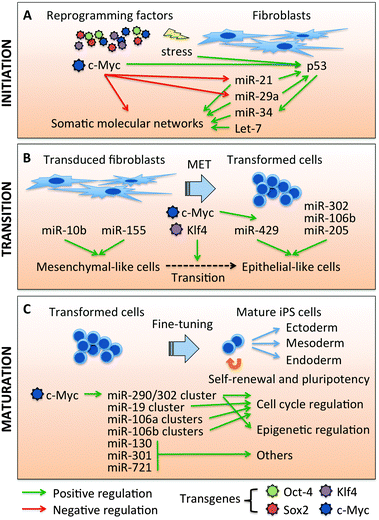 | ||
| Fig. 1 MicroRNAs play important roles to support a reprogramming progress. (A) MicroRNAs in the initial step of reprogramming. c-Myc has been shown to disrupt the molecular networks of somatic cells to promote the initiation of reprogramming. c-Myc is also the main regulator of miRNA expression early in reprogramming with miR-21 and miR-29a being the main targets of c-Myc at this stage. Let-7 is repressed by c-Myc through Lin-28b-mediated regulation, but this is not detected during early reprogramming. c-Myc also induces the oncogenic stress response by activating p53/miR-34. (B) MicroRNAs in mesenchymal-to-epithelial transition (MET) of reprogramming. miR-10b and miR-155 promote mesenchymal-like properties, while miR-205 and the miR-200 family (miR-429) promote epithelial-like characteristics. c-Myc coordinates with Klf4, to induce miR-429 upon reprogramming, which accelerates the transition step. miR-290/302 seed family, miR-302 cluster and miR-106b cluster, have been shown to enhance epithelial properties. (C) MicroRNAs in the late stage of reprogramming. In this last step of iPSC maturation, the ES-specific molecular signatures are reactivated, including ES-specific miRNAs, miR-290, and miR-302 clusters. c-Myc plays major roles in inducing miR-290 and miR-302 clusters. miR-290, miR-302, and other miR-290/302 seed family miRNAs also play important roles to restore the ESC properties. | ||
The MEF-enriched miRNAs, miR-21, miR-29a, and let-7, act as barriers to the initial stage of reprogramming
We previously demonstrated that the miRNA expression profile changes dramatically upon OSKM introduction into MEFs, with c-Myc playing the dominant regulatory role in this process.29 Furthermore, we have shown that c-Myc decreases the expression of MEF-enriched miRNAs, such as miR-21 and miR-29a (Fig. 1). c-Myc transcriptionally suppresses miR-29 expression by binding to its promoter,80 while the molecular mechanism by which c-Myc regulates miR-21 expression is still unclear. miR-21 positively regulates the TGFβ181 and MAP kinase82 pathways, which have been shown to act as roadblocks to reprogramming.27,28,31,34,69,83–86 miR-29a has been shown to indirectly induce p53 protein levels by post-transcriptionally inhibiting CDC42 and p85α.87 Consistent with these observations, depleting miR-21 or miR-29a dramatically (2- to 3-fold) increases reprogramming efficiency, suggesting that MEF-enriched miRNAs act as barriers to reprogramming.29 We also showed that miR-21 and miR-29a modulate reprogramming by regulating phosphorylation of ERK1/2 by 45–60% through Spry1 protein expression. In addition, depletion of miR-21 and miR-29a downregulates p53 protein levels by 25–40% through elevation of CDC42/p85α expression, which consequently enhances reprogramming efficiency. These data provide evidence for new regulatory networks during reprogramming involving c-Myc, miR-21, and miR-29a.Another abundant miRNA in MEFs, let-7 (Fig. 1), has been shown to act as a barrier to reprogramming, since depleting let-7 enhanced the reprogramming efficiency by 4.3-fold with only the OSK reprogramming factors.32 Ectopic expression of c-Myc reduces let-7a expression in MEFs during reprogramming, although to a relatively modest degree.29 It has been reported that c-Myc represses let-7 through Lin-28b transactivation;75 however, Lin-28b mRNA is undetectable during the early stage of reprogramming,29 suggesting other indirect mechanisms may be involved. The let-7 family may exert negative effects on reprogramming, because they are known to repress numerous pluripotent regulators, including Myc, Hmga2, Lin-28, and Sall4.32,88–91 To summarize, the MEF-enriched miRNAs, miR-21, miR-29a, and let-7a modulate various pathways to antagonize the reprogramming process. Furthermore, c-Myc has an intrinsic ability to initiate the reprogramming transition, not only by targeting the promoter regions of numerous genes, but also by inhibiting MEF-enriched miRNAs in the initial stage of reprogramming.
p53-regulated miRNAs miR-34 and miR145 play important roles in reprogramming
The introduction of reprogramming factors into somatic cells initiates the cellular stress response to viral infection and oncogenes. Among the stress response molecules, transformation-related protein 53 (Trp53 or p53) plays a critical role as a gate-keeper to ensure that only cells with genomic integrity will survive to reach the pluripotent status, while stochastic nuclear reprogramming is induced.21–26 miR-34 (Fig. 1) has been identified as a downstream target of p53 and contributes significantly to p53-mediated cell cycle arrest and apoptosis.92–94 miR-34a deficiency in murine somatic cells improves the efficiency (>4-fold) and kinetics (by two days) of reprogramming.95 Consistent with this observation, p53-induced miR-34a/b/c act as negative regulators of reprogramming, in part through the repression of pluripotency genes such as Lin28a,96 Nanog, Sox2, N-Myc,95 and c-Myc.97After initiation of reprogramming, mesenchymal-to-epithelial transition (MET) is the next step towards pluripotency.69,70 Approximately 5 days after OSKM induction, transformed cells undergo dramatic morphological changes from mesenchymal-like (polarized and mono-adherent) to epithelial-like (densely packed) cells. MET is critical for somatic cells to complete the first step of de-differentiation. Interestingly, miR-34a/b/c have been shown to compromise Snail1-dependent EMT (the reverse transition to MET) in cancer cells by targeting the 3′ untranslated region (UTR) of Snail1,98 while Snail1 and ZEB1 impose a negative feedback loop on miR-34a/b/c by binding the E-boxes of the miR-34a/b/c promoters.97 miR-34a also decreases other EMT factors, such as β-catenin, LEF1, Axin2,98 Slug, and ZEB1,97 and ectopic expression of miR-34a also prevents TGF-β induced EMT.97 Therefore, short-term introduction of miR-34a/b/c may suppress the EMT in the early reprogramming process, while reprogramming factors coordinately affect MET to de-differentiate somatic cells. Since miR-34 modulates various functional pathways, miR-34 may play dual roles to secure cell integrity and promote MET in the cell-context-dependent manner.
miR-145, suggested to be the direct target of p53,96 plays critical roles in direct differentiation of ES cells.99 miR-145 has been reported to downregulate Sox2,100 Klf4,96 and Oct4 to promote mesoderm and ectoderm differentiation in ES cells.99 Furthermore, the miR-145 promoter region is bound and repressed by Oct4 in ES cells,99 suggesting Oct4 may lift the suppression of endogenous OSK by miR-145 during reprogramming. However, regulatory networks of miR-145 during reprogramming need to be vigorously interrogated, since this has not been tested during the process of reprogramming.
EMT/MET-associated miRNAs, miR-155, miR-10b, miR-205, and miR-429, play important roles in modulating the transitional stage of reprogramming
During the MET stage of reprogramming (Fig. 1), pro-mesenchymal miRNAs miR-10b101 and miR-155102 decrease,69 and pro-epithelial miRNAs miR-205103,104 and miR-429 (miR-200 family)49,105–107 increase.69,70 miR-10b promotes EMT in cancer metastases by targeting homeobox D10,101 and miR-10b antagomirs suppress metastases in vivo.108 miR-155 plays an important role in TGF-β-induced EMT by targeting RhoA,102 one of the key factors maintaining junction formation and stabilization.109 However, a recent report showed that miR-155 may have dual functions in modulating EMT depending on the microenvironment of the tumor,110 suggesting that the functions of miRNAs are cell and/or tissue context-dependent.Conversely, the miR-200 family and miR-205 have been shown to positively regulate MET by targeting ZEB1 and ZEB2,107,111–113 while ZEB1 reciprocally represses the miR-200 family.105,106 In addition, bone morphogenetic protein (BMP), which is required for efficient reprogramming with OSKM, promotes MET and also induces expression of miR-205 and the miR-200 family during OSKM-induced reprogramming.70 Introduction of miR-200b/c mimics synergize with OSKM to promote more efficient reprogramming.70 Notably, c-Myc (Fig. 1) may boost reprogramming efficiency by directly inducing expression of the miR-200 family (miR-200, miR-141, and miR-429), and possibly coordinating with Klf4 to initiate MET.69,114
The miR-290/302 seed family plays significant roles during the programming progress
It has been shown that microRNAs maintain the murine ES property by promoting the G1–S transition of the cell cycle and that aberrant miRNA biogenesis impairs proliferation of ES cells, which accumulate in the G1 phase.115–117 The key miRNAs for these functions are the miR-290 and miR-302 clusters, which are the most abundant miRNAs (Table 1) in mouse and human ES cells, respectively.118–120 These two clusters have almost identical seed regions (miR-290/302 seed family; also see Table 2), suggesting they have highly similar target and/or regulatory networks. In human ES cells, the miR-302 cluster is regulated by Oct4/Sox2 to post-transcriptionally modulate cyclin D1, a key regulator of cell cycle progression.121,122 The miR-290/302 seed family modulates cell cycle progression by targeting diverse regulators of the cell cycle, including p21 and other inhibitors of the cyclin E/Cdk2 pathway.115 Numerous targets of the miR-302 cluster were uncovered using the photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation method (PAR-CLIP); these included modulation of BMP signaling through suppression of Tob2, Dazap2, and Slain1.30 miR-302a is known to target Lefty1/2, which is an agonist of the TGF-β/Nodal signaling pathway in embryogenesis.123 In mouse ES cells, the miR-290 family controls de novo methylation through Rbl2-dependent regulation of DNA methyltransferase (Dnmts).124,125 Based on these findings, it appears that the main function of the miR-290/302 seed family is to shorten the G1 phase of the cell cycle to support self-renewal, and to secure the epigenetic status that maintains the pluripotency of ES cells.| Both in ES and iPS cells | Only in ES cells | |
|---|---|---|
| hsa-miR-17 | hsa-miR-205 | hsa-miR-96 |
| hsa-miR-18a | hsa-miR-302a | hsa-miR-222* |
| hsa-miR-18b | hsa-miR-302a* | hsa-miR-371-5p |
| hsa-miR-19a | hsa-miR-302b | hsa-miR-372 |
| hsa-miR-19b | hsa-miR-302b* | hsa-miR-373 |
| hsa-miR-20a | hsa-miR-302c | hsa-miR-512-3p |
| hsa-miR-20b | hsa-miR-302c* | hsa-miR-515-5p |
| hsa-miR-20b* | hsa-miR-302d | hsa-miR-516a-5p |
| hsa-miR-25 | hsa-miR-302d* | hsa-miR-516b |
| hsa-miR-30e | hsa-miR-363 | hsa-miR-518b |
| hsa-miR-92a | hsa-miR-363* | hsa-miR-518d-5p |
| hsa-miR-92b | hsa-miR-421 | hsa-miR-518e* |
| hsa-miR-93 | hsa-miR-486-5p | hsa-miR-518f* |
| hsa-miR-106a | hsa-miR-489 | hsa-miR-519c-3p |
| hsa-miR-106b | hsa-miR-498 | hsa-miR-520f |
| hsa-miR-140a | hsa-miR-517a | hsa-miR-520g |
| hsa-miR-182 | hsa-miR-517b | hsa-miR-520h |
| hsa-miR-183 | hsa-miR-638 | hsa-miR-525-5p |
| hsa-miR-187 | hsa-miR-663 | hsa-miR-629 |
| hsa-miR-200c | hsa-miR-923 | |
| miRNAs | 5′–3′ Guide strand sequencea |
|---|---|
| a Nucleotides in bold represent conserved regions. | |
| miR-290 cluster | |
| mmu-miR-290-3p | aaagugccgccuaguuuuaagccc |
| mmu-miR-291a-3p | aaagugcuuccacuuugugugc |
| mmu-miR-292-3p | aaagugccgccagguuuugagugu |
| mmu-miR-291b-3p | aaagugcauccauuuuguuugu |
| mmu-miR-294-3p | aaagugcuucccuuuugugugu |
| mmu-miR-295-3p | aaagugcuacuacuuuugagucu |
| miR-302b cluster | |
| mmu-miR-302b-3p | uaagugcuuccauguuuuaguag |
| mmu-miR-302c-3p | aagugcuuccauguuucagugg |
| mmu-miR-302a-3p | uaagugcuuccauguuuugguga |
| mmu-miR-302d-3p | uaagugcuuccauguuugagugu |
| miR-17 cluster | |
| mmu-miR-17-5p | caaagugcuuacagugcagguag |
| mmu-miR-20a-5p | uaaagugcuuauagugcagguag |
| miR-106b cluster | |
| mmu-miR-106b-5p | uaaagugcugacagugcagau |
| mmu-miR-93-5p | caaagugcuguucgugcagguag |
| miR-106a cluster | |
| mmu-miR-106a | caaagugcuaacagugcagguag |
| mmu-miR-20b | caaagugcucauagugcagguag |
| miR-130/301/721 family | |
| mmu-miR-130a | cagugcaauguuaaaagggcau |
| mmu-miR-130b | cagugcaaugaugaaagggcau |
| mmu-miR-301a | cagugcaauaguauugucaaagc |
| mmu-miR-301b | cagugcaaugguauugucaaagc |
| mmu-miR-721 | cagugcaauuaaaagggggaa |
During MET stage of OSKM-induced reprogramming, aggregates and colonies of reprogrammed cells become visible under low magnification microscopy as reprogrammed cells acquire epithelial cell features. These cells begin to express pluripotency markers, of which SSEA-1 is the earliest surface marker to indicate the potential iPS cells.17,18 As the reprogrammed cells move towards a state of pluripotency, Nanog, Esrrb, Lin28, Dppa4, Tert, Sox2, and Oct4 are endogenously expressed, demonstrating that the core circuit of pluripotency has been established.17–19,70 The embryonic stem cell cell-cycle-regulating (ESCC) miRNAs, the miR-302 clusters (Fig. 1), are also expressed during the transition stage day 4 to day 7 post induced reprogramming,34,126 mainly induced by the reprogramming factor c-Myc.31,37 Recent study has shown that the promoter of the miR-302 cluster can be bound and that the expression of miR-302 can be induced by vitamin C-dependent Oct4/Jhdm1b cooperation during reprogramming.127 The biogenesis of miRNAs has been shown to be critical to efficient reprogramming, because Ago2 downregulation reduces the number of iPSC colonies.34 Depletion of the miR-302 family reduces reprogramming efficiency in response to OSK or OSKM,31,127 suggesting that the miR-302 family plays essential roles in the reprogramming process. Ectopic expression of the miR-290 or miR-302 clusters has been shown to improve OSKM- or OSK-reprogramming36,37 by promoting MET through inhibition of the TGF-β receptor.31,34,36 Recent finding128 also demonstrated that the miR-290 cluster maintains pluripotency by repressing the nuclear factor kappa B (NF-κB) signaling pathway, which in turn restricts epithelial to mesenchymal transition in ES cells. Furthermore, the miR-290/302 seed family targets diverse functional groups to positively enhance induced reprogramming, including cell cycle regulation (Cdkn1a, Rbl2, and Cdc2l6) and epigenetic regulation (Aof1, Aof2, Mecp1-p66, MECP2, Mbd2, and Smarcc2).13,36
Other miRNAs have been identified to enhance reprogramming. For example, miR-17/92, miR-106b/25, and miR-106a/363 clusters boost reprogramming by targeting Tgfbr2 and p21.34 Notably, miR-17, miR-93, and miR-106a have also been induced during the MET stage (day 4 post reprogramming).34,49 Moreover, introduction of miR-106b and miR-93 miRNA mimics can promote MET (Fig. 1) to boost reprogramming efficiency.34 The miR-130/301/721 family (Fig. 1), identified by miRNA screening, targets the homeobox transcription factor Meox2 (also known as Gax) to achieve ∼2-fold increase in reprogramming.35 All those miRNAs (miR-17, 106a, 106b clusters, and miR-130/301/721 family) share a similar seed region with the miR-290/302 family (Table 2), suggesting that an abundance of miRNAs containing the miR-290/302 seed region play significant roles in various biological functions and intrinsically act as a positive regulator to reprogramming.
The miR-290/302 seed family plays multiple roles at the ES stage121 and during the reprogramming process, and the miR-290 and miR-302 clusters are the predominant miRNA population expressed in mammalian ES cells. Therefore, the miR-290/302 seed family may have the potential to induce somatic cell reprogramming in the absence of transgenes OSKM. The reprogramming potential of the miR-302 cluster was first tested in human cancer cells, which showed that the signature properties of pluripotent stem cells are acquired in miR-iPSCs.14 Following this finding, several reports demonstrated that the miR-302 cluster could reprogram various primary cell types into pluripotent stem cells.13,15,16 Among these, Anokye-Danso et al. demonstrated that miRNA-mediated reprogramming is more efficient than the transcription factor-mediated method in both mouse (81.5% versus 17.9%) and human somatic cells (10% versus 0.004%).15 But to achieve this striking efficiency, suppression or low level of Hdac2 seems to be required in both mouse and human cells.15,16 Miyoshi et al. further demonstrated that clinically-applicable iPS cells can be generated by introducing only a group of mature miRNAs (miR-200c, miR-302 family, and miR-369s), without retro-viral integration in genome.16 However, this transfection-base reprogramming can only reach 0.01% reprogramming efficiency in mouse cells, and even lower (0.001%) in human cells.16 The methodology of administrating miRNAs may be one of the main reasons to cause the difference in miR-induced reprogramming efficiency between these two reports, Anokye-Danso et al. (virus transduction)15 and Miyoshi et al. (small RNA transfection).16 In addition, distinct combinations of miRNAs were employed for the reprogramming process: the miR-302 cluster combined with valproic acid (VPA) treatment vs. miR-200c, miR-302abcd, and miR-369s. Interestingly, VPA for Hdac2 suppression and miR-367 are required in the viral transduction method, but both are dispensable in the miR transfection method. These protocol variations may need to be vigorously tested to further improve the efficiency of reprogramming and clinical applicability of iPS cells. Despite the phenomenon of miR-induced somatic reprogramming, how microRNAs can perturb somatic molecular networks and then launch pluripotent regulatory networks remains unknown.15,16
The mechanisms of miRNA-only reprogramming
As described above, numerous miR-290/302 seed family targets have been identified, but these molecular networks have only been shown in the ES cell context or with forced expression of reprogramming factors OSKM. Previous studies showed that the miR-302 cluster reactivates Oct4 and Nanog through releasing epigenetic repression on the promoter regions by targeting lysine-specific histone demethylases 1 and 2 (AOF1/2), which regulate the histone lysine 3 methylation level, and also by targeting methyl-CpG binding proteins 1 and 2 (MECP1/2), which coordinate with DNMT1-mediated gene regulations.13 However, Subramanyam et al. showed that only Tgfb2r was detectably changed under the same miR-only reprogramming conditions.36 A change in expression of MECP2 and other known targets of miR-302 could only be detected in the presence of three or four reprogramming factors,36 suggesting that the reprogramming effect of miR-302 is due to modulating regulatory networks in a cell context-dependent manner. Therefore, the miR-302 cluster may exert its reprogramming potential through distinct routes from those of Yamanaka's factors, but can induce reprogrammed cells to eventually reach a similar, if not identical, stage of pluripotency.Can miRNAs bring us toward the next level of regenerative medicine?
Creating iPS cells by using miRNAs represent a great advance in stem cell biology and regenerative medicine. Inspired by induced reprogramming technology, numerous laboratories have shown that forced expression of specific transcription factors can also transform one cell type into the desired one.129–132 In near future, we may be able to alter the cell identities with high efficiency by transient introduction of tissue-specific miRNAs only. Since miRNAs can be easily introduced into a variety of cell types by transfection similar to siRNAs, using only mature miRNAs without viral integration can significantly reduce the technical requirement. Oncogene introduction and genomic integration of transgenes can also be eliminated, because transfected miRNAs are transiently expressed in cells with specific half-lives. With efficiency of two orders of magnitude increase relative to OSKM-induced reprogramming,15 utilizing miRNAs has provided the best way to date to induce pluripotent stem cells. Despite the great advance of reprogramming technologies, we are only about to understand the molecular basis underneath the reprogramming process. It could be challenging to decipher the molecular regulations in miRNA-mediated reprogramming because miRNAs usually target a large number of genes to modulate biological networks. New high throughput RNA capturing and sequencing technologies may assist in identifying miRNA targets and their roles in reprogramming.133,134Given the reprogramming process takes place in a stochastic manner, it is likely that only part of modulating forces from each reprogramming factor affect positively toward inducing pluripotency. Upon introduction of four reprogramming factors into somatic cells, transcriptional networks are disturbed by forced-expressed transcription factors. On the other hand, those factors bind and regulate hundreds of targets to rebuild distinct networks. The types of reconstructed transcriptional networks are possibly decided by what combinations are from the contribution of each reprogramming factor. Since pluripotency is the only expected phenotype, most of the reprogrammed cell types fail and only a few cells working a way out of this chaos to reach the end point, bearing ES-like transcriptome. And since it is a random event upon induced reprogramming, it is unlikely to only have a single and well-defined molecular transition under the coordination of four reprogramming factors, which may vary cell to cell. A secondary tet-inducible reprogramming system may serve this purpose better to shed light on the transcriptome transitions, because the defined dosage of reprogramming factors135 may reduce the noise compared with random combinations of OSKM. miRNAs may serve as another tools for deciphering the cloud of the reprogramming process, since the targets of miRNAs are directed by seed region sequences. However, the progression of miR-induced reprogramming is unknown and the molecular transitions are likely distinct from the ones induced with OSKM, while critical molecular signatures may still conserve. Furthermore, extensive examinations are required to establish standard protocols to create miR-induced pluripotent stem cells, since several discrepancies on miRNA combinations, administering methods, and reprogramming efficiency need to be clarified. Therefore, step-wise dissection of miR-induced reprogramming is required to elucidate the critical features, which are involved in both transcriptional factor-induced and miRNA-induced reprogramming.
Acknowledgements
We are grateful to members of the Rana laboratory for helpful discussions. This work was supported in part by grants from the National Institutes of Health to T.M.R.References
- K. Takahashi and S. Yamanaka, Cell, 2006, 126, 663–676 CrossRef CAS.
- J. Yu, M. A. Vodyanik, K. Smuga-Otto, J. Antosiewicz-Bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, I. I. Slukvin and J. A. Thomson, Science, 2007, 318, 1917–1920 CrossRef CAS.
- M. Wernig, A. Meissner, R. Foreman, T. Brambrink, M. Ku, K. Hochedlinger, B. E. Bernstein and R. Jaenisch, Nature, 2007, 448, 318–324 CrossRef CAS.
- K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda and S. Yamanaka, Cell, 2007, 131, 861–872 CrossRef CAS.
- A. Meissner, M. Wernig and R. Jaenisch, Nat. Biotechnol., 2007, 25, 1177–1181 CrossRef CAS.
- I. H. Park, R. Zhao, J. A. West, A. Yabuuchi, H. Huo, T. A. Ince, P. H. Lerou, M. W. Lensch and G. Q. Daley, Nature, 2008, 451, 141–146 CrossRef CAS.
- S. Yamanaka, Cell Stem Cell, 2007, 1, 39–49 CrossRef CAS.
- M. Stadtfeld and K. Hochedlinger, Genes Dev., 2010, 24, 2239–2263 CrossRef CAS.
- K. Plath and W. E. Lowry, Nat. Rev. Genet., 2011, 12, 253–265 CrossRef CAS.
- R. Ho, C. Chronis and K. Plath, J. Cell. Physiol., 2011, 226, 868–878 CrossRef CAS.
- K. Hochedlinger and K. Plath, Development, 2009, 136, 509–523 CrossRef CAS.
- L. Warren, P. D. Manos, T. Ahfeldt, Y. H. Loh, H. Li, F. Lau, W. Ebina, P. K. Mandal, Z. D. Smith, A. Meissner, G. Q. Daley, A. S. Brack, J. J. Collins, C. Cowan, T. M. Schlaeger and D. J. Rossi, Cell Stem Cell, 2010, 7, 618–630 CrossRef CAS.
- S. L. Lin, D. C. Chang, C. H. Lin, S. Y. Ying, D. Leu and D. T. Wu, Nucleic Acids Res., 2011, 39, 1054–1065 CrossRef CAS.
- S. L. Lin, D. C. Chang, S. Chang-Lin, C. H. Lin, D. T. Wu, D. T. Chen and S. Y. Ying, RNA, 2008, 14, 2115–2124 CrossRef CAS.
- F. Anokye-Danso, C. M. Trivedi, D. Juhr, M. Gupta, Z. Cui, Y. Tian, Y. Zhang, W. Yang, P. J. Gruber, J. A. Epstein and E. E. Morrisey, Cell Stem Cell, 2011, 8, 376–388 CrossRef CAS.
- N. Miyoshi, H. Ishii, H. Nagano, N. Haraguchi, D. L. Dewi, Y. Kano, S. Nishikawa, M. Tanemura, K. Mimori, F. Tanaka, T. Saito, J. Nishimura, I. Takemasa, T. Mizushima, M. Ikeda, H. Yamamoto, M. Sekimoto, Y. Doki and M. Mori, Cell Stem Cell, 2011, 8, 633–638 CrossRef CAS.
- M. Stadtfeld, N. Maherali, D. T. Breault and K. Hochedlinger, Cell Stem Cell, 2008, 2, 230–240 CrossRef CAS.
- T. Brambrink, R. Foreman, G. G. Welstead, C. J. Lengner, M. Wernig, H. Suh and R. Jaenisch, Cell Stem Cell, 2008, 2, 151–159 CrossRef CAS.
- R. Sridharan, J. Tchieu, M. J. Mason, R. Yachechko, E. Kuoy, S. Horvath, Q. Zhou and K. Plath, Cell, 2009, 136, 364–377 CrossRef CAS.
- C. S. Yang, C. G. Lopez and T. M. Rana, Stem Cells, 2011, 29, 1528–1536 CrossRef CAS.
- H. Hong, K. Takahashi, T. Ichisaka, T. Aoi, O. Kanagawa, M. Nakagawa, K. Okita and S. Yamanaka, Nature, 2009, 460, 1132–1135 CrossRef CAS.
- H. Li, M. Collado, A. Villasante, K. Strati, S. Ortega, M. Canamero, M. A. Blasco and M. Serrano, Nature, 2009, 460, 1136–1139 CrossRef CAS.
- R. M. Marion, K. Strati, H. Li, M. Murga, R. Blanco, S. Ortega, O. Fernandez-Capetillo, M. Serrano and M. A. Blasco, Nature, 2009, 460, 1149–1153 CrossRef CAS.
- T. Kawamura, J. Suzuki, Y. V. Wang, S. Menendez, L. B. Morera, A. Raya, G. M. Wahl and J. C. Izpisua Belmonte, Nature, 2009, 460, 1140–1144 CrossRef CAS.
- A. Banito, S. T. Rashid, J. C. Acosta, S. Li, C. F. Pereira, I. Geti, S. Pinho, J. C. Silva, V. Azuara, M. Walsh, L. Vallier and J. Gil, Genes Dev., 2009, 23, 2134–2139 CrossRef CAS.
- J. Utikal, J. M. Polo, M. Stadtfeld, N. Maherali, W. Kulalert, R. M. Walsh, A. Khalil, J. G. Rheinwald and K. Hochedlinger, Nature, 2009, 460, 1145–1148 CrossRef CAS.
- J. K. Ichida, J. Blanchard, K. Lam, E. Y. Son, J. E. Chung, D. Egli, K. M. Loh, A. C. Carter, F. P. Di Giorgio, K. Koszka, D. Huangfu, H. Akutsu, D. R. Liu, L. L. Rubin and K. Eggan, Cell Stem Cell, 2009, 5, 491–503 CrossRef CAS.
- N. Maherali and K. Hochedlinger, Curr. Biol., 2009, 19, 1718–1723 CrossRef CAS.
- C. S. Yang, Z. Li and T. M. Rana, RNA, 2011, 17, 1451–1460 CrossRef CAS.
- I. Lipchina, Y. Elkabetz, M. Hafner, R. Sheridan, A. Mihailovic, T. Tuschl, C. Sander, L. Studer and D. Betel, Genes Dev., 2011, 25, 2173–2186 CrossRef CAS.
- B. Liao, X. Bao, L. Liu, S. Feng, A. Zovoilis, W. Liu, Y. Xue, J. Cai, X. Guo, B. Qin, R. Zhang, J. Wu, L. Lai, M. Teng, L. Niu, B. Zhang, M. A. Esteban and D. Pei, J. Biol. Chem., 2011, 286, 17359–17364 CrossRef CAS.
- C. Melton, R. L. Judson and R. Blelloch, Nature, 2010, 463, 621–626 CrossRef CAS.
- S. L. Lin, D. C. Chang, S. Y. Ying, D. Leu and D. T. Wu, Cancer Res., 2010, 70, 9473–9482 CrossRef CAS.
- Z. Li, C. S. Yang, K. Nakashima and T. M. Rana, EMBO J., 2011, 30, 823–834 CrossRef.
- N. Pfaff, J. Fiedler, A. Holzmann, A. Schambach, T. Moritz, T. Cantz and T. Thum, EMBO Rep., 2011, 12, 1153–1159 CrossRef CAS.
- D. Subramanyam, S. Lamouille, R. L. Judson, J. Y. Liu, N. Bucay, R. Derynck and R. Blelloch, Nat. Biotechnol., 2011, 29, 443–448 CrossRef CAS.
- R. L. Judson, J. E. Babiarz, M. Venere and R. Blelloch, Nat. Biotechnol., 2009, 27, 459–461 CrossRef CAS.
- H. Cao, C. S. Yang and T. M. Rana, PLoS One, 2008, 3, e2820 Search PubMed.
- T. M. Rana, Nat. Rev. Mol. Cell Biol., 2007, 8, 23–36 CrossRef CAS.
- Z. Li and T. M. Rana, Acc. Chem. Res., 2012, 45, 1122–1131 CrossRef CAS.
- V. Ambros, Curr. Opin. Genet. Dev., 2011, 21, 511–517 CrossRef CAS.
- G. Tiscornia and J. C. Izpisua Belmonte, Genes Dev., 2010, 24, 2732–2741 CrossRef CAS.
- D. Subramanyam and R. Blelloch, Curr. Opin. Genet. Dev., 2011, 21, 498–503 CrossRef CAS.
- M. Kim, A. L. Kasinski and F. J. Slack, Lancet Oncol., 2011, 12, 319–321 CrossRef CAS.
- A. L. Kasinski and F. J. Slack, Curr. Opin. Mol. Ther., 2010, 12, 147–157 CAS.
- M. Esteller, Nat. Rev. Genet., 2011, 12, 861–874 CrossRef CAS.
- T. A. Farazi, J. I. Spitzer, P. Morozov and T. Tuschl, J. Pathol., 2011, 223, 102–115 CrossRef CAS.
- M. van Kouwenhove, M. Kedde and R. Agami, Nat. Rev. Cancer, 2011, 11, 644–656 CrossRef CAS.
- J. Chen, G. Wang, C. Lu, X. Guo, W. Hong, J. Kang and J. Wang, PLoS One, 2012, 7, e40849 CAS.
- J. H. Hanna, K. Saha and R. Jaenisch, Cell, 2010, 143, 508–525 CrossRef CAS.
- S. H. Orkin and K. Hochedlinger, Cell, 2011, 145, 835–850 CrossRef CAS.
- M. I. Lai, W. Y. Wendy-Yeo, R. Ramasamy, N. Nordin, R. Rosli, A. Veerakumarasivam and S. Abdullah, J. Assisted Reprod. Genet., 2011, 28, 291–301 CrossRef.
- J. W. Han and Y. S. Yoon, Antioxid. Redox Signaling, 2012, 17, 205–223 CrossRef CAS.
- F. Gonzalez, S. Boue and J. C. Izpisua Belmonte, Nat. Rev. Genet., 2011, 12, 231–242 CrossRef CAS.
- G. Tiscornia, E. L. Vivas and J. C. Izpisua Belmonte, Nat. Med. (N. Y.), 2011, 17, 1570–1576 CAS.
- D. A. Robinton and G. Q. Daley, Nature, 2012, 481, 295–305 CrossRef CAS.
- M. Grskovic, A. Javaherian, B. Strulovici and G. Q. Daley, Nat. Rev. Drug Discov., 2011, 10, 915–929 CAS.
- I. Lipchina, L. Studer and D. Betel, Cell Cycle, 2012, 11, 1517–1523 CrossRef CAS.
- S. K. Mallanna and A. Rizzino, Dev. Biol., 2010, 344, 16–25 CrossRef CAS.
- C. H. Kuo, J. H. Deng, Q. Deng and S. Y. Ying, Biochem. Biophys. Res. Commun., 2012, 417, 11–16 CrossRef CAS.
- Y. Wang and R. Blelloch, Results Probl. Cell Differ., 2011, 53, 459–472 CrossRef CAS.
- E. M. Heinrich and S. Dimmeler, Circ. Res., 2012, 110, 1014–1022 CrossRef CAS.
- M. A. Li and L. He, BioEssays, 2012, 34, 670–680 CrossRef CAS.
- N. Pfaff, T. Moritz, T. Thum and T. Cantz, J. Mol. Med., 2012, 90, 747–752 CrossRef CAS.
- S. Yamanaka, Nature, 2009, 460, 49–52 CrossRef CAS.
- J. Hanna, K. Saha, B. Pando, J. van Zon, C. J. Lengner, M. P. Creyghton, A. van Oudenaarden and R. Jaenisch, Nature, 2009, 462, 595–601 CrossRef CAS.
- B. D. MacArthur, C. P. Please and R. O. Oreffo, PLoS One, 2008, 3, e3086 Search PubMed.
- R. P. Koche, Z. D. Smith, M. Adli, H. Gu, M. Ku, A. Gnirke, B. E. Bernstein and A. Meissner, Cell Stem Cell, 2011, 8, 96–105 CrossRef CAS.
- R. Li, J. Liang, S. Ni, T. Zhou, X. Qing, H. Li, W. He, J. Chen, F. Li, Q. Zhuang, B. Qin, J. Xu, W. Li, J. Yang, Y. Gan, D. Qin, S. Feng, H. Song, D. Yang, B. Zhang, L. Zeng, L. Lai, M. A. Esteban and D. Pei, Cell Stem Cell, 2010, 7, 51–63 CrossRef CAS.
- P. Samavarchi-Tehrani, A. Golipour, L. David, H. K. Sung, T. A. Beyer, A. Datti, K. Woltjen, A. Nagy and J. L. Wrana, Cell Stem Cell, 2010, 7, 64–77 CrossRef CAS.
- M. Wanzel, S. Herold and M. Eilers, Trends Cell Biol., 2003, 13, 146–150 CrossRef CAS.
- T. C. Chang, D. Yu, Y. S. Lee, E. A. Wentzel, D. E. Arking, K. M. West, C. V. Dang, A. Thomas-Tikhonenko and J. T. Mendell, Nat. Genet., 2008, 40, 43–50 CrossRef CAS.
- C. D. Lotterman, O. A. Kent and J. T. Mendell, Cell Cycle, 2008, 7, 2493–2499 CrossRef CAS.
- P. Gao, I. Tchernyshyov, T. C. Chang, Y. S. Lee, K. Kita, T. Ochi, K. I. Zeller, A. M. De Marzo, J. E. Van Eyk, J. T. Mendell and C. V. Dang, Nature, 2009, 458, 762–765 CrossRef CAS.
- T. C. Chang, L. R. Zeitels, H. W. Hwang, R. R. Chivukula, E. A. Wentzel, M. Dews, J. Jung, P. Gao, C. V. Dang, M. A. Beer, A. Thomas-Tikhonenko and J. T. Mendell, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3384–3389 CrossRef CAS.
- C. H. Lin, C. Lin, H. Tanaka, M. L. Fero and R. N. Eisenman, PLoS One, 2009, 4, e7839 Search PubMed.
- C. H. Lin, A. L. Jackson, J. Guo, P. S. Linsley and R. N. Eisenman, EMBO J., 2009, 28, 3157–3170 CrossRef CAS.
- K. Smith and S. Dalton, Regener. Med., 2010, 5, 947–959 CrossRef CAS.
- K. N. Smith, A. M. Singh and S. Dalton, Cell Stem Cell, 2010, 7, 343–354 CrossRef CAS.
- J. L. Mott, S. Kurita, S. C. Cazanave, S. F. Bronk, N. W. Werneburg and M. E. Fernandez-Zapico, J. Cell. Biochem., 2010, 110, 1155–1164 CrossRef CAS.
- G. Liu, A. Friggeri, Y. Yang, J. Milosevic, Q. Ding, V. J. Thannickal, N. Kaminski and E. Abraham, J. Exp. Med., 2010, 207, 1589–1597 CrossRef CAS.
- T. Thum, C. Gross, J. Fiedler, T. Fischer, S. Kissler, M. Bussen, P. Galuppo, S. Just, W. Rottbauer, S. Frantz, M. Castoldi, J. Soutschek, V. Koteliansky, A. Rosenwald, M. A. Basson, J. D. Licht, J. T. Pena, S. H. Rouhanifard, M. U. Muckenthaler, T. Tuschl, G. R. Martin, J. Bauersachs and S. Engelhardt, Nature, 2008, 456, 980–984 CrossRef CAS.
- J. Yang, A. L. van Oosten, T. W. Theunissen, G. Guo, J. C. Silva and A. Smith, Cell Stem Cell, 2010, 7, 319–328 CrossRef CAS.
- J. Wray, T. Kalkan and A. G. Smith, Biochem. Soc. Trans., 2010, 38, 1027–1032 CrossRef CAS.
- J. Nichols, J. Silva, M. Roode and A. Smith, Development, 2009, 136, 3215–3222 CrossRef CAS.
- Q. L. Ying, J. Wray, J. Nichols, L. Batlle-Morera, B. Doble, J. Woodgett, P. Cohen and A. Smith, Nature, 2008, 453, 519–523 CrossRef CAS.
- S. Y. Park, J. H. Lee, M. Ha, J. W. Nam and V. N. Kim, Nat. Struct. Mol. Biol., 2009, 16, 23–29 CAS.
- H. H. Kim, Y. Kuwano, S. Srikantan, E. K. Lee, J. L. Martindale and M. Gorospe, Genes Dev., 2009, 23, 1743–1748 CrossRef CAS.
- A. Rybak, H. Fuchs, L. Smirnova, C. Brandt, E. E. Pohl, R. Nitsch and F. G. Wulczyn, Nat. Cell Biol., 2008, 10, 987–993 CrossRef CAS.
- V. B. Sampson, N. H. Rong, J. Han, Q. Yang, V. Aris, P. Soteropoulos, N. J. Petrelli, S. P. Dunn and L. J. Krueger, Cancer Res., 2007, 67, 9762–9770 CrossRef CAS.
- S. M. Park, S. Shell, A. R. Radjabi, R. Schickel, C. Feig, B. Boyerinas, D. M. Dinulescu, E. Lengyel and M. E. Peter, Cell Cycle, 2007, 6, 2585–2590 CrossRef CAS.
- L. He, X. He, L. P. Lim, E. de Stanchina, Z. Xuan, Y. Liang, W. Xue, L. Zender, J. Magnus, D. Ridzon, A. L. Jackson, P. S. Linsley, C. Chen, S. W. Lowe, M. A. Cleary and G. J. Hannon, Nature, 2007, 447, 1130–1134 CrossRef CAS.
- N. Raver-Shapira, E. Marciano, E. Meiri, Y. Spector, N. Rosenfeld, N. Moskovits, Z. Bentwich and M. Oren, Mol. Cell, 2007, 26, 731–743 CrossRef CAS.
- T. C. Chang, E. A. Wentzel, O. A. Kent, K. Ramachandran, M. Mullendore, K. H. Lee, G. Feldmann, M. Yamakuchi, M. Ferlito, C. J. Lowenstein, D. E. Arking, M. A. Beer, A. Maitra and J. T. Mendell, Mol. Cell, 2007, 26, 745–752 CrossRef CAS.
- Y. J. Choi, C. P. Lin, J. J. Ho, X. He, N. Okada, P. Bu, Y. Zhong, S. Y. Kim, M. J. Bennett, C. Chen, A. Ozturk, G. G. Hicks, G. J. Hannon and L. He, Nat. Cell Biol., 2011, 13, 1353–1360 CrossRef CAS.
- A. K. Jain, K. Allton, M. Iacovino, E. Mahen, R. J. Milczarek, T. P. Zwaka, M. Kyba and M. C. Barton, PLoS Biol., 2012, 10, e1001268 CAS.
- H. Siemens, R. Jackstadt, S. Hunten, M. Kaller, A. Menssen, U. Gotz and H. Hermeking, Cell Cycle, 2011, 10, 4256–4271 CrossRef CAS.
- N. H. Kim, H. S. Kim, X. Y. Li, I. Lee, H. S. Choi, S. E. Kang, S. Y. Cha, J. K. Ryu, D. Yoon, E. R. Fearon, R. G. Rowe, S. Lee, C. A. Maher, S. J. Weiss and J. I. Yook, J. Cell Biol., 2011, 195, 417–433 CrossRef CAS.
- N. Xu, T. Papagiannakopoulos, G. Pan, J. A. Thomson and K. S. Kosik, Cell, 2009, 137, 647–658 CrossRef CAS.
- T. Liu, W. Cheng, Y. Huang, Q. Huang, L. Jiang and L. Guo, Exp. Cell Res., 2012, 318, 424–434 CrossRef CAS.
- L. Ma, J. Teruya-Feldstein and R. A. Weinberg, Nature, 2007, 449, 682–688 CrossRef CAS.
- W. Kong, H. Yang, L. He, J. J. Zhao, D. Coppola, W. S. Dalton and J. Q. Cheng, Mol. Cell. Biol., 2008, 28, 6773–6784 CrossRef CAS.
- P. A. Gregory, A. G. Bert, E. L. Paterson, S. C. Barry, A. Tsykin, G. Farshid, M. A. Vadas, Y. Khew-Goodall and G. J. Goodall, Nat. Cell Biol., 2008, 10, 593–601 CrossRef CAS.
- E. D. Wiklund, V. S. Catts, S. V. Catts, T. F. Ng, N. J. Whitaker, A. J. Brown and L. H. Lutze-Mann, Int. J. Cancer, 2010, 126, 28–40 CrossRef CAS.
- U. Burk, J. Schubert, U. Wellner, O. Schmalhofer, E. Vincan, S. Spaderna and T. Brabletz, EMBO Rep., 2008, 9, 582–589 CrossRef CAS.
- U. Wellner, J. Schubert, U. C. Burk, O. Schmalhofer, F. Zhu, A. Sonntag, B. Waldvogel, C. Vannier, D. Darling, A. zur Hausen, V. G. Brunton, J. Morton, O. Sansom, J. Schuler, M. P. Stemmler, C. Herzberger, U. Hopt, T. Keck, S. Brabletz and T. Brabletz, Nat. Cell Biol., 2009, 11, 1487–1495 CrossRef CAS.
- J. Chen, L. Wang, L. V. Matyunina, C. G. Hill and J. F. McDonald, Gynecol. Oncol., 2011, 121, 200–205 CrossRef CAS.
- L. Ma, F. Reinhardt, E. Pan, J. Soutschek, B. Bhat, E. G. Marcusson, J. Teruya-Feldstein, G. W. Bell and R. A. Weinberg, Nat. Biotechnol., 2010, 28, 341–347 CrossRef CAS.
- H. R. Wang, Y. Zhang, B. Ozdamar, A. A. Ogunjimi, E. Alexandrova, G. H. Thomsen and J. L. Wrana, Science, 2003, 302, 1775–1779 CrossRef CAS.
- X. Xiang, X. Zhuang, S. Ju, S. Zhang, H. Jiang, J. Mu, L. Zhang, D. Miller, W. Grizzle and H. G. Zhang, Oncogene, 2011, 30, 3440–3453 CrossRef CAS.
- P. A. Gregory, C. P. Bracken, A. G. Bert and G. J. Goodall, Cell Cycle, 2008, 7, 3112–3118 CrossRef CAS.
- E. D. Wiklund, J. Kjems and S. J. Clark, Epigenomics, 2010, 2, 823–840 CrossRef CAS.
- S. M. Park, A. B. Gaur, E. Lengyel and M. E. Peter, Genes Dev., 2008, 22, 894–907 CrossRef CAS.
- J. Chen, J. Liu, J. Yang, Y. Chen, S. Ni, H. Song, L. Zeng, K. Ding and D. Pei, Cell Res., 2011, 21, 205–212 CrossRef CAS.
- Y. Wang, S. Baskerville, A. Shenoy, J. E. Babiarz, L. Baehner and R. Blelloch, Nat. Genet., 2008, 40, 1478–1483 CrossRef CAS.
- C. Kanellopoulou, S. A. Muljo, A. L. Kung, S. Ganesan, R. Drapkin, T. Jenuwein, D. M. Livingston and K. Rajewsky, Genes Dev., 2005, 19, 489–501 CrossRef CAS.
- E. P. Murchison, J. F. Partridge, O. H. Tam, S. Cheloufi and G. J. Hannon, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12135–12140 CrossRef CAS.
- H. B. Houbaviy, M. F. Murray and P. A. Sharp, Dev. Cell, 2003, 5, 351–358 CrossRef CAS.
- M. R. Suh, Y. Lee, J. Y. Kim, S. K. Kim, S. H. Moon, J. Y. Lee, K. Y. Cha, H. M. Chung, H. S. Yoon, S. Y. Moon, V. N. Kim and K. S. Kim, Dev. Biol., 2004, 270, 488–498 CrossRef CAS.
- P. Landgraf, M. Rusu, R. Sheridan, A. Sewer, N. Iovino, A. Aravin, S. Pfeffer, A. Rice, A. O. Kamphorst, M. Landthaler, C. Lin, N. D. Socci, L. Hermida, V. Fulci, S. Chiaretti, R. Foa, J. Schliwka, U. Fuchs, A. Novosel, R. U. Muller, B. Schermer, U. Bissels, J. Inman, Q. Phan, M. Chien, D. B. Weir, R. Choksi, G. De Vita, D. Frezzetti, H. I. Trompeter, V. Hornung, G. Teng, G. Hartmann, M. Palkovits, R. Di Lauro, P. Wernet, G. Macino, C. E. Rogler, J. W. Nagle, J. Ju, F. N. Papavasiliou, T. Benzing, P. Lichter, W. Tam, M. J. Brownstein, A. Bosio, A. Borkhardt, J. J. Russo, C. Sander, M. Zavolan and T. Tuschl, Cell, 2007, 129, 1401–1414 CrossRef CAS.
- A. Marson, S. S. Levine, M. F. Cole, G. M. Frampton, T. Brambrink, S. Johnstone, M. G. Guenther, W. K. Johnston, M. Wernig, J. Newman, J. M. Calabrese, L. M. Dennis, T. L. Volkert, S. Gupta, J. Love, N. Hannett, P. A. Sharp, D. P. Bartel, R. Jaenisch and R. A. Young, Cell, 2008, 134, 521–533 CrossRef CAS.
- D. A. Card, P. B. Hebbar, L. Li, K. W. Trotter, Y. Komatsu, Y. Mishina and T. K. Archer, Mol. Cell. Biol., 2008, 28, 6426–6438 CrossRef.
- A. Rosa, F. M. Spagnoli and A. H. Brivanlou, Dev. Cell, 2009, 16, 517–527 CrossRef CAS.
- R. Benetti, S. Gonzalo, I. Jaco, P. Munoz, S. Gonzalez, S. Schoeftner, E. Murchison, T. Andl, T. Chen, P. Klatt, E. Li, M. Serrano, S. Millar, G. Hannon and M. A. Blasco, Nat. Struct. Mol. Biol., 2008, 15, 268–279 CAS.
- L. Sinkkonen, T. Hugenschmidt, P. Berninger, D. Gaidatzis, F. Mohn, C. G. Artus-Revel, M. Zavolan, P. Svoboda and W. Filipowicz, Nat. Struct. Mol. Biol., 2008, 15, 259–267 CAS.
- M. Kamata, M. Liang, S. Liu, Y. Nagaoka and I. S. Chen, PLoS One, 2010, 5, e11834 Search PubMed.
- T. Wang, K. Chen, X. Zeng, J. Yang, Y. Wu, X. Shi, B. Qin, L. Zeng, M. A. Esteban, G. Pan and D. Pei, Cell Stem Cell, 2011, 9, 575–587 CrossRef CAS.
- P. Luningschror, B. Stocker, B. Kaltschmidt and C. Kaltschmidt, Stem Cells, 2012, 30, 655–664 CrossRef.
- R. Ambasudhan, M. Talantova, R. Coleman, X. Yuan, S. Zhu, S. A. Lipton and S. Ding, Cell Stem Cell, 2011, 9, 113–118 CrossRef CAS.
- A. S. Yoo, A. X. Sun, L. Li, A. Shcheglovitov, T. Portmann, Y. Li, C. Lee-Messer, R. E. Dolmetsch, R. W. Tsien and G. R. Crabtree, Nature, 2011, 476, 228–231 CrossRef CAS.
- C. Jopling, S. Boue and J. C. Izpisua Belmonte, Nat. Rev. Mol. Cell Biol., 2011, 12, 79–89 CrossRef CAS.
- S. M. Chambers and L. Studer, Cell, 2011, 145, 827–830 CrossRef CAS.
- H. Baigude, Ahsanullah, Z. Li, Y. Zhou and T. M. Rana, Angew. Chem., Int. Ed., 2012, 51, 5880–5883 CrossRef CAS.
- H. Zhou, M. L. Arcila, Z. Li, E. J. Lee, C. Henzler, J. Liu, T. M. Rana and K. S. Kosik, Nucleic Acids Res., 2012, 40, 5864–5875 CrossRef CAS.
- S. Markoulaki, J. Hanna, C. Beard, B. W. Carey, A. W. Cheng, C. J. Lengner, J. A. Dausman, D. Fu, Q. Gao, S. Wu, J. P. Cassady and R. Jaenisch, Nat. Biotechnol., 2009, 27, 169–171 CrossRef CAS.
- K. D. Wilson, S. Venkatasubrahmanyam, F. Jia, N. Sun, A. J. Butte and J. C. Wu, Stem Cells Dev., 2009, 18, 749–758 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |